


Abstract
The RNA exosome is a conserved multi-subunit complex essential for processing and degradation of several types of RNAs. Although many of the functions of the RNA exosome are well established, whether the activity of this complex is regulated remains unclear. Here we performed a proteomic analysis of the RNA exosome complex purified from Schizosaccharomyces pombe and identified 39 post-translational modifications (PTMs), including phosphorylation, methylation, and acetylation sites. Interestingly, most of the modifications were identified in Dis3, a catalytic subunit of the RNA exosome, as well as in the exosome-associated RNA helicase, Mtr4. Functional analysis of selected PTM sites using modification-deficient and -mimetic versions of exosome subunits revealed substitutions that affected cell growth and exosome functions. Notably, our results suggest that site-specific phosphorylation in the catalytic center of Dis3 and in the helical bundle domain of Mtr4 control their activity. Our findings support a view in which post-translational modifications fine-tune exosome activity and add a layer of regulation to RNA degradation.
INTRODUCTION
Gene expression is a complex process that demands accurate transcription initiation, precise splicing, efficient nuclear export, error-free translation, and timely RNA decay. Rather than being independent, it has become evident that many of the steps underlying gene expression are intimately connected (1). Mechanisms involved in the timely regulation of gene expression thus usually modulate the equilibrium between RNA synthesis and degradation. Accordingly, the accumulation of properly processed mRNAs as well as many classes of noncoding RNAs (ncRNAs) is controlled by nuclear and cytosolic RNA quality control machineries that monitor various steps of gene expression (2).
A central player involved in eukaryotic RNA surveillance is the exosome complex of 3′–5′ exonucleases. Essential for cellular viability, the RNA exosome complex is conserved in most eukaryotic species, including yeast, flies, plants, and mammals (3–6). A primitive version of the eukaryotic exosome is found in bacteria and archaebacteria, and promotes RNA degradation through a combination of hydrolytic and phosphorolytic nucleases (7). Interestingly, whereas yeast and mammalian exosomes appear to have lost the phosphorolytic activity, a combination of hydrolytic and phosphorolytic activities contribute to RNA degradation by the Arabidopsis exosome (8). The core of the eukaryotic exosome complex adopts a barrel-like structure consisting of two stacked rings with a prominent central channel that is wide enough to accommodate single-stranded RNA (9). The bottom ring is composed of six RNase PH-like proteins (Rrp41, Rrp42, Rrp43, Rrp45, Rrp46 and Mtr3), while three S1/KH RNA-binding proteins (Rrp4, Rrp40 and Csl4) form the top ring, which is often referred as the exosome cap structure (10). Two additional subunits provide the catalytic activity of the eukaryotic exosome: Rrp6 exhibits distributive 3′-5′ exonucleolytic activity and is attached to the cap structure, whereas Dis3 is a processive 3′-5′ exoribonuclease that is anchored to the bottom PH-like ring (11). In addition, Dis3 has a non-essential endoribonuclease active site in its N-terminal PIN domain (12,13), but it remains unclear what substrates are targeted by this activity in vivo. Although Rrp6 and Dis3 have independent catalytic activities, structural data suggest functional linkage between both nucleases. Accordingly, when an RNA substrate is engaged in the exosome central channel path to Dis3, Rrp6 is unable to bind another substrate (14). In addition, Rrp6 appears to contribute to efficient elimination of polyadenylated substrates by Dis3 (15,16).
Although the RNA exosome has been investigated in a wide range of organisms, it has been most extensively studied in the model organism Saccharomyces cerevisiae. In this organism, the 10-subunit exosome (core exosome with Dis3) is found in both the nucleus and cytoplasm, whereas the 11-subunit complex, which includes the exosome-associated exonuclease Rrp6, is restricted to the nucleus (17). One of the first roles ascribed to the RNA exosome was in 3′ end processing of ribosomal RNA (rRNA) precursors during ribosome biogenesis in budding yeast (6). Since then, several transcriptome-wide analyses have led to the conclusion that the RNA exosome is required for 3′ end trimming and/or complete degradation of an array of protein-coding and ncRNAs (18). In the fission yeast Schizosaccharomyces pombe, the RNA exosome has been shown to contribute to timely meiotic differentiation by targeting meiosis-specific transcripts for polyadenylation-dependent RNA decay during the mitotic cell cycle (19,20). In humans, the RNA exosome is important for antibody diversification in B cells by promoting the recruitment of the activation-induced cytidine deaminase (AID) complex to sites of transcriptional arrest/pausing, such as Ig loci (21,22). Intriguingly, congenital mutations in human exosome subunits that affect exosome activity are linked to cognitive impairment and neurodegenerative disorders (23,24). However, the molecular basis of how mutations in exosome subunits cause these tissue-specific clinical features remains poorly understood.
To assist the RNA exosome in this broad range of functions, several protein complexes associate with the exosome to facilitate substrate recognition. One of the best characterized exosome cofactor is the Trf4/5-Air1/2-Mtr4 polyadenylation (TRAMP) complex. By adding short oligo(A) tails, TRAMP provides an unstructured extension to the 3′ end of RNAs, which facilitates processing or degradation by the RNA exosome (25). The Mtr4 RNA helicase also contributes to exosome-mediated RNA processing independently of the TRAMP complex. Accordingly, structural studies of the budding yeast exosome complex indicate that the cofactor protein Mpp6 transiently recruits Mtr4 to the exosome where the helicase activity of Mtr4 is required for rRNA maturation (26–28). Notably, S. pombe also expresses an Mtr4-like helicase (Mtl1) in addition to Mtr4. Fission yeast Mtl1 physically associates with the zinc finger protein Red1 to form the Mtl1-Red1 core (MTREC)/nuclear RNA silencing (NURS) complex, which is involved in targeting meiotic differentiation transcripts, cryptic unstable RNAs, and unspliced pre-mRNAs to the nuclear exosome (29–31). In humans, Mtr4 is present in at least two independent exosome cofactor complexes in addition to TRAMP, the nuclear exosome targeting complex (NEXT) and the poly(A) tail exosome targeting complex (PAXT), which use different RNA features for targeting substrates to the exosome (32,33).
The RNA exosome thus exploits a number of different adaptor complexes to target a broad range of transcripts for either maturation or complete degradation. Yet, the underlying basis for the recruitment of one cofactor complex versus another still remains poorly understood. Furthermore, little is known about mechanisms that control the activity of the RNA exosome. We approached these questions with the view that post-translational modifications (PTMs) could potentially contribute to exosome function and regulation. For instance, it was shown that a cold stress induces SUMOylation of human Rrp6 (EXOSC10), correlating with reduced EXOSC10 accumulation and defective ribosome biogenesis (34). Several exosome subunits have also been found to contain phosphorylation sites in S. cerevisiae (35). However, the functional relevance of these modifications on exosome function was not investigated. In this study, we used mass spectrometry to disclose a catalog of phosphorylation, methylation, and acetylation sites on subunits of the S. pombe RNA exosome and its conserved cofactor, Mtr4. We show that the presence of negative charges that effectively mimic the phosphorylated state of specific residues in Dis3 and Mtr4 can inhibit their activity. Our findings support that site-specific phosphorylation of core exosome subunits can control the function of the RNA exosome.
MATERIALS AND METHODS
Yeast strains and DNA constructs
A list of all S. pombe strains and DNA constructs used in this study is provided in Supplementary Table 1. Cells were grown at 30°C in yeast extract medium with supplements (YES) or Edinburgh minimum medium (EMM) containing appropriate supplements. Conditional strains in which the genomic copy of dis3 and mtr4 is expressed from the thiamine-repressible nmt promoter (Pnmt) were generated as described previously (36). Pnmt-dependent expression was repressed by the addition of 60 μM thiamine to the growth medium for 12–15 h. Gene disruptions and C-terminal tagging were performed by PCR-mediated gene targeting (37), using the lithium acetate method for cell transformation. Conditional or knockout strains were complemented with either wild type or mutant alleles of dis3, mtr4 and rrp6 that were expressed from their endogenous promoter using the ade6 integration vector (38).
TAP purification and preparation for LC–MS/MS
Yeast were grown in yeast extract media supplemented with appropriate amino acids (YES) at 30°C until OD600 = 1.0 and then pelleted. Cells were resuspended in lysis buffer (50 mM HEPES–KOH, pH 7.5, 100 mM KCl, 0.25% NP-40, 10% glycerol, 1 mM EDTA, 5 mM MgCl2, 0.5 mM DTT, PhosStop (Roche), 1 mM PMSF and Complete protease cocktail (Roche)) and were then pelleted in liquid nitrogen. The frozen pellets were milled using a Freezer Mill grinder (Freezer/Mill 6870, SPEX SamplePrep) and the resulting powder was resolubilized in IP buffer (same as lysis buffer but without DTT). The lysate was cleared with centrifugation and incubated 2 h at 4°C with pre-equilibrated Pan Mouse IgG beads (Dynabeads, Invitrogen). After extensive washing with IP buffer and TEV buffer (10 mM Tris–HCl, pH 8.0, 150 mM NaCl, 0.1% NP-40, 0.5 mM EDTA, 1.0 mM DTT), proteins were eluted from the beads using the AcTEV protease (Novex, Life Technologies). Three volumes of calmodulin-binding buffer (CalBB) (10 mM Tris–HCl, pH 8.0, 150 mM NaCl, 1 mM magnesium acetate, 1 mM imidazole, 2 mM CaCl2, 10 mM β-mercaptoethanol, 0.1% NP-40) was added to the TEV eluate and incubated 2 h at 4°C with pre-equilibrated calmodulin-binding resin (Agilent Technologies). The beads were then washed twice with CalBB+0.1% NP-40, three times with CalBB + 0.02% NP-40, and five times with 20 mM ammonium bicarbonate in MS grade water. Proteins were then reduced in 10 mM DTT before being alkylated with 15mM of iodoacetamide (IAA). Protein-bound beads were subjected to trypsin (Promega) or chymotrypsin (ThermoScientific) digestion at 37°C overnight and stopped by the addition of formic acid (final concentration of 1%). Peptides were then extracted using acetonitrile, lyophilized, and reconstituted in 1% formic acid.
LC–MS/MS analysis
Following trypsin digestion, peptides were sorted using a Dionex Ultimate 3000 nanoHPLC system. Approximately 2 μg (10 μl) of peptides in 1% (vol/vol) formic acid were injected with a flow of 4 μl/min on an Acclaim PepMap100 C18 column (0.3 mm id × 5 mm, Dionex Corporation). Peptides were eluted in a PepMap C18 nanocolumn (75 μm × 50 cm, Dionex Corporation) over 240 minutes with a flow of 200 nl/min using a gradient of 5–35% solvent B (90% acetonitrile with 0.1% formic acid). Through an EasySpray source, the HPLC system was combined to an OrbiTrap QExactive mass spectrometer (Thermo Fisher Scientific). The spray voltage was set to 2.0 kV and the column temperature was set to 40°C. With a resolution of 70000 after the accumulation of 1000000 ions, full scan MS overall spectra (m/z 350–1600) in profile mode were acquired in the Orbitrap. After 50000 ions accumulated, fragmentation by collision induced dissociation (resolution of 17 500, normalised energy 35%) of the 10 strongest peptide ions from the preview scan in the Orbitrap occurred. Top filling times were 250 ms for the whole scans and 60 ms for the MS/MS scans. We enabled precursor ion charge state screening and rejected all unassigned charge states as well as singly, seven and eight charged species. We limited to a maximum of 500 entries the dynamic exclusion list with a maximum retention length of 40 s and a relative mass window of 10 ppm. To improve mass accuracy, the lock mass option was enabled. The Xcalibur software was used to acquire data (39).
MS data analysis and bioinformatics
The MaxQuant software package version 1.5.1.2 was used to process, search, and quantify the data collected as described previously (40), employing the S. pombe Uniprot proteome with 5142 protein annotations (Proteome ID: UP000002485). The settings used for the MaxQuant analysis were: two miscleavages were allowed; fixed modification was carbamidomethylation on cysteine; enzymes were trypsin or chymotrypsin; variable modifications included in the analysis were methionine oxidation and protein N-terminal acetylation; variable modifications were Phospho (STY), Methyl (KR), Dimethyl (KR), Acetyl (K) and GlyGly (K). Variable modifications were analysed independently and sequentially from one another. For precursor ions, 7 ppm was used as mass tolerance and for fragment ions, 20 ppm was used as tolerance threshold. To obtain candid identifications with a false discovery rate (FDR) of <1%, every protein was considered based on the criterion that the amount of forward hits in the database was minimally 100-fold higher than the amount of reverse database hits. Each protein had a minimum of two peptides quantified. Isoforms and protein indistinguishable based on their identified peptides were grouped and organised in a single line with various accession numbers. The mass spectrometry raw files have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD009529.
Growth assay
Liquid growth assays were performed as previously described (36). Briefly, triplicates of 50 μl of culture were diluted into 50 μl of fresh EMM supplemented with thiamine at a final concentration of 60 μM to a final OD600 = 0.02 or 0.1 in 96-well plates. Growth was monitored by cultivating cells in 100 μl of medium with shaking at 30°C in a BioTek microplate reader instrument and measuring the OD600 every 10 min over a period of 48h. Calculation of doubling times using the resulting growth curves was done as previously described (41).
RNA analysis
RNA extraction was done using a hot phenol-chloroform protocol as previously described (42). Reverse transcription and quantitative PCR analysis of the meiRNA was performed as described previously (19). For northern blot analysis of 5.8S rRNA processing, RNA was extracted from mid-log phase cultures using the hot acidic phenol-chloroform method. 10 μg of total RNA was loaded on 6% acrylamide–8 M urea gels and migrated in TBE buffer. After transfer onto a nylon membrane and UV crosslinking, the blot was pre-incubated in Church buffer at 42°C. Specific DNA probes were 5′-end radiolabeled using the T4 Polynucleotide kinase (New England Biolabs) and [γ-32P]-ATP (Perkin Elmer) and hybridized overnight at 42°C. Two 5 minutes washes in 2× SSC-0.1% SDS, and two 15 min washes in 0.1× SSC-0.1% SDS were performed before exposing the membrane to a phosphor screen. Visualization and quantification was carried out using a Typhoon Trio instrument and ImageQuant TL, respectively (GE Healthcare).
Protein analysis
Total cell extracts were prepared by either mechanical disruption using bead-beating in a Fastprep instrument or cryogenic cell lysis with a freezer mill grinder. After lysate clarification by centrifugation, protein concentration was normalised using the Bradford protein assay. Proteins were resolved on SDS-Page and transferred onto nitrocellulose membrane using a Tran-Blot® Turbo™ system (BioRad). Immunoblotting was done using a rabbit polyclonal antibody specific to Dis3 (B-Bridge, #63-123; 1:500 (v/v) dilution), a rabbit polyclonal antibody specific to Mtr4 (1:3000; generous gift from Marc Bühler), a mouse monoclonal antibody specific to α-tubulin (Sigma-Aldrich, T5168; 1:1000 (v/v) dilution), and a rabbit polyclonal antibody specific to protein A (Sigma-Aldrich, P3775; 1:10 000 (v/v) dilution) for TAP-tagged proteins. Blot were then probed with donkey anti-rabbit antibody conjugated to IRDye 800CW (LI-COR, 926-32213; 1:15000 (v/v) dilution) and a goat anti-mouse antibody conjugated to AlexaFluor 680 (Life Technologies, A-21057; 1:15000 (v/v) dilution). Protein detection was achieved using an Odyssey infrared imaging system (LI-COR).
Csl4-TAP purifications
Yeast cells were grown to mid-log phase in 50 ml of EMM supplemented with 60 μM thiamine, pelleted, washed and dropped in liquid nitrogen. Frozen cells were lysed using a freezer mill grinder (Freezer/Mill 6870, SPEX SamplePrep) and the resulting lysate was resuspended in IP buffer (50 mM HEPES–KOH, pH 7.5, 100 mM KCl, 0.25% NP-40, 10% glycerol, 1 mM EDTA, 5 mM MgCl2, 0.5 mM DTT, PMSF and Complete protease cocktail (Roche)). Cell extracts were incubated with pre-equilibrated Pan Mouse IgG beads for 2 h at 4°C, and the beads were washed three times with IP buffer and denatured using 2× SDS-PAGE loading buffer. Protein samples were resolved on a 10% SDS-PAGE gel and analyzed by Western blotting.
RNA decay assay
The protocol designed to measure the ribonucleolytic activity of the fission yeast exosome complex was established based on a previously described procedure (43). Briefly, total cell extracts were prepared from frozen cells using freezer mill grinder and submitted to Csl4-TAP purification using magnetic Pan Mouse IgG beads (Dynabeads, Invitrogen). After extensive washing, purified exosome complexes were incubated at 30°C in a thermomixer with a specific RNA substrate (5′-AAUUAUUUAUUAUUUAUUUAUUAUUUAUUUAUUUAUUAUUUAUUUAUUA-PO4-3′) (44) that was 5′-end labeled with [γ-32P]-ATP using T4 Polynucleotide kinase-3′ phosphatase minus (New England Biolabs). RNA decay reactions were stopped by adding 2× formamide RNA loading dye containing 20mM EDTA. Samples were loaded on a pre-run 20% acrylamide-8M urea/TBE gel and ran at 10W for 20 minutes. The gel was exposed on Phosphor screen for 20 min before visualization and quantification with Typhoon Trio instrument and ImageQuant TL, respectively (GE Healthcare).
RESULTS
Affinity purification of the fission yeast RNA exosome complex
To identify post-translational modification (PTMs) in core subunits of the fission yeast RNA exosome, we tagged the genomic copy of dis3 and rrp4 to express carboxy (C)-terminal fusions for tandem affinity purification (45). The results of tandem affinity purifications (TAP) of Dis3-TAP and Rrp4-TAP are shown in Figure 1A. As a consequence of C-terminal tagging, we observed an expected change in the molecular weight of Dis3 and Rrp4 after affinity purification of Dis3-TAP and Rrp4-TAP, respectively (Figure 1A, compare lanes 1–2). Importantly, TAP purification of Dis3 and Rrp4 primarily isolated 11-subunits exosome complexes, as revealed by the near stoichiometric amounts of exosome subunits (Figure 1A). Next, the whole purification mix was subjected to proteolytic digestion using either trypsin or chymotrypsin and analyzed by liquid chromatography-tandem mass spectrometry (LC–MS/MS). In total, 324 and 250 proteins were identified with a minimum of two unique peptides in at least two biological replicates in the purification of TAP-tagged Dis3 and Rrp4, respectively (Supplementary Tables S2 and S3). Several of the known nuclear exosome-associated proteins that are part of the MTREC/NURS complex (29–31) were recovered among the top-15% of Dis3 and Rrp4 interactors, including Mtl1, Red1, Red5, and Iss10 (Supplementary Tables S2 and S3).
Figure 1.

Affinity purification of the S. pombe RNA exosome complex. (A) SYPRO RUBY-stained SDS-PAGE of affinity purified Dis3-TAP (lane 1) and Rrp4-TAP (lane 2). Subunits of the RNA exosome identified by mass spectrometry are indicated on the right. (B and C) Enrichment of RNA exosome subunits after affinity purification of Dis3-TAP (B) and Rrp4-TAP (C) as plotted by relative protein abundance (total peptide intensity) up the y-axis and percentage sequence coverage (amino acids) out the x-axis. Components of the RNA exosome complex are identified by green (B) and purple (C) squares. (D) MS determination of peptide number and sequence coverage for the indicated exosome subunits in Dis3-TAP and Rrp4-TAP purifications.
To determine the relative abundance of the co-purified proteins in each purification, we used a label-free intensity-based quantification method (46) that calculates the sum of all peptide peak intensities matching to a specific protein. When the relative peptide intensity was plotted against the protein sequence coverage (percentage of amino acids of a specific protein identified by MS), our results showed a clear enrichment of all 11 exosome subunits in both Dis3 and Rrp4 purifications (Figure 1B and C), consistent with the recovery of mostly integral complexes. Furthermore, the enrichment of RNA exosome complexes via affinity purification of Dis3 and Rrp4 allowed for high sequence coverage of core exosome subunits, ranging between 73% and 98% among biological replicates (Figure 1D). The reason for the slightly lower sequence coverage of Rrp41 and Rrp42 relative to other exosome subunits is the reduced number of protease cleavage sites, which yields longer peptides that are more challenging to detect by LC–MS/MS. In addition to core exosome subunits, two proteins were prominently enriched in independent TAP purifications of Dis3 and Rrp4: SPCC1827.03c and Mpp6 (Figure 1B and C). SPCC1827.03c is a predicted acetyl-CoA ligase whose S. cerevisiae homolog, Psc60p, was shown to have RNA-binding activity (47), and Mpp6 is the homolog of a well characterized S. cerevisiae nuclear exosome-associated protein (Mpp6p) whose recent structures reveal how it stimulates the nuclease activity of the nuclear exosome via coordination with the Mtr4 helicase (26,28). Collectively, affinity purifications of S. pombe Dis3 and Rrp4 recovered RNA exosome complex with unambiguous identification of all 11 protein subunits.
Identification of post-translational modifications in the fission yeast RNA exosome
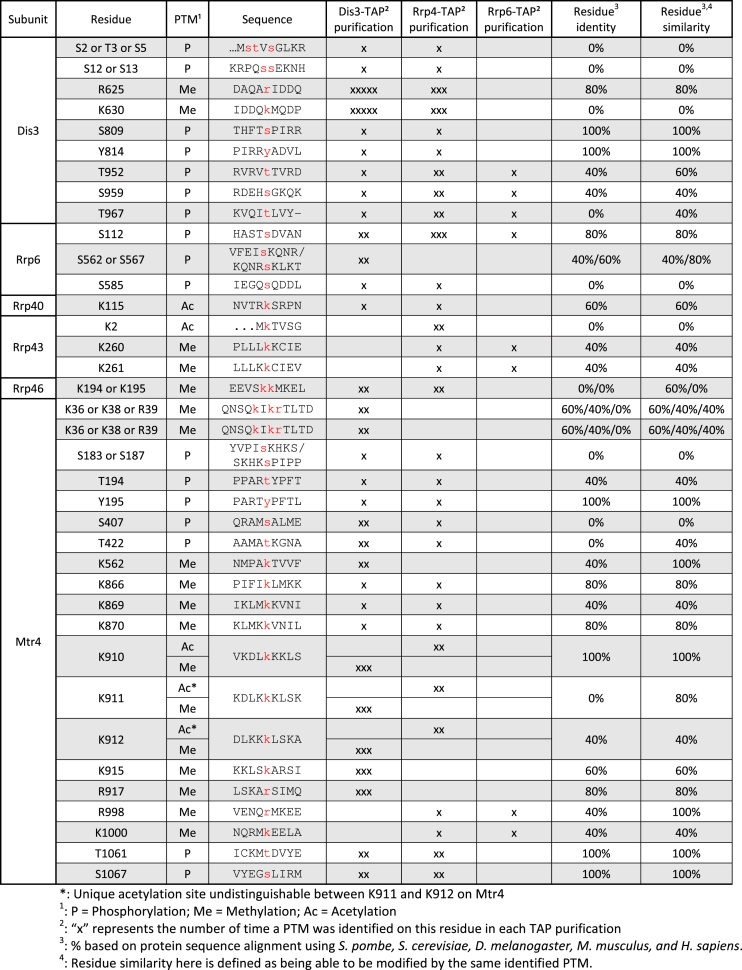
The extensive sequence coverage obtained from the multiple TAP purifications of Dis3 and Rrp4 was an important consideration for the characterization of post-translational modifications (PTMs) in core subunits of the RNA exosome. To provide greater depth in sequence coverage for the identification of exosome PTMs, we also included the MS data from a single affinity purification of Rrp6-TAP. For the analysis of PTMs, we also considered Mtr4, which is an evolutionarily conserved co-factor of the nuclear exosome (48). For a PTM to be considered, a modification had to be mapped on the same residue in at least two independent TAP purifications of either Dis3, Rrp4, or Rrp6. Collectively, we identified a total of 39 PTMs, including acetylation, phosphorylation, and methylation, on core exosome subunits and the RNA helicase Mtr4 of S. pombe (Table 1). For each detected PTMs, modified and non-modified peptides were detected, and semiquantitative analysis indicated that modified peptides were generally in substoichiometric amounts relative to the non-modified peptides. A schematic view of the modifications mapped on exosome subunits and Mtr4 is presented in Figure 2A. In addition, the position of the identified PTMs on resolved structures of the 11-subunit exosome and Mtr4 of S. cerevisiae is illustrated in Supplementary Figure 1. Interestingly, ubiquitination sites were not detected on any exosome subunits. In addition, Rrp4, Rrp42, Rrp45, Mtr3, Rrp41, and Csl4 did not appear to be modified by either acetylation, phosphorylation, methylation, or ubiquitination. The two catalytic subunits of the RNA exosome, Dis3 and Rrp6, were primarily modified by phosphorylation. In contrast, structural subunits Rrp43, Rrp40, and Rrp46 did not contain phosphorylation sites, but were found to be acetylated and methylated (Figure 2A). Our data also revealed that Mtr4 is heavily modified in S. pombe, as a variety of PTMs were identified across the Mtr4 helicase (Figure 2A) despite a lower sequence coverage (39%) as compared to core exosome subunits.
Table 1.
Summary of PTM sites identified in S. pombe RNA exosome subunits and Mtr4 helicase
|
Figure 2.

Repertoire of post-translational modifications (PTMs) identified on subunits of the fission yeast RNA exosome and Mtr4. (A) Schematic representation of the identified PTMs on the indicated exosome subunits and the Mtr4 helicase. Known domains in the indicated proteins: CR3, three cysteine residues motif; PIN, PilT N-terminal domain that catalyze endonuclease activity, CSD, cold-shock domains; RNB, RNase II catalytic domain that catalyses 3′-5′ exonuclease activity; S1, RNA-binding domain; PMC2NT, polycystin 2 N-terminal domain; EXO, 3′-5′ exonuclease domain; HRDC, helicase and RNase D C-terminal domain; ExoBD, exosome-binding domain; PH, ribonuclease PH-like domains; KH, K homology RNA-binding domain; PNPase, Polyribonucleotide nucleotidyltransferase domain; RecA, RecA-like domain. (B) Pie chart showing the distribution of PTMs detected in RNA exosome subunits and Mtr4 by MS. (C) Pie chart showing the distribution of the three types of phosphorylation sites identified in subunits of the RNA exosome and Mtr4. Phospho-peptides for which serine or threonine phosphorylation could not be distinguished are classified in light gray. (D) Pie chart showing the distribution of the two types of methylation sites identified in this study. Methylated-peptides for which arginine and lysine methylation could not be distinguished are classified in light gray.
Globally, most of the modifications identified on RNA exosome subunits and Mtr4 were methylation (46%) and phosphorylation (43%) sites, whereas acetylation accounted for ∼10% of the identified PTMs (Figure 2B). Not surprisingly, about half of the phosphorylation sites were on serine residues, while phospho-threonine and phospho-tyrosine accounted for 29% and 12%, respectively, of the phosphorylation sites identified on exosome subunits and Mtr4 (Figure 2C). This phosphorylation pattern slightly deviates from the global fission yeast phosphoproteome. Accordingly, the general features of large-scale phosphorylation data using total extracts of S. pombe revealed a 84%/13%/3% distribution for Ser/Thr/Tyr phosphorylation, respectively (49). Given that our study focused on the RNA exosome complex and identified a total of 17 phosphorylation sites, a divergence in the distribution of Ser/Thr/Tyr phosphorylation from the global S. pombe phosphoproteome is not unexpected. As for protein methylation of RNA exosome subunits, most of the methylated residues were lysines (72%), while arginine methylation was substantially less frequent (Figure 2D). In the case of 7 PTMs, analysis of mass spectra did not unambiguously discriminate between alternative positions for a single modification on the same peptide. We nevertheless included this information in our analysis (Table 1 and Figure 2A) since the evidence supporting the presence of the corresponding PTMs is reliable and restricts possible positions of putative modifications. We also performed multiple sequence alignment to determine the extent to which the identified PTMs were located in evolutionarily conserved regions of RNA exosome subunits or Mtr4 (see Table 1). Together, our MS analysis of purified exosome complex provides a detailed list of modified residues on several subunits of the fission yeast RNA exosome.
Functional analysis of selected sites of post-translational modification in Dis3, Mtr4, and Rrp6
To begin a functional assessment of PTM sites in RNA exosome subunits, we prioritized evolutionarily conserved PTM sites on catalytically active proteins. In total, we selected six phosphorylation sites, two sites of arginine methylation, and three sites of lysine methylation/acetylation in Dis3 (R625, K630, S809, and Y814), Mtr4 (Y195, K910, K911, K912, T1061 and S1067), and Rrp6 (S112). We first addressed how substitutions of modified residues affected the ability of Dis3, Mtr4, and Rrp6 to support normal growth of S. pombe. As dis3 and mtr4 are essential genes, we used previously described conditional strains (36,38) in which endogenous dis3 and mtr4 promoters have been replaced by the thiamine-repressible nmt promoter (Pnmt-dis3 and Pnmt-mtr4). Accordingly, addition of thiamine to the growth medium resulted in a significant increase in the doubling time of both Pnmt-dis3 and Pnmt-mtr4 strains relative to the wild-type control (Figure 3A and B, respectively). For Rrp6 variants, we used a rrp6Δ mutant that also significantly affects growth rate as compared to a control strain (Figure 3C). Next, we generated mutant alleles of dis3, mtr4, and rrp6 that expressed phosphorylation-deficient (S/T/Y > A) and methylation/acetylation-deficient (K/R > A) versions with substitutions at the aforementioned selected residues. Wild-type and mutant alleles of dis3, mtr4, and rrp6 were chromosomally integrated as a single copy into the Pnmt-dis3, Pnmt-mtr4, and rrp6Δ strains, respectively, and the extent to which the mutant versions restored the growth defect induced by depletion of endogenous Dis3, Mtr4, and Rrp6 was examined by liquid growth assays. All of the phosphorylation-deficient and methylation/acetylation-deficient alanine substitutions fully rescued the growth defects observed for Pnmt-dis3, Pnmt-mtr4 and rrp6Δ strains (Figure 3A–C). We thus generated a panel of phospho-mimetic (S/T/Y > D, negatively charged aspartate) mutants of Dis3, Mtr4 and Rrp6, in which the introduced aspartic acid residue mimics the phosphorylated state of the protein (50). As shown in Figure 3A, Dis3-depleted cells that expressed S809D and Y814D phospho-mimetic versions of Dis3 showed increased doubling time as compared to wild-type Dis3 despite normal protein expression (see Figure 4B). In the case of Mtr4, Y195D, T1061D, and S1067D single mutants all rescued the growth defects of Mtr4-deficient cells similar to the wild-type version (Figure 3B). However, a T1061D–S1067D double mutant showed a significant increase in the doubling time, which was comparable to Pnmt-mtr4 cells containing empty vector control (Figure 3B). In contrast, the corresponding Mtr4 T1061A–S1067A phosphorylation-deficient mutant fully complemented Mtr4-depleted cells (Figure 3B). Both T1061D–S1067D and T1061A–S1067A Mtr4 mutants were expressed at similar levels (Figure 4C). As for Rrp6, expression of the Rrp6 S112D phospho-mimetic mutant rescued the defective growth rate of the rrp6Δ mutant (Figure 3C).
Figure 3.

Functional characterization of Dis3, Mtr4, and Rrp6 variants with substitutions at selected modified residues. (A–C) Doubling time calculated from growth curves of Pnmt-dis3 (A), Pnmt-mtr4 (B), and rrp6Δ (C) strains complemented with either the wild-type (WT) version of Dis3 (A), Mtr4 (B), and Rrp6 (C), or the indicated variants with substitutions at modified residues as determined by MS analysis. EV, empty vector control. In B, the 3KA version of Mtr4 corresponds to the multi-site substitution K910A-K911A-K912A. (D–F) RT-qPCR analysis of sme2 (meiRNA) expression using total RNA prepared from the strains described in panels A–C. RT-qPCR data were normalized to the housekeeping nda2 mRNA, and fold changes expressed relative to Pnmt-dis3 (D), Pnmt-mtr4 (E), and rrp6Δ (F) strains complemented with the wild-type version of Dis3, Mtr4, and Rrp6, respectively. Pnmt-dis3 and Pnmt-mtr4 strains were cultured in thiamine-supplemented medium to deplete endogenous Dis3 and Mtr4, respectively. The data and error bars represent the average and standard deviation from at least three independent experiments. P-values * ≤0.05, ** ≤0.01, *** ≤0.001, **** ≤0.0001; Student's t-test.
Figure 4.

Phospho-mimetic versions of Dis3 and Mtr4 result in defective 5.8S rRNA synthesis. (A) Schematic of 7S pre-rRNA processing into mature 5.8S rRNA. Following endonucleolytic cleavage of the 27S rRNA precursor at site C2, the 7S pre-rRNA is trimmed 3′-5′ by the nuclear exosome assisted by the helicase activity of Mtr4 (green pacman). The resulting 6S intermediate is further processed by cytoplasmic ribonucleases (purple pacman) to generate the mature 5.8S rRNA. (B) Western blot (WB) and Northern blot (NB) analysis of wild-type (lane 1) and Pnmt-dis3 (lane 2–8) strains expressing the indicated phospho-deficient (lanes 5 and 7) and phospho-mimetic (lanes 6 and 8) versions of Dis3. EV, empty vector control. The position of the 7S pre-rRNA and 5.8S rRNA are indicated on the right. (C) Western blot (WB) and Northern blot (NB) analysis of wild-type (lane 1) and Pnmt-mtr4 (lane 2–6) strains expressing the indicated phospho-deficient (TASA; lane 5) and phospho-mimetic (TDSD; lane 6) versions of Mtr4. EV, empty vector control. The position of the 7S pre-rRNA and 5.8S rRNA are indicated on the right. The asterisk (*) shows the 5′-extended product specifically detected in the phospho-mimetic version of Mtr4. (B-C) The 5S rRNA and Tubulin were used as loading controls for northern and Western blots, respectively. (D and E) Quantification of 7S/5.8S ratios for the indicated versions of Dis3 (D) and Mtr4 (E). The calculated 7S/5.8S ratio was normalized to the 5S rRNA and expressed relative to Pnmt-dis3 (D) and Pnmt-mtr4 (E) strains complemented with the wild-type version of Dis3 and Mtr4, respectively. (B–E) Pnmt-dis3 and Pnmt-mtr4 strains were cultured in thiamine-supplemented medium to deplete endogenous Dis3 and Mtr4, respectively. The data and error bars represent the average and standard deviation from at least three independent experiments. P-values * ≤0.05, ** ≤0.01, *** ≤0.001; Student's t-test.
We next examined our different PTM-deficient and phospho-mimetic exosome mutants for their ability to function in the repression of meiotic differentiation genes during mitotic growth of S. pombe. We therefore analyzed the meiosis-specific ncRNA expressed from the sme2 gene, meiRNA, which is rapidly degraded by the nuclear exosome during the mitotic cell cycle (19,20,51). Accordingly, cells deficient for Dis3, Mtr4, and Rrp6 showed a significant accumulation of meiRNA, a phenotype that was rescued by exogenous expression of wild-type versions of Dis3, Mtr4, and Rrp6, respectively (Figure 3D–F). As shown in Figure 3D, S809D and Y814D phospho-mimetic versions of Dis3 resulted in meiRNA accumulation, consistent with the growth defects observed for these mutants (Figure 3A). Expression of the Mtr4 double phospho-mimetic mutant (T1061D–S1067D) in the Pnmt-mtr4 strain showed accumulation of the meiRNA at levels similar to the empty vector control (Figure 3E). All of the other phosphorylation-deficient, acetylation/methylation-deficient, and phospho-mimetic mutants of Dis3, Mtr4 and Rrp6 demonstrated normal repression of meiRNA accumulation (Figure 3D–F), coinciding with their ability to fully complement the growth defects resulting from the depletion of endogenous Dis3, Mtr4, and Rrp6. Collectively, our results suggest that Dis3 phosphorylation on Ser-809 and Tyr-814 alters nuclear exosome function, and that multi-site phosphorylation of both Thr-1061 and Ser-1067 on Mtr4 impairs its ability to promote RNA degradation by the RNA exosome.
Phospho-mimetic mutants of Dis3 and Mtr4 impair synthesis of the 5.8S rRNA
As Mtr4 is not a central player in the negative regulation of meiotic differentiation genes in fission yeast (compare fold increase of meiRNA in Mtr4-deficient cells relative to exosome mutants in Figure 3D–F), we examined the processing of the 7S pre-rRNA into mature 5.8S rRNA (Figure 4A), a maturation step for which the activity of Mtr4 is key to exosome-mediated 3′ end trimming (27,52). Accordingly, depletion of Dis3 and Mtr4 from S. pombe cells resulted in a marked accumulation of the 7S precursor together with reduced levels of the mature 5.8S rRNA (Figure 4B and C, compare lanes 1–2). Cells expressing the Dis3 Y814D phosphorylation-mimicking mutant showed accumulation of 7S pre-rRNA at levels similar to Pnmt-dis3 cells with the empty vector control (Figure 4B, compare lane 8 to lane 3; quantification shown in Figure 4D). The S809D phospho-mimetic mutant also impaired 5.8S rRNA production, but resulted in reduced levels of 7S pre-rRNA accumulation as compared to the Y814D mutant (Figure 4B, compare lane 6 to lane 8; Figure 4D). In contrast, both S809A and Y814A phospho-deficient versions of Dis3 showed normal levels of 7S processing into mature 5.8S rRNA (Figure 4B, compare lanes 5 and 7 to lane 4; Figure 4D), consistent with data obtained for the negative regulation of the meiRNA (Figure 3D).
As for analysis of meiRNA accumulation, the single Mtr4 phosphorylation-mimicking mutants (T1061D and S1067D) did not show reduction of 5.8S rRNA production (Supplementary Figure S2A). However, the double phospho-mimetic mutant of Mtr4 (T1061D–S1067D) showed a marked defect in 5.8S rRNA synthesis; yet, these defects appeared slightly different than for cells deficient of Mtr4 (Figure 4C). Specifically, whereas the reduction of 5.8S rRNA production in the T1061D–S1067D mutant was comparable to Mtr4-deficient cells (Figure 4C, compare lanes 6 to 3), expression of the Mtr4 double phospho-mimetic mutant resulted in the accumulation of an rRNA intermediate larger than the 7S pre-rRNA (Figure 4C, lane 6). As a probe specific to a sequence located in internal transcribed spacer 1 (ITS1) detected this RNA (Supplementary Figure S2B), this 5.8S rRNA-containing intermediate does not appear to result from defects in the endonucleolytic cleavage at site C2 in ITS2 (Figure 4A), but from a defective processing step that occurs prior to the generation of the 27S pre-rRNA. Intriguingly, this RNA product was not detected in Mtr4-deficient cells (Figure 4C, compare lane 6 to lanes 2–3; quantification Figure 4E), which primarily accumulated the 7S pre-rRNA. The double T1061A–S1067A phosphorylation-deficient mutant of Mtr4 showed normal 5.8S rRNA production, consistent with results obtained for the analysis of the meiRNA (Figure 3B). Our results thus suggest that the growth and RNA processing defects of the Dis3 S809D and Y814D mutants as well as the Mtr4 T1061D–S1067D mutant are due to the presence of negative charges that mimic the phosphorylated state of Dis3 and Mtr4.
Phosphorylation mimic versions of Dis3 Ser-809 and Tyr-814 show reduced catalytic activity
We next focused on Dis3 to gain molecular insights into the mechanism by which the S809D and Y814D phospho-mimetic mutants exhibited reduced exosome function and impaired growth. One possibility was that phosphorylation of Ser-809 and Tyr-814 residues in Dis3 could control assembly of the core exosome complex. To assess this possibility, we tested whether wild-type and mutant versions of Dis3 can be equally recovered after the purification of Csl4, which is a non-catalytic subunit of the heterotrimeric cap structure (with Rrp4 and Rrp40) that is located above the hexameric ring in the core exosome complex (7). As expected, affinity purification of Csl4-TAP copurified endogenous Dis3 (Figure 5A, lane 2) as well as the wild-type version of Dis3 expressed from a transgene in Pnmt-dis3 cells cultured in the presence of thiamine to deplete endogenous Dis3 (Figure 5A, lane 4). In contrast, Dis3 was not detected after TAP purification using extracts prepared from a control untagged strain (Figure 5A, lane 1). Notably, we found that both phospho-deficient and phospho-mimetic versions of Dis3 copurified with Csl4-TAP at levels similar to wild-type Dis3 (Figure 5A, compare lanes 5–8 to lane 4). These results suggest that phosphorylation of Ser-809 and Tyr-814 are unlikely to impair stable incorporation of Dis3 into the exosome complex.
Figure 5.

The presence of a negative charge that mimic the phosphorylated state of Ser-809 and Tyr-814 of Dis3 inhibits its catalytic activity. (A) Western blot analysis of total cell extracts (Input, bottom three panels) and affinity purifications (IP, top two panels) prepared from wild-type (lanes 1–2) and Pnmt-dis3 (lanes 3–8) strains that expressed either TAP-tagged (lanes 2–8) or untagged (lane 1) versions of Cls4 as well as the indicated versions of Dis3 (lanes 4–8). Pnmt-dis3 strains were cultured in thiamine-supplemented medium to deplete endogenous Dis3. (B) Analysis of RNA degradation kinetics of a 3′-phosphate AU-rich 49-nt RNA using affinity purified exosome complex prepared from extracts of Csl4-TAP strains that expressed wild-type (lanes 1–5), Endo/Exo-deficient (lanes 6–10), S809D (lanes 11–15), and Y814D (lanes 16–20) versions of Dis3. (C) Quantitative analysis of RNA decay assays as shown in panel B. The data and error bars represent the average and standard deviation from at least three independent experiments. ****P ≤ 0.0001, two-way ANOVA test.
Next, we addressed whether the catalytic activity of the phophorylation-mimetic versions of Dis3 was affected by using an in vitro RNA degradation assay. To prevent catalytic activity from the exosome-associated Rrp6 exoribonuclease, we used a previously described 3′ phosphate RNA substrate that can be degraded by the 3′-5′ exonucleolytic activity of Dis3, but not of Rrp6 (44). Lysates from Csl4-TAP and untagged control strains were subjected to TAP purifications and the purified exosome was analyzed for Dis3-mediated RNA degradation activity after incubation with a 49-nt-long AU-rich 3′ phosphate RNA. Precipitates prepared from the Csl4-TAP purification efficiently degraded the 49-nt substrate into small RNA decay products (Supplementary Figure S3, lanes 4–5). In contrast, TAP precipitates from an untagged control strain did not degrade the 3′ phosphate RNA (Supplementary Figure S3, lanes 2–3). To confirm that the RNA degradation activity associated with Csl4-TAP precipitates was mediated by Dis3 and not Rrp6, we purified Csl4-TAP using extracts of a Pnmt-dis3 conditional strain that expressed a version of Dis3 with single amino acid substitutions at conserved aspartate residues that were previously shown to be devoid of exonucleolytic or endonucleolytic activities (12,36). Consistent with Dis3-mediated RNA degradation of the 3′ phosphate RNA, Csl4-TAP precipitates from the strains expressing the catalytically-inactive version of Dis3 showed roughly 75% of the full-length RNA substrate remaining after 20 min of incubation (Figure 5B, lanes 6–10; quantifications shown in Figure 5C), whereas less than 5% of the substrate RNA was detected after 20 min using Csl4-TAP precipitates from a strain expressing wild-type Dis3 (Figure 5B, lanes 1–5; Figure 5C). Since we are using a Dis3 depletion system, the faint degradation of the 3′ phosphate RNA using the catalytically-inactive version of Dis3 is likely the consequence of residual endogenous wild-type Dis3 present in Csl4-TAP purifications (see Figure 5A, lane 3). Notably, Csl4-TAP precipitates prepared from strains expressing the phospho-mimetic versions of Dis3 showed a significant reduction in RNA degradation activity (Figure 5B, lanes 11–20). Quantification from independent RNA decay assays revealed that the degradation kinetics were not significantly different between the Dis3 mutants (Figure 5C), indicating a robust inactivation of exosome RNA degradation activity. RNA decays assays using the corresponding phospho-deficient versions of Dis3 (S809A and S814A) showed degradation kinetics of the 3′ phosphate RNA comparable to wild-type Dis3, consistent with normal exosome function in vivo (Figures 3 and 4). Together, these results indicate that the introduction of a negative charge that mimic the effect of phosphorylation at residues 809 and 814 inhibits the RNA degradation activity of Dis3.
Ser-809 and Tyr-814 are located near the exoribonuclease catalytic center of Dis3
Given that phospho-mimetic substitutions at Ser-809 and Tyr-814 residues impaired the catalytic activity of Dis3, we examined the localization of these residues in the 3D structure of the S. cerevisiae exosome (10). Interestingly, these two phosphorylation sites were found to be located in the vicinity of the Dis3 catalytic center (Figure 6A). Notably, Ser-809 is adjacent to one of the key aspartate residues (Asp-516 in S. pombe and Asp-551 in S. cerevisiae) that is responsible for coordinating magnesium ions in the catalytic site of Dis3 (10,53). Tyr-814 is located at the amino-terminal end of an alpha helical structure that is positioned next to two consecutive arginine residues (Figure 6A–B) that make polar interactions with phosphate groups along the RNA backbone (10). Sequence alignment also indicate that these two phosphorylation sites are located within a highly conserved region of the Dis3 RNB domain (Figure 6B). The structural analysis of Dis3 supports the idea that the addition of a negative charge on Ser-809 and Tyr-814 by phosphorylation could alter the alignment of active site residues with RNA, which ultimately translates to effects on RNA degradation.
Figure 6.

Ser-809 and Tyr-814 of S. pombe Dis3 are evolutionarily conserved residues located near the exosome catalytic center. (A) Ribbon diagram representation of S. cerevisiae Dis3 (PDB: 4IFD) with a close-up view of the position of residues corresponding to Ser-809 (green) and Tyr-814 (magenta) in S. pombe Dis3. The position of the Asp-516 in S. pombe Dis3 (corresponding to Asp-551 in S. cerevisiae), which is critical for coordinating magnesium ions in the catalytic center of Dis3, is shown in blue. Single-stranded RNA is shown in orange. (B) Amino acid sequence alignment of Dis3 from fission and budding yeasts, Drosophila, mouse, and humans. Numbers on the right correspond to the position of the last amino acid shown for each sequence. Identical amino acids are shown in black outlined and similar amino acids are shown in gray outline. Ser-809 and Tyr-814 phosphorylation sites in S. pombe Dis3 are underlined.
DISCUSSION
In this study, a post-translational modification (PTM) map of the RNA exosome and its adaptor helicase Mtr4 was obtained by using a proteomics approach on purified S. pombe exosome complex. A total of 39 new and high-confidence PTM sites were identified here, which were not detected in previous global proteomic data sets from fission yeast (49,54–56). By analyzing the distribution of the different modifications identified on subunits of the RNA exosome and Mtr4, we observed that similar types of PTMs were frequently clustered in specific regions (Figure 2A and Supplementary Figure S1). Studies of protein phosphorylation using large-scale proteomics data have in fact shown that phospho-sites are not distributed randomly on target proteins, but rather concentrated on surface patches (57,58). Such closely positioned phosphorylation sites tend to be activated by the same kinase (57,59). Notably, DNA- and RNA-binding proteins are a particularly enriched group of proteins that show phosphorylation sites clustering (60), as modulation of DNA/RNA–protein interactions may benefit more from having a gradual and additive change in electrostatic charge rather than a switch in structure elicited by phosphorylation of a single residue.
A mass spectrometry analysis of the exosome complex enriched via affinity purification has been previously described in S. cerevisiae, identifying 8 phosphorylation sites on six different subunits of the core exosome (35). Surprisingly, none of the 8 phospho-sites reported on S. cerevisiae exosome subunits matched any of the 10 phosphorylation sites identified here on the S. pombe exosome. Notably, phosphorylation sites were not detected on S. cerevisiae Dis3 (35), whereas we mapped 7 phospho-sites on Dis3 of S. pombe. There are a number of reasons that could explain the apparent absence of overlap between phospho-sites of budding and fission yeast exosome. First, given that the exosome has evolved the use of species-specific cofactor complexes, such as NNS in S. cerevisiae and MTREC/NURS in S. pombe, it is possible that the regulation of exosome functions/interactions by PTMs will be largely different between budding and fission yeasts. Different sets of proteolytic enzymes were also used for the analysis of peptides by LC–MS/MS, which is known to introduce bias into peptide length and sequence coverage, thereby affecting PTM identification (61,62). In Drosophila, Ser-786 of Dis3 was shown to be phosphorylated, a modification that inhibits its exonucleolytic activity (63). Notably, Ser-786 in Drosophila Dis3 corresponds to Ser-809 in S. pombe Dis3, indicating that phosphorylation of Dis3 in its exonuclease domain is conserved evolutionarily. Analyses of databases that collect data from high-throughput proteomics experiments using human cells (64) revealed that five residues modified in the exosome and Mtr4 of S. pombe show similar modifications in human cells (Supplementary Table S4). As yet, however, it remains to be determined whether the modified residues shown to have functional consequence on Dis3 (Ser-809 and Tyr-814) and Mtr4 (Thr-1061 and Ser-1067) in fission yeast are phosphorylated in humans. Future studies will therefore be required to assess the conservation of PTM sites on subunits of the RNA exosome complex.
Our functional analysis of Dis3, Rrp6 and Mtr4 mutants on specific phosphorylation, methylation, and acetylation sites implied roles for site-specific phosphorylation of Dis3 and Mtr4 in the control of exosome function. Notably, our findings suggest that phosphorylation of Dis3 on Ser-809 and Tyr-814 negatively control its activity. This conclusion is supported by the identification of peptides containing phosphorylated Ser-809 and Tyr-814 by mass spectrometry from exosome complexes that were purified independently using Dis3-TAP and Rrp4-TAP strains (Table 1). Importantly, phospho-mimetic versions of Dis3 that simulate a constitutive state of phosphorylation on Ser-809 and Tyr-814 showed impaired exosome functions in vivo (Figures 3 and 4) and a marked reduction in exonucleolytic activity in vitro (Figure 5). In contrast, phospho-deficient versions of Dis3 in which Ser-809 and Tyr-814 were substituted to alanine showed wild-type exosome function (Figures 3 and 4). These results indicate that the side chain of these highly conserved residues is not critical for Dis3 function, but that the presence of a negative charge that mimic a phosphorylated version of Ser-809 or Tyr-814 inhibits Dis3. As these residues lie near the catalytic center of Dis3 (Figure 6), it is tempting to speculate that phosphorylation of Ser-809 and Tyr-814 could repel – by virtue of their negative charges – the negatively charged RNA, causing misalignment of the RNA substrate in relation to the catalytic site. It remains formally possible that the introduced phospho-mimetic substitutions simply reflect inherent sensitivity of Dis3 at these sites, and thus, are not imitating a constitutive state of phosphorylation. We believe this to be unlikely however, as phosphorylation of Ser-786 (equivalent to S. pombe Ser-809) by CDK1 has been shown to inhibit Dis3 in Drosophila (63), findings that support the existence of an evolutionarily conserved mechanism of Dis3 regulation.
The functional relevance of Ser-809 and Tyr-814 phosphorylation on Dis3 as well as the kinase (or kinases) responsible for these modifications remain to be determined. One possible role of Ser-809/Tyr-814 phosphorylation could be to keep the free Dis3 protein in a catalytically inactive state until incorporation into the exosome core, a step that has been shown to downregulate the exoribonuclease activity of Dis3 (65–67). Still, little is known about how and where the core exosome complex is assembled in the cell. Alternatively, inhibition of Dis3 exonucleolytic activity by Ser-809/Tyr-814 phosphorylation could induce conformational changes in the exosome core that would favor RNA path selection toward the Rrp6 active site. In either scenario, a phosphatase and a kinase need to gain access to Ser-809 and Tyr-814 in the catalytic site of Exo11 to activate Dis3 by dephosphorylation and inhibit Dis3 by phosphorylation, respectively. Interestingly, phospho-site Ser-809 in Dis3 is part of a common proline-directed phosphorylation motif (Ser-Pro) that is frequently seen for Cyclin-dependent kinase (Cdk)-mediated cell cycle control (49), suggesting potential mitotic regulation of exosome activity in S. pombe, as reported in Drosophila (63).
Our data also point to the regulation of the Mtr4 helicase by phosphorylation. In the case of Mtr4, our results suggest that phosphorylation of both Thr-1061 and Ser-1067 is required to control Mtr4, as cell growth and rRNA processing were not affected in the single phospho-mimetic versions of Mtr4 (Figure 3 and Supplementary Figure S2). These results support the view that a change in charge imparted by the multi-site phosphorylation of Mtr4 is more relevant as a regulatory mechanism than a possible change in structure. Consistent with this idea, analysis of a recent Mtr4 structure (27) indicates that Thr-1061 and Ser-1067 are both positioned in the vicinity of key sites involved in RNA-protein interactions (Supplementary Figure S4). Specifically, Ser-1067 occupies the same face of the ratchet helix of Mtr4 than Arg-1074, which has been shown in S. cerevisiae (Arg-1030 in budding yeast Mtr4) to contribute to RNA recognition and unwinding (68). Thr-1061 is located in a loop structure N-terminal to the Mtr4 ratchet helix (Supplementary Figure S4) that is positioned opposite to the β-harpin loop proposed to promote strand separation within the second RecA domain of Mtr4 (69). Intriguingly, the double T1061D/S1067D phosho-mimetic mutant resulted in the accumulation of an undefined rRNA product that was detected using both 5.8S- and ITS1-specific probes (Supplementary Figure S2), suggesting incomplete processing of the 5′ end of a 5.8S rRNA precursor. Given that this RNA was detected neither in the phosphorylation-deficient double mutant nor in Mtr4-depleted cells (Figure 4C), we suspect that this RNA corresponds to an aberrant rRNA intermediate destined for degradation but trapped in a catalytically inactive state with Mtr4.
Collectively, our findings argue against a model in which once the exosome is assembled, it remains a static and uniform complex with spontaneous and constitutive activity for RNA degradation. Accordingly, dynamic and reversible phosphorylation events have been shown to play important roles in the regulation of analogous multi-subunit complexes, including the spliceosome and the proteasome, contributing to complex assembly, catalytic activity, localization, and stability (70,71). Interestingly, several exosome cofactor systems show similarities between S. pombe and humans, such as the Pab2/PABPN1-mediated RNA decay pathway (5,33,72,73), as well as between the S. pombe MTREC/NURS and the human NEXT-CBC complexes (29–32,74). Given the notable conservation of cofactors used by the S. pombe and human exosomes, the characterization of PTM sites in the fission yeast exosome complex will provide powerful biochemical and genetic tools to unveil the regulatory steps important for the assembly and function of this RNA decay machinery.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dominique Lévesque for help with the use of MS instruments, Louis-Philippe Morency and Pierre Lavigne for advices on using PyMOL, and Marc Larochelle for critical reading of the manuscript.
Author contributions: C.T. and F.B. conceived the study and the experimental frame, while F.-M.B supervised the MS analyses and the pipelines for the identification of PTMs. C.T. performed the TAP purifications, the generation of conditional yeast strains and DNA constructs, as well as all of the functional assays. C.T. and F.B. prepared and finalized the figures as well as wrote the manuscript, which was reviewed by all authors.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Discovery Grants from the Natural Sciences and Engineering Research Council of Canada (NSREC) [05482 to F.B., 418404 to F.-M.B.]; Canada Research Chair in Quality Control of Gene Expression [230977 to F.B.]. Funding for open access charge: Natural Sciences and Engineering Research Council of Canada (NSREC).
Conflict of interest statement. None declared.
REFERENCES
- 1. Bentley D.L. Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet. 2014; 15:163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bresson S., Tollervey D.. Surveillance-ready transcription: nuclear RNA decay as a default fate. Open Biol. 2018; 8:170270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andrulis E.D., Werner J., Nazarian A., Erdjument-Bromage H., Tempst P., Lis J.T.. The RNA processing exosome is linked to elongating RNA polymerase II in Drosophila. Nature. 2002; 420:837–841. [DOI] [PubMed] [Google Scholar]
- 4. Chekanova J.A., Gregory B.D., Reverdatto S.V., Chen H., Kumar R., Hooker T., Yazaki J., Li P., Skiba N., Peng Q. et al. Genome-wide high-resolution mapping of exosome substrates reveals hidden features in the Arabidopsis transcriptome. Cell. 2007; 131:1340–1353. [DOI] [PubMed] [Google Scholar]
- 5. Lemay J.F., D’Amours A., Lemieux C., Lackner D.H., St-Sauver V.G., Bahler J., Bachand F.. The nuclear poly(A)-binding protein interacts with the exosome to promote synthesis of noncoding small nucleolar RNAs. Mol. Cell. 2010; 37:34–45. [DOI] [PubMed] [Google Scholar]
- 6. Mitchell P., Petfalski E., Shevchenko A., Mann M., Tollervey D.. The exosome: a conserved eukaryotic RNA processing complex containing multiple 3′–>5′ exoribonucleases. Cell. 1997; 91:457–466. [DOI] [PubMed] [Google Scholar]
- 7. Januszyk K., Lima C.D.. The eukaryotic RNA exosome. Curr. Opin. Struct. Biol. 2014; 24:132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sikorska N., Zuber H., Gobert A., Lange H., Gagliardi D.. RNA degradation by the plant RNA exosome involves both phosphorolytic and hydrolytic activities. Nat. Commun. 2017; 8:2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu Q., Greimann J.C., Lima C.D.. Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell. 2006; 127:1223–1237. [DOI] [PubMed] [Google Scholar]
- 10. Makino D.L., Baumgartner M., Conti E.. Crystal structure of an RNA-bound 11-subunit eukaryotic exosome complex. Nature. 2013; 495:70–75. [DOI] [PubMed] [Google Scholar]
- 11. Zinder J.C., Lima C.D.. Targeting RNA for processing or destruction by the eukaryotic RNA exosome and its cofactors. Genes Dev. 2017; 31:88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lebreton A., Tomecki R., Dziembowski A., Seraphin B.. Endonucleolytic RNA cleavage by a eukaryotic exosome. Nature. 2008; 456:993–996. [DOI] [PubMed] [Google Scholar]
- 13. Schneider C., Leung E., Brown J., Tollervey D.. The N-terminal PIN domain of the exosome subunit Rrp44 harbors endonuclease activity and tethers Rrp44 to the yeast core exosome. Nucleic Acids Res. 2009; 37:1127–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wasmuth E.V., Januszyk K., Lima C.D.. Structure of an Rrp6-RNA exosome complex bound to poly(A) RNA. Nature. 2014; 511:435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mukherjee K., Gardin J., Futcher B., Leatherwood J.. Relative contributions of the structural and catalytic roles of Rrp6 in exosomal degradation of individual mRNAs. RNA. 2016; 22:1311–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wasmuth E.V., Lima C.D.. The Rrp6 C-terminal domain binds RNA and activates the nuclear RNA exosome. Nucleic Acids Res. 2017; 45:846–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Allmang C., Petfalski E., Podtelejnikov A., Mann M., Tollervey D., Mitchell P.. The yeast exosome and human PM-Scl are related complexes of 3′ –>5′ exonucleases. Genes Dev. 1999; 13:2148–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kilchert C., Wittmann S., Vasiljeva L.. The regulation and functions of the nuclear RNA exosome complex. Nat. Rev. Mol. Cell Biol. 2016; 17:227–239. [DOI] [PubMed] [Google Scholar]
- 19. St-Andre O., Lemieux C., Perreault A., Lackner D.H., Bahler J., Bachand F.. Negative regulation of meiotic gene expression by the nuclear poly(a)-binding protein in fission yeast. J. Biol.Chem. 2010; 285:27859–27868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamanaka S., Yamashita A., Harigaya Y., Iwata R., Yamamoto M.. Importance of polyadenylation in the selective elimination of meiotic mRNAs in growing S. pombe cells. EMBO J. 2010; 29:2173–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Basu U., Meng F.L., Keim C., Grinstein V., Pefanis E., Eccleston J., Zhang T., Myers D., Wasserman C.R., Wesemann D.R. et al. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell. 2011; 144:353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pefanis E., Wang J., Rothschild G., Lim J., Chao J., Rabadan R., Economides A.N., Basu U.. Noncoding RNA transcription targets AID to divergently transcribed loci in B cells. Nature. 2014; 514:389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boczonadi V., Muller J.S., Pyle A., Munkley J., Dor T., Quartararo J., Ferrero I., Karcagi V., Giunta M., Polvikoski T. et al. EXOSC8 mutations alter mRNA metabolism and cause hypomyelination with spinal muscular atrophy and cerebellar hypoplasia. Nat. Commun. 2014; 5:4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wan J., Yourshaw M., Mamsa H., Rudnik-Schoneborn S., Menezes M.P., Hong J.E., Leong D.W., Senderek J., Salman M.S., Chitayat D. et al. Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nat. Genet. 2012; 44:704–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schmidt K., Butler J.S.. Nuclear RNA surveillance: role of TRAMP in controlling exosome specificity. Wiley Interdiscipl. Rev. RNA. 2013; 4:217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Falk S., Bonneau F., Ebert J., Kogel A., Conti E.. Mpp6 Incorporation in the nuclear exosome contributes to RNA channeling through the Mtr4 helicase. Cell Rep. 2017; 20:2279–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schuller J.M., Falk S., Fromm L., Hurt E., Conti E.. Structure of the nuclear exosome captured on a maturing preribosome. Science. 2018; 360:219–222. [DOI] [PubMed] [Google Scholar]
- 28. Wasmuth E.V., Zinder J.C., Zattas D., Das M., Lima C.D.. Structure and reconstitution of yeast Mpp6-nuclear exosome complexes reveals that Mpp6 stimulates RNA decay and recruits the Mtr4 helicase. eLife. 2017; 6:e29062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Egan E.D., Braun C.R., Gygi S.P., Moazed D.. Post-transcriptional regulation of meiotic genes by a nuclear RNA silencing complex. RNA. 2014; 20:867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee N.N., Chalamcharla V.R., Reyes-Turcu F., Mehta S., Zofall M., Balachandran V., Dhakshnamoorthy J., Taneja N., Yamanaka S., Zhou M. et al. Mtr4-like protein coordinates nuclear RNA processing for heterochromatin assembly and for telomere maintenance. Cell. 2013; 155:1061–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou Y., Zhu J., Schermann G., Ohle C., Bendrin K., Sugioka-Sugiyama R., Sugiyama T., Fischer T.. The fission yeast MTREC complex targets CUTs and unspliced pre-mRNAs to the nuclear exosome. Nat. Commun. 2015; 6:7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lubas M., Christensen M.S., Kristiansen M.S., Domanski M., Falkenby L.G., Lykke-Andersen S., Andersen J.S., Dziembowski A., Jensen T.H.. Interaction profiling identifies the human nuclear exosome targeting complex. Mol. Cell. 2011; 43:624–637. [DOI] [PubMed] [Google Scholar]
- 33. Meola N., Domanski M., Karadoulama E., Chen Y., Gentil C., Pultz D., Vitting-Seerup K., Lykke-Andersen S., Andersen J.S., Sandelin A. et al. Identification of a nuclear exosome decay pathway for processed transcripts. Mol. Cell. 2016; 64:520–533. [DOI] [PubMed] [Google Scholar]
- 34. Knight J.R., Bastide A., Peretti D., Roobol A., Roobol J., Mallucci G.R., Smales C.M., Willis A.E.. Cooling-induced SUMOylation of EXOSC10 down-regulates ribosome biogenesis. RNA. 2016; 22:623–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Synowsky S.A., van den Heuvel R.H., Mohammed S., Pijnappel P.W., Heck A.J.. Probing genuine strong interactions and post-translational modifications in the heterogeneous yeast exosome protein complex. Mol. Cell Proteomics. 2006; 5:1581–1592. [DOI] [PubMed] [Google Scholar]
- 36. Lemay J.F., Larochelle M., Marguerat S., Atkinson S., Bahler J., Bachand F.. The RNA exosome promotes transcription termination of backtracked RNA polymerase II. Nat. Struct. Mol. Biol. 2014; 21:919–926. [DOI] [PubMed] [Google Scholar]
- 37. Bahler J., Wu J.Q., Longtine M.S., Shah N.G., McKenzie A. 3rd, Steever A.B., Wach A., Philippsen P., Pringle J.R.. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998; 14:943–951. [DOI] [PubMed] [Google Scholar]
- 38. Lemieux C., Marguerat S., Lafontaine J., Barbezier N., Bahler J., Bachand F.. A Pre-mRNA degradation pathway that selectively targets intron-containing genes requires the nuclear poly(A)-binding protein. Mol. Cell. 2011; 44:108–119. [DOI] [PubMed] [Google Scholar]
- 39. Mathieu A.A., Ohl-Seguy E., Dubois M.L., Jean D., Jones C., Boudreau F., Boisvert F.M.. Subcellular proteomics analysis of different stages of colorectal cancer cell lines. Proteomics. 2016; 16:3009–3018. [DOI] [PubMed] [Google Scholar]
- 40. Boisvert F.M., Lam Y.W., Lamont D., Lamond A.I.. A quantitative proteomics analysis of subcellular proteome localization and changes induced by DNA damage. Mol. Cell Proteomics. 2010; 9:457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Toussaint M., Conconi A.. High-throughput and sensitive assay to measure yeast cell growth: a bench protocol for testing genotoxic agents. Nat. Protoc. 2006; 1:1922–1928. [DOI] [PubMed] [Google Scholar]
- 42. Lemay J.F., Marguerat S., Larochelle M., Liu X., van Nues R., Hunyadkurti J., Hoque M., Tian B., Granneman S., Bahler J. et al. The Nrd1-like protein Seb1 coordinates cotranscriptional 3′ end processing and polyadenylation site selection. Genes Dev. 2016; 30:1558–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Domanski M., LaCava J.. RNA degradation assay using RNA exosome complexes, Affinity-purified from HEK-293 cells. Bio. Protoc. 2017; 7:e2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zinder J.C., Wasmuth E.V., Lima C.D.. Nuclear RNA exosome at 3.1 A reveals substrate specificities, RNA paths, and allosteric inhibition of Rrp44/Dis3. Mol. Cell. 2016; 64:734–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tasto J.J., Carnahan R.H., McDonald W.H., Gould K.L.. Vectors and gene targeting modules for tandem affinity purification in Schizosaccharomyces pombe. Yeast. 2001; 18:657–662. [DOI] [PubMed] [Google Scholar]
- 46. Schwanhausser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M.. Global quantification of mammalian gene expression control. Nature. 2011; 473:337–342. [DOI] [PubMed] [Google Scholar]
- 47. Tsvetanova N.G., Klass D.M., Salzman J., Brown P.O.. Proteome-wide search reveals unexpected RNA-binding proteins in Saccharomyces cerevisiae. PLoS One. 2010; 5:e12671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Khemici V., Linder P.. RNA helicases in RNA decay. Biochem. Soc. Trans. 2018; 46:163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wilson-Grady J.T., Villen J., Gygi S.P.. Phosphoproteome analysis of fission yeast. J. Proteome Res. 2008; 7:1088–1097. [DOI] [PubMed] [Google Scholar]
- 50. Pearlman S.M., Serber Z., Ferrell J.E. Jr. A mechanism for the evolution of phosphorylation sites. Cell. 2011; 147:934–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen H.M., Futcher B., Leatherwood J.. The fission yeast RNA binding protein Mmi1 regulates meiotic genes by controlling intron specific splicing and polyadenylation coupled RNA turnover. PLoS One. 2011; 6:e26804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. de la Cruz J., Kressler D., Tollervey D., Linder P.. Dob1p (Mtr4p) is a putative ATP-dependent RNA helicase required for the 3′ end formation of 5.8S rRNA in Saccharomyces cerevisiae. EMBO J. 1998; 17:1128–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lorentzen E., Basquin J., Tomecki R., Dziembowski A., Conti E.. Structure of the active subunit of the yeast exosome core, Rrp44: diverse modes of substrate recruitment in the RNase II nuclease family. Mol. Cell. 2008; 29:717–728. [DOI] [PubMed] [Google Scholar]
- 54. Carpy A., Krug K., Graf S., Koch A., Popic S., Hauf S., Macek B.. Absolute proteome and phosphoproteome dynamics during the cell cycle of Schizosaccharomyces pombe (Fission Yeast). Mol. Cell Proteomics. 2014; 13:1925–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kettenbach A.N., Deng L., Wu Y., Baldissard S., Adamo M.E., Gerber S.A., Moseley J.B.. Quantitative phosphoproteomics reveals pathways for coordination of cell growth and division by the conserved fission yeast kinase pom1. Mol. Cell Proteomics. 2015; 14:1275–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Koch A., Krug K., Pengelley S., Macek B., Hauf S.. Mitotic substrates of the kinase aurora with roles in chromatin regulation identified through quantitative phosphoproteomics of fission yeast. Sci. Signal. 2011; 4:rs6. [DOI] [PubMed] [Google Scholar]
- 57. Schweiger R., Linial M.. Cooperativity within proximal phosphorylation sites is revealed from large-scale proteomics data. Biol. Direct. 2010; 5:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yachie N., Saito R., Sugahara J., Tomita M., Ishihama Y.. In silico analysis of phosphoproteome data suggests a rich-get-richer process of phosphosite accumulation over evolution. Mol. Cell Proteomics. 2009; 8:1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Amoutzias G.D., He Y., Lilley K.S., Van de Peer Y., Oliver S.G.. Evaluation and properties of the budding yeast phosphoproteome. Mol. Cell Proteomics. 2012; 11:doi:10.1074/mcp.M111.009555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li Y., Zhou X., Zhai Z., Li T.. Co-occurring protein phosphorylation are functionally associated. PLoS Comput. Biol. 2017; 13:e1005502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Daniels C.M., Ong S.E., Leung A.K.. The promise of proteomics for the study of ADP-ribosylation. Mol. Cell. 2015; 58:911–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tran D.T., Cavett V.J., Dang V.Q., Torres H.L., Paegel B.M.. Evolution of a mass spectrometry-grade protease with PTM-directed specificity. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:14686–14691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Snee M.J., Wilson W.C., Zhu Y., Chen S.Y., Wilson B.A., Kseib C., O’Neal J., Mahajan N., Tomasson M.H., Arur S. et al. Collaborative control of cell cycle progression by the RNA exonuclease Dis3 and Ras is conserved across species. Genetics. 2016; 203:749–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hornbeck P.V., Zhang B., Murray B., Kornhauser J.M., Latham V., Skrzypek E.. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015; 43:D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bonneau F., Basquin J., Ebert J., Lorentzen E., Conti E.. The yeast exosome functions as a macromolecular cage to channel RNA substrates for degradation. Cell. 2009; 139:547–559. [DOI] [PubMed] [Google Scholar]
- 66. Dziembowski A., Lorentzen E., Conti E., Seraphin B.. A single subunit, Dis3, is essentially responsible for yeast exosome core activity. Nat. Struct. Mol. Biol. 2007; 14:15–22. [DOI] [PubMed] [Google Scholar]
- 67. Wasmuth E.V., Lima C.D.. Exo- and endoribonucleolytic activities of yeast cytoplasmic and nuclear RNA exosomes are dependent on the noncatalytic core and central channel. Mol. Cell. 2012; 48:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Taylor L.L., Jackson R.N., Rexhepaj M., King A.K., Lott L.K., van Hoof A., Johnson S.J.. The Mtr4 ratchet helix and arch domain both function to promote RNA unwinding. Nucleic Acids Res. 2014; 42:13861–13872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Weir J.R., Bonneau F., Hentschel J., Conti E.. Structural analysis reveals the characteristic features of Mtr4, a DExH helicase involved in nuclear RNA processing and surveillance. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:12139–12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Guo X., Huang X., Chen M.J.. Reversible phosphorylation of the 26S proteasome. Protein Cell. 2017; 8:255–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Will C.L., Luhrmann R.. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011; 3:a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Beaulieu Y.B., Kleinman C.L., Landry-Voyer A.M., Majewski J., Bachand F.. Polyadenylation-dependent control of long noncoding RNA expression by the poly(A)-binding protein nuclear 1. PLoS Genet. 2012; 8:e1003078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bresson S.M., Conrad N.K.. The human nuclear poly(a)-binding protein promotes RNA hyperadenylation and decay. PLoS Genet. 2013; 9:e1003893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Andersen P.R., Domanski M., Kristiansen M.S., Storvall H., Ntini E., Verheggen C., Schein A., Bunkenborg J., Poser I., Hallais M. et al. The human cap-binding complex is functionally connected to the nuclear RNA exosome. Nat. Struct. Mol. Biol. 2013; 20:1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.