

Abstract
Objective:
To characterize the features associated with PCDH19-related epilepsy, also known as “female-limited epilepsy.”
Methods:
We analyzed data from participants enrolled in the PCDH19 Registry, focusing on the seizure-related, developmental, neurobehavioral, and sleep-related features. We evaluated variants for pathogenicity based on previous reports, population databases, and in silico predictions, and included individuals with pathogenic or potentially pathogenic variants. We performed a retrospective analysis of medical records and administered a targeted questionnaire to characterize current or past features in probands and genotype-positive family members.
Results:
We included 38 individuals with pathogenic or potentially pathogenic variants in PCDH19: 21 de novo, 5 maternally inherited, 7 paternally inherited, and 5 unknown. All 38 had epilepsy; seizure burden varied, but typical features of clustering of seizures and association with fever were present. Thirty individuals had intellectual disability (ID), with a wide range of severity reported; notably, 8/38 (22%) had average intellect. Behavioral and sleep dysregulation were prominent, in 29/38 (76%) and 20/38 (53%), respectively. Autistic features were present in 22/38 (58%), of whom 12 had a formal diagnosis of autism spectrum disorder (ASD). We had additional data from 5 genotype-positive mothers, all with average intellect and 3 with epilepsy and from 1 genotype-positive father.
Keywords: PCDH19, female-limited epilepsy, seizure, autism spectrum disorder, sleep disorder
Introduction
Recent advances in next-generation sequencing have led to the discovery of many monogenic causes of epilepsy 1–3 accounting for a major portion of otherwise unexplained epilepsies 4. Along with SCN1A, PCDH19 (protocadherin-19) is among the most commonly implicated epilepsy genes 2–15. There are over 150 cases of PCDH19-related epilepsy reported in the literature 9,16,17 and PCDH19 has recently been recognized as an important genetic cause of sporadic focal epilepsy 18.
PCDH19 was first identified as a cause of epilepsy and intellectual disability in 2008 8,19. Pathogenic variants in PCDH19 have been associated with a complex syndrome characterized by medically refractory seizures 5,8 intellectual disability (ID) 5,15, autism spectrum disorder (ASD) 17 and behavioral dysregulation 7,8. Patients often display fever-related seizures which typically cluster, with multiple seizures in a given episode. The condition follows a unique X-linked pattern of expression in which females manifest core symptoms while carrier males typically do not, although carrier males have been reported to manifest some neuropsychiatric features 19. It has been postulated that natural mosaicism created by X-inactivation in females accounts for the wide phenotypic variability observed in females with pathogenic PCDH19 variants. Interestingly, some males with mosaic PCDH19 variants present similarly to affected females 20–23, which further suggests that the mosaic state may be required for expression of many of the core features of this syndrome. The number of diagnosed cases is increasing as recognition of the gene has grown since 2008, and continues to grow as clinical genetic testing becomes more routine. Already, the phenotypic spectrum related to PCDH19 is broadening: patients with early onset epilepsy and ID with a phenotype milder than initially reported, and patients with sporadic focal epilepsy undergoing testing with epilepsy gene panels, have recently been found to harbor likely pathogenic variants in PCDH19 12,24. Our series highlights the under recognition of many behavioral aspects of this condition.
The development of gene- or mechanism-specific treatments for monogenic epilepsies requires robust genotype and phenotype characterization in humans. We aim to characterize the PCDH19-related spectrum of seizures and neurodevelopmental disorders in order to assess for genotype-phenotype correlations as well as to identify key features that can be targeted in future treatment trials for this disorder. Identifying and characterizing gene-specific patient populations are essential steps for new trials of targeted treatments. Here, we present formal phenotyping of 38 individuals with PCDH19-related epilepsy, including previously under-reported behavioral problems, such that future clinical trials can focus on these features in addition to the classic epilepsy phenotype associated with PCDH19.
Methods
Participant Ascertainment
Our analysis included participants who are enrolled in the PCDH19 Registry, which was established by the Boston Children’s Hospital (BCH) Epilepsy Genetics Program in partnership with the University of California, San Francisco (UCSF) Pediatric Epilepsy Center. The PCDH19 Registry is a repository of genotypic and phenotypic data collected from individuals who have been diagnosed with PCDH19-related epilepsy as well as their genotype-positive family members. The PCDH19 Registry has been approved by the Institutional Review Boards of BCH and UCSF and we obtained informed consent from all participants or their parents. Our analysis included participants enrolled between October 2014 and April 2017 who had clinical genetic testing performed in a CLIA-certified laboratory and for whom clinical data were available at the time of review. We excluded participants with likely benign variants and participants with an alternative genetic variant that better explained their phenotype.
Data Collection
We collected medical records for each proband, including clinical genetic test reports, neurology notes, neuropsychology notes, electroencephalograph (EEG) recordings and reports and magnetic resonance (MRI) images and reports. We created and administered a detailed, 25 page medical and family history questionnaire that we mailed to participants or completed over the phone through a structured interview, based on participant choice. The questionnaire was administered for each proband as well as any enrolled genotype-positive family member. The questionnaire targeted seizure (onset, types, medication response), developmental and neuropsychiatric histories (Supplement 1). We encouraged families to elaborate on any of these features as well as freely describe personality traits, behavior and sleep patterns (features anecdotally reported to be a major concern for families) as well as any other medical concerns, reflecting past and current issues. Based on initial feedback from families, we administered a validated survey, the Children’s Sleep Habits Questionnaire-Abbreviated 25 to attempt to further describe sleep disturbances
Data Analysis
We performed a retrospective analysis on all medical records. We mapped all genetic variants to the canonical transcript (NM_001184880.1). For each variant, we began with the pathogenicity classification provided by the clinical laboratory and performed additional systematic analyses. We determined whether the variant had been previously reported in the literature or identified through clinical testing and reported to ClinVar 26. We determined population frequency using gnomAD 27. Finally, we incorporated the in silico prediction tools of SIFT, PolyPhen2 and MutationTaster. We combined data from medical records and the questionnaires to detail seizure histories, medication response, and presence or absence of other core features such as autistic features, behavioral dysregulation and sleep challenges throughout the probands’ lifetimes as the severity of these features may fluctuate over time. The presence of clusters of seizures was determined by review of the clinic notes and interviews with the families, and definitions for what constituted clusters varied across individuals. We used the current definition for epilepsy resolution according to the International League Against Epilepsy (ILAE).28 We had sufficient data to compare severity of epilepsy as measured by response to medications, and presence of other core features between individuals with different variant types. We used a Chi-square test of proportions to compare each feature between individuals with missense variants and those with predicted loss of function variants (nonsense, frameshift, or gene deletion).
Results
Patient Ascertainment
We enrolled 50 probands with variants in PCDH19, as well as 8 genotype-positive mothers and 3 genotype-positive fathers in the PCDH19 Registry. We excluded 8 enrolled probands because of insufficient medical records. We restricted our analysis to carefully vetted cases based on laboratory classification of variants in addition to population data, in silico predictions and clinical correlation of the phenotype based on available records. As such, we excluded an additional 4 probands because their variants are likely benign. Our analysis included 38 of the 50 enrolled probands, all female, as well as 5 of 8 enrolled genotype positive mothers and 1 of 3 genotype-positive fathers, corresponding to the included probands. Two of the 38 probands, #10 and #20, have previously been reported29. Of the 38 included participants, the parents of 29 participants completed the questionnaire, 12 of whom did so by telephone interview. The average age of participants at the time of enrollment was 7.6 years (range 1.4–25.0 years).
Genetics
Figure 1 shows the variants mapped on to the PCDH19 protein. The genetic variants and inheritance patterns are listed in Table 1. Three were whole gene deletions, 17 were loss-of-function (LOF) variants (8 frameshift, 9 nonsense), and 18 were missense variants. Twenty-one of the 38 variants were de novo, 5/38 were maternally inherited, 7/38 were paternally inherited and 5/38 were of unknown inheritance. Notably, of the 5 variants with unknown inheritance, 2 were present in a pair of sisters (variant p.K331*, participants #16 and #18), so we conclude that the variant was either inherited from an unaffected parent or the result of gonadal or parental somatic mosaicism. There was one recurrent variant (c.1091dupC; p.Y366Lfs*10) present in two unrelated individuals and another frameshift variant occurring at the same amino acid (c.1095_1101del; p.Y366fs*201). The three genotype-positive mothers of participants #13, #25 and #28 (harboring variants p.Y166*, p.D448H, and p.R625G, respectively) and the genotype-positive father of participant #29 were reportedly symptomatic, as shown in Figure 2 and further detailed below.
Figure 1.
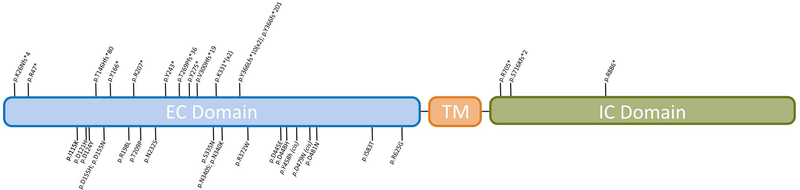
PCDH19 protein depicting variants present in our cohort. Missense variants are noted below the protein while nonsense and frameshift variants are noted above.
EC: Extracellular, TM: Transmembrane, IC: Intracellular
Table 1.
Genotype and corresponding phenotypes of probands, organized by variant type then by age at enrollment.
| ID | Age @ Enrollment (Years) | cDNA | Variant | Inheritance | Age Seizure Onset (Months) | Seizure Types | Medication Response | Development | Autism Spectrum Disorder | Behavior | Sleep | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Deletion | 1 | 2.8 | chrX del | − | D | 18 | F | U ≥ 3 | M | + | + | + |
| 2 | 6.6 | chrX del | − | D | 11 | F | U ≥ 3 | L, R | + | + | + | |
| 3 | 7.9 | chrX del | − | U | 7 | NK | U ≥ 3 | L, M, R | + + | + | + | |
| Frameshift | 4 | 1.4 | c.1091dupC | p.Y366Lfs*10 | D | 6 | F | U ≥ 3 | L, P | − | + | + |
| 5 | 2.5 | c.898_899delGT | p.V300Hfs*19 | D | 12 | F | U < 3 | L, M, R | + | − | + | |
| 6 | 3.3 | c.805delA | p.T269Pfs*36 | D | 9 | F, G | U < 3 | L | ++ | + | + | |
| 7 | 5.0 | c.2146dupA | p.S716Kfs*2 | UK | 11 | F, G | C ≥ 3 | L | − | + | − | |
| 8 | 7.0 | c.434_435InsG | p.T146Hfs*80 | D | 8 | F | U < 3 | L, R | − | − | − | |
| 9 | 9.6 | c.1091dupC | p.Y366Lfs*10 | P | 10 | NK | U ≥ 3 | L, M, R | ++ | + | + | |
| 10¥ | 13.0 | c.78delG | p.K26Nfs*4 | D | 6 | F | U ≥ 3 | L, M | ++ | + | − | |
| 11 | 15.5 | c.1095_1101del | p.Y366fs*201 | D | 4.5 | NK | U ≥ 3 | L, M, R | ++ | − | + | |
| Nonsense | 12 | 3.0 | c.139C>T | p.R47* | D | 15 | F | U ≥ 3 | − | − | − | − |
| 13 | 3.5 | c.498C>G | p.Y166* | M# | 10 | F | U ≥ 3 | L, M, R | + | + | + | |
| 14 | 5.0 | c.2656C>T | p.R886* | M | 22 | F, G | C < 3 | − | − | + | − | |
| 15 | 6.0 | c.2113C>T | p.R705* | D | 12 | F, G | C < 3 | L | − | − | − | |
| 16+ | 8.5 | c.991A>T | p.K331* | UK | 4 | F | U ≥ 3 | L, M | − | + | − | |
| 17 | 9.0 | c.619 C>T | p.R207* | D | 15 | G | C < 3 | L | − | + | − | |
| 18+ | 11.2 | c.991A>T | p.K331* | UK | 6 | F | U ≥ 3 | L, M | ++ | + | − | |
| 19 | 18.1 | c.825C>A | p.Y275* | U | 10 | F, G | U ≥ 3 | L, M | ++ | + | + | |
| 20¥ | 25.0 | c.729C>A | p.Y243* | D | 9 | F | U ≥ 3 | L | + | + | − | |
| Missense | 21 | 1.4 | c.695A>G | p.N232S | D | 4 | F | U ≥ 3 | L, M, P | + | − | + |
| 22 | 2.0 | c.1114C>T | p.R372W | D | 6 | F, G | U ≥ 3 | − | − | − | + | |
| 23 | 2.0 | c.344T>A | p.I115K | D | 8 | F | U ≥ 3 | − | − | + | − | |
| 24 | 2.3 | c.1441G>A | p.D481N | D | 7 | F | U ≥ 3 | − | − | + | + | |
| 25 | 2.3 | c.1342G>A | p.D448H | M# | 24 | NK | U < 3 | − | − | − | + | |
| 26 | 4.0 | c.1019A>G | p.N340S | D | 14 | G | U < 3 | L, P | ++ | + | + | |
| 27 | 4.2 | c.1335C>A | p.D445E | M | 6 | F, G | U ≥ 3 | L, M | − | + | + | |
| 28 | 4.5 | c.1873A>G | p.R625G | M# | 6 | F | U ≥ 3 | − | + | + | − | |
| 29 | 4.9 | c.1004G>A | p.S335N | P# | <23 | F | U < 3 | L, M | − | + | + | |
| 30 | 6.8 | c.625A>C | p.T209P | P | 36 | G | C ≥ 3 | − | − | + | − | |
| 31 | 7.3 | c.370G>T | p.D124Y | D | 10 | F | U ≥ 3 | L | ++ | + | − | |
| 32 | 8.5 | c.593G>T | p.R198L | D | 8 | F | C ≥ 3 | L, M, R | + | + | + | |
| 33 | 9.0 | c.463G>C | p.D155H | P | 3 | F, G | U < 3 | L, M, R | ++ | + | + | |
| 34 | 9.8 | c.631G>C | p.D121H | D | 15 | F | U ≥ 3 | − | − | + | + | |
| 35 | 11.3 | c.463G>A | p.D155N | P | 6 | F | U ≥ 3 | L, M, P | ++ | + | + | |
| 36 | 12.0 | c.1020T>G | p.N340K | P | 15 | F | U ≥ 3 | L, M | ++ | + | + | |
| 37 | 13.8 | c.1372T>C◦; c.1435G>A◦ | p.Y458H; p.D479N | D | 15 | F | C ≥ 3 | L, M | ++ | + | − | |
| 38 | 17.5 | c.1748T>C | p.I583T | P | 48 | G | C ≥ 3 | L, M | + | − | − | |
ID: ¥ = Previously reported by Jamal et al (2010), + = Sisters
cDNA: ◦ = Variants in cis, both de novo
Inheritance: D= de novo, M=Maternally-inherited, P= Paternally-inherited, UK= Unknown inheritance, #Parent affected
Seizure Types: F=Focal, G=Generalized, NK=Not Known
Medication Response: U= Uncontrolled, C= Controlled (currently seizure-free for >1 year), <3= Less than 3 medications, ≥ 3= Greater than or equal to 3 medications
Development: L=Language delay, M=Motor delay, P=Plateau, R=Regression
Autism Spectrum Disorder: +=features, ++ = formal ASD diagnosis
Figure 2.
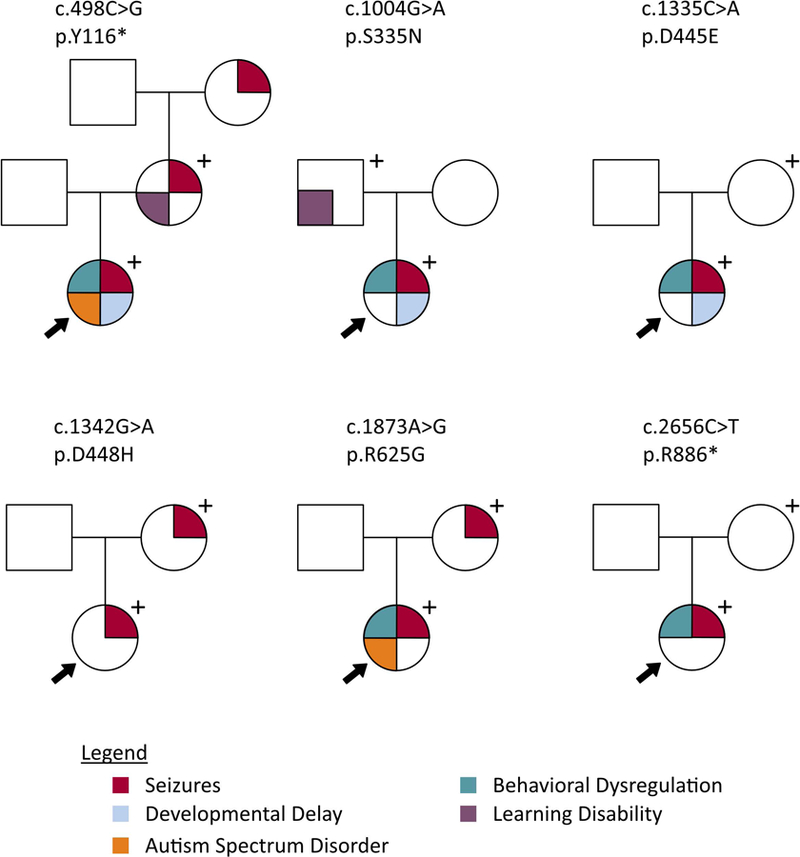
Pedigrees displaying phenotypes of probands and genotype-positive parents.
Seizure History
All probands had epilepsy with a mean age of onset of 11.8 months (range 4–48 months). Pertinent features are summarized in Table 1. MRI images did not show any structural abnormalities. Of the 38 probands, 22 (58%) had focal seizures, 4 (11%) had generalized seizures and 8 (21%) had both focal and generalized seizures. For the remaining 4 probands we had insufficient data to determine when seizure onset was focal or generalized. Thirty-two probands (84%) had seizures that were triggered by fever. Thirty-five probands (92%) had seizures that occurred in clusters, a hallmark feature of PCDH19-related epilepsy. Cluster frequency was variable among probands and ranged from daily to yearly, with nearly half reporting clusters every 1–3 months. The majority of clusters were reported to last hours to days in duration. However, frequency and severity varied over time with each individual.
Response to Medication
Participants in our cohort were treated with a wide array of standard anti-seizure medications, with no preferred anti-seizure medication class or mechanism of action. Most frequently used medications include benzodiazepines such as clobazam, oxcarbazepine, sodium valproate, and levetiracetam. Proband response to medication is listed in Table 1. Only the affected mothers of participants had reached epilepsy resolution (>10 years seizure free including the last 5 years off medication). Eight (21%) probands were seizure-free for at least one year (range 13 months-3 years) on medication at the time of enrollment. Twenty-two (58%) of probands in our cohort remained refractory to 3 or more medications.
Developmental History
Eight of the 38 probands (22%) had typical development. Of the 30 probands who had developmental delay in at least one domain, 4 had delays prior to seizure onset. The most prominent developmental challenge was in the speech and language domain. Of the 30 probands with developmental delay, there was only 1 who did not have delays in language development. Developmental histories are summarized in Table 1.
Neuropsychiatric / Behavioral Features
Twelve probands (32%) carried a formal diagnosis of ASD, while an additional 10 probands were reported to have autistic features but did not have a formal diagnosis of ASD, as shown in Table 1. These features included different deficits in social communication and interaction, restricted or repetitive behavioral patterns or abnormal reactivity to sensory input. Twelve of the 38 (32%) had a formal neuropsychiatric evaluation. An additional 3 probands received an evaluation through Early Intervention. Fifteen participants (age range 1.4–13.8 years) did not have a neuropsychiatric evaluation. We had incomplete data to determine whether such evaluations occurred on the remaining 8 probands. Several families reported that their child was never referred for formal evaluation because of their PCDH19 diagnosis. Twenty-nine probands (76%) were reported to have notable behavioral dysregulation, which are delineated in Table 2. Behavioral challenges were present across all age groups, as shown in Table 1. Out of all participants (N=38), 9 (24%) were reported to have obsessive-compulsive types of behaviors, including 2 who had a formal diagnosis of obsessive compulsive disorder (OCD). Parents described ritualized behavioral patterns and insistence on a particular order of actions or object arrangement. While we asked whether the above features were ever present in their child, we did not collect sufficient data to correlate the any fluctuation in severity with seizures, treatment or other factors. During the phone interviews, many parents anecdotally reported that the neuropsychiatric and behavioral manifestations were often more challenging than seizure management. This information was not quantified as part of as part of this study.
Table 2:
Detailed behavior characterization for those with reported behavioral dysregulation (N=29). Parents could select more than one response
| Behavior | ||
| Aggressive/ Violent behavior | 15 | |
| Stubborness/ Definance/ Strong-Willed | 17 | |
| Crying/ Irritability/ Outbursts | 15 | |
| Impulsivity | 4 | |
| Dysregulated NOS | 3 | |
| Difficulty with Transitions | 9 |
Sleep
The parents of 20 probands reported abnormal sleeping patterns during their child’s lifetime, which are detailed in Figure 3. The most prominent feature was consistent with sleep maintenance insomnia, with many children waking up too early (3:00 to 5:00 am) and struggling to return to sleep. The parents of 9 probands completed the additional sleep survey. Qualitatively, they reported that this instrument did not capture the sleep behaviors of their child. Specifically, they felt it did not capture the fluctuations over time, and how seizure clusters and behavioral challenges surrounding bedtime routines affected sleep.
Figure 3.
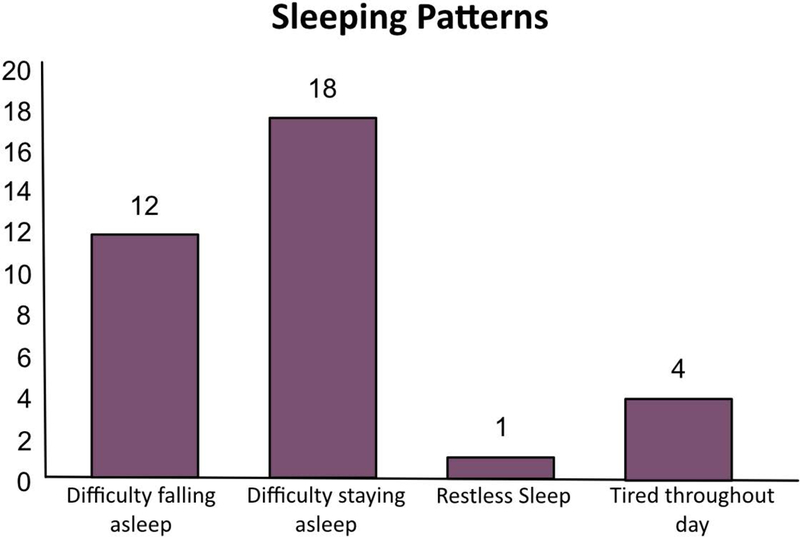
Abnormal sleep patterns for those with reported sleep dysregulation (N=22), participants could select more than one response.
Genotype-Positive Parents
Three genotype-positive mothers had a history of seizures. The mother of participant #13 (p.Y166*) had 2 convulsive seizures at approximately 5 years of age in the setting of fever. Notably, her mother (the maternal grandmother of participant #13) reportedly had a history of febrile seizures, although she has not had genetic testing to date. The mother of participant #25 (p.D448H) began having seizures at 15 months of age. Semiologies were both focal and generalized and occurred in clusters. Her seizure frequency reportedly increased drastically around puberty and she had her last seizure at 12.5 years of age. The mother of participant #28 (p.R625G) began having seizures at the age of 4 months, described as staring spells with unresponsiveness that resolved around 5 or 6 years of age. Additional details on seizure histories and data on medication(s) for the genotype-positive parents were unavailable. All 5 genotype-positive mothers had typical development. The mother of participant #25 (p.D448H) self-reported a learning disability in reading and writing. None of the other genotype-positive mothers reported learning disabilities or formal developmental testing. The father of participant #29 (p.S335N) also self-reported a learning disability in visual processing.
Genotype-Phenotype Correlation
Seizures were uncontrolled by medication in 14/18 (77.8%) probands with missense variants and 16/20 (80%) with gene deletions and predicted LOF variants (p=0.88). In comparing the presence or absence of features of ASD, 10/18 (55.6%) with missense variants and 12/20 (60%) with gene deletions and predicted LOF variants (p= 0.79) displayed these features. Behavioral dysregulation was present in 14/18 (77.8%) with missense variants and 15/20 (75%) with gene deletions and LOF variants (p=0.84). Sleep challenges were present in 12/18 (66.6%) with missense variants and 10/20 (50%) with gene deletions and LOF variants (p=0.31). In addition, typical development was present in 6/18 (33.3%) probands with missense variants and 2/20 (10%) probands with gene deletions predicted LOF variants (p=0.08). The 3 individuals with whole gene deletion of PCDH19 had all symptoms present and uncontrolled seizures.
Discussion
We present the genotypes and corresponding phenotypic information for a well-curated cohort of patients with PCDH19-related epilepsy. The phenotypic spectrum within this population is broad. While the basis for this remains elusive, an attractive theory is that some combination of the variant’s effect on the protein and the pattern of X-inactivation are likely contributors.
As illustrated and previously reported, seizures are a core feature in our cohort, often presenting in clusters and characterized by temperature sensitivity. A majority of affected individuals have focal seizures, consistent with what has been reported previously 30. Despite the preponderance of focal seizures in this population, one specific or a specific combination of anti-seizure agent(s) has not yet been identified to be particularly successful in reducing seizure frequency or intensity in this population, in contrast to that reported in a prior study of patients with PCDH19–related epilepsy 31. The role of other anti-seizure treatments such as ketogenic diet or VNS is not yet well described in this condition. Development in our cohort ranged from typical, as seen in the genotype-positive mothers and some of the affected probands, to quite significantly delayed. In fact, 22% percent of the girls within our cohort had average intellect, which is in contrast to the prevailing view that PCDH19–related epilepsy is nearly always associated with ID. We found no significant correlation between variant type and severity of phenotype, though there was a trend toward typical development occurring in individuals with missense variants compared to those with loss-of-function variants. The small group of 3 individuals with whole gene deletion had all features and uncontrolled seizures, representing the most severely affected variant type. The discrepancy in severity between genotype-positive mothers and their affected daughters further supports the hypothesis that X-inactivation patterns influence phenotype, and raises the possibility of a contribution from genetic modifiers. Comparative X-inactivation studies within this population could provide insight into these hypotheses.
The one included genotype-positive father did not have epilepsy, which is consistent with the established inheritance pattern for pathogenic variants in PCDH19, although he did report a learning disability which has not been reported in male carriers to our knowledge; it is unknown if this is related to the PCDH19 variant. It would be of interest to further characterize the neuropsychiatric phenotype of all male carriers to see if there is a significant prevalence of these features, which has been suggested in previous reports 32.
In addition to epilepsy, affected individuals display a complex neuropsychiatric syndrome in which the behavioral and sleep dysregulation are prominent. In our cohort, 32% of individuals had a diagnosis of ASD and 5% had a diagnosis of OCD. Of the remaining 25 individuals without formal ASD or OCD diagnoses, the parents of over two-thirds (17) of those probands report deficits in social communication, stereotypies, ritualized behaviors, and difficulty with transitions. We acknowledge the discrepancy between reported symptoms and formal diagnoses; this may be related to inadequate recognition or perception of symptom severity, but also to inadequate access to formal evaluation. Additionally, this could represent a behavioral phenotype outside the strictly-defined ASD or OCD diagnoses, which requires further study and individualized clinical diagnosis and therapy. It is also important to note that certain features of ASD can resemble those of OCD and it is difficult to distinguish the cause of these features based on the available data. Our unique approach of incorporating parental input allows our series to highlight the behavioral and neuropsychiatric features. Interestingly, many parents stated that these features presented more of a challenge than the seizures. While we did not collect quantitative data or utilize any validated quality of life measures to draw formal conclusions, based on these anecdotal comments in addition to the prevalence of these features it would be reasonable to conclude that these features are of great concern to families. Referrals and access to appropriate evaluations and therapies for children with PCDH19-related epilepsy, as well as support services for parents, may be of particular importance with this population.
Similarly, a majority have sleep dysregulation, representing an area that requires deeper characterization. Sleep disturbances manifest in our cohort primarily as difficulty staying asleep rather than falling asleep. This may have implications for seizure control and behavior regulation. This particular feature of impaired sleep regulation in PCDH19-related epilepsy is not well-described in the literature and warrants further study, perhaps with novel survey instruments to capture the day to day variability and the correlation with seizure frequency and severity. The impact of improved sleep on seizure cluster frequency and severity, behavior and psychiatric symptomatology would be of interest.
We acknowledge that there are limitations to our study, including that it was retrospective and included parent-reported data. We obtained medical records to confirm as much of the history as possible but acknowledge that the histories reported in medical records are, too, parent-reported in large part. While parents were offered the option to complete the questionnaire either in writing or by telephone, it is possible that due to the length of the questionnaire not all information was captured. In particular, the questionnaire utilized non-standardized tools to measure the neuropsychiatric and developmental features, which could be more comprehensively addressed through structured interviews or formal testing in future studies. We had data on age of seizure onset but no similar data for other core features. Not all participants completed the questionnaire, so some neurodevelopmental features may have been missed if they were not included in the clinical notes. Regarding symptomatic parents, histories were self-reported, and we did not have access to medical records to corroborate reported features or obtain additional details. Further, the modest size of each of the subgroups (e.g., missense vs. LOF, de novo vs. inherited) limited our ability to detect strong genotype-phenotype correlations.
Despite these limitations, we have added to the characterization of PCDH19-related epilepsy. In a cohort of patients with PCDH19 variants, we demonstrate not only refractory epilepsy in clusters in the majority of patients, but also prevalence of neuropsychiatric and behavioral challenges, principally, autistic features, disordered sleep, and obsessive-compulsive/anxiety symptoms. As such, we strongly recommend early referral to behavioral therapists, psychiatrists, sleep specialists, and counselors for all patients with PCDH19-related epilepsy, in anticipation of these challenges. Our data suggest that further research is needed to more systematically approach and understand the many facets to this condition. We note that average intelligence is possible in the setting of PCDH19-related epilepsy, which is an important point for prognosis, particularly in young patients who appear to be developing typically. We also identified a major gap in formal neuropsychiatric evaluation in this cohort, which further research and awareness in the clinical community should address. Regarding seizures, a large portion of individuals with PCDH19-related epilepsy have refractory epilepsy, as reported in the literature and supported by data in our cohort, for which further research into more targeted therapies would be beneficial. Some patients did achieve seizure freedom, however, indicating that this goal is not necessarily unattainable in patients with PCDH19 variants, and a proactive approach to attempt to control seizures is warranted. Ultimately, a prospective natural history study of individuals with PCDH19–related epilepsy in addition to standardized neuropsychiatric evaluations will be required to fully capture the full range of clinical conditions and course related to this gene. Gene discovery in epilepsy has raised the promise that targeted therapies can be developed 33. Given the contribution of PCHD19 in epilepsy and its association with difficult to control seizures and challenging behavior manifestations, PCDH19 should be prioritized in the conversation around precision medicine in epilepsy.
Supplementary Material
Significance:
Our series represents a robust cohort with carefully curated PCDH19 variants. We observed seizures as a core feature with a range of seizure types and severity. While the majority of individuals had ID, we highlight the possibility of average intellect in the setting of PCDH19-related epilepsy. We also note the high prevalence and severity of neurobehavioral phenotypes associated with likely pathogenic variants in PCDH19. Sleep dysregulation was also a major area of concern. Our data emphasize the importance of appropriate referrals for formal neuropsychological evaluations as well as the need for formal prospective studies to characterize the PCDH19-related neurodevelopmental syndrome in children and their genotype-positive parents.
Key Points:
-
#1
Seizure phenotype of PCDH19-related epilepsy is broad.
-
#2
Social immaturity, obsessive-compulsive and aggressive behaviors are prevalent.
-
#3
Behavioral and sleep features are of high concern to parents.
Acknowledgements
We thank the PCDH19 Alliance, led by President and Co-founder Julie Walters, for initial funding to launch the PCDH19 Registry, as well as the PCDH19 research community for valuable discussions. We are grateful to all of the patients and families who have enrolled and participated in our on-going research projects. We thank the referring neurologists and neuropsychologists whose clinical notes were used in the phenotyping. We thank Dr. Kiran Maski for advising us on the sleep survey instrument. AP has received support from the BCH Translational Research Program. This work was also supported by the NIH (NINDS R01NS100766).
Footnotes
Disclosure of Conflicts of Interest
None of the authors has any conflicts of interest to disclose.
Ethical Publication Statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References:
- 1.Epi KC, Epilepsy Phenome/Genome P, Allen AS, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Euro E-RESC, Epilepsy Phenome/Genome P, Epi KC. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet 2014;95:360–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Veeramah KR, Johnstone L, Karafet TM, et al. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia 2013;54:1270–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas RH, Berkovic SF. The hidden genetics of epilepsy-a clinically important new paradigm. Nat Rev Neurol 2014;10:283–92. [DOI] [PubMed] [Google Scholar]
- 5.Depienne C, Bouteiller D, Keren B, et al. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet 2009;5:e1000381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Depienne C, LeGuern E. PCDH19-related infantile epileptic encephalopathy: an unusual X-linked inheritance disorder. Human mutation 2012;33:627–34. [DOI] [PubMed] [Google Scholar]
- 7.Depienne C, Trouillard O, Bouteiller D, et al. Mutations and deletions in PCDH19 account for various familial or isolated epilepsies in females. Human mutation 2011;32:E1959–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dibbens LM, Tarpey PS, Hynes K, et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet 2008;40:776–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gagliardi M, Annesi G, Sesta M, et al. PCDH19 mutations in female patients from Southern Italy. Seizure 2015;24:118–20. [DOI] [PubMed] [Google Scholar]
- 10.Higurashi N, Nakamura M, Sugai M, et al. PCDH19-related female-limited epilepsy: further details regarding early clinical features and therapeutic efficacy. Epilepsy Res 2013;106:191–9. [DOI] [PubMed] [Google Scholar]
- 11.Higurashi N, Shi X, Yasumoto S, et al. PCDH19 mutation in Japanese females with epilepsy. Epilepsy Res 2012;99:28–37. [DOI] [PubMed] [Google Scholar]
- 12.Hildebrand MS, Myers CT, Carvill GL, et al. A targeted resequencing gene panel for focal epilepsy. Neurology 2016;86:1605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hynes K, Tarpey P, Dibbens LM, et al. Epilepsy and mental retardation limited to females with PCDH19 mutations can present de novo or in single generation families. J Med Genet 2010;47:211–6. [DOI] [PubMed] [Google Scholar]
- 14.Marini C, Darra F, Specchio N, et al. Focal seizures with affective symptoms are a major feature of PCDH19 gene-related epilepsy. Epilepsia 2012;53:2111–9. [DOI] [PubMed] [Google Scholar]
- 15.Marini C, Mei D, Parmeggiani L, et al. Protocadherin 19 mutations in girls with infantile-onset epilepsy. Neurology 2010;75:646–53. [DOI] [PubMed] [Google Scholar]
- 16.Leonardi E, Sartori S, Vecchi M, et al. Identification of four novel PCDH19 Mutations and prediction of their functional impact. Ann Hum Genet 2014;78:389–98. [DOI] [PubMed] [Google Scholar]
- 17.van Harssel JJ, Weckhuysen S, van Kempen MJ, et al. Clinical and genetic aspects of PCDH19-related epilepsy syndromes and the possible role of PCDH19 mutations in males with autism spectrum disorders. Neurogenetics 2013;14:23–34. [DOI] [PubMed] [Google Scholar]
- 18.Kc Epi, Epilepsy Phenome/Genome P. Ultra-rare genetic variation in common epilepsies: a case-control sequencing study. Lancet Neurol 2017;16:135–43. [DOI] [PubMed] [Google Scholar]
- 19.Scheffer IE, Turner SJ, Dibbens LM, et al. Epilepsy and mental retardation limited to females: an under-recognized disorder. Brain 2008;131:918–27. [DOI] [PubMed] [Google Scholar]
- 20.de Lange IM, Rump P, Neuteboom RF, et al. Male patients affected by mosaic PCDH19 mutations: five new cases. Neurogenetics 2017;18:147–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Depienne C, Bouteiller D, Keren B, et al. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet 2009;5:e1000381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Terracciano A, Trivisano M, Cusmai R, et al. PCDH19-related epilepsy in two mosaic male patients. Epilepsia 2016. [DOI] [PubMed]
- 23.Thiffault I, Farrow E, Smith L, et al. PCDH19-related epileptic encephalopathy in a male mosaic for a truncating variant. American journal of medical genetics Part A 2016. [DOI] [PubMed]
- 24.Trump N, McTague A, Brittain H, et al. Improving diagnosis and broadening the phenotypes in early-onset seizure and severe developmental delay disorders through gene panel analysis. J Med Genet 2016;53:310–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Owens JA, Spirito A, McGuinn M. The Children’s Sleep Habits Questionnaire (CSHQ): psychometric properties of a survey instrument for school-aged children. Sleep 2000;23:1043–51. [PubMed] [Google Scholar]
- 26.Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016;44:D862–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia 2014;55:475–82. [DOI] [PubMed] [Google Scholar]
- 29.Jamal SM, Basran RK, Newton S, et al. Novel de novo PCDH19 mutations in three unrelated females with epilepsy female restricted mental retardation syndrome. American journal of medical genetics Part A 2010;152A:2475–81. [DOI] [PubMed] [Google Scholar]
- 30.Trivisano M, Pietrafusa N, Ciommo V, et al. PCDH19-related epilepsy and Dravet Syndrome: Face-off between two early-onset epilepsies with fever sensitivity. Epilepsy Res 2016;125:32–6. [DOI] [PubMed] [Google Scholar]
- 31.Lotte J, Bast T, Borusiak P, et al. Effectiveness of antiepileptic therapy in patients with PCDH19 mutations. Seizure 2016;35:106–10. [DOI] [PubMed] [Google Scholar]
- 32.Scheffer IE, Turner SJ, Dibbens LM, et al. Epilepsy and mental retardation limited to females: an under-recognized disorder. Brain 2008;131:918–27. [DOI] [PubMed] [Google Scholar]
- 33.Epi PMC. A roadmap for precision medicine in the epilepsies. Lancet Neurol 2015;14:1219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.