

Abstract
The pleiotropic functions of circulating high density lipoprotein (HDL) on peripheral vascular health are well established. HDL plays a pivotal role in reverse cholesterol transport and is also known to suppress inflammation, endothelial activation and apoptosis in peripheral vessels. Although not expressed in the central nervous system, HDL has nevertheless emerged as a potential resilience factor for dementia in multiple epidemiological studies. Animal model data specifically support a role for HDL in attenuating the accumulation of β-amyloid within cerebral vessels concomitant with reduced neuroinflammation and improved cognitive performance. As the vascular contributions to dementia are increasingly appreciated, this review seeks to summarize recent literature focused on the vasoprotective properties of HDL that may extend to cerebral vessels, discuss potential roles of HDL in dementia relative to brain-derived lipoproteins, identify gaps in current knowledge, and highlight new opportunities for research and discovery.
Keywords: high density lipoprotein, vascular function, vascular disease, alzheimer disease, HDL-proteome, HDL-lipidome
The pleiotropic functions of high density lipoprotein (HDL)
Like other mature lipoproteins, HDL consists of a core of hydrophobic lipids surrounded by a phospholipid and free cholesterol monolayer studded by proteins (Fig. 1)[1]. A key protein found in most HDL particles is apolipoprotein A-I (apoA-I), which makes up 70% of its protein content[2]. The major lipid classes found on HDL include cholesterol and other steroids, phospholipids, cholesteryl esters, sphingolipids, and triglycerides[3]. Overall, HDL particles consist of approximately 85-95 distinct proteins[4] and hundreds of lipid subtypes[5] that together mediate diverse functions including lipid transport and metabolism, anti-oxidation, immune response, hemostasis, metal binding, and vitamin transport[5–7].
Fig.1. Pleiotropic contents and functions of HDL.
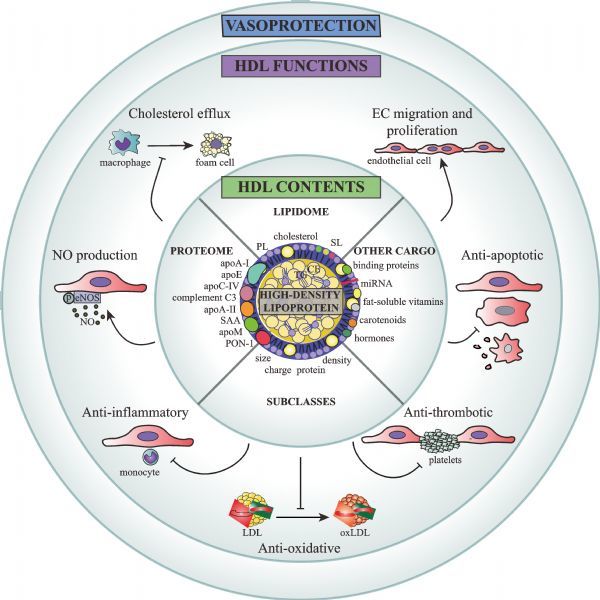
apoA-I: apolipoprotein A-I; apoA-II: apolipoprotein A-II; apoC-IV: apolipoprotein C-IV; apoE: apolipoprotein E; apoM: apolipoprotein M; EC: endothelial cell; eNOS: endothelial nitric oxide synthase; LDL: low density lipoprotein; miR-223: micro RNA 223; NO: nitric oxide; oxLDL: oxidized LDL; p: phosphate group; PL: phospholipid; PON-1: paraoxonase 1; S1P: sphingosine-1-phosphate; SAA: serum amyloid A; SM: sphingomyelin.
HDL is the smallest and densest of the plasma lipoproteins and contains an estimated 85-95 distinct proteins, 200 lipid species and several other nonpolar cargo molecules. HDL components and subclass distribution can vary between individuals and is altered by diseased states. The compositional profile of HDL confers pleiotropic functions to the population of circulating particles.
HDL-C and cardiovascular disease
The association between low HDL cholesterol (HDL-C) levels and elevated cardiovascular disease (CVD) was first suggested in the 1960s in the Framingham Heart Study[8]. Since then, a multitude of clinical studies have strengthened this relationship[9–11]. Although Mendelian randomization studies have now demonstrated that HDL-C levels per se have no causal relationship with CVD[12–14], the question remains as to whether HDL-C, a static measure of HDL's cholesterol content, adequately reflects the beneficial functions of HDL on vascular health.
In humans, genetic deficiency of APOA-I or the ATP binding cassette transporter 1 (ABCA1) leads to very low levels of HDL-C that can be associated with increased risk of and accelerated onset of coronary artery disease (CAD)[15]. Similar outcomes are observed in some but not all cases of lecithin cholesterol acyltransferase (LCAT) deficiency[15]. However, other forms of genetically altered HDL-C levels suggest that there is more complexity to the role of HDL in health and disease. For example, carriers of the APOA-I mutation known as apoA-I Milano have very low HDL-C levels but similar levels of atherosclerosis and CAD as controls with normal HDL-C levels[16]. Conversely, carriers of mutations in SCARB1, the gene encoding HDL receptor scavenger receptor class B type I (SR-B1), have abnormally high HDL-C levels and yet are at increased risk for CAD[17].
Unlike humans where the major plasma lipoprotein is low-density lipoprotein (LDL), the major plasma lipoprotein in mice is HDL. The innately high HDL:LDL ratio in mice renders them generally resistant to CVD and advanced atherosclerosis compared to humans[18]. As a result, atherosclerosis studies in mice overwhelmingly depend on genetically modified models, including deficiency of either apoE or low-density lipoprotein receptor (LDLR), to allow the effects of HDL on atherosclerosis to be studied[19]. For example, genetic deletion of either apoA-I, ABCA1, LCAT and SR-B1 in apoE −/− or LDLR −/− animals alters murine HDL-C levels. In addition, the effects on atherosclerosis can also vary with strain background and animal diet[20]. Nonetheless, HDL-targeted therapies in murine models of atherosclerosis appear, overall, to be beneficial. For example, transgenic overexpression of apoA-I, gene transfer of human apoA-I, adenoviral transfer of apoA-I, and infusion with recombinant apoA-I or HDL can reduce or stabilize atherosclerotic plaques in apoE −/− or LDLR −/− mice[21].
Changes in HDL cholesterol efflux capacity
The cholesterol efflux capacity (CEC) of HDL is modified in CVD[22–26], metabolic syndrome[27], and during acute inflammation[28]. How distinct CEC and HDL-C are as biomarkers of disease has been debated, with some studies observing reduced CEC independent of changes in HDL-C[22-23,25,28–30], while others find that CEC and HDL-C changes correlate[24,26-27]. Possible mechanisms to explain diminished CEC in most of these studies is increased HDL-associated serum amyloid A (SAA)[28] and reduced HDL-associated paraoxonase 1 (PON-1)[26] in inflammatory states, which will be discussed below. Importantly, despite its obvious implication for reverse cholesterol transport (RCT), CEC is not the only known function of HDL. Here, we will discuss additional vasoprotective properties of HDL, with a focus on HDL's anti-inflammatory, anti-oxidative, vasodilatory and anti-apoptotic functions.
Anti-inflammatory effects of HDL
Several mechanisms by which HDL exerts anti-inflammatory effects on endothelial cells have been described. These include pathways dependent on the HDL receptor SR-B1[31–33] as well as through vascular sphingosine 1 phosphate (S1P) receptor 1 and 3[34], which trigger a signaling cascade through the PI3K/Akt pathway leading to phosphorylation of endothelial nitric oxide synthase (eNOS). The vasoprotective effects of nitric oxide produced by eNOS phosphorylation are well-established, and include vasodilation, reduced endothelial cell permeability, and inhibition of vascular cell adhesion molecule-1 (VCAM-1) expression via downregulation of the pro-inflammatory NFκB signaling pathway[35]. HDL-S1P action can also directly inhibit NFκB signaling to suppress adhesion molecule expression[34], reduce endothelial exocytosis[31], and maintain annexin-1 expression[33]. Additionally, HDL can indirectly increase eNOS activity via actions of the lipid transporter ABCG1 to maintain proper membrane fluidity for eNOS function[36,37]. The HDL-associated protein PON-1 prevents lipid and LDL oxidation, thereby protecting endothelial cells from oxidative damage, pro-inflammatory signaling, and apoptosis[38-39].
Many disease states, particularly those with an inflammatory component, can affect HDL's vasoprotective functions (Table 1). For example, HDL isolated from CVD patients exhibits reduced ability to phosphorylate eNOS[6,40-41] and displays a distinct repertoire of immune cell trafficking proteins[41]. HDL isolated from children with chronic kidney disease exhibits reduced ability to protect from endothelial cell activation [42-43]. Acute inflammation, as in the case of periodontal therapy, can also alter the ability of HDL to induce eNOS phosphorylation[44]. In abdominal aortic aneurysm (AAA), a quantitative reduction in apoA-I-mediated vasoprotection may result from decreased levels of circulating small HDL, a process which itself may partially be due to the sequestering of apoA-I at the site of inflammation in thrombotic aortic tissue[45-46]. Moreover, inflammatory-remodelling of HDL composition during AAA by the inclusion of pro-oxidant proteins may further reduce HDL quality and contribute to the observed loss of anti-thrombotic and anti-oxidative capacity[46]. In contrast, exercise training improves the ability of HDL to protect endothelial cells from tumor necrosis factor-α induced injury, monocyte adhesion, and VCAM-1 expression in metabolic syndrome while also elevating eNOS activation[47]. Importantly, changes to the anti-inflammatory functions of HDL have even been observed in disease in the absence of changes in circulating HDL-C levels[43-44].
Tab.1.
HDL compositional and functional heterogeneity in disease
| Disease | Proteome | Lipidome | Size | Function |
|---|---|---|---|---|
| Cardiovascular | ↓/--PON-1[26,63,71] | ↓S1P[90], SM[240] | ↓/↑ large HDL[23,59-61,64] | ↓ cholesterol efflux[25,26] |
| disease | ↑ SAA[41, 84, 239] | ↑/↓PL[239-241] | ↓ small HDL[63] | ↓ eNOS phosphorylation[23,41,75] |
| ↓ apoA-I, apoA-II, apoE[84] | ↑TG[239,240] | ↑ inflammatory activity[242] | ||
| Acute inflammation | ↓ PON-1[28,44,74] ↑ SAA, apoA-II, complement C3[44,74] ↓/↑apoA-I[44,74] | ↓S1P, SM[74,243] ↑TG, FFA[243] | ↓/-- cholesterol efflux[28,44,74] ↓ NO production[44,74] ↑ inflammatory activity[44] ↑ oxidative activity[44] | |
| Chronic kidney disease | ↓/--PON-1[76,77,82] ↑SAA, SDMA, apoC-II[43,75,81,98] ↓ apoA-I, apoA-II[75,81] | ↑ TG[81] ↓ PL[81] | ↓ small HDL[244] | ↓ NO production[43] ↓ cholesterol efflux[43,81] ↑ inflammatory activity[42,43] ↑ oxidative activity[43,98] ↓ EC proliferation and migration[42] |
| Cirrhosis | ↓/↑ PON-1[58,73] ↓ apoA-I, -II, apoC-II, -III[72] ↑ SAA, apoE[72] | ↑ PL[73] | ↑ large HDL[72,73] | ↓ cholesterol efflux[72] ↓ PON1 activity[73] |
| Aging | -- PON-1[78,79] ↓ apoE[78] ↑ SAA, complement C3[78] | ↑ SM[245] | ↑ oxidative activity[78,79] ↓/-- cholesterol efflux[78,245] ↓ PON1 activity[78,79] | |
| Arthritis | ↓ PON-1[77] ↑ SAA[77] | ↑ SM, PL[246] | ↑ inflammatory activity[247] | |
| Type 2 diabetes mellitus | ↓ apoA-I[85] ↑ apoA-II[85] | ↑TG, S1P[85,90,248] | -- cholesterol efflux[27] ↓ NO production[27] ↑ inflammatory activity[248] | |
| Alzheimer's disease | ? | ? | ? | ↓ cholesterol efflux[249] ↑ inflammatory activity[249] |
| Age-related macular degeneration | ↑ SAA[250] | ? | ? | ↑ anti-inflammatory activity[250] |
apoA-I: apolipoprotein A-I; apoA-II: apolipoprotein A-II; apoC-II: apolipoprotein C-II; apoC-III: apolipoprotein C-III; apoE: apolipoprotein E; EC: endothelial cell; eNOS: endothelial nitric oxide synthase; FFA: free fatty acid; NO: nitric oxide; PL: phospholipid; PON-1: paraoxonase 1; S1P: sphingosine-1-phosphate; SAA: serum amyloid A; SM: sphingomyelin; SMDA: symmetrical dimethylarginine; TG: triglyceride; ↓: decrease; ↑: increase; --: no change; ?: unknown in literature.
HDL-C is an imperfect marker
The disassociation between HDL functions and HDL-C levels may help to explain why the epidemiological associations of HDL and CVD risk are not easily captured by mere HDL-C measures. HDL functional assays may also be more informative than HDL-C measures in clinical trials. For example, efforts to use statins, niacin, and cholesteryl ester transfer protein (CETP) inhibitors to raise HDL-C levels and protect against CVD have been disappointing[48–50]. Although statins consistently reduce CVD events, the prognostic utility of HDL-C in statin users is unclear[51–54]. Two large randomized control trials to test the effect of niacin on CVD showed no statistically significant reduction in CVD despite elevated HDL-C levels[42,55–57]. CETP inhibitors also failed to reduce CVD events despite raised HDL-C levels[49]. A meta-analysis of randomized controlled trials for niacin, fibrates, and CETP inhibitor therapies in conjunction with statin therapy found no change to mortality, CAD mortality and myocardial infarction compared to patients treated with statins alone[58].
HDL heterogeneity and modification in disease
HDL can be classified by a variety of schemes including apolipoprotein content, size, surface charge, and density (Fig. 1)[2]. The distribution of HDL-C among different sizes has been observed to vary with exercise, CVD risk factors, and CVD disease status and lipid-lowering medications. However, the direction of association between HDL subclass and disease outcomes has been controversial. Many studies have found that large HDL subclasses appear to be beneficial for cardiovascular health with stronger associations with disease than total HDL-C[23,59–62]. By contrast, other studies have observed that CAD patients have lower levels of small HDL[63] and elevated levels of large HDL[64]. Investigations on the effect of statins on HDL subclass is also not as clearly defined as their well-established ability to elevate total HDL-C levels[65]. For example, statins have been found to exert no effect[66] or to lead to elevated levels of the large HDL2 subclass while decreasing levels of small HDL3 levels[67-68], although contrasting reports indicate an increase in HDL3 levels[69]. Fibrates increase HDL3 levels while decreasing those of HDL2[65] whereas niacin has the opposite effect on HDL subclass by promoting conversion to mature HDL2 particles[65]. Combination treatments of lipid lowering drugs present no net change in HDL subclass compared to monotherapy[67] or have reported an additive effect with improved HDL functionality[70]. Variations in cohort, drug regimen and experimental techniques likely contribute to the varying observations of medication use on HDL subclass distribution and net function.
To add further complexity to the matter of HDL subclass, a recent report has found that HDL appears to be secreted from the liver in all of its unique sizes and remains in those size classes for several days before excretion[71]. This is contrary to the traditional view of how HDL subclasses are formed, which posits that HDL is first secreted from the liver as small, lipid-poor, discoidal HDL that is lipidated to evolve into the larger spherical forms over time.
Another measure of interest is the heterogeneity of the HDL proteome. The HDL proteome varies considerably between individuals based on disease, diet, age, and inflammatory status (Table 1). For example, PON-1 content or activity on HDL is reduced in patients with CVD[26,72] liver cirrhosis[73-74], acute inflammation[28,44,75], chronic kidney disease[76-77], rheumatoid arthritis[78], in the elderly[79-80], and is elevated with exercise[47] or a diet rich in olive oil[81]. Conversely, the SAA content on HDL has been found to increase in chronic kidney disease[76,82], aging[79], acute inflammation[44,75], rheumatoid arthritis[78] and cirrhosis[73]. Proteomic analysis of plasma specimens of AAA patients identify disease-associated reductions in HDL's major lipoproteins, namely apoA-I[45,83] and apoA-II[45]. Contrasting reports, however, observe an upregulation in apoA-I and apo-J levels in this patient population[84]. Importantly, in several cases, changes to the HDL proteome can be observed in inflammatory or disease states without a change in total plasma HDL-C[44,79], again highlighting the importance of looking beyond HDL-C when considering lipoprotein function in the etiology of disease. Other alterations to the HDL proteome that have been observed in vascular and inflammatory pathologies include reduced or elevated apoA-I, apoA-II, apoC-II and apoE, and elevated complement C3 and apoC-III[44,73,75-76,79,82,85-86].
The HDL proteome, and by implication HDL function, is also subject to change by hypolipidemic agents. Green et al.[87] report that CAD-associated changes in the HDL3 apolipoprotein profile, including increased levels of apoE coupled with decreased levels of apoF and phospholipid transfer protein, are reversed by combination therapy of atorvastatin and niacin. Niacin is also shown in a separate study to exhibit a synergistic enhancement of apoA-I in concert with atorvastatin[70]. Fibrates increase apoA-I levels and, to a greater extent, boost apoA-II levels[88]. The recent study by Gordon et al.[89] reports that rosuvastatin dramatically increases the levels of alpha-1-antitrypsin in the large HDL fraction which in turn enhances HDL's anti-inflammatory properties. Additionally, lipid lowering drugs also alter the activity of HDL-associated anti-oxidant proteins such as PON-1 to augment its vasoprotective function[65].
The HDL lipidome is another field of emerging interest. While most of the work thus far has investigated the lipidome of HDL from healthy subjects, it is becoming clear that changes to the proportions of HDL lipids in disease can have functional consequences. For example, the CEC, antioxidant, and anti-inflammatory activities of HDL are impaired with excess triglyceride, cholesteryl ester, oxidized lipids, and sphingomyelin[90]. A well-studied bioactive lipid on HDL is S1P, which is well known to be at least partially responsible for the anti-inflammatory actions of HDL. S1P on HDL can be reduced in CVD[91] and acute inflammation[75] resulting in impaired signaling to eNOS. In type 2 diabetes mellitus (T2DM), S1P has been observed to be elevated on HDL possibly as a compensatory mechanism[92]. Many other changes to the HDL lipidome have been observed including changes to triglyceride, phospholipid, and sphingomyelin content (Table 1).
Among other cargo carried on HDL are small non-coding RNAs including tRNA-derived RNA fragments, RNase P-derived RNA fragments, and microRNA (miRNA)[93]. MiRNAs in particular have emerged as an exciting topic in lipid research for their potential as biomarkers and in therapeutic approaches. HDL has been found to be regulated by and to carry a number of miRNAs that vary between individuals according to a number of factors including diet[94], weight loss[95], and CAD[72]. As with changes to the HDL proteome, changes to the miRNA profile of HDL can be observed even when there is no change in total plasma HDL-C[72,95] or apoA-I levels[94]. Interestingly, HDL-associated miR-223, which can be altered with diet or weight loss[94-95], has even been found to be transferred to endothelial cells[96] and to alter gene expression of intercellular adhesion molecule-1 in those cells[97].
HDL also carries a number of other nonpolar molecules including fat-soluble vitamins, vitamin binding proteins, carotenoids, steroids and other hormones, potentially serving as a transporter for delivery to other tissues[93]. Polar metabolites have also been found on HDL, some of which correlate with the insulin resistance[98]. An additional molecule of interest on HDL is symmetric dimethylarginine, a metabolite that is increased in children with chronic kidney disease and may be partially responsible for impaired vasoprotective actions of HDL in these patients[99].
A further layer of complexity to HDL heterogeneity includes modifications to its protein components including the addition of aldehydes such as acrolein[100], modifications of apoA-I by myeloperoxidase[101], or carbamylation of HDL-associated proteins[77]. These protein modifications are associated with CVD and compromise HDL functions including cholesterol efflux, antioxidant properties, and promotion of endothelial cell migration and proliferation[77,100-101].
Cerebral vessel disease and dementia
The brain comprises only 2% of total body mass but consumes approximately 12% of total cardiac output[102]. The intimate association of neurons with vessels via neurovascular coupling regulates cerebral blood flow (CBF) in response to changes in neuronal activity. This coupling also maintains the necessary influx of oxygen, glucose and ions balanced by homeostatic clearance of neurotoxic by-products from the brain throughout the lifespan. As the brain cannot always easily or quickly compensate for restricted blood supply, structural and functional impairments in the cerebrovasculature can profoundly impact brain function. Central nervous system (CNS) microvessels are distinguished by the presence of the blood brain barrier (BBB), which stringently controls the movement of solutes into the brain to maintain a CNS ionic environment optimal for neuronal activity.
Cerebral vessel disease (CeVD) is one of the most common vascular pathologies of the aging brain with heterogeneous changes that undermine the integrity and function of cerebral vessels (arteries, arterioles, venules and capillaries) including atherosclerosis, arteriosclerosis, lipohyalinosis, and cerebral amyloid angiopathy (CAA)[103-104]. Increased by CVD risk factors[105], CeVD can restrict CBF causing local or global ischemia in the brain through narrowing of the vessel lumen, arterial occlusion, loss of cerebrovascular resistance and micro and macro hemorrhage. Resulting brain damage from this process can lead to vascular cognitive impairment or vascular dementia (VaD), which is clinically identified by impaired locomotor function in addition to memory loss and executive dysfunction[104]. In particular, cerebral small vessel disease (CSVD), which manifests as white matter lesions (i.e. lacunes, lacunar infarcts, and leukoaraiosis), is a leading contributor to VaD[106]. How impaired cerebrovascular function may relate to cognitive decline and dementia is an area of intense interest.
Alzheimer's disease (AD), the most common form of dementia[107], is clinically characterized by memory loss and conclusively diagnosed by the presence of β-amyloid (Aβ) plaques and neurofibrillary tangles in brain tissue[108]. The amyloid hypothesis of AD, which proposes that accumulation of Aβ aggregates is the primary pathogenic factor that initiates and drives neurodegeneration in AD, was founded on the discovery of genetic mutations that cause aberrant overproduction of Aβ in familial early onset AD (EOAD) (<65 years old) and evidence that Aβ is neurotoxic[108]. However, as only 1-3% of AD cases are attributed to causal mutations[109] and Aβ aggregates can be present in elderly people who show no signs of cognitive decline[110], major efforts are being deployed to understand the etiology of sporadic or late onset AD (LOAD) (>65 years old). Cardiovascular risk factors including hypertension, T2DM and mid-life dyslipidemia are all associated with increased AD risk[111]. The majority of AD patients possess extensive damage to their cerebral blood vessels[112] and exhibit mixed vascular pathology with CeVD (atherosclerosis of the circle of Willis and its branches) as well as cerebrovascular lesions including leukoaraiosis, and lacunar infarcts, microbleeds, microinfarcts, and CAA[113]. Importantly, increased severity of CeVD in subjects over 65 years of age is associated with lower scores across several cognitive domains including episodic memory and perceptual speed, the respective neuropsychological hallmarks of AD and VaD, which remain even after adjusting for established genetic AD or vascular risk factors[114]. Intriguingly, white matter hyperintensities indicative of CSVD were recently found to be elevated in EOAD mutation carriers (mean age 39 years) that were detectable at least 6 years prior to clinical onset[115]. That these EOAD subjects were too young to exhibit classical age-related cardiovascular risk factors provides compelling support that cerebrovascular dysfunction may be an important driver of neuronal compromise and cognitive decline.
The brains of AD patients show a plethora of structural and functional vascular abnormalities that correlate with severity of neurodegeneration, including reduced microvascular density with remaining vessels appearing tortuous and string-like[112,116]. One potential cause of this brain vessel atrophy may be the Aβ-driven vascular pathology of CAA, in which Aβ is deposited in the walls of the arteries, arterioles and capillaries in the leptomeninges and cerebral cortex[117]. CAA prevalence in AD is 80-90%[118] and CAA may contribute to degeneration of mural cells in arteries, arterioles, and capillaries, leading to vessel stiffening and impaired vasomotor function[117]. Notably, weakening of cerebral vessels increases their susceptibility to rupture and CAA is also associated with ischemic lesions, micro- and macro-hemorrhages, and impaired CBF[118]. Microscopic cerebral hemorrhages, also known as cerebral microbleeds (CMBs) are a type of CSVD that indicates weakness within the microvascular system[119]. Studies show that CAA-associated vasculopathies lead to the development of CMBs in the lobar temporal and parietal cortex although there is some debate as to the direct association between CAA and CMBs[120]. In contrast, CMBs in non-lobar deep white matter regions are associated with vascular risk factors such as stroke and hypertension[121]. Recent findings from a prospective analysis of the Rotterdam cohort suggest CMBs may prove to be useful predictors of future cognitive decline and pre-clinical dementia regardless of their location in the brain[122]. Whether microhemorrhages affect cognitive status and are a reliable biomarker of cognitive decline is a key debate, with one study reporting no effect of CMBs on cognitive function[123] contrasting with another study that found an association between frontal lobe lacunar infarcts and pre-dementia[124].
The close relationship between AD and cerebral vascular pathology raises the hypothesis that vascular damage may plays a considerable role in precipitating and driving AD pathogenesis. The two-hit vascular hypothesis proposes that vascular risk factors (hit one) leads to the cerebrovascular dysfunction (i.e. BBB dysfunction, oligaemia) that precedes cognitive impairment[116]. This vascular damage induces early neuronal dysfunction due to the accumulation of neurotoxic molecules, capillary hypoperfusion and altered Aβ metabolism that accelerates Aβ retention and accumulation in the brain[116]. Increased cerebral Aβ represents hit two, which amplifies neuronal dysfunction leading to a self-propagating acceleration of neurodegeneration, cognitive decline and ultimately dementia[116].
VaD is the second most common dementia[125], wherein, unlike AD, various types of vascular injury including ischemic, hemorrhagic, or hypoperfusion directly causes cognitive impairment. It is increasingly appreciated that AD and VaD share considerable overlap in clinical[126], pathological[103] and epidemiological features[127]. Interestingly, up to 45% of clinical dementia cases have evidence of mixed neuropathology for both AD and VaD[128] and the prevalence of mixed dementia (AD and VaD) increases with age[129]. This lends support to the argument that vascular dysfunction interacts synergistically with other pathogenic neurodegenerative pathways to promote various forms of dementia. Mirroring the worsening cognitive decline seen in humans exhibiting cerebral hypoperfusion[103], experimental restriction of CBF in animals recapitulates both the amyloid and vascular neuropathology of mixed dementia[130]. Chronic cerebral hypoperfusion as a result of cerebrovascular dysfunction may therefore serve as a common catalyst for the development of CAA and subsequent Aβ-associated pathologies[130]. Stroke is an established risk factor for AD[105], and while an initial report of increased cerebral Aβ levels in ischemic stroke patients[131] failed to reproduce in a subsequent larger cohort[132], new evidence that cerebral hypoxia may diminish enzymatic Aβ-degradation[133] offers a potential mechanism by which cerebrovascular incidents may accelerate AD pathogenesis.
An important point for potential therapeutic considerations is that cerebrovascular lesions may correlate with more severe cognitive dysfunction in early AD rather than late in progression[103]. As 20 years of research on the amyloid hypothesis and Aβ- targeting therapies have not yet produced an approved treatment for AD, it is imperative that the multifactorial aspects of AD be addressed in the future. Given the beneficial vasoprotective roles of HDL in peripheral vessels, expanding HDL research toward the cerebrovasculature and neurodegeneration may be highly promising.
Lipoproteins and cognitive function
The brain is the most cholesterol-rich reservoir in the body, containing 25% of the body's total cholesterol content[134]. The connection of lipid metabolism to AD was first noted in Dr. Alois Alzheimer's characterization of the disease in 1906 that described lipid deposits in the brain[135]. Today, genome wide association studies confirm this connection, with the identification of confirmed AD risk genes that function in various aspects of lipid metabolism[136]. Of these, genetic variation in apoE is the strongest genetic risk factor for AD in humans, with APOE4 [137-138], APOE3 neutral[139], and APOE2 protective[140]. Over 60% of AD cases possess at least one APOE4 allele[141], and carriers of the APOE4 allele show increased risk, earlier onset, and exacerbated cognitive decline[137-138,141]. Despite this strong association, the exact mechanisms by which apoE, which is produced by both astrocytes and microglia, modifies AD risk in an isoform-specific manner are not yet completely defined. One undisputed function of apoE relates to its role in Aβ deposition, as APOE4 carriers consistently develop greater Aβ burden at an earlier age compared to non-APOE4 carriers[142–146]. APOE4 is the strongest genetic risk factor of LOAD[137,147], and moreover predisposes carriers to cardiovascular disease[148], reinforcing the importance of cholesterol metabolism in the vascular-mediated pathogenesis of sporadic LOAD. Importantly, apoE may also impair cerebrovascular function, as apoE4 is associated with aberrant binding and cell signaling at the neurovascular unit resulting in diminished Aβ clearance[149-150], reduced eNOS expression[151], vascular inflammation, and BBB dysfunction[152]. Exacerbation of vascular dysfunction by apoE4 would be expected to aggravate cognitive decline in APOE4 carriers, and while there is a suggestion of such a relationship[153], others find a lack of association of APOE4 and cognitive impairment with concurrent CeVD[154]. How APOE4 may play a multifaceted role in AD pathogenesis remains to be fully elucidated.
Apo J, or clusterin, is the other major lipoprotein besides apoE that is abundantly produced within the CNS[155]. Genetic mutations in the clusterin gene (CLU) were identified by two independent GWAS studies as risk factors for LOAD[156-157]. Clusterin is elevated in AD brains[158], present within Aβ plaques[158-159] and co-localizes with Aβ deposits in CAA-affected leptomenigeal arteries[160]. Clusterin has been shown to facilitate Aβ egress from the brain when co-injected into mice[161]. Recent findings by Miners et al.[162] also support a role for clusterin in regional Aβ clearance in humans. Intriguingly, they showed that although clusterin levels are highest in brain regions with plaque pathology, the molar ratio of clusterin: Aβ42 surprisingly declines with insoluble Aβ42 levels in a region-dependent manner, suggesting that rising Aβ42 levels outstrip increased clusterin levels, thereby decreasing Aβ clearance and promoting its region-specific deposition. In vitro studies suggest that clusterin acts as a chaperone protein facilitating Aβ egress at the BBB[161,163] and Aβ transport to microglia for degradation via interactions with the microglial receptor TREM-2[164]. Clusterin also interferes with Aβ peptide aggregation and neutralizes Aβ oligomer neurotoxicity[165], and deficiency of clusterin signaling through its receptor, plexin A4, leads to memory and learning deficits[166]. Clusterin has also been linked to accelerated atrophy in brain regions first affected by AD through an unknown interaction with Aβ[167]. Further work is needed to determine the dynamics of this glycoprotein in the context of health and neurodegeneration.
In addition to brain-derived apoE and apoJ, circulating lipoproteins may also be important to cerebrovascular health. Along with its defined association with CVD, high levels of plasma HDL-C in elderly people are associated with better memory[168-169], lower Aβ burden[170], and less cognitive decline[171]. Conversely, HDL-C has been found to be reduced in AD subjects who have vascular risk factors[172], and plasma apoA-I has been reported to be reduced in AD patients[173] and negatively associates with cognitive decline independent of Aβ, indicating a protective homeostatic role for apoA-I against cognitive decline in the elderly[174]. In symptomatic AD patients, plasma apoA-I levels negatively correlate with hippocampal and whole brain volume as well as mean entorhinal cortical thickness[175]. Compared to age-matched cognitively healthy controls, levels of cerebrospinal fluid (CSF) apoA-I increase during aging but are significantly lower in AD and mild cognitive impairment patients compared to age-matched cognitively normal controls[176]. Additionally, AD patients were found to have significantly lower gene expression of APOA1, APOC3 and APOA4, which correlated with AD severity[177]. However, controversy exists in the relationship between HDL-C levels and cognition, as other studies report no relationship between HDL-C and dementia[178] nor a link between genetically altered HDL-C and AD using Mendelian randomization approaches[179-180]. In a Japanese cohort, the previously reported positive associations of APOA1 polymorphisms and AD[181-182] were not reproduced, but a single nucleotide polymorphism, Rs7659 in apoD, was correlated to EOAD after stratification against APOE4 genotype[183]. The discovery that methylation of an APOA1 CpG site increases protein levels of plasma apoA-I and negatively correlates with episodic memory in an older population again suggests a complex relationship between HDL and cognition[184]. Notably, this study identifies epigenetic regulation of cholesterol metabolism and the impact of environmental and lifestyle influences as new areas of interest in dementia research. As HDL functions can be discordant with HDL-C levels, the usefulness of HDL-C as a parameter of HDL effectiveness in CNS disorders remains to be determined. On the other hand, changes in HDL's functional antioxidant activity, as estimated by reduced activity of serum PON-1, correlates with cognitive decline particularly in mixed AD-VaD dementia[185-186].
Associations of HDL reach beyond AD to include other neurodegenerative diseases. A protective association of HDL has been found in multiple sclerosis in which HDL-C inversely correlates with BBB damage and leukocyte extravasation[187]. Similarly, in Parkinson's Disease, plasma HDL-C levels are reduced especially in the early stage of the disease[188–190]. Intriguingly, HDL-C levels and RCT function are only diminished in cases of AD with cardiovascular co-morbidity when compared to AD without co-cardiovascular morbidities or to age-matched, healthy cognitively normal controls[172].
HDL and dementia: mechanisms and therapeutic potential
Mechanisms by which circulating HDL affects the CNS likely involve the cerebral vasculature, particularly with respect to CAA and inflammation. Studies in experimental models allow such mechanisms to be explored. For example, genetic deletion of apoA-I in AD mice[191] worsens CAA and neuroinflammation and exacerbates cognitive function without an overall change in parenchymal amyloid. In harmony with this finding, transgenic overexpression of human apoA-I from its endogenous promoter that drives expression in liver and intestine in AD mice selectively ameliorates CAA and neuroinflammation and partially restores memory[192]. More recently, intravenous injection of reconstituted human HDL into AD mice was found to acutely reduce soluble amyloid levels in the brain[193], consistent with a previous rodent study in which oral administration of an apoA-I mimetic reduces Aβ burden in the brain[194]. As apoA-I is not synthesized in the brain by glial or neuronal cells yet is present in the CSF at levels similar to that of its brain-derived apoE counterpart[195], and lipid-poor plasma apoA-I gains accesses to the CSF via the choroid plexus in mice[196], an emerging question is whether apoA-I may affect cerebral vessels from the “blood side” or “brain side.” This question has important implications for possible therapeutic opportunities that could involve systemically acting agents that do not necessarily need to cross the BBB. Equally importantly, such therapeutic options could leverage on the considerable investments already made to develop cardiovascular therapies. Both in vitro and in vivo pre-clinical studies report that administration of plasma-isolated human HDL after cerebrovascular insult improves BBB integrity[197] and limits neuroinflammation by inhibiting neutrophil extravasation into the brain[198-199], offering one explanation of the observed preservation of cognition post-stroke with this HDL therapy. Conclusive investigation into whether these improvements in brain microvasculature directly translate into cognitive enhancement or stabilization has yet to be thoroughly explored and warrants further research.
While several epidemiological and animal model studies suggest that statins may protect from dementia, comprehensive analysis of randomized clinical trials have found no beneficial effect when statin use was initiated in late life[200-201]. Studies in the Taiwanese population suggest the time of drug administration, drug dosage and duration are important factors for statins to affect dementia outcome. Lin et al. found that use of statins prior to definite AD diagnosis associates with delayed disease progression in mild-moderate AD patients[202]. Chen et al. observed that dementia risk is decreased by high dose and long-term use of statins, an effect that is not observed with fibrates or other lipid-lowering drugs (acipimox, cholestyramine, niceritrol, nicofuranose, nicomol, and probucol[203]. Conclusively determining whether statins are effective for delaying or treating dementia will require further attention to the timing, dosage and duration of statin use. Fibrates, another class of lipid-lowering agents in clinical use, also fails to show any benefit to prevent cognitive decline in older populations[204] and those at risk for CVD[205]. Whether dementia risk or progression may be affected by cardiovascular therapies that alter circulating lipoprotein levels and functions, including fibrates, niacin, CETP inhibitors and the recently released PCSK9 inhibitors[206], has yet to be systematically tested. Despite the controversial benefit of statins supplemented with niacin on CAD[207], the effect of combination treatments on cognition is still relatively unknown and may prove to be a more potent option to limit dementia risk.
Considerable investment has been made in testing pharmacological agents that affect Liver-X-Receptor (LXR), Retinoid-X-Receptor (RXR) and peroxisome proliferator-activated receptor gamma pathways, as these are master regulators of both lipid metabolism and inflammation[208] and influence key pathogenic pathways in neurodegenerative disease. ABCA1, a downstream gene target of these nuclear receptor pathways, has been consistently and independently shown to protect against AD phenotypes in animals[209–212] and an epidemiological study in Denmark identified a loss of function ABCA1 gene as a risk factor for both AD and cerebrovascular disease[213]. In pre-clinical studies pharmacological activation of LXR/RXR effectively ameliorates AD phenotypes, although with varying changes in cerebral Aβ pathology[214–221]. There is mounting evidence in both AD and experimental stroke models that LXR/RXR agonists mediate improvements in cerebrovascular health via preservation of BBB integrity, which is one potential mechanism by which this drug class may exert neurological and cognitive benefits[222–227]. Efficacy of LXR/RXR agonists in ameliorating AD pathology and memory loss in mice is dependent on ABCA1[228], and although not a direct gene target of the LXRs, CSF levels of apoA-I are increased with an oral regimen of an LXR agonist[229]. Further studies are needed to delineate the contribution of peripheral lipoproteins as mediators of the protective action of LXR agonists against neurodegeneration.
Bexarotene, a USA Food and Drug Administration approved anti-cancer drug and RXR agonist, was reported to rapidly decrease Aβ pathology and significantly ameliorate cognitive decline in AD mice[230]. This study spearheaded several investigations into the potential therapeutic benefit of bexarotene, which is currently being examined in AD clinical trials despite mixed data on its effectiveness in AD animal models[218,231–236]. The 2016 phase 2a clinical trial in which bexarotene was administered over four weeks to early AD patients failed to reduce brain amyloid levels as measured by positron emission tomography[237]. Although post-hoc analysis suggests potential effects in non-APOE4 carriers[237], larger numbers will be needed to decisively determine the efficacy of bexarotene in subjects of each APOE genotype. A case report of improved cognition in an AD patient with no concurrent changes in Aβ neuropathology leaves room for cautious optimism[238]. A major drawback to further development of LXR/RXR agonists is their undesirable side effects, namely hyperlipidemia caused by increased fatty acid synthesis in the liver[208]. New evidence that statins may interfere with ABCA1 expression[70,239] may also necessitate the consideration of alternative cholesterol management plans to traditional statin use.
Gaps and opportunities
Taken together, much remains to be discovered about HDL's role in health and disease. In particular, despite its association with dementia, the mechanisms by which HDL may promote healthy aging and protect from neurodegeneration remain far from clear. Studies investigating the role of HDL in ameliorating cerebral vessel disease could be expanded to include cognitive aspects. Other unanswered questions include what compositional changes of HDL occur throughout neurodegeneration and dementia and how these compare to other chronic inflammatory states such as in metabolic syndrome, T2DM, chronic kidney disease, or CVD. Whether dementia-specific compositional changes of HDL may impair CNS function and potentially lead to new therapeutic targets remains to be determined. Filling these knowledge gaps will improve our understanding of the dynamic nature of HDL for vascular physiology and neurodegeneration.
Contributor Information
Guilaine Boyce, Department of Pathology and Laboratory Medicine, Djavad Mowafaghian Centre for Brain Health, University of British Columbia, Vancouver, BC V6T 1Z3, Canada..
Emily Button, Department of Pathology and Laboratory Medicine, Djavad Mowafaghian Centre for Brain Health, University of British Columbia, Vancouver, BC V6T 1Z3, Canada..
Sonja Soo, Department of Pathology and Laboratory Medicine, Djavad Mowafaghian Centre for Brain Health, University of British Columbia, Vancouver, BC V6T 1Z3, Canada..
Cheryl Wellington, Email: wcheryl@mail.ubc.ca, Department of Pathology and Laboratory Medicine, Djavad Mowafaghian Centre for Brain Health, University of British Columbia, Vancouver, BC V6T 1Z3, Canada..
References
- 1. Rye KA. High density lipoprotein structure, function, and metabolism: a new Thematic Series[J]. J Lipid Res, 2013, 54(8): 2031–2033 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Murphy AJ. High Density Lipoprotein: Assembly, Structure, Cargo, and Functions[J]. ISRN Physiol, 2013; 2013(5): 1–20. [Google Scholar]
- 3. Kontush A, Lhomme M. Lipidomics of plasma high-density lipoprotein: insights into anti-atherogenic function[J]. J Glycomics Lipidomics, 2015, 5: 3–6. [Google Scholar]
- 4. Davidson. HDL Proteome Watch[J]. http://homepages.uc.edu/~davidswm/HDLproteome.html. 2015, accessed 1 July 2016.
- 5. Shah AS, Tan L, Long JL, et al. Proteomic diversity of high density lipoproteins: our emerging understanding of its importance in lipid transport and beyond[J]. J Lipid Res, 2013, 54(10): 2575–2585 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lüscher TF, Landmesser U, von Eckardstein A, et al. High-density lipoprotein: vascular protective effects, dysfunction, and potential as therapeutic target[J]. Circ Res, 2014, 114(1): 171–182 . [DOI] [PubMed] [Google Scholar]
- 7. Riwanto M, Rohrer L, Roschitzki B, et al. Altered activation of endothelial anti- and proapoptotic pathways by high-density lipoprotein from patients with coronary artery disease: role of high-density lipoprotein-proteome remodeling[J]. Circulation, 2013, 127(8): 891–904 . [DOI] [PubMed] [Google Scholar]
- 8. Kannel WB, Dawber TR, Friedman GD, et al. Risk factors in coronary heart disease: an evaluation of several serum lipids as predictors of coronary heart disease: the framingham study[J]. Ann Intern Med, 1964, 61: 888–899 . [DOI] [PubMed] [Google Scholar]
- 9. Gordon T, Castelli WP, Hjortland MC, et al. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study[J]. Am J Med, 1977, 62(5): 707–714 . [DOI] [PubMed] [Google Scholar]
- 10. Gordon DJ, Probstfield JL, Garrison RJ, et al. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies[J]. Circulation, 1989, 79(1): 8–15 . [DOI] [PubMed] [Google Scholar]
- 11. Rasmussen KL, Tybjærg-Hansen A, Nordestgaard BG, et al. Data on plasma levels of apolipoprotein E, correlations with lipids and lipoproteins stratified by APOE genotype, and risk of ischemic heart disease[J]. Data Brief, 2016, 6: 923–932 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Swerdlow DI, Kuchenbaecker KB, Shah S, et al. Selecting instruments for Mendelian randomization in the wake of genome-wide association studies[J]. Int J Epidemiol, 2016, 45(5): 1600–1616 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Holmes MV, Asselbergs FW, Palmer TM, et al. , and the UCLEB consortium. Mendelian randomization of blood lipids for coronary heart disease[J]. Eur Heart J, 2015, 36(9): 539–550 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Voight BF, Peloso GM, Orho-Melander M, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study[J]. Lancet, 2012, 380(9841): 572–580 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weissglas-Volkov D, Pajukanta P. Genetic causes of high and low serum HDL-cholesterol[J]. J Lipid Res, 2010, 51(8): 2032–2057 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sirtori CR, Calabresi L, Franceschini G, et al. Cardiovascular status of carriers of the apolipoprotein A-I(Milano) mutant: the Limone sul Garda study[J]. Circulation, 2001, 103(15): 1949–1954 . [DOI] [PubMed] [Google Scholar]
- 17. Zanoni P, Khetarpal SA, Larach DB, et al. , and the CHD Exome+ Consortium, and the CARDIoGRAM Exome Consortium, and the Global Lipids Genetics Consortium. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease[J]. Science, 2016, 351(6278): 1166–1171 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gordon SM, Li H, Zhu X, et al. A comparison of the mouse and human lipoproteome: suitability of the mouse model for studies of human lipoproteins[J]. J Proteome Res, 2015, 14(6): 2686–2695 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Getz GS, Reardon CA. Animal models of atherosclerosis[J]. Arterioscler Thromb Vasc Biol, 2012, 32(5): 1104–1115 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. von Eckardstein A, Kardassis D. High density lipoproteins: From biological understanding to clinical exploitation[J]. In: Handbook of Experimental Pharmacology. 2015, pp. 593–615. 25523003
- 21. Feig JE, Hewing B, Smith JD, et al. High-density lipoprotein and atherosclerosis regression: evidence from preclinical and clinical studies[J]. Circ Res, 2014, 114(1): 205–213 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Potočnjak I, Degoricija V, Trbušić M, et al. Metrics of High-Density Lipoprotein Function and Hospital Mortality in Acute Heart Failure Patients[J]. PLoS One, 2016, 11(6): e0157507 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Monette JS, Hutchins PM, Ronsein GE, et al. Patients With Coronary Endothelial Dysfunction Have Impaired Cholesterol Efflux Capacity and Reduced HDL Particle Concentration[J]. Circ Res, 2016, 119(1): 83–90 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Norimatsu K, Kuwano T, Miura S, et al. Significance of the percentage of cholesterol efflux capacity and total cholesterol efflux capacity in patients with or without coronary artery disease[J]. Heart Vessels; [ Epub 2016 Apr 22]. [DOI] [PubMed] [Google Scholar]
- 25. Ishikawa T, Ayaori M, Uto-Kondo H, et al. High-density lipoprotein cholesterol efflux capacity as a relevant predictor of atherosclerotic coronary disease[J]. Atherosclerosis, 2015, 242(1): 318–322 . [DOI] [PubMed] [Google Scholar]
- 26. Bounafaa A, Berrougui H, Ikhlef S, et al. Alteration of HDL functionality and PON1 activities in acute coronary syndrome patients[J]. Clin Biochem, 2014, 47(18): 318–325 . [DOI] [PubMed] [Google Scholar]
- 27. Annema W, Dikkers A, de Boer JF, et al. Impaired HDL cholesterol efflux in metabolic syndrome is unrelated to glucose tolerance status: the CODAM study[J]. Sci Rep, 2016, 6: 27367 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vaisar T, Tang C, Babenko I, et al. Inflammatory remodeling of the HDL proteome impairs cholesterol efflux capacity[J]. J Lipid Res, 2015, 56(8): 1519–1530 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Agarwal R, Talwar P, Kushwaha SS, et al. Effect of apolipoprotein E (APO E) polymorphism on leptin in Alzheimer’s disease[J]. Ann Indian Acad Neurol, 2015, 18(3): 320–326 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rohatgi A. High-Density Lipoprotein Function Measurement in Human Studies: Focus on Cholesterol Efflux Capacity[J]. Prog Cardiovasc Dis, 2015, 58(1): 32–40 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cameron SJ, Morrell CN, Bao C, et al. A novel anti-inflammatory effect for high density lipoprotein[J]. PLoS One, 2015, 10(12): e0144372 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fruhwürth S, Krieger S, Winter K, et al. I nhibition of mTOR down-regulates scavenger receptor, class B, type i (SR-BI) expression, reduces endothelial cell migration and impairs nitric oxide production[J]. Biochim Biophys Acta- Mol Cell Biol Lipids 2014; 1841: 944–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pan B, Kong J, Jin J, et al. A novel anti-inflammatory mechanism of high density lipoprotein through up-regulating annexin A1 in vascular endothelial cells[J]. Biochim Biophys Acta 2016; 1861: 501–512. [DOI] [PubMed] [Google Scholar]
- 34. Galvani S, Sanson M, Blaho VA, et al. HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation[J]. Sci Signal, 2015, 8(389): ra79 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kimura T, Tomura H, Mogi C, et al. Role of scavenger receptor class B type I and sphingosine 1-phosphate receptors in high density lipoprotein-induced inhibition of adhesion molecule expression in endothelial cells[J]. J Biol Chem, 2006, 281(49): 37457–37467 . [DOI] [PubMed] [Google Scholar]
- 36. Terasaka N, Yu S, Yvan-Charvet L, et al. ABCG1 and HDL protect against endothelial dysfunction in mice fed a high-cholesterol diet[J]. J Clin Invest, 2008, 118(11): 3701–3713 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Uittenbogaard A, Shaul PW, Yuhanna IS, et al. High density lipoprotein prevents oxidized low density lipoprotein-induced inhibition of endothelial nitric-oxide synthase localization and activation in caveolae[J]. J Biol Chem, 2000, 275(15): 11278–11283 . [DOI] [PubMed] [Google Scholar]
- 38. Mackness MI, Arrol S, Abbott C, et al. Protection of low-density lipoprotein against oxidative modification by high-density lipoprotein associated paraoxonase[J]. Atherosclerosis, 1993, 104(1-2): 129–135 . [DOI] [PubMed] [Google Scholar]
- 39. Garcia-Heredia A, Marsillach J, Rull A, et al. Paraoxonase-1 inhibits oxidized low-density lipoprotein-induced metabolic alterations and apoptosis in endothelial cells: A nondirected metabolomic study[J]. Mediators Inflamm 2013; 2013: 156053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sattler K, Gräler M, Keul P, et al. Defects of High-Density Lipoproteins in Coronary Artery Disease Caused by Low Sphingosine-1-Phosphate Content: Correction by Sphingosine-1-Phosphate-Loading[J]. J Am Coll Cardiol, 2015, 66(13): 1470–1485 . [DOI] [PubMed] [Google Scholar]
- 41. Oberbach A, Adams V, Schlichting N, et al. Proteome profiles of HDL particles of patients with chronic heart failure are associated with immune response and also include bacteria proteins[J]. Clin Chim Acta, 2016, 453: 114–122 . [DOI] [PubMed] [Google Scholar]
- 42. Kaseda R, Jabs K, Hunley TE, et al. Dysfunctional high-density lipoproteins in children with chronic kidney disease[J]. Metabolism, 2015, 64(2): 263–273 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shroff R, Speer T, Colin S, et al. HDL in children with CKD promotes endothelial dysfunction and an abnormal vascular phenotype[J]. J Am Soc Nephrol, 2014, 25(11): 2658–2668 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. O’Neill F, Riwanto M, Charakida M, et al. Structural and functional changes in HDL with low grade and chronic inflammation[J]. Int J Cardiol, 2015, 188: 111–116 . [DOI] [PubMed] [Google Scholar]
- 45. Burillo E, Lindholt JS, Molina-Sánchez P, et al. ApoA-I/HDL-C levels are inversely associated with abdominal aortic aneurysm progression[J]. Thromb Haemost, 2015, 113(6): 1335–1346 . [DOI] [PubMed] [Google Scholar]
- 46. Delbosc S, Diallo D, Dejouvencel T, et al. Impaired high-density lipoprotein anti-oxidant capacity in human abdominal aortic aneurysm[J]. Cardiovasc Res, 2013, 100(2): 307–315 . [DOI] [PubMed] [Google Scholar]
- 47. Sang H, Yao S, Zhang L, et al. Walk-run training improves the anti-inflammation properties of high-density lipoprotein in patients with metabolic syndrome[J]. J Clin Endocrinol Metab, 2015, 100(3): 870–879 . [DOI] [PubMed] [Google Scholar]
- 48. Rosenson RS. The High-Density Lipoprotein Puzzle: Why Classic Epidemiology, Genetic Epidemiology, and Clinical Trials Conflict?[J] Arterioscler Thromb Vasc Biol, 2016, 36(5): 777–782 . [DOI] [PubMed] [Google Scholar]
- 49. Kingwell BA, Chapman MJ, Kontush A, et al. HDL-targeted therapies: progress, failures and future[J]. Nat Rev Drug Discov, 2014, 13(6): 445–464 . [DOI] [PubMed] [Google Scholar]
- 50. Heinecke JW. The not-so-simple HDL story: A new era for quantifying HDL and cardiovascular risk[J]? Nat Med, 2012, 18(9): 1346–1347 . [DOI] [PubMed] [Google Scholar]
- 51. Barter P, Gotto AM, LaRosa JC, et al. , and the Treating to New Targets Investigators. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events[J]. N Engl J Med, 2007, 357(13): 1301–1310 . [DOI] [PubMed] [Google Scholar]
- 52. Ridker PM, Genest J, Boekholdt SM, et al. , and the JUPITER Trial Study Group. HDL cholesterol and residual risk of first cardiovascular events after treatment with potent statin therapy: an analysis from the JUPITER trial[J]. Lancet, 2010, 376(9738): 333–339 . [DOI] [PubMed] [Google Scholar]
- 53. Duggal JK, Singh M, Attri N, et al. Effect of niacin therapy on cardiovascular outcomes in patients with coronary artery disease[J]. J Cardiovasc Pharmacol Ther, 2010, 15(2): 158–166 . [DOI] [PubMed] [Google Scholar]
- 54. Bruckert E, Labreuche J, Amarenco P. Meta-analysis of the effect of nicotinic acid alone or in combination on cardiovascular events and atherosclerosis[J]. Atherosclerosis, 2010, 210(2): 353–361 . [DOI] [PubMed] [Google Scholar]
- 55.] Sorrentino SA, Besler C, Rohrer L, et al. Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy[J]. Circulation, 2010, 121(1): 110–122 . [DOI] [PubMed] [Google Scholar]
- 56. Landray MJ, Haynes R, Hopewell JC, et al. , and the HPS2-THRIVE Collaborative Group. Effects of extended-release niacin with laropiprant in high-risk patients[J]. N Engl J Med, 2014, 371(3): 203–212 . [DOI] [PubMed] [Google Scholar]
- 57. Wheeler AP, Bernard GR, Thompson BT, et al. , and the National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Pulmonary-artery versus central venous catheter to guide treatment of acute lung injury[J]. N Engl J Med, 2006, 354(21): 2213–2224 . [DOI] [PubMed] [Google Scholar]
- 58. Keene D, Price C, Shun-Shin MJ, et al. Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: meta-analysis of randomised controlled trials including 117,411 patients[J]. BMJ, 2014, 349: g4379 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Musunuru K, Orho-Melander M, Caulfield MP, et al. Ion mobility analysis of lipoprotein subfractions identifies three independent axes of cardiovascular risk[J]. Arterioscler Thromb Vasc Biol, 2009, 29(11): 1975–1980 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Phillips CM, Perry IJ. Lipoprotein particle subclass profiles among metabolically healthy and unhealthy obese and non-obese adults: does size matter?[J] Atherosclerosis, 2015, 242(2): 399–406 . [DOI] [PubMed] [Google Scholar]
- 61. Xu RX, Zhang Y, Ye P, et al. Analysis of Lipoprotein Subfractions in Chinese Han Patients with Stable Coronary Artery Disease[J]. Heart Lung Circ, 2015, 24(12): 1203–1210 . [DOI] [PubMed] [Google Scholar]
- 62. Li JJ, Zhang Y, Li S, et al. Large HDL Subfraction But Not HDL-C Is Closely Linked With Risk Factors, Coronary Severity and Outcomes in a Cohort of Nontreated Patients With Stable Coronary Artery Disease: A Prospective Observational Study[J]. Medicine (Baltimore), 2016, 95(4): e2600 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bostan M, Uydu HA, Yildirmiş S, et al. Pleiotropic effects of HDL subfractions and HDL-associated enzymes on protection against coronary artery disease[J]. Acta Cardiol, 2015, 70(3): 333–340 . [DOI] [PubMed] [Google Scholar]
- 64. McGarrah RW, Craig DM, Haynes C, et al. High-density lipoprotein subclass measurements improve mortality risk prediction, discrimination and reclassification in a cardiac catheterization cohort[J]. Atherosclerosis, 2016, 246: 229–235 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tian L, Chen Y, Li C, et al. Statin treatment improves plasma lipid levels but not HDL subclass distribution in patients undergoing percutaneous coronary intervention[J]. Lipids, 2013, 48(2): 127–137 . [DOI] [PubMed] [Google Scholar]
- 66. Berthold HK, Rizzo M, Spenrath N, et al. Effects of lipid-lowering drugs on high-density lipoprotein subclasses in healthy men-a randomized trial[J]. PLoS One, 2014, 9(3): e91565 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Asztalos BF, Le Maulf F, Dallal GE, et al. Comparison of the effects of high doses of rosuvastatin versus atorvastatin on the subpopulations of high-density lipoproteins[J]. Am J Cardiol, 2007, 99(5): 681–685 . [DOI] [PubMed] [Google Scholar]
- 68. Harangi M, Mirdamadi HZ, Seres I, et al. Atorvastatin effect on the distribution of high-density lipoprotein subfractions and human paraoxonase activity[J]. Transl Res, 2009, 153(4): 190–198 . [DOI] [PubMed] [Google Scholar]
- 69. Ronsein GE, Hutchins PM, Isquith D, et al. Niacin Therapy Increases High-Density Lipoprotein Particles and Total Cholesterol Efflux Capacity But Not ABCA1-Specific Cholesterol Efflux in Statin-Treated Subjects[J]. Arterioscler Thromb Vasc Biol, 2016, 36(2): 404–411 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mendivil CO, Furtado J, Morton AM, et al. Novel Pathways of Apolipoprotein A-I Metabolism in High-Density Lipoprotein of Different Sizes in Humans[J]. Arterioscler Thromb Vasc Biol, 2016, 36(1): 156–165 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Niculescu LS, Simionescu N, Sanda GM, et al. MiR-486 and miR-92a identified in circulating HDL discriminate between stable and vulnerable coronary artery disease patients[J]. PLoS One, 2015, 10(10): e0140958 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Trieb M, Horvath A, Birner-Gruenberger R, et al. Liver disease alters high-density lipoprotein composition, metabolism and function[J]. Biochim Biophys Acta 2016; 1861: 630–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.[ Marsillach J, Aragonès G, Mackness B, et al. Decreased paraoxonase-1 activity is associated with alterations of high-density lipoprotein particles in chronic liver impairment[J]. Lipids Health Dis, 2010, 9: 46 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gomaraschi M, Ossoli A, Favari E, et al. Inflammation impairs eNOS activation by HDL in patients with acute coronary syndrome[J]. Cardiovasc Res, 2013, 100(1): 36–43 . [DOI] [PubMed] [Google Scholar]
- 75. Shao B, de Boer I, Tang C, et al. A Cluster of Proteins Implicated in Kidney Disease Is Increased in High-Density Lipoprotein Isolated from Hemodialysis Subjects[J]. J Proteome Res, 2015, 14(7): 2792–2806 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sun JT, Yang K, Lu L, et al. Increased carbamylation level of HDL in end-stage renal disease: carbamylated-HDL attenuated endothelial cell function[J]. Am J Physiol Renal Physiol, 2016, 310(6): F511–F517 . [DOI] [PubMed] [Google Scholar]
- 77. Gómez Rosso L, Lhomme M, Meroño T, et al. Altered lipidome and antioxidative activity of small, dense HDL in normolipidemic rheumatoid arthritis: relevance of inflammation[J]. Atherosclerosis, 2014, 237(2): 652–660 . [DOI] [PubMed] [Google Scholar]
- 78. Holzer M, Trieb M, Konya V, et al. Aging affects high-density lipoprotein composition and function[J]. Biochim Biophys Acta- Mol Cell Biol Lipids 2013; 1831: 1442–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jaouad L, de Guise C, Berrougui H, et al. Age-related decrease in high-density lipoproteins antioxidant activity is due to an alteration in the PON1's free sulfhydryl groups[J]. Atherosclerosis, 2006, 185(1): 191–200 . [DOI] [PubMed] [Google Scholar]
- 80. Pedret A, Catalán Ú, Fernández-Castillejo S, et al. Impact of virgin olive oil and phenol-enriched virgin olive oils on the HDL proteome in hypercholesterolemic subjects: A double blind, randomized, controlled, cross-over clinical trial (VOHF study)[J]. PLoS One, 2015, 10(6): e0129160 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Holzer M, Birner-Gruenberger R, Stojakovic T, et al. Uremia alters HDL composition and function[J]. J Am Soc Nephrol, 2011, 22(9): 1631–1641 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ahnström J, Gottsäter A, Lindblad B, et al. Plasma concentrations of apolipoproteins A-I, B and M in patients with abdominal aortic aneurysms[J]. Clin Biochem, 2010, 43(4-5): 407–410 . [DOI] [PubMed] [Google Scholar]
- 83. Gamberi T, Puglia M, Guidi F, et al. A proteomic approach to identify plasma proteins in patients with abdominal aortic aneurysm[J]. Mol Biosyst, 2011, 7(10): 2855–2862 . [DOI] [PubMed] [Google Scholar]
- 84. Krishnan S, Huang J, Lee H, et al. Combined High-Density Lipoprotein Proteomic and Glycomic Profiles in Patients at Risk for Coronary Artery Disease[J]. J Proteome Res, 2015, 14(12): 5109–5118 . [DOI] [PubMed] [Google Scholar]
- 85. Sánchez-Quesada JL, Vinagre I, De Juan-Franco E, et al. Impact of the LDL subfraction phenotype on Lp-PLA2 distribution, LDL modification and HDL composition in type 2 diabetes[J]. Cardiovasc Diabetol, 2013, 12: 112 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Green PS, Vaisar T, Subramaniam P, et al. Combined Statin and Niacin Therapy Remodels the HDL Proteome[J]. Circulation, 2008, 118(12): 1259–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yetukuri L, Huopaniemi I, Koivuniemi A, et al. High density lipoprotein structural changes and drug response in lipidomic profiles following the long-term fenofibrate therapy in the FIELD substudy[J]. PLoS One, 2011, 6(8): e23589 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gordon SM, McKenzie B, Kemeh G, et al. Rosuvastatin Alters the Proteome of High Density Lipoproteins: Generation of alpha-1-antitrypsin Enriched Particles with Anti-inflammatory Properties[J]. Mol Cell Proteomics, 2015, 14(12): 3247–3257 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kontush A, Lhomme M, Chapman MJ. Unraveling the complexities of the HDL lipidome[J]. J Lipid Res, 2013, 54(11): 2950–2963 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Argraves KM, Sethi AA, Gazzolo PJ, et al. S1P, dihydro-S1P and C24:1-ceramide levels in the HDL-containing fraction of serum inversely correlate with occurrence of ischemic heart disease[J]. Lipids Health Dis, 2011, 10: 70 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tong X, Lv P, Mathew AV, et al. The compensatory enrichment of sphingosine-1- phosphate harbored on glycated high-density lipoprotein restores endothelial protective function in type 2 diabetes mellitus[J]. Cardiovasc Diabetol, 2014, 13: 82 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Vickers KC, Remaley AT. Functional Diversity of HDL Cargo[J]. J Lipid Res 2013; jlr.R035964v2. [Google Scholar]
- 93. Desgagné V, Guay SP, Guérin R, et al. Variations in HDL-carried miR-223 and miR-135a concentrations after consumption of dietary trans fat are associated with changes in blood lipid and inflammatory markers in healthy men- an exploratory study[J]. Epigenetics, 2016, 11(6): 438–448 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tabet F, Cuesta Torres LF, Ong KL, et al. High-Density Lipoprotein-Associated miR-223 Is Altered after Diet-Induced Weight Loss in Overweight and Obese Males[J]. PLoS One, 2016, 11(3): e0151061 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Vickers KC, Palmisano BT, Shoucri BM, et al. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins[J]. Nat Cell Biol, 2011, 13(4): 423–433 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Tabet F, Vickers KC, Cuesta Torres LF, et al. HDL-transferred microRNA-223 regulates ICAM-1 expression in endothelial cells[J]. Nat Commun, 2014, 5: 3292 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hyötyläinen T, Mattila I, Wiedmer SK, et al. Metabolomic analysis of polar metabolites in lipoprotein fractions identifies lipoprotein-specific metabolic profiles and their association with insulin resistance[J]. Mol Biosyst, 2012, 8(10): 2559–2565 . [DOI] [PubMed] [Google Scholar]
- 98. Speer T, Rohrer L, Blyszczuk P, et al. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of Toll-like receptor-2[J]. Immunity, 2013, 38(4): 754–768 . [DOI] [PubMed] [Google Scholar]
- 99. Chadwick AC, Holme RL, Chen Y, et al. Acrolein impairs the cholesterol transport functions of high density lipoproteins[J]. PLoS One, 2015, 10(4): e0123138 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Fonslow BR, Stein BD, Webb KJ, et al. Digestion and depletion of abundant proteins improves proteomic coverage[J]. Nat Methods, 2013, 10(1): 54–56 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Williams LR, Leggett RW. Reference values for resting blood flow to organs of man[J]. Clin Phys Physiol Meas, 1989, 10(3): 187–217 . [DOI] [PubMed] [Google Scholar]
- 102. Attems J, Jellinger KA. The overlap between vascular disease and Alzheimer’s disease--lessons from pathology[J]. BMC Med, 2014, 12: 206 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Iadecola C. The pathobiology of vascular dementia[J]. Neuron, 2013, 80(4): 844–866 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Pendlebury ST, Rothwell PM. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: a systematic review and meta-analysis[J]. Lancet Neurol, 2009, 8(11): 1006–1018 . [DOI] [PubMed] [Google Scholar]
- 105. Consortium M. METACOHORTS for the study of vascular disease and its contribution to cognitive decline and neurodegeneration: An initiative of the Joint Programme for Neurodegenerative Disease Research[J]. Alzheimer’s Dement 2016; pii: S1552: 30064–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. World Health Organization[J]. Fact Sheet: Dementia http://www.who.int/mediacentre/factsheets/fs362/en/ (accessed 1 July 2016).
- 107. De Strooper B, Karran E. The Cellular Phase of Alzheimer’s Disease[J]. Cell, 2016, 164(4): 603–615 . [DOI] [PubMed] [Google Scholar]
- 108. Bird TD. Early-Onset Familial Alzheimer Disease[J]. In: Pagon R, Adam M, Ardinger H, et al. (eds) GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle, 2012. [Google Scholar]
- 109. Mathis CA, Kuller LH, Klunk WE, et al. In vivo assessment of amyloid-β deposition in nondemented very elderly subjects[J]. Ann Neurol, 2013, 73(6): 751–761 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Meng XF, Yu JT, Wang HF, et al. Midlife vascular risk factors and the risk of Alzheimer’s disease: a systematic review and meta-analysis[J]. J Alzheimers Dis, 2014, 42(4): 1295–1310 . [DOI] [PubMed] [Google Scholar]
- 111. Toledo JB, Arnold SE, Raible K, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre[J]. Brain, 2013, 136(Pt 9): 2697–2706 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kalaria RN. Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer’s disease[J]. Acta Neuropathol, 2016, 131(5): 659–685 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Arvanitakis Z, Capuano AW, Leurgans SE, et al. Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: a cross-sectional study. Lancet Neurol, 2016, 15(9): 934–943 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Lee S, Viqar F, Zimmerman ME, et al. , and the Dominantly Inherited Alzheimer Network. White matter hyperintensities are a core feature of Alzheimer’s disease: Evidence from the dominantly inherited Alzheimer network[J]. Ann Neurol, 2016, 79(6): 929–939 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci, 2011, 12(12): 723–738 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum[J]. J Neurol Neurosurg Psychiatry, 2012, 83(2): 124–137 . [DOI] [PubMed] [Google Scholar]
- 117. Yamada M. Cerebral amyloid angiopathy: emerging concepts[J]. J Stroke, 2015, 17(1): 17–30 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Yates PA, Villemagne VL, Ellis KA, et al. Cerebral microbleeds: a review of clinical, genetic, and neuroimaging associations[J]. Front Neurol, 2014, 4: 205 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kövari E, Charidimou A, Herrmann FR, et al. No neuropathological evidence for a direct topographical relation between microbleeds and cerebral amyloid angiopathy[J]. Acta Neuropathol Commun, 2015, 3: 49 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Martinez-Ramirez S, Greenberg SM, Viswanathan A. Cerebral microbleeds: overview and implications in cognitive impairment[J]. Alzheimers Res Ther, 2014, 6(3): 33 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Akoudad S, Wolters FJ, Viswanathan A, et al. Association of Cerebral Microbleeds With Cognitive Decline and Dementia[J]. JAMA Neurol, 2016, 73(8): 934–943 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. van der Vlies AE, Goos JDC, Barkhof F, et al. Microbleeds do not affect rate of cognitive decline in Alzheimer disease[J]. Neurology, 2012, 79(8): 763–769 . [DOI] [PubMed] [Google Scholar]
- 123. Wang N, Allali G, Kesavadas C, et al. Cerebral Small Vessel Disease and Motoric Cognitive Risk Syndrome: Results from the Kerala-Einstein Study[J]. J Alzheimers Dis, 2016, 50(3): 699–707 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. O’Brien JT, Thomas A. Vascular dementia[J]. Lancet, 2015, 386(10004): 1698–1706 . [DOI] [PubMed] [Google Scholar]
- 125. Mathias JL, Burke J. Cognitive functioning in Alzheimer’s and vascular dementia: a meta-analysis[J]. Neuropsychology, 2009, 23(4): 411–423 . [DOI] [PubMed] [Google Scholar]
- 126. Chui HC, Zheng L, Reed BR, et al. Vascular risk factors and Alzheimer’s disease: are these risk factors for plaques and tangles or for concomitant vascular pathology that increases the likelihood of dementia? An evidence-based review[J]. Alzheimers Res Ther, 2012, 4(1): 1 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Jellinger KA. Pathology and pathogenesis of vascular cognitive impairment-a critical update[J]. Front Aging Neurosci, 2013, 5: 17 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Jellinger KA, Attems J. Prevalence of dementia disorders in the oldest-old: an autopsy study[J]. Acta Neuropathol, 2010, 119(4): 421–433 . [DOI] [PubMed] [Google Scholar]
- 129. Zhai Y, Yamashita T, Nakano Y, et al. Chronic Cerebral Hypoperfusion Accelerates Alzheimer’s Disease Pathology with Cerebrovascular Remodeling in a Novel Mouse Model[J]. J Alzheimers Dis, 2016, 53(3): 893–905 . [DOI] [PubMed] [Google Scholar]
- 130. Ly JV, Rowe CC, Villemagne VL, et al. Subacute ischemic stroke is associated with focal 11C PiB positron emission tomography retention but not with global neocortical Aβ deposition[J]. Stroke, 2012, 43(5): 1341–1346 . [DOI] [PubMed] [Google Scholar]
- 131. Sahathevan R, Linden T, Villemagne VL, et al. Positron emission tomographic imaging in stroke: Cross-sectional and follow-up assessment of amyloid in ischemic stroke[J]. Stroke, 2016, 47(1): 113–119 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Kerridge C, Kozlova DI, Nalivaeva NN, et al. Hypoxia affects neprilysin expression through caspase activation and an APP intracellular domain-dependent mechanism[J]. Front Neurosci, 2015, 9: 426 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Dietschy JM. Central nervous system: cholesterol turnover, brain development and neurodegeneration[J]. Biol Chem, 2009, 390(4): 287–293 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Foley P. Lipids in Alzheimer’s disease: A century-old story[J]. Biochimica et Biophysica Acta- Molecular and Cell Biology of Lipids 2010; 1801: 750–753. [DOI] [PubMed] [Google Scholar]
- 135. Jones L, Harold D, Williams J. Genetic evidence for the involvement of lipid metabolism in Alzheimer’s disease[J]. Biochim Biophys Acta 2010; 1801: 754–761. [DOI] [PubMed] [Google Scholar]
- 136. Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families[J]. Science, 1993, 261(5123): 921–923 . [DOI] [PubMed] [Google Scholar]
- 137. Farrer LA, Cupples LA, Haines JL, et al. , and the APOE and Alzheimer Disease Meta Analysis Consortium. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis.[J] JAMA, 1997, 278(16): 1349–1356 . [PubMed] [Google Scholar]
- 138. Liu CC, Kanekiyo T, Xu H, et al. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy[J]. Nat Rev Neurol, 2013, 9(2): 106–118 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Shinohara M, Kanekiyo T, Yang L, et al. APOE2 eases cognitive decline during aging: clinical and preclinical evaluations[J]. Ann Neurol; [Epub 2016 Mar 2]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Rebeck GW, Reiter JS, Strickland DK, et al. Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions[J]. Neuron, 1993, 11(4): 575–580 . [DOI] [PubMed] [Google Scholar]
- 141. Kok E, Haikonen S, Luoto T, et al. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age[J]. Ann Neurol, 2009, 65(6): 650–657 . [DOI] [PubMed] [Google Scholar]
- 142. Hultman K, Strickland S, Norris EH. The APOE 4/4 genotype potentiates vascular fibrin(ogen) deposition in amyloid-laden vessels in the brains of Alzheimer’s disease patients[J]. J Cereb Blood Flow Metab, 2013, 33(8): 1251–1258 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Polvikoski T, Sulkava R, Haltia M, et al. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein[J]. N Engl J Med, 1995, 333(19): 1242–1247 . [DOI] [PubMed] [Google Scholar]
- 144. Schmechel DE, Saunders AM, Strittmatter WJ, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease[J]. Proc Natl Acad Sci U S A, 1993, 90(20): 9649–9653 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Tiraboschi P, Hansen LA, Masliah E, et al. Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease[J]. Neurology, 2004, 62(11): 1977–1983 . [DOI] [PubMed] [Google Scholar]
- 146. Strittmatter WJ, Saunders AM, Schmechel D, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease[J]. Proc Natl Acad Sci U S A, 1993, 90(5): 1977–1981 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Bennet AM, Di Angelantonio E, Ye Z, et al. Association of apolipoprotein E genotypes with lipid levels and coronary risk[J]. JAMA, 2007, 298(11): 1300–1311 . [DOI] [PubMed] [Google Scholar]
- 148. Castellano JM, Kim J, Stewart FR, et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance[J]. Sci Transl Med, 2011, 3(89): 89ra57 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Tai LM, Thomas R, Marottoli FM, et al. The role of APOE in cerebrovascular dysfunction[J]. Acta Neuropathol, 2016, 131(5): 709–723 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Ulrich V, Konaniah ES, Herz J, et al. Genetic variants of ApoE and ApoER2 differentially modulate endothelial function[J]. Proc Natl Acad Sci U S A, 2014, 111(37): 13493–13498 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Bell RD, Winkler EA, Singh I, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A[J]. Nature, 2012, 485(7399): 512–516 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Bangen KJ, Beiser A, Delano-Wood L, et al. APOE genotype modifies the relationship between midlife vascular risk factors and later cognitive decline[J]. J Stroke Cerebrovasc Dis, 2013, 22(8): 1361–1369 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Chai YL, Yeo HK, Wang J, et al. Apolipoprotein 4 is Associated with Dementia and Cognitive Impairment Predominantly Due to Alzheimer’s Disease and Not with Vascular Cognitive Impairment: A Singapore-Based Cohort[J]. J Alzheimers Dis, 2016, 51(4): 1111–1118 . [DOI] [PubMed] [Google Scholar]
- 154. Roheim PS, Carey M, Forte T, et al. Apolipoproteins in human cerebrospinal fluid[J]. Proc Natl Acad Sci U S A, 1979, 76(9): 4646–4649 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease[J]. Nat Genet, 2009, 41(10): 1088–1093 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Lambert JC, Heath S, Even G, et al. , and the European Alzheimer’s Disease Initiative Investigators. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease[J]. Nat Genet, 2009, 41(10): 1094–1099 . [DOI] [PubMed] [Google Scholar]
- 157. Lidström AM, Bogdanovic N, Hesse C, et al. Clusterin (apolipoprotein J) protein levels are increased in hippocampus and in frontal cortex in Alzheimer’s disease[J]. Exp Neurol, 1998, 154(2): 511–521 . [DOI] [PubMed] [Google Scholar]
- 158. Del Valle E, Navarro A, Martínez-Pinilla E, et al. Apo J and Apo D: Complementary or Antagonistic Roles in Alzheimer’s Disease?[J] J Alzheimers Dis, 2016, 53(2): 639–650 . [DOI] [PubMed] [Google Scholar]
- 159. Manousopoulou A, Gatherer M, Smith C, et al. Systems proteomic analysis reveals that Clusterin and Tissue Inhibitor of Metalloproteinases 3 increase in leptomeningeal arteries affected by cerebral amyloid angiopathy[J]. Neuropathol Appl Neurobiol; [ Epub 2016 Oct 5]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Bell RD, Sagare AP, Friedman AE, et al. Transport pathways for clearance of human Alzheimer’s amyloid β-peptide and apolipoproteins E and J in the mouse central nervous system[J]. J Cereb Blood Flow Metab, 2007, 27(5): 909–918 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Miners JS, Clarke P, Love S. Clusterin levels are increased in Alzheimer’s disease and influence the regional distribution of A[J]. Brain Pathol; [Epub 2016 Jun 1]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Merino-Zamorano C, Fernández-de Retana S, Montañola A, et al. Modulation of Amyloid-β1-40 Transport by ApoA1 and ApoJ Across an in vitro Model of the Blood-Brain Barrier[J]. J Alzheimers Dis, 2016, 53(2): 677–691 . [DOI] [PubMed] [Google Scholar]
- 163. Yeh FL, Wang Y, Tom I, et al. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia[J]. Neuron, 2016, 91(2): 328–340 . [DOI] [PubMed] [Google Scholar]
- 164. Beeg M, Stravalaci M, Romeo M, et al. Clusterin binds to A??1–42 Oligomers with high affinity and interferes with peptide aggregation by inhibiting primary and secondary nucleation[J]. J Biol Chem, 2016, 291(13): 6958–6966 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Kang SS, Kurti A, Wojtas A, et al. Identification of plexin A4 as a novel clusterin receptor links two Alzheimer’s disease risk genes[J]. Hum Mol Genet, 2016, 25(16): 3467–3475 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Desikan RS, Thompson WK, Holland D, et al. , and the Alzheimer’s Disease Neuroimaging Initiative Group. The role of clusterin in amyloid-β-associated neurodegeneration[J]. JAMA Neurol, 2014, 71(2): 180–187 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Bates KA, Sohrabi HR, Rainey-Smith SR, et al. Serum high-density lipoprotein is associated with better cognitive function in a cross-sectional study of aging women[J]. Int J Neurosci; [Epub 2016 May 15]. [DOI] [PubMed] [Google Scholar]
- 168. Crichton GE, Elias MF, Davey A, et al. Higher HDL cholesterol is associated with better cognitive function: the Maine-Syracuse study[J]. J Int Neuropsychol Soc, 2014, 20(10): 961–970 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Reed B, Villeneuve S, Mack W, et al. Associations between serum cholesterol levels and cerebral amyloidosis[J]. JAMA Neurol, 2014, 71(2): 195–200 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Taniguchi Y, Shinkai S, Nishi M, et al. Nutritional biomarkers and subsequent cognitive decline among community-dwelling older Japanese: a prospective study[J]. J Gerontol A Biol Sci Med Sci, 2014, 69(10): 1276–1283 . [DOI] [PubMed] [Google Scholar]
- 171. Dias IHK, Polidori MC, Li L, et al. Plasma levels of HDL and carotenoids are lower in dementia patients with vascular comorbidities[J]. J Alzheimers Dis, 2014, 40(2): 399–408 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Shih YH, Tsai KJ, Lee CW, et al. Apolipoprotein C-III is an amyloid-β-binding protein and an early marker for Alzheimer’s disease[J]. J Alzheimers Dis, 2014, 41(3): 855–865 . [DOI] [PubMed] [Google Scholar]
- 173. Choi HJ, Seo EH, Yi D, et al. Amyloid-Independent Amnestic Mild Cognitive Impairment and Serum Apolipoprotein A1 Levels[J]. Am J Geriatr Psychiatry, 2016, 24(2): 144–153 . [DOI] [PubMed] [Google Scholar]
- 174. Hye A, Riddoch-Contreras J, Baird AL, et al. Plasma proteins predict conversion to dementia from prodromal disease[J]. Alzheimers Dement, 2014, 10(6): 799–807.e2 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Yang Y, Keene CD, Peskind ER, et al. Cerebrospinal Fluid Particles in Alzheimer Disease and Parkinson Disease[J]. J Neuropathol Exp Neurol, 2015, 74(7): 672–687 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Lin Q, Cao Y, Gao J. Decreased expression of the APOA1-APOC3-APOA4 gene cluster is associated with risk of Alzheimer’s disease[J]. Drug Des Devel Ther, 2015, 9: 5421–5431 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Batty GD, Russ TC, Starr JM, et al. Modifiable cardiovascular disease risk factors as predictors of dementia death: pooling of ten general population-based cohort studies[J]. J Negat Results Biomed, 2014, 13: 8 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Østergaard SD, Mukherjee S, Sharp SJ, et al. , and the Alzheimer’s Disease Genetics Consortium, and the GERAD1 Consortium, and the EPIC-InterAct Consortium. Associations between Potentially Modifiable Risk Factors and Alzheimer Disease: A Mendelian Randomization Study[J]. PLoS Med, 2015, 12(6): e1001841 , discussion e1001841 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Proitsi P, Lupton MK, Velayudhan L, et al. , and the Alzheimer’s Disease Neuroimaging Initiative, and the GERAD1 Consortium. Genetic predisposition to increased blood cholesterol and triglyceride lipid levels and risk of Alzheimer disease: a Mendelian randomization analysis[J]. PLoS Med, 2014, 11(9): e1001713 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Vollbach H, Heun R, Morris CM, et al. APOA1 polymorphism influences risk for early-onset nonfamiliar AD[J]. Ann Neurol, 2005, 58(3): 436–441 . [DOI] [PubMed] [Google Scholar]
- 181. Song F, Poljak A, Crawford J, et al. Plasma apolipoprotein levels are associated with cognitive status and decline in a community cohort of older individuals[J]. PLoS One, 2012, 7(6): e34078 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Shibata N, Nagata T, Shinagawa S, et al. Genetic association between APOA1 and APOD polymorphisms and Alzheimer’s disease in a Japanese population[J]. J Neural Transm (Vienna), 2013, 120(11): 1599–1603 . [DOI] [PubMed] [Google Scholar]
- 183. Lazarus J, Mather KA, Armstrong NJ, et al. DNA methylation in the apolipoprotein-A1 gene is associated with episodic memory performance in healthy older individuals[J]. J Alzheimers Dis, 2015, 44(1): 175–182 . [DOI] [PubMed] [Google Scholar]
- 184. Cervellati C, Trentini A, Romani A, et al. Serum paraoxonase and arylesterase activities of paraoxonase-1 (PON-1), mild cognitive impairment, and 2-year conversion to dementia: A pilot study[J]. J Neurochem, 2015, 135(2): 395–401 . [DOI] [PubMed] [Google Scholar]
- 185. Castellazzi M, Trentini A, Romani A, et al. Decreased Arylesterase activity of Paraoxonase-1 (PON-1) might be a common denominator of neuroinflammatory and neurodegenerative diseases[J]. Int J Biochem Cell Biol 2016; S1357–2725: 30148–0. [DOI] [PubMed] [Google Scholar]
- 186. Fellows K, Uher T, Browne RW, et al. Protective associations of HDL with blood-brain barrier injury in multiple sclerosis patients[J]. J Lipid Res, 2015, 56(10): 2010–2018 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Swanson CR, Berlyand Y, Xie SX, et al. Plasma apolipoprotein A1 associates with age at onset and motor severity in early Parkinson’s disease patients[J]. Mov Disord, 2015, 30(12): 1648–1656 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Zhang X, Yin X, Yu H, et al. Quantitative proteomic analysis of serum proteins in patients with Parkinson’s disease using an isobaric tag for relative and absolute quantification labeling, two-dimensional liquid chromatography, and tandem mass spectrometry[J]. Analyst, 2012, 137(2): 490–495 . [DOI] [PubMed] [Google Scholar]
- 189. Qiang JK, Wong YC, Siderowf A, et al. Plasma apolipoprotein A1 as a biomarker for Parkinson disease[J]. Ann Neurol, 2013, 74(1): 119–127 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Lefterov I, Fitz NF, Cronican AA, et al. Apolipoprotein A-I deficiency increases cerebral amyloid angiopathy and cognitive deficits in APP/PS1DeltaE9 mice[J]. J Biol Chem, 2010, 285(47): 36945–36957 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Lewis TL, Cao D, Lu H, et al. Overexpression of human apolipoprotein A-I preserves cognitive function and attenuates neuroinflammation and cerebral amyloid angiopathy in a mouse model of Alzheimer disease[J]. J Biol Chem, 2010, 285(47): 36958–36968 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Robert J, Stukas S, Button E, et al. Reconstituted high-density lipoproteins acutely reduce soluble brain A b levels in symptomatic APP / PS1 mice[J]. Biochim Biophys Acta 2016; 1862: 1027–36. [DOI] [PubMed] [Google Scholar]
- 193. Handattu SP, Garber DW, Monroe CE, et al. Oral apolipoprotein A-I mimetic peptide improves cognitive function and reduces amyloid burden in a mouse model of Alzheimer’s disease[J]. Neurobiol Dis, 2009, 34(3): 525–534 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.] Koch S, Donarski N, Goetze K, et al. Characterization of four lipoprotein classes in human cerebrospinal fluid[J]. J Lipid Res, 2001, 42(7): 1143–1151 . [PubMed] [Google Scholar]
- 195. Stukas S, Robert J, Lee M, et al. Intravenously injected human apolipoprotein A-I rapidly enters the central nervous system via the choroid plexus[J]. J Am Heart Assoc, 2014, 3(6): e001156 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Lapergue B, Dang BQ, Desilles JP, et al. High-density lipoprotein-based therapy reduces the hemorrhagic complications associated with tissue plasminogen activator treatment in experimental stroke[J]. Stroke, 2013, 44(3): 699–707 . [DOI] [PubMed] [Google Scholar]
- 197. Lapergue B, Moreno JA, Dang BQ, et al. Protective effect of high-density lipoprotein-based therapy in a model of embolic stroke[J]. Stroke, 2010, 41(7): 1536–1542 . [DOI] [PubMed] [Google Scholar]
- 198. Bao Dang Q, Lapergue B, Tran-Dinh A, et al. High-density lipoproteins limit neutrophil-induced damage to the blood-brain barrier in vitro[J]. J Cereb Blood Flow Metab, 2013, 33(4): 575–582 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. McGuinness B, Craig D, Bullock R, et al. Statins for the prevention of dementia[J]. Cochrane Database Syst Rev, 2016, (1): CD003160 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Power MC, Weuve J, Sharrett AR, et al. Statins, cognition, and dementia—systematic review and methodological commentary[J]. Nat Rev Neurol, 2015, 11(4): 220–229 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Lin FC, Chuang YS, Hsieh HM, et al. Early Statin Use and the Progression of Alzheimer Disease: A Total Population-Based Case-Control Study[J]. Medicine (Baltimore), 2015, 94(47): e2143 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Chen PY, Liu SK, Chen CL, et al. Long-term statin use and dementia risk in Taiwan[J]. J Geriatr Psychiatry Neurol, 2014, 27(3): 165–171 . [DOI] [PubMed] [Google Scholar]
- 203. Ancelin ML, Carrière I, Barberger-Gateau P, et al. Lipid lowering agents, cognitive decline, and dementia: the three-city study[J]. J Alzheimers Dis, 2012, 30(3): 629–637 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Williamson JD, Launer LJ, Bryan RN, et al. , and the Action to Control Cardiovascular Risk in Diabetes Memory in Diabetes Investigators. Cognitive function and brain structure in persons with type 2 diabetes mellitus after intensive lowering of blood pressure and lipid levels: a randomized clinical trial[J]. JAMA Intern Med, 2014, 174(3): 324–333 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Swiger KJ, Martin SS. PCSK9 inhibitors and neurocognitive adverse events: exploring the FDA directive and a proposal for N-of-1 trials[J]. Drug Saf, 2015, 38(6): 519–526 . [DOI] [PubMed] [Google Scholar]
- 206. Wang H, Blumberg JB, Chen CY, et al. Dietary modulators of statin efficacy in cardiovascular disease and cognition[J]. Mol Aspects Med, 2014, 38: 1–53 . [DOI] [PubMed] [Google Scholar]
- 207. Hong C, Tontonoz P. Liver X receptors in lipid metabolism: opportunities for drug discovery[J]. Nat Rev Drug Discov, 2014, 13(6): 433–444 . [DOI] [PubMed] [Google Scholar]
- 208. Hirsch-Reinshagen V, Maia LF, Burgess BL, et al. The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease[J]. J Biol Chem, 2005, 280(52): 43243–43256 . [DOI] [PubMed] [Google Scholar]
- 209. Koldamova R, Staufenbiel M, Lefterov I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice[J]. J Biol Chem, 2005, 280(52): 43224–43235 . [DOI] [PubMed] [Google Scholar]
- 210. Wahrle SE, Jiang H, Parsadanian M, et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease[J]. J Clin Invest, 2008, 118(2): 671–682 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Wahrle SE, Jiang H, Parsadanian M, et al. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE[J]. J Biol Chem, 2004, 279(39): 40987–40993 . [DOI] [PubMed] [Google Scholar]
- 212. Nordestgaard LT, Tybjærg-Hansen A, Nordestgaard BG, et al. Loss-of-function mutation in ABCA1 and risk of Alzheimer’s disease and cerebrovascular disease[J]. Alzheimers Dement, 2015, 11(12): 1430–1438 . [DOI] [PubMed] [Google Scholar]
- 213. Cui X, Chopp M, Zacharek A, et al. The neurorestorative benefit of GW3965 treatment of stroke in mice[J]. Stroke, 2013, 44(1): 153–161 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Fitz NF, Cronican A, Pham T, et al. Liver X receptor agonist treatment ameliorates amyloid pathology and memory deficits caused by high-fat diet in APP23 mice[J]. J Neurosci, 2010, 30(20): 6862–6872 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Jiang Q, Lee CYD, Mandrekar S, et al. ApoE promotes the proteolytic degradation of Abeta[J]. Neuron, 2008, 58(5): 681–693 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Koldamova RP, Lefterov IM, Staufenbiel M, et al. The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer’s disease[J]. J Biol Chem, 2005, 280(6): 4079–4088 . [DOI] [PubMed] [Google Scholar]
- 217. LaClair KD, Manaye KF, Lee DL, et al. Treatment with bexarotene, a compound that increases apolipoprotein-E, provides no cognitive benefit in mutant APP/PS1 mice[J]. Mol Neurodegener, 2013, 8: 18 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Skerrett R, Pellegrino MP, Casali BT, et al. Combined liver X receptor/peroxisome proliferator-activated receptor γ agonist treatment reduces amyloid β levels and improves behavior in amyloid precursor protein/presenilin 1 mice[J]. J Biol Chem, 2015, 290(35): 21591–21602 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Sun Y, Fan J, Zhu Z, et al. Small molecule TBTC as a new selective retinoid X receptor α agonist improves behavioral deficit in Alzheimer’s disease model mice[J]. Eur J Pharmacol, 2015, 762: 202–213 . [DOI] [PubMed] [Google Scholar]
- 220. Vanmierlo T, Rutten K, Dederen J, et al. Liver X receptor activation restores memory in aged AD mice without reducing amyloid[J]. Neurobiol Aging, 2011, 32(7): 1262–1272 . [DOI] [PubMed] [Google Scholar]
- 221. Certo M, Endo Y, Ohta K, et al. Activation of RXR/PPAR γ underlies neuroprotection by bexarotene in ischemic stroke[J]. Pharmacol Res, 2015, 102: 298–307 . [DOI] [PubMed] [Google Scholar]
- 222. Kim E, Woo MS, Qin L, et al. Daidzein Augments Cholesterol Homeostasis via ApoE to Promote Functional Recovery in Chronic Stroke[J]. J Neurosci, 2015, 35(45): 15113–15126 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. ElAli A, Hermann DM. Liver X receptor activation enhances blood-brain barrier integrity in the ischemic brain and increases the abundance of ATP-binding cassette transporters ABCB1 and ABCC1 on brain capillary cells[J]. Brain Pathol, 2012, 22(2): 175–187 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Sironi L, Mitro N, Cimino M, et al. Treatment with LXR agonists after focal cerebral ischemia prevents brain damage[J]. FEBS Lett, 2008, 582(23-24): 3396–3400 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Morales JR, Ballesteros I, Deniz JM, et al. Activation of liver X receptors promotes neuroprotection and reduces brain inflammation in experimental stroke[J]. Circulation, 2008, 118(14): 1450–1459 . [DOI] [PubMed] [Google Scholar]
- 226. Sandoval-Hernández AG, Restrepo A, Cardona-Gómez GP, et al. LXR activation protects hippocampal microvasculature in very old triple transgenic mouse model of Alzheimer’s disease[J]. Neurosci Lett, 2016, 621: 15–21 . [DOI] [PubMed] [Google Scholar]
- 227. Donkin JJ, Stukas S, Hirsch-Reinshagen V, et al. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice[J]. J Biol Chem, 2010, 285(44): 34144–34154 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Stukas S, May S, Wilkinson A, et al. The LXR agonist GW3965 increases apoA-I protein levels in the central nervous system independent of ABCA1. Biochim Biophys Acta- Mol Cell Biol Lipids 2012; 1821: 536–546. [DOI] [PubMed] [Google Scholar]
- 229. Cramer PE, Cirrito JR, Wesson DW, et al. ApoE-Directed Therapeutics Rapidly Clear b-Amyloid and Reverse Deficits in AD Mouse Models[J]. Science (80-) 2012; 335: 1503–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. O’Hare E, Jeggo R, Kim EM, et al. Lack of support for bexarotene as a treatment for Alzheimer’s disease[J]. Neuropharmacology, 2016, 100: 124–130 . [DOI] [PubMed] [Google Scholar]
- 231. Bomben V, Holth J, Reed J, et al. Bexarotene reduces network excitability in models of Alzheimer’s disease and epilepsy[J]. Neurobiol Aging, 2014, 35(9): 2091–2095 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Savage JC, Jay T, Goduni E, et al. Nuclear receptors license phagocytosis by trem2+ myeloid cells in mouse models of Alzheimer’s disease[J]. J Neurosci, 2015, 35(16): 6532–6543 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Kuntz M, Candela P, Saint-Pol J, et al. Bexarotene Promotes Cholesterol Efflux and Restricts Apical-to-Basolateral Transport of Amyloid-β Peptides in an In Vitro Model of the Human Blood-Brain Barrier[J]. J Alzheimers Dis, 2015, 48(3): 849–862 . [DOI] [PubMed] [Google Scholar]
- 234. Schweinzer C, Kober A, Lang I, et al. Processing of endogenous AβPP in blood-brain barrier endothelial cells is modulated by liver-X receptor agonists and altered cellular cholesterol homeostasis[J]. J Alzheimers Dis, 2011, 27(2): 341–360 . [DOI] [PubMed] [Google Scholar]
- 235. Wang S, Wen P, Wood S. Effect of LXR/RXR agonism on brain and CSF Aβ40 levels in rats[J]. F1000Res, 2016, 5: 138 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Cummings JL, Zhong K, Kinney JW, et al. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene Xin moderate Alzheimer’s disease[J]. Alzheimers Res Ther, 2016, 8: 4 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Pierrot N, Lhommel R, Quenon L, et al. Targretin improves cognitive and biological markers in a patient with Alzheimer’s disease[J]. J Alzheimers Dis, 2016, 49(2): 271–276 . [DOI] [PubMed] [Google Scholar]
- 238. Niesor EJ, Schwartz GG, Perez A, et al. Statin-induced decrease in ATP-binding cassette transporter A1 expression via microRNA33 induction may counteract cholesterol efflux to high-density lipoprotein[J]. Cardiovasc Drugs Ther, 2015, 29(1): 7–14 . [DOI] [PubMed] [Google Scholar]
- 239. Rached F, Lhomme M, Camont L, et al. Defective functionality of small, dense HDL3 subpopulations in ST segment elevation myocardial infarction: Relevance of enrichment in lysophosphatidylcholine, phosphatidic acid and serum amyloid A[J]. Biochim Biophys Acta- Mol Cell Biol Lipids 2015; 1851: 1254–1261. [DOI] [PubMed] [Google Scholar]
- 240. Papathanasiou A, Kostara C, Cung MT, et al. Analysis of the composition of plasma lipoproteins in patients with extensive coronary heart disease using 1H NMR spectroscopy[J]. Hellenic J Cardiol, 2008, 49(2): 72–78 . [PubMed] [Google Scholar]
- 241. Ozerova IN, Perova NV, Shchel’tsyna NV, et al. Parameters of high-density lipoproteins in patients with arterial hypertension in combination with other components of metabolic syndrome[J]. Bull Exp Biol Med, 2007, 143(3): 320–322 . [DOI] [PubMed] [Google Scholar]
- 242. Ross DJ, Hough G, Hama S, et al. Proinflammatory high-density lipoprotein results from oxidized lipid mediators in the pathogenesis of both idiopathic and associated types of pulmonary arterial hypertension[J]. Pulm Circ, 2015, 5(4): 640–648 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Pruzanski W, Stefanski E, de Beer FC, et al. Comparative analysis of lipid composition of normal and acute-phase high density lipoproteins[J]. J Lipid Res, 2000, 41(7): 1035–1047 . [PubMed] [Google Scholar]
- 244. Honda H, Hirano T, Ueda M, et al. High-Density Lipoprotein Subfractions and Their Oxidized Subfraction Particles in Patients with Chronic Kidney Disease[J]. J Atheroscler Thromb, 2016, 23(1): 81–94 . [DOI] [PubMed] [Google Scholar]
- 245. Berrougui H, Isabelle M, Cloutier M, et al. Age-related impairment of HDL-mediated cholesterol efflux[J]. J Lipid Res, 2007, 48(2): 328–336 . [DOI] [PubMed] [Google Scholar]
- 246. Parra S, Castro A, Masana L. The pleiotropic role of HDL in autoimmune diseases[J]. Clin Investig Arterioscler, 2015, 27(2): 97–106 . [DOI] [PubMed] [Google Scholar]
- 247. McMahon M, Grossman J, FitzGerald J, et al. Proinflammatory high-density lipoprotein as a biomarker for atherosclerosis in patients with systemic lupus erythematosus and rheumatoid arthritis[J]. Arthritis Rheum, 2006, 54(8): 2541–2549 . [DOI] [PubMed] [Google Scholar]
- 248. Perségol L, Vergès B, Gambert P, et al. Inability of HDL from abdominally obese subjects to counteract the inhibitory effect of oxidized LDL on vasorelaxation[J]. J Lipid Res, 2007, 48(6): 1396–1401 . [DOI] [PubMed] [Google Scholar]
- 249. Khalil A, Berrougui H, Pawelec G, et al. Impairment of the ABCA1 and SR-BI-mediated cholesterol efflux pathways and HDL anti-inflammatory activity in Alzheimer’s disease[J]. Mech Ageing Dev, 2012, 133(1): 20–29 . [DOI] [PubMed] [Google Scholar]
- 250. Pertl L, Kern S, Weger M, et al. High-density lipoprotein function in exudative age-related macular degeneration[J]. PLoS One, 2016, 11: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]