


Abstract
Contact domains are closely linked to gene regulation and lineage commitment, while current understanding of contact domains and their boundaries is still limited. Here, we present a novel method HiCDB, which is constructively based on local relative insulation metric and multi-scale aggregation approach to detect contact domain boundaries (CDBs) on Hi-C maps. Compared with other ‘state-of-art’ methods, HiCDB shows improved sensitivity and specificity in determining CDBs at various Hi-C resolutions. The superiority of HiCDB enabled us to study the epigenetic features of detected CDBs and showed enrichment of architectural proteins and cell-type-specific transcription factor binding sites at CDBs. The further comparison of GM12878 and IMR90 Hi-C datasets suggested that cell-type-specific CDBs are marked by active regulatory signals and correlate with activation of nearby cell identity genes.
INTRODUCTION
Chromatin organization and its functions in both gene regulation and cell identity have drawn great attention in cell biology researches. Recent developments in sequencing and imaging technologies have led to unprecedented progresses toward understanding chromatin organization (1–5). One of the most striking features of chromatin configuration is the squares with enhanced contact frequencies tiling the diagonal of chromatin interaction matrixes observed in Hi-C data (6–9). These squares were originally observed in the 40-kb resolution Hi-C maps and referred as topologically associating domains (TADs) by Dixon et al. (7). With increased sequencing depth, Rao et al. showed that there are contact domains within the megabase-sized chromatin domains (8). Phillips-Cremins et al. elucidated that cell-type-specific chromatin organization occurs at this sub-megabase scale by looking into the chromosome conformation around six key developmentally regulated genes based on chromosome conformation capture carbon copy (5C) data (10). These cell-type-specific contact domains were also reported in regulation of HoxA genes in limbs development (11). It has also been demonstrated that changes of contact domains are accompanied by alternations in histone modifications and long-term contact pattern (8,12). However, few studies have compared the contact domain boundaries (CDBs) across cell types systemically or uncovered the association between CDBs and genome-wide histone modifications as well as transcription. Herein, sensitive and robust CDB detection methods are of great demand to reveal the function of the CDBs. In particular, deep-sequencing data are preferred for detecting more CDBs, which require the CDB detection methods to be computationally efficient in processing high-resolution Hi-C data.
Several computational methods have been proposed to detect chromatin domains or their boundaries on Hi-C maps (7,8,13–23). These methods can be categorized into 1D statistic-based methods and 2D contact matrix-based methods. The 1D statistic-based methods, such as directionality index (DI), Insulation score and TopDom, calculated a 1D statistic for each bin by averaging interaction frequencies in sliding windows on the original contact matrix (7,15,16). In the DI method, first, a metric called DI was proposed to define the direction preference of each bin in contact with 2 Mb upstream and 2 Mb downstream; then, a hidden Markov model was used to determine the domain boundaries by identifying interaction transitions from the upstream to the downstream (7). The Insulation score method assigned an insulation score to each bin by aggregating interactions of nearby regions. The local minimums of the insulation profile were identified as TAD boundaries (15). As a modification of Insulation score, the TopDom method fitted a piecewise linear function to the insulation profile and conducted a statistical test to reduce false positives (16). On the contrary, the 2D contact matrix-based methods utilized global information of the contact matrix instead of the local information captured by 1D statistic. Armatus quantified the domain quality by a scoring function and identified consistent domain pattern across several resolutions (14). HiCseg formulated the TAD detection problem into a 2D segmentation problem and computed the segmentation via the maximum likelihood, which has a high computational complexity (13). IC-Finder performed hierarchical clustering on the whole Hi-C map to partition the genome into a hierarchical organization, leading to results affected by long-term interaction patterns (20). The Arrowhead method transformed the original contact matrix into an arrowhead-shaped matrix that exaggerated the original edges of the domains and then identified hierarchical domains by heuristically searching for the arrowhead corner pattern (8). The DI, Insulation score and TopDom methods were initially designed to detect TAD boundaries on relatively low-resolution Hi-C data. It has been suggested that they could be applied to detect smaller scale contact domains by tuning parameters such as the size of the insulation or DI windows (24). However, their performances in detecting smaller scale CDBs have not been tested on high-resolution data.
In general, most of the methods were troubled by heuristic tuning parameters and were not tested on data of different sequencing depths. Meanwhile, the lack of robust tools restricts systematic detection of differential CDBs across cell types and sequential functional analysis. Hence, we introduce HiCDB, a local insulation metric based approach that was designed to fill the gaps with improved abilities in (i) accurate CDB detection, (ii) less parameter tuning, (iii) handling Hi-C data with different resolutions, (iv) sufficient robustness to facilitate differential CDB detection, (v) lower computational cost and (vi) a convenient visualization interface.
In addition to overwhelming performance improvement of HiCDB, we were able to further investigate several biological questions tightly related to CDBs, namely: what are the functional features of CDBs? How do these CDBs vary across different cell types? What is the function of differential CDBs? By systematically relating CDBs to several kinds of epigenetic and transcriptional data, the results yielded many insights into these questions.
MATERIALS AND METHODS
Overview of the HiCDB method
The rationale behind HiCDB is that CDBs are local peaks with high insulation strength. To measure the insulation strength, a metric called the local relative insulation (LRI), which converts 2D Hi-C maps into a 1D vector, was proposed in HiCDB. HiCDB goes through the following steps to identify the CDBs (Figure 1A). First, to avoid massive parameter tuning, HiCDB calculates the relative insulation 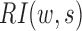 under different window sizes instead of using only one specific window size parameter, which occurs in other methods. Mathematically, for each genomic locus
under different window sizes instead of using only one specific window size parameter, which occurs in other methods. Mathematically, for each genomic locus 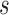 between bins
between bins 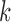 and
and 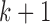 , given the window size
, given the window size 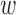 ,
, 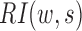 is defined as follows:
is defined as follows:
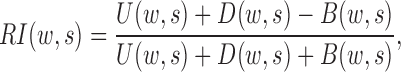 |
where U, D and B are the total interaction frequencies of the upstream, downstream and intermediate regions, respectively (Figure 1A, see the upper right quadrant):
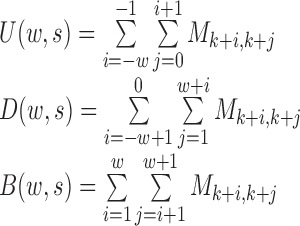 |
Figure 1.

Overview of the HiCDB method. (A) HiCDB method workflow. This region is extracted from the 10-kb GM12878 Hi-C matrix. The detected CDBs are shown as blue dots in the heatmap. The upper right quadrant shows how to calculate the relative insulation in detail. The panels under the Hi-C contact map show the process of how to calculate the RI and LRI. (B) Schematic representation of the difference between AI, RI and LRI. CDB1 represents a CDB with high AI and high LRI. CDB2 represents a CDB with low AI but high LRI in a highly connected region. (C) A summary of the HiCDB analysis pipeline. This flowchart presents the main features and the sequential operations of HiCDB software. The yellow squares are input data, and green squares represent optional outputs. Curves with arrowheads represent optional steps.
Compared with the self-association of the upstream or downstream regions, the rarer the intermediate interactions (insulation) are, the larger 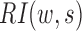 will be. Then, HiCDB averages the RI under different window sizes to facilitate robust detection of the CDBs:
will be. Then, HiCDB averages the RI under different window sizes to facilitate robust detection of the CDBs:
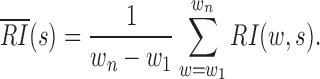 |
The average RI makes the original domain borders more pronounced (Figure 1A). Thus, the local maximum peaks of the average RI are detected as the candidate CDBs by the MATLAB built-in function findpeaks. A genomic locus is declared to be a local peak if its average RI value is greater than or unilaterally equal to its left and right neighbors. With the minimum peak distance d specified, HiCDB will ignore small peaks that occur within d bins of a larger peak. At last, HiCDB calculates the LRI by subtracting the local background of the average RI to further enhance robust CDB signals:
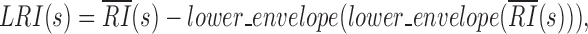 |
where lower_envelope is the local minimum peak envelope of a signal determined with linear interpolation over the local minima. The LRI threshold can be manually adjusted to obtain the final CDB outputs from the candidate CDBs or kept as the default HiCDB cutoff option based on CCCTC-binding factor (CTCF) motif enrichment.
The LRI metric combines both the self-association and the insulation properties of contact domains to detect CDBs, while the previous methods utilized only a single property. For instance, Insulation score and TopDom measured the absolute insulation (AI) of domain boundaries without referring to the local backgrounds, which had the tendency to underestimate the insulation strength of the CDBs in active regions (15,16). In these models, for each genomic locus 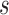 , the average AI was calculated by averaging only the between interactions (B):
, the average AI was calculated by averaging only the between interactions (B):
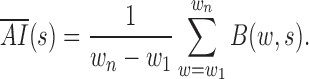 |
A schematic representation of the differences between the AI and LRI is shown in Figure 1B. CDB1 represents a CDB with high AI and high LRI, whereas CDB2 represents a CDB with low AI but high LRI in a densely self-associated contact domain. The LRI metric is preferred over the AI, in terms of making the insulation strength metric comparable across the genome and differentiating more CDBs from noise, including the CDBs within regions of overall higher contact frequencies.
In summary, HiCDB tries to improve its sensitivity in CDB detection by considering both self-association and insulation property of contact domains, as well as its specificity by applying multi-scale aggregation and background removal.
HiCDB cutoff option
Domain boundaries were reported to be occupied by the architectural protein CTCF (8,25,26). Two recent studies have further revealed that the degradation of endogenous CTCF or cohesin eliminates domain insulation and chromatin loops (27,28). Therefore, HiCDB also provides a biologically meaningful CDB cutoff option that takes the CTCF motif enrichment into consideration based on a method adapted from gene set enrichment analysis (29,30). When the HiCDB cutoff option is declared, HiCDB will first rank the candidate CDBs according to their LRI. Then, an enrichment score 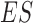 is calculated by going through the list, which reflects the CTCF motif enrichment at the top of the candidate CDB list.
is calculated by going through the list, which reflects the CTCF motif enrichment at the top of the candidate CDB list. 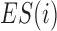 is defined as follows:
is defined as follows:
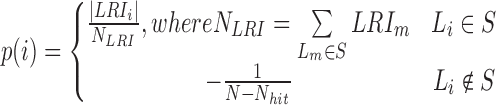 |
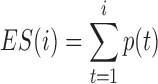 |
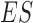 is a running-sum statistic, which increases when it encounters a peak with the CTCF motif and decreases otherwise.
is a running-sum statistic, which increases when it encounters a peak with the CTCF motif and decreases otherwise. 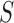 represents the set of candidate CDBs with the CTCF motif.
represents the set of candidate CDBs with the CTCF motif. 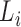 denotes the
denotes the 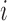 th candidate CDBs.
th candidate CDBs. 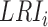 represents the LRI of the
represents the LRI of the 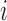 th candidate CDBs.
th candidate CDBs. 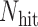 is the number of candidate CDBs in
is the number of candidate CDBs in 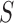 , while
, while 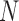 is the total number of candidate CDBs. The LRI at the maximum
is the total number of candidate CDBs. The LRI at the maximum 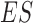 is chosen as the CDB detection cutoff since the candidate CDBs are less reliable after the maximum
is chosen as the CDB detection cutoff since the candidate CDBs are less reliable after the maximum 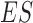 (Supplementary Figure S1). The candidate CDBs with a higher LRI than the cutoff are all kept in the final results including those without CTCF motif. This cutoff option keeps a balance between the CDB detection number and the CTCF enrichment, but it does not bias the CTCF enrichment on the HiCDB-detected CDBs (as seen in Supplementary Figure S3). The whole genome CTCF motif loci are obtained by using HOMER motif analysis with the CTCF PWM matrix from the JASPAR database (31,32).
(Supplementary Figure S1). The candidate CDBs with a higher LRI than the cutoff are all kept in the final results including those without CTCF motif. This cutoff option keeps a balance between the CDB detection number and the CTCF enrichment, but it does not bias the CTCF enrichment on the HiCDB-detected CDBs (as seen in Supplementary Figure S3). The whole genome CTCF motif loci are obtained by using HOMER motif analysis with the CTCF PWM matrix from the JASPAR database (31,32).
Differential CDB detection option
To identify differential boundaries on Hi-C data with replicates, we calculate the CDBs on the merged raw Hi-C matrix of each condition and pool the resulting CDBs together first. The CDBs within one bin of each other are merged. Then, HiCDB calculates the average RIs across the replicates for each genomic bin after the in-sample, library size and between-replicate normalizations of Hi-C maps. KR normalization is used on each replicates to correct the in-sample bias (33). Then a size factor for correcting the library size difference is multiplied to each of the replicates, which is defined as the average matrix sum of all replicates divided by the matrix sum of each of the Hi-C replicates. MA normalization, derived from the MA-plot for genomic data, is further applied to correct the system bias between the replicates of the same condition as previously defined (34). To control the false positives, only the CDBs detected in one condition are tested for significantly differential or not with their average RIs across samples. A CDB is considered to be differential if the difference of its average RI values between two conditions is above the 90% quantile of the all CDB average RI differences, or its average RI values are significantly different between conditions (P-value <0.05, t test) and the difference is above the 50% quantile of the all CDBs. For Hi-C datasets without replicates, HiCDB detects the CDBs on the library size-normalized matrix of each condition and determines the differential CDBs by intersection.
HiCDB as a CDB analysis pipeline
In practical applications, we implemented HiCDB as a CDB analysis pipeline with additional features (Figure 1C). First, KR normalization is performed on the raw Hi-C matrix in either a dense or sparse format. Then, the CDBs are detected on the KR normalized Hi-C map by applying HiCDB. The CDBs pre-calculated for 21 cell types with Hi-C data generated from Schmitt et al. (35) and their consistency across cell types are packaged in HiCDB, which can serve as a reference to annotate the detected CDBs in new samples. In addition, HiCDB provides the differential CDB detections for Hi-C data with or without replicates. At last, HiCDB implements the visualizations for single Hi-C map and comparison between two Hi-C maps with annotated CDBs.
Data sources
The raw matrixes of medium resolution (40-kb) Hi-C data used to compare the CDB detection methods were obtained from http://chromosome.sdsc.edu/mouse/hi-c/download.html (7). The higher resolution (10-kb) Hi-C datasets and Hi-C loops detected by HiCCUPS were obtained from NCBI, with accession number GSE63525 (8). For IMR90, Hi-C matrixes of two replicates were not available, thus the calculated results of Juicer were used (36). The Hi-C matrixes of 21 human cell lines and primary tissues were obtained from NCBI, with accession number GSE87112 (35). CTCF and RNA polymerase II (POLR2A) ChIA-PET data of GM12878 cell line were downloaded from NCBI with accession number GSE72816 (26). All the ChIP-seq and RNA-seq data were downloaded from the ENCODE database (37). Differential genes were recognized by DESeq2 with adjusted P-value < 0.01 and log2-fold change >1 (38).
RESULTS
Comparison between HiCDB and existing methods in detecting CDBs
We compared the performance of methods in detecting CDBs using both medium resolution (40-kb) and higher resolution (10-kb) original Hi-C data. Several quantitative standards were measured, including the CDB number, consistency, protein binding enrichment, robustness and time complexity. HiCDB was compared with Armatus, DI, HiCseg, IC-Finder, Insulation and TopDom on the 40-kb datasets. On the 10-kb dataset, DI and HiCseg were excluded from the comparison for their high-computational time complexity, and Arrowhead was included in the comparison because it was designed for loop-resolution Hi-C experiments and called a significantly smaller number of domain boundaries than other methods in 40-kb datasets (36,39). For each of these methods except Arrowhead and IC-Finder, we manually fine-tuned the parameters following the instructions to optimize the detection of CDBs (see Supplementary Table S1).
First, method consistency was analyzed to reflect the accuracy of CDB detection (Figure 2A and B). HiCDB detected 5768 CDBs, which was the highest in actual counts on the 40-kb IMR90 dataset, with 76% of them reported by the other methods. On the 10-kb dataset, Arrowhead-identified CDBs yielded the highest consistency ratio of 86%, while HiCDB got a comparable consistency ratio of 85%. Even though Armatus and IC-Finder identified the most CDBs on the 40- and 10-kb datasets, respectively, their consistency ratios and numbers were lower than those of HiCDB.
Figure 2.

Comparison between HiCDB and existing methods in detecting CDBs. (A and B) Consistency between different methods. The purple bar represents the total number of CDBs detected by each method. The blue bar represents the CDB number confirmed by any other methods with the proportion above it. The consistency is calculated permitting a one bin error in the 10-kb data. (C and D) Aggregation of peak numbers per 40 kb/10 kb at the CDBs identified by different methods in the 40- and 10-kb datasets. (E and F) Aggregation of peak numbers per 40 kb/10 kb at the CDBs uniquely predicted by different methods. (G) Reproducibility of different methods. The CDB reproducibility was calculated under different cutoffs if a method had a ranked CDB output, otherwise, the methods were shown as a single dot. The reproducibility is calculated by dividing the overlapping CDB number by the average CDB number detected on two Hi-C replicates. The reproducibility is calculated permitting a one bin error in the 10-kb data. (H) Average run time for different chromosomes (computer configuration: CPU 24 2.6 GHz).
2.6 GHz).
CTCF and cohesin enrichments were further used to compare the different methods because they are widely accepted characteristics of domain boundaries and are frequently used to compare domain detection methods (14,16,17,19,40). POLR2A binding at the CDBs was also considered, as the results showed that active transcription correlates with CDB formation. The proportions of the HiCDB-detected CDBs that overlapped with the CTCF, cohesin and POLR2A-binding sites were all the highest among the methods on both datasets (Supplementary Figure S2). Notably, the CTCF binding percentages of the HiCDB-detected CDBs were always the highest for different cutoffs (Supplementary Figure S3). This superiority could be achieved over a wide configuration range of the parameters (Supplementary Figure S4).
Furthermore, we inspected the distributions of architectural proteins or histone modification signals at the CDBs via aggregation plot (Figure 2C and D). Both the architectural proteins and active transcription signals concentrated more at the center of the HiCDB-detected CDBs especially on the 10-kb dataset, whereas the other methods possessed a wider enrichment region, indicating that HiCDB might detect the exact functional loci.
Apart from the high consistency between the detected CDBs of HiCDB and those of the other methods, the CDBs uniquely detected by HiCDB showed a clear insulation border (Supplementary Figure S5) and the strongest enrichment of the architectural and regulatory signals on both Hi-C datasets (Figure 2E and F). Lower enrichments of the architectural and regulatory signals coinciding with a fuzzy insulation border were observed at the flanking regions of the uniquely predicted CDBs of TopDom, IC-Finder and DI on the 40-kb IMR90 dataset, which means these CDBs were not predicted precisely at their exact loci. Moreover, only HiCDB uniquely predicted CDBs exhibited clear insulation and highly enriched architectural and regulatory signals on the 10-kb GM12878 dataset. Similar results were also found for the 40-kb hESC and 10-kb IMR90 Hi-C datasets (Supplementary Figure S6).
Meanwhile, HiCDB is quite robust and fast. Reproducibility with replicated datasets is important for evaluating robustness. All methods were applied on the replicates of 40-kb hESC and 10-kb GM12878 Hi-C datasets to obtain their reproducibility ratios. HiCDB outperformed the other methods in terms of the reproducibility ratio under different cutoffs on both resolution (Figure 2G). In addition, the time complexity of HiCDB is O(n), where n is the row/column number of the Hi-C contact matrix. HiCDB took ∼2 min to compute the whole genome CDBs, which was two and a half times faster than the second fastest method Insulation score when analyzing 40-kb data. It took ∼10 min to analyze the 10-kb data, making it two times faster than Arrowhead and Insulation score (Figure 2H).
HiCDB can identify smaller-scale CDBs accurately
Next, we inspected the CDB distance distributions of different methods (Figure 3A). Armatus tended to detect many small regions clustering together on both datasets, as also seen in Figure 3C. The mean HiCDB-detected CDB distances was 505 kb, the smallest among all the methods except for Armatus on the 40-kb dataset. With the 10-kb data, HiCDB, Arrowhead, TopDom and IC-Finder found comparable CDB distances of ∼200 kb. Notably, the CDB distance distributions of Arrowhead and TopDom had two peaks, which means that a fraction of the CDBs detected by these two methods located closely to each other, as also seen in Figure 3C.
Figure 3.

HiCDB can identify smaller scale CDBs accurately. (A) CDB distance distributions of different methods. (B) Performance of different methods in detecting CDBs on the 40-kb IMR90 Hi-C maps using the CDBs detected by at least two methods on the deep-sequencing IMR90 sample as the gold standard. (C) Method comparison at a representative region (chr21: 32.30–34.30 Mb) in the deep-sequencing GM12878 sample.
In addition to the aforementioned evidence that HiCDB might have the best performance in detecting CDBs, we took the CDBs detected by more than two methods in the 10-kb IMR90 Hi-C matrix as the ‘truth’ to assess the specificity and sensitivity of the different methods quantitatively at the 40-kb resolution (Figure 3B). The rationale is that the CDBs detected based on the 10-kb contact matrix are more accurate and complete than those based on the 40-kb matrix due to the higher signal-to-noise ratio in deep-sequencing data. HiCDB outperformed the other methods tested with the highest sensitivity (34.1%) and specificity (69.0%). This again proves that HiCDB can detect smaller scale CDBs more accurately than other methods on 40-kb datasets. The next best performing method was TopDom, with 26.7% sensitivity and 67.5% specificity, followed by IC-Finder. The remaining methods, DI, Insulation score and HiCseg, were initially designed for TAD boundary detection in low-resolution Hi-C data, which caused their relatively low sensitivities.
Due to the lack of an appropriate reference to evaluate the performance for the 10-kb dataset, we examined the CDBs detected by different methods in light of other independent epigenetic annotations. A representative two-megabase region of the GM12878 genome (chr21: 32.30–34.30 Mb) is shown (Figure 3C), containing 15, 13, 9, 7, 7 and 6 CDBs detected by HiCDB, Arrowhead, Armatus, IC-Finder, TopDom and Insulation score, respectively. HiCDB detected five more CDBs besides the accurate identification of the major structures in this region, namely, B1-B5. These uniquely detected CDBs all showed high LRI under the intensely self-associated domains like CDB2 mentioned in the ‘Materials and Methods’ section. B1, B2 and B3 were located closely to CTCF-mediated loop anchors, whereas B4 and B5 appeared to be the boundary of the POLR2A-mediated loop clusters covered by active histone markers. In addition, Hi-C loops detected in this region tended to be the strong CTCF-mediated loops and failed to predict the loops with anchors like B1-B7 in the intense self-associated domains. In general, HiCDB can detect smaller scale CDBs accurately from Hi-C data under different resolutions. These detected CDBs are accurately located around the well-known architectural protein (such as CTCF and cohesin)-binding sites and well-documented active regulatory signals. Further, the reproducibility ratio of HiCDB also outperforms the other methods tested, facilitating detection of both the consistent and differential CDBs.
Epigenetic features of CDBs and their relation to both Hi-C loops and ChIA-PET loops
In this section, we verified the functional relations between the predicted CDBs and multiple epigenetic annotations, such as Hi-C loops, ChIA-PET loops and the binding sites of extensive transcription factors (TFs). All the analyses were performed on the GM12878 cell line.
First, the predicted CDBs were overlapped with the chromHMM annotation to reveal their relationship to the chromatin states (41). In the 40-kb dataset, the CDBs were significantly were enriched with insulators (2.11-fold) and promoters (1.75-fold). Meanwhile, the CDBs detected in the 10-kb dataset were enriched with active promoters (5.86-fold), insulators (3.36-fold) and enhancers (3.23-fold) (Supplementary Table S2).
Then, we compared the CDBs with the Hi-C loops defined by HiCCUPs, another structural feature observed in Hi-C maps. Whereas Rao et al. found that 39% of the contact domains were demarcated by the Hi-C loops (8), we also found that 56% of our CDBs were consistent with the Hi-C loop anchors (Supplementary Figure S7). Based on this observation, we wondered what those CDBs that did not overlap with Hi-C loops were. Results showed that the vast majority (88%) of the specific Hi-C loop anchors and 57% of the specific CDBs were bounded by CTCF loop anchors, while 25% of the specific CDBs were bounded by POLR2A loop anchors independent of CTCF (Supplementary Figure S7). These analyses suggest that the CDBs and the Hi-C loops might represent distinct fractions of the looping events on Hi-C maps, where the CDBs reflect more POLR2A-mediated loops. As an example, the CDBs detected on a four-megabase region (chr21: 42,50–46,50 M) of GM12878 are presented with multiple annotations in Figure 4A. It is shown that the 40-kb CDBs and Hi-C loops mainly overlapped CTCF ChIA-PET anchors, while the 10-kb CDBs also sketched the anchors of POLR2A interaction clusters. There were 11 CDBs that overlapped with POLR2A loop anchors not predicted as Hi-C loops in this region.
Figure 4.

Epigenetic features of CDBs. (A) The snapshot of chr21: 42,50–46,50M in GM12878. HiCDB-detected CDBs in the 40-kb and 10-kb GM12878 datasets are both illustrated in the snapshot. The 10-kb CDBs that did not overlap with the Hi-C loop anchors are marked in red. (B) TF enrichments at the CDBs of GM12878 detected at both resolutions. The x-axis shows the percentages of CDBs overlapping with certain TF-binding sites. The y-axis shows the TF-binding site enrichment at CDBs compared with random regions.
In addition, we wondered whether other proteins would be associated with the CDBs apart from CTCF and POLR2A. Consequently, the CDBs were compared with 229 ChIP-seq datasets of TFs and histone modifications from ENCODE database (37). Besides architectural proteins, such as CTCF and cohesin, the binding sites of ZNF143, YY1, TRIM22 and additional TFs, such as IKZF1, RUNX3 and BHLHE40, occupied a large percentage of the predicted CDBs (Figure 4B and Supplementary Table S3). The binding sites of TFs, such as RXRA, IRF3, MYC and BRCA1, occupied a relatively low fraction of the predicted CDBs. However, these TFs were enriched by more than 2- and 6-fold at the CDBs than expected under the 40- and 10-kb resolutions, respectively. It is noteworthy that the enrichment folds of many cell-type-specific TFs at the CDBs were higher than those at Hi-C loop anchors (Supplementary Figure S8). Impressively, TRIM22 ChIP-positive sites were found in half of the CDBs in the 10-kb data and 61% of the CDBs in the 40-kb data, comparable with the structural regulator cohesin and YY1 (42). Interestingly, TRIM22 was also reported to be one of the most informative proteins for predicting human chromosome structures (43). TRIM22 is probably a structural regulator and its functions require further investigation.
In summary, the enrichment of CTCF independent loops and cell-type-specific TFs on CDBs strongly suggests that the CDBs are not only structurally but also functionally related to transcription regulation.
Cell-type-specific CDBs correlate with cell-type-specific histone modification and gene activation
In this section, we described how epigenetic information and transcription patterns might be associated with the differential CDBs. We applied HiCDB with the differential CDB detection option to the 10-kb GM12878 and IMR90 datasets and predicted the GM12878-specific CDBs and IMR90-specific CDBs. The Hi-C aggregation heat maps confirmed the evident gain or loss of insulation at the differential CDBs (Figure 5A).
Figure 5.

Comparison of GM12878 and IMR90 from the CDB view. (A) Aggregation of Hi-C maps centered on the differential CDBs. Hi-C maps show a gain of insulation at the cell-type-specific CDBs. (B) Cell-type-specific CDBs enriched in cell-type-specific active regulatory signals. (C) The fold change distributions of the differential expressed genes near differential CDBs. P value is calculated using Wilcoxon rank-sum test. (D) A differential region (chr9:36.50–37.50 Mb) between GM12878 and IMR90 detected by HiCDB. This region possesses a B cell important regulator, PAX5, which is not expressed in IMR90. E1-E3 marks three potential enhancers of PAX5 detected by HiCDB other than HiCCUPs. The IMR90 POLR2A- and CTCF-mediated loops are not shown because the ChIA-PET data are not currently available, while the Hi-C loops are not shown because no Hi-C loops were detected on the IMR90 Hi-C map in this region.
First, we investigated how regulatory elements were enriched at the differential CDBs by aggregating POLR2A, H3K4me3, H3K27ac and H3K27me3 ChIP-seq signals at the CDBs in GM12878 and IMR90 (Figure 5B). In GM12878, the active regulatory signals, especially enhancer marker H3K27ac, were found to be more enriched on the GM12878-specifc CDBs than on the IMR90-specifc CDBs. In contrast, H3K27me3, as a repressive histone marker, was depleted at the GM12878-specifc CDBs but more enriched at the IMR90-specifc CDBs (44). Corresponding results were also observed in IMR90, which indicates that cell-type-specific CDBs correlate with cell-type-specific histone modifications.
To examine the functions of these differential CDBs, we performed a gene ontology (GO) terms analysis of genes located within 500 kb of these CDBs (45). The results showed that the genes nearest to the GM12878-specific CDBs were strongly associated with B cell-related biological processes, such as regulation of B-cell activation and the interferon-γ-mediated signaling pathway, while the genes nearest to the IMR90-specific CDBs were strongly associated with lung development (Supplementary Figure S9). The Genes around IMR90-specific CDBs tended to be upregulated in IMR90, while genes around GM12878-specific CDBs tended to be activated in GM12878 and downregulated in IMR90 (Figure 5C). These results indicate that the emergence of CDBs are frequently associated with the activation of nearby cell-type-specific genes.
PAX5 is an important regulator of B-cell differentiation, which encodes the B-cell lineage-specific activator protein in early development (46,47). PAX5 hypermutation in diffuse large-cell lymphomas as well as PAX5 overexpression associated with the translocation t(9;14) suggest that PAX5 may act as a dominant oncogene in tumorigenesis. HiCDB found extensive CDB changes in the PAX5-nearby region (chr9:36.50–37.50 Mb) in GM12878 compared with IMR90 (Figure 5D). Most of the CDBs detected in this region exhibited enhanced active regulatory signals. In addition to the promoter (P) and long-range enhancer of PAX5 discovered by the Hi-C loop, three other enhancers (E1-E3) overlapped the CDBs detected in GM12878. However, P, E1 and E3 were not detectable in IMR90, and PAX5 is not expressed. Other six examples of the cell-type-specific CDBs supported by multiple epigenetic signals further confirmed that cell-type-specific CDBs are marked by cell-type-specific active histone modifications and are associated with the upregulation of nearby cell identity genes (Supplementary Figure S10).
In addition, we have also applied our method to 40-kb Hi-C data from 21 human cell lines and primary tissues (35). A total 37 518 bins were detected as CDBs in at least one cell type and 6615 bins were predicted as CDBs in each cell type on average. Hierarchical clustering analysis of the CDB average RI value revealed similarities among several cell types (Figure 6). The results showed that tissues from the same organ clustered together, for example, the cortex and hippocampus from the brain as well as the aorta, left ventricle and right ventricle from the heart. The cluster of human embryonic stem (ES) cells and other four human ES-cell-derived lineages in this paper was comparable with the clustering results of the A/B compartments in Dixon et al. (48).
Figure 6.

Estimated interrelationship for 21 human cell lines and primary tissues based on relative insulation of all identified CDBs. The hierarchical clustering is calculated from the pairwise similarity measured by Spearman’s correlation of average RI values.
DISCUSSION
Tremendous efforts have been made to understand the chromatin conformation in development and disease with Hi-C technology over the last decade. However, limited sequencing depth and computational tools restrict most of the studies at TAD and compartment level, which cannot reveal exact regulatory elements or enhancer–promoter loops. Researches of Hi-C data at kilobase resolution focused more on Hi-C loops to reveal the enhancer–promoter loops and treated contact domains as units to study their relations with histone modification (8). Less attention has been paid to the smaller scale CDBs and their relation to regulatory elements or enhancer–promoter loops.
In this study, we developed an efficient CDB detection method named HiCDB. It can predict more CDBs specifically and robustly from Hi-C maps under multiscale resolutions. These CDBs are mainly CTCF- or POLR2A-mediated loop anchors and enrich both architectural proteins and cell-type-specific TFs, which are different from Hi-C loops bounded mainly by architectural proteins, such as CTCF. We suppose that the protein binding difference between CDBs and Hi-C loops is caused by the different natures of loops mediated by CTCF and POLR2A. Hi-C loops are the local peaks on Hi-C maps, representing the stable loops in bulk cells, which thus enrich CTCF-mediated architectural loops. However, POLR2A-mediated loops are not as stable as CTCF-mediated loops and are generally smaller in size. Some of the POLR2A-mediated loops are not presented as local peaks on Hi-C maps but can be detected as CDBs with limited resolution.
Our results also show that the CDB emergences are not deterministic but are correlated with the upregulation of nearby genes. Whereas TAD boundaries are enriched with housekeeping genes (7), the CDBs are relatively more dynamic. The differential CDBs tend to locate closer to actively transcribed cell identity genes and enrich cell-type-specific active histone modifications. This finding suggests that such CDBs may serve as epigenetic feature representing cell-type-specific local chromatin structure. While our paper was in preparation, a separate group reported an ultra-high-resolution Hi-C map of mouse neural differentiation and independently confirmed that the local insulation correlates to active transcription by inspecting the insulation changes at gene promoters (49).
Although HiCDB can detect small scale CDBs accurately and give insights into their potential function, there are still some limitations. The functional epigenetic annotations for the detected CDBs are limited. It is possible to detect hierarchical domains based on HiCDB. However, even if we had detected all the hierarchical domains, we would still not be able to reveal the underlying enhancer–promoter relations because of the so-called ‘extrusion domain’. ‘Extrusion domain’ forms as a by-product of the nearby looping events instead of the interactions between its two anchors, as demonstrated by the loop extrusion model and confirmed by the wild-type genome (50).
At last, there are probably other finer and more sophisticated structures in deep-sequencing Hi-C data that need to be further explored. For example, we noted that many regions showing an LRI lower than the CDB cutoff are also associated with active histone modifications. As more Hi-C datasets become available (51,52), we believe that HiCDB could help gain more insight into 3D chromatin organization and its functional impacts.
DATA AVAILABILITY
This HiCDB method and its related CDB analysis pipeline were implemented as both MATLAB scripts and R package that are available at https://github.com/ChenFengling/HiCDB and https://github.com/ChenFengling/RHiCDB, respectively.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge Bo Yuan of Tsinghua University for the help on R coding. We thank for Minping Qian of Peking University and Zhaofeng Ye, Xianglin Zhang of Tsinghua University for helpful discussion and suggestion.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31871343, 31671384, 91729301]; National Key Research and Development Program of China [2017YFA0505503, 2018YFB0704304]; Cross-discipline Foundation of Tsinghua University [20161080043]. Funding for open access charge: National Natural Science Foundation of China [31871343, 31671384, 91729301]; National Key Research and Development Program of China [2017YFA0505503, 2018YFB0704304]; Cross-discipline Foundation of Tsinghua University [20161080043].
Conflict of interest statement. None declared.
REFERENCES
- 1. Gorkin D.U., Leung D., Ren B.. The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell. 2014; 14:762–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bonev B., Cavalli G.. Organization and function of the 3D genome. Nat. Rev. Genet. 2016; 17:661–678. [DOI] [PubMed] [Google Scholar]
- 3. Krijger P.H.L., De Laat W.. Regulation of disease-associated gene expression in the 3D genome. Nat. Rev. Mol. Cell Bio. 2016; 17:771–782. [DOI] [PubMed] [Google Scholar]
- 4. Dekker J., Mirny L.. The 3D genome as moderator of chromosomal communication. Cell. 2016; 164:1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Davies J.O., Oudelaar A.M., Higgs D.R., Hughes J.R.. How best to identify chromosomal interactions: a comparison of approaches. Nat. Methods. 2017; 14:125–134. [DOI] [PubMed] [Google Scholar]
- 6. Lieberman-Aiden E., Van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O.. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009; 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B.. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S.. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014; 159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dixon J.R., Gorkin D.U., Ren B.. Chromatin domains: the unit of chromosome organization. Mol. Cell. 2016; 62:668–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Phillips-Cremins J.E., Sauria M.E., Sanyal A., Gerasimova T.I., Lajoie B.R., Bell J.S., Ong C.-T., Hookway T.A., Guo C., Sun Y.. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013; 153:1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Berlivet S., Paquette D., Dumouchel A., Langlais D., Dostie J., Kmita M.. Clustering of tissue-specific sub-TADs accompanies the regulation of HoxA genes in developing limbs. PLoS Genet. 2013; 9:e1004018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wijchers P.J., Krijger P.H., Geeven G., Zhu Y., Denker A., Verstegen M.J., Valdes-Quezada C., Vermeulen C., Janssen M., Teunissen H.. Cause and consequence of tethering a subTAD to different nuclear compartments. Mol. Cell. 2016; 61:461–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lévy-Leduc C., Delattre M., Mary-Huard T., Robin S.. Two-dimensional segmentation for analyzing Hi-C data. Bioinformatics. 2014; 30:i386–i392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Filippova D., Patro R., Duggal G., Kingsford C.. Identification of alternative topological domains in chromatin. Algorithms Mol. Biol. 2014; 9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Crane E., Bian Q., McCord R.P., Lajoie B.R., Wheeler B.S., Ralston E.J., Uzawa S., Dekker J., Meyer B.J.. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature. 2015; 523:240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shin H., Shi Y., Dai C., Tjong H., Gong K., Alber F., Zhou X.J.. TopDom: An efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res. 2015; 44:e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen J., Hero III A.O., Rajapakse I.. Spectral identification of topological domains. Bioinformatics. 2016; 32:2151–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weinreb C., Raphael B.J.. Identification of hierarchical chromatin domains. Bioinformatics. 2015; 32:1601–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yan K.-K., Lou S., Gerstein M.. MrTADFinder: A network modularity based approach to identify topologically associating domains in multiple resolutions. PLoS Comput. Biol. 2017; 13:e1005647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Haddad N., Vaillant C., Jost D.. IC-Finder: inferring robustly the hierarchical organization of chromatin folding. Nucleic Acids Res. 2017; 45:e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang X.-T., Cui W., Peng C.. HiTAD: detecting the structural and functional hierarchies of topologically associating domains from chromatin interactions. Nucleic Acids Res. 2017; 45:e163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu W., He B., Tan K.. Identifying topologically associating domains and subdomains by Gaussian Mixture model And Proportion test. Nat. Commun. 2017; 8:535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhan Y., Mariani L., Barozzi I., Schulz E.G., Blüthgen N., Stadler M., Tiana G., Giorgetti L.. Reciprocal insulation analysis of Hi-C data shows that TADs represent a functionally but not structurally privileged scale in the hierarchical folding of chromosomes. Genome Res. 2017; 27:479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schmitt A.D., Hu M., Ren B.. Genome-wide mapping and analysis of chromosome architecture. Nat. Rev. Mol. Cell Biol. 2016; 17:743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rudan M.V., Barrington C., Henderson S., Ernst C., Odom D.T., Tanay A., Hadjur S.. Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep. 2015; 10:1297–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang Z., Luo O.J., Li X., Zheng M., Zhu J.J., Szalaj P., Trzaskoma P., Magalska A., Wlodarczyk J., Ruszczycki B.. CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell. 2015; 163:1611–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nora E.P., Goloborodko A., Valton A.-L., Gibcus J.H., Uebersohn A., Abdennur N., Dekker J., Mirny L.A., Bruneau B.G.. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell. 2017; 169:930–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rao S.S., Huang S.-C., St Hilaire B.G., Engreitz J.M., Perez E.M., Kieffer-Kwon K.-R., Sanborn A.L., Johnstone S.E., Bascom G.D., Bochkov I.D.. Cohesin loss eliminates all loop domains. Cell. 2017; 171:305–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Clark N.R., Ma’ayan A.. Introduction to statistical methods for analyzing large data sets: gene-set enrichment analysis. Sci. Signal. 2011; 4:tr4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S.. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bryne J.C., Valen E., Tang M.-H.E., Marstrand T., Winther O., da Piedade I., Krogh A., Lenhard B., Sandelin A.. JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic Acids Res. 2007; 36:D102–D106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kalhor R., Tjong H., Jayathilaka N., Alber F., Chen L.. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat. Biotechnol. 2012; 30:90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Djekidel M.N., Chen Y., Zhang M.Q.. FIND: differential chromatin interactions detection using a spatial Poisson process. Genome Res. 2018; 28:412–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schmitt A.D., Hu M., Jung I., Xu Z., Qiu Y., Tan C.L., Li Y., Lin S., Lin Y., Barr C.L.. A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep. 2016; 17:2042–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Durand N.C., Shamim M.S., Machol I., Rao S.S., Huntley M.H., Lander E.S., Aiden E.L.. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Sys. 2016; 3:95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Neph S., Vierstra J., Stergachis A.B., Reynolds A.P., Haugen E., Vernot B., Thurman R.E., John S., Sandstrom R., Johnson A.K.. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012; 489:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Forcato M., Nicoletti C., Pal K., Livi C.M., Ferrari F., Bicciato S.. Comparison of computational methods for Hi-C data analysis. Nat. Methods. 2017; 14:679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dali R., Blanchette M.. A critical assessment of topologically associating domain prediction tools. Nucleic Acids Res. 2017; 45:2994–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ernst J., Kellis M.. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods. 2012; 9:215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Weintraub A.S., Li C.H., Zamudio A.V., Sigova A.A., Hannett N.M., Day D.S., Abraham B.J., Cohen M.A., Nabet B., Buckley D.L.. YY1 is a structural regulator of enhancer-promoter loops. Cell. 2017; 171:1573–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Di Pierro M., Cheng R.R., Aiden E.L., Wolynes P.G., Onuchic J.N.. De novo prediction of human chromosome structures: Epigenetic marking patterns encode genome architecture. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:12126–12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen T., Dent S.Y.. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat. Rev. Genet. 2014; 15:93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G.. GREAT improves functional interpretation of cis-regulatory regions. Nature Biotechnol. 2010; 28:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cobaleda C., Schebesta A., Delogu A., Busslinger M.. Pax5: The guardian of B cell identity and function. Nat. Immunol. 2007; 8:463–470. [DOI] [PubMed] [Google Scholar]
- 47. Medvedovic J., Ebert A., Tagoh H., Busslinger M.. Advances in immunology. Elsevier. 2011; 111:179–206. [DOI] [PubMed] [Google Scholar]
- 48. Dixon J.R., Jung I., Selvaraj S., Shen Y., Antosiewicz-Bourget J.E., Lee A.Y., Ye Z., Kim A., Rajagopal N., Xie W.. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015; 518:331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bonev B., Cohen N.M., Szabo Q., Fritsch L., Papadopoulos G.L., Lubling Y., Xu X., Lv X., Hugnot J.-P., Tanay A.. Multiscale 3D genome rewiring during mouse neural development. Cell. 2017; 171:557–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sanborn A.L., Rao S.S., Huang S.-C., Durand N.C., Huntley M.H., Jewett A.I., Bochkov I.D., Chinnappan D., Cutkosky A., Li J.. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E6456–E6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lin D., Hong P., Zhang S., Xu W., Jamal M., Yan K., Lei Y., Li L., Ruan Y., Fu Z.F.. Digestion-ligation-only Hi-C is an efficient and cost-effective method for chromosome conformation capture. Nat. Genet. 2018; 50:754–763. [DOI] [PubMed] [Google Scholar]
- 52. Liang Z., Li G., Wang Z., Djekidel M.N., Li Y., Qian M.-P., Zhang M.Q., Chen Y.. BL-Hi-C is an efficient and sensitive approach for capturing structural and regulatory chromatin interactions. Nat. Commun. 2017; 8:1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This HiCDB method and its related CDB analysis pipeline were implemented as both MATLAB scripts and R package that are available at https://github.com/ChenFengling/HiCDB and https://github.com/ChenFengling/RHiCDB, respectively.