


Abstract
DYRK1A, dual-specificity tyrosine phosphorylation-regulated kinase 1A, which is linked to mental retardation and microcephaly, is a member of the CMGC group of kinases. It has both cytoplasmic and nuclear functions, however, molecular mechanisms of how DYRK1A regulates gene expression is not well understood. Here, we identify two histone acetyltransferases, p300 and CBP, as interaction partners of DYRK1A through a proteomics study. We show that overexpression of DYKR1A causes hyperphosphorylation of p300 and CBP. Using genome-wide location (ChIP-sequencing) analysis of DYRK1A, we show that most of the DYRK1A peaks co-localize with p300 and CBP, at enhancers or near the transcription start sites (TSS). Modulation of DYRK1A, by shRNA mediated reduction or transfection mediated overexpression, leads to alteration of expression of downstream located genes. We show that the knockdown of DYRK1A results in a significant loss of H3K27acetylation at these enhancers, suggesting that DYRK1A modulates the activity of p300/CBP at these enhancers. We propose that DYRK1A functions in enhancer regulation by interacting with p300/CBP and modulating their activity. Overall, DYRK1A function in the regulation of enhancer activity provides a new mechanistic understanding of DYRK1A mediated regulation of gene expression, which may help in better understanding of the roles of DYRK1A in human pathologies.
INTRODUCTION
DYRK1A is a highly conserved protein kinase of the CMGC group of proline-directed kinases (1). The DYRK1A gene is located on chromosome 21 in the Down Syndrome Critical Region (DSCR), a region associated with Down syndrome phenotype in human trisomy. Overexpression of DYRK1A in human trisomy is considered to be one of the leading causes of development of Down Syndrome phenotype. Children with Down Syndrome exhibit a 20-fold higher incidence of leukemia, and a link between overexpression of DYRK1A and development of megakaryoblastic leukemia in mouse has been established (2). DYRK1A mutations in humans have been associated with general growth retardation, reduced brain volume (3,4), craniofacial abnormality, behavior and motor alterations (5). Studies with knockout mice have shown that Dyrk1a is critical for development, and homozygotes die at embryonic stages. Heterozygote mice are smaller, and exhibit alterations in behavior, with structural defects in brain (6,7). In Drosophila, mutations in DYRK1A homologue, minibrain(mnb), leads to reduced brain volume and behavioral defects (8). Moreover, RNAi mediated tissue-specific reduction of mnb leads to smaller legs and wings (9). Therefore, DYRK1A is considered to be a critical regulator of brain growth (10), and based on growth retardation in Dyrk1a heterozygous mice and mutant flies, DYRK1A could be a much broader regulator for growth.
A number of interaction partners of DYRK1A has been identified in the past decade, including DCAF7, ARIP4, NFATc1, GSK3B, Lin52, p53, Tau and RNA polymerase II (RNA pol II) C terminal domain (CTD) (11). However, we know little about the functions of DYRK1A within the nucleus and how it may regulate transcription. Two recent reports revealed chromatin functions of DYRK1A and showed that DYRK1A localizes at TSS of its target genes. Di Vona et al. reported that DYRK1A interacted with RNA pol II and phosphorylates CTD and thus promoted transcription (12). Jang et al. demonstrated that DYRK1A phosphorylated histone 3 (H3T45 and H3S57) at promoters of inducible genes (13). However, a detailed analysis of DYRK1A localization on chromatin and its target genes is needed.
CBP (CREBBP) and p300 (EP300) are two closely related Histone acetyltransferases (HAT) required for acetylation of multiple residues on histones, including H3K27acetylation. CBP/p300 function as transcriptional coactivators and promote transcription through relaxing chromatin structure at promoters and recruiting transcription machinery (14). Majority of CBP and p300 binding sites localize at enhancers of their target genes, and currently, p300/CBP localization along with H3K27 acetylation and H3K4 monomethylation (H3K4me1) are considered to be markers of active enhancers (15). Both CBP and p300 are known to interact with a vast repertoire of transcription factors and function as coactivators for a broad range of genes involved in cellular processes including cell proliferation, differentiation, and various signaling pathways (16). These transcription factors recruit CBP/p300 to their respective target enhancers and mediate activation of transcription.
Here, in this study, we have identified CBP and p300 as interaction partners of DYRK1A and, using ChIP-seq analysis, show that DYRK1A co-localizes with CBP and p300 on enhancers or promoters of its target sites. We propose that DYRK1A may serve as transcription factor to promote the HAT activity of p300/CBP at the enhancers and thus regulate the target gene expression.
MATERIALS AND METHODS
Expression plasmids and cell culture
HA-CBP and Myc-p300 plasmids were gifts from Dr Hou Zhaoyuan. Dyrk1a cDNA (BC129888.1) was from K.K. DNAFORM (Japan) and cloned into pCDNA5/FRT-TO vector (Invitrogen) with an N-terminal Flag tag. All constructs were confirmed by DNA sequencing. To generate inducible cell lines, pcDNA-Flag-Dyrk1a or pcDNA-Flag-Dyrk1a-KD (K188R) were transfected into 293 Flp-in-TRex cells and selected for integration with hygromycin. Expression of Flag-tagged proteins was induced with 1 μg/ml Doxycycline for 36 to 48 h. HEK293, HEK293T, 293 Flp-in-TRex cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% FBS. T98G cells were cultured in MEM. All cell lines were obtained from the American Type Culture Collection (ATCC). All cell lines were maintained at 37°C under 5% CO2.
Antibodies
Antibodies against Actin, Tubulin and Flag were from Sigma, CBP from Diagenode (C15410224) for western blots, Novus (NB100-381) and ThermoFisher (PA5-27369) for ChIP, p300 from Santa Cruz (SC585) or from Invitrogen (PA1-848), H3K27ac was from Abcam (ab4729). Pan-phospho antibody was from Abcam (ab17464). Rabbit antisera were raised against two different Dyrk1a peptides—1598-HSHQYSDRRQPNISDQQVSALSYSD QIQQPLTNQ, 1602-TYQFSANTGPAHYMTEGHLTM RQGADREES. Antibodies were validated by shRNA mediated knockdown of DYRK1A (Supplemental Figure S1).
Immunoblotting
For immunoblotting, total cell extracts were prepared. Briefly, collected cells were washed once with PBS, and lysed in lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% TritonX 100, 0.25% sodium deoxycholate, 1 mM EDTA + fresh protease inhibitor Sigma P8340) for 15 min on ice. Lysates were cleared by centrifugation, and protein concentration was measured using BCA kit (Pierce). Western blot signals were quantitated using ImageJ software.
Flag purification and MudPIT analysis
Approximately 109 cells for each cell line were collected and washed with PBS. Cells were swollen for 15 min in hypertonic buffer (Buffer A: 10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT) freshly supplemented with protease inhibitor cocktail (Sigma P8340). Swollen cells were dounced 20 times in a Wheaton dounce homogenizer till about 90% cells were lysed (as observed in a microscope). Lysate is then centrifuged at 5 Kg for 20 min, to pellet Nuclei. For nuclear extract, the pellet is extracted in Buffer C (20 mM HEPES pH 7.9, 10% glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA and 0.5 mM DTT + fresh protease inhibitors). After incubation for 30 min at 4°C, nuclear extract was cleared at ∼100 kg for 1 h. Salt concentration in nuclear lysate was brought down to 300 mM, by addition of Buffer C without salt. Balanced nuclear extract is then incubated in presence of Benzonase (Sigma) with Flag beads for 3–4 h. Beads were then washed with Flag wash buffer (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 300 mM NaCl, 10 mM KCl, 0.2% TritonX-100) three times, and then eluted two times in elution buffer (200 μg/ml Flag peptide, 10 mM HEPES pH 7.9, 0.1 M NaCl, 1.5 mM MgCl2, 0.05% TritonX-100). Eluates were analysed by silver staining and Western blotting. Trichloroacetic acid-precipitated protein mixtures from the Flag purifications were digested with endoproteinase Lys-C and trypsin (Roche) and analysed by MudPIT as previously described (17).
Immunoprecipitations
HEK293T cells were washed once with PBS and lysed in a lysis buffer (20 mM HEPES [pH 7.4], 150 mM NaCl, 1 mM MgCl2, 0.5% Triton X-100, 1 mM dithiothreitol [DTT]) containing proteinase inhibitors (Sigma) for 30 min at 4°C. After centrifugation at 17 Kg for 15 min, cleared lysate were incubated with antibodies and protein A beads (Invitrogen) for 3 h at 4°C with gentle rotation. Beads were washed three times with wash buffer (10 mM HEPES [pH 7.4], 1 mM MgCl2, 150 mM NaCl, 10 mM KCl, 0.2% Triton X-100) and eluted in SDS loading buffer.
Chromatin immunoprecipitation (ChIP) and ChIP-Seq analyses
ChIP was performed as previously described (18). Briefly, cells were cross-linked with 1% formaldehyde for 10 min at room temperature, followed by quenching with 125 mM glycine. After washing with PBS three times, cells were flash frozen in liquid nitrogen and stored at –80°C till processed. Cells were thawed at 4°C, and sonicated in lysis buffer using probe sonicator, and fragment length determined by agarose gel electrophoresis. After incubation with beads and antibodies, and washes, ChIP DNA was eluated with 50μl Elution buffer. Eluate was further diluted to 100 μl in water. ChIP products were (2 μl in each reaction mixture) analysed by qPCR using SYBR green on Roche qPCR machine as detailed in the RT-qPCR section, except that primer concentrations were 0.2 μM. The comparative cycle threshold method was used to determine enrichment relative to the level of input. Antibodies used in ChIP are—H3K27ac from Abcam (ab4729) and DYRK1A (1598). For CBP ChIP, we combined antibodies from Novus (NB100-381; Lot A1) and ThermoFisher (PA5-27369; Lot No. TG259977913) (12 and 8 μl, respectively for each ChIP).
ChIP-Seq analyses was performed using publicly available programs. After quality control and data filtering, sequences were mapped to hg19 using Bowtie1 alignment algorithm. ChIP-enriched regions were determined by the MACS1.4 (19) peak-finding program. Sequence reads for each ChIP-Seq data set and their associated IgG control were used for the input and control file, respectively. VENN diagram was generated using ‘bedtools intersect’. Gene ontology analysis and KEGG pathway analysis were performed using DAVID 6.7.
shRNA mediated knockdown
Short hairpin RNA (shRNA) targeting p300, DYRK1A, and CBP were purchased from ThermoFisher. RT-qPCR primers and shRNA sequences are provided in the supplemental. Lentiviral particle preparation and infection were performed as previously described (18). Infected T98G or HEK293 cells were selected with 1 μg/ml puromycin for 4 days before harvesting.
Quantitative reverse transcription-polymerase chain reaction (RT-qPCR) assays
Total RNA from fresh cells were extracted using TRIzol (Invitrogen) as per the manufacturer's instructions. Fresh RNA was digested with RNase-free DNase I (NEB) to remove residual DNA. After determining the total RNA concentration and quality using a Nanodrop (Agilent), 1 μg RNA was used in 10 μl reaction mixture to reverse transcribe RNA using TOYOBO ReverTra Ace Kit (FSQ-101) kit and stored at –20°C. After 10-fold dilution of synthesized cDNA, 2 μl was used to perform qPCR using SYBR® Green Realtime PCR Master Mix (TOYOBO QPK-201) with 0.1 μM forward and reverse gene specific primers. The cycling consisted of 2 min at 95°C, followed by 40 cycles of 5 s at 95°C and 10 s at 60°C. Following completion of the final cycle, a melting curve analysis was performed to monitor the purity of the PCR products. Each sample was analyzed in triplicate. Quantitation was performed by means of the comparative Ct method (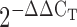 ). The mRNA expression of target genes were normalized to that of GAPDH, and the data are represented as the mean ± SD of biological replicates.
). The mRNA expression of target genes were normalized to that of GAPDH, and the data are represented as the mean ± SD of biological replicates.
Statistical analysis
All statistical data were presented as the mean ± SD of at least three independent experiments. All the analysis was performed using GraphPad Prism software version 7. The significance of differences between the experimental group and the control group was analyzed using Student's t test. Summary of P value = ns (P > 0.05), * (P < 0.05).
Cell proliferation assays
To monitor the number of live THP1 cells (suspension culture) in cell culture, live cells were counted by using a hemocytometer. To measure cell proliferation in HEK293 cells, a Cell Counting Kit-8 (Dojin Laboratories) was used.
Mass spectrometry dataset accessibility
Original data underlying this manuscript can be accessed after publication from the Stowers Original Data Repository at http://www.stowers.org/research/publications/libpb-1222
GEO accession numbers
DYRK1A ChIP-seq data (T98G and HeLa) are from GEO accession number GSE63712 (12); p300 and CBP ChIP-seq data (T98G) are from GEO accession number GSE21026 (20).
RESULTS
DYRK1A interacts with histone acetyltransferases CBP and p300
To investigate the interactions of DYRK1A in the nucleus and provide a better mechanistic understanding of DYRK1A functions at the chromatin (12), we generated an inducible Flag-DYRK1A expression HEK293 cell line. We performed Flag-affinity purification from the nuclear lysate and analysed the Flag-DYRK1A eluate by SDS-PAGE. Silver stained gel showed a strong enrichment of large number of proteins in the WT-DYRK1A flag eluates, compared to parental cell line eluates (Figure 1A). Eluates were then subjected to MudPIT analysis for identification of proteins. A number of known DYRK1A interaction partners were identified, including DDB1 and CUL4 Associated Factor 7 (DCAF7), Androgen receptor-interacting protein 4 (ARIP4), and Retinoblastoma 1 (RB1), demonstrating the efficacy of our affinity purification in identifying interaction partners of DYRK1A. Most interestingly, we identified a large number of peptides from CBP and p300, two histone acetyltransferases (Figure 1B). Mapping of these peptides to the CBP and p300 proteins revealed that peptides span the entire length of the proteins, indicating the validity of the interaction between DYRK1A and CBP/p300 (not shown). Moreover, affinity purification of catalytically inactive Flag-DYRK1A (K188R mutant), hereafter F-DYRK1A-KD, also identified a large number of p300 and CBP peptides. To confirm the interaction, we performed co-immunoprecipitation experiments, and found that indeed endogenous CBP and p300 are pulled down by F-DYRK1A (Figure 1C) and F-DYRK1A-KD (Figure 1D). Taken together, our data suggests that DYRK1A interacts with CBP and p300.
Figure 1.

DYRK1A co-purifies with p300 and CBP. (A) A clonal HEK293 cell line inducibly expressing Flag-DYRK1A was generated, and nuclear extracts prepared followed by affinity purification. Eluates were analysed by SDS-PAGE and mass spectrometry. Arrow marks the location of DYRK1A in the silver stained gel. (B) MudPIT analyses of the relative abundance of DYRK1A and highly enriched proteins are shown. The distributed Normalized Spectral Abundance Factor (dNSAF) reflects the relative abundance of the identified proteins in the samples (44). Many of the previously identified DYRK1A interacting proteins were reproducibly identified, including DCAF7 and ARIP4. These interactions were also observed with the kinase-dead DYRK1A-KD mutant. (C) Flag affinity pull-down of Flag-DYRK1A and 293T control whole cell extracts were probed with CBP and p300 antibodies, confirming interaction between DYRK1A and CBP/p300. (D) Flag affinity pull-down of Flag-DYRK1A-WT, Flag-DYRK1A-KD and 293T control whole cell extracts were probed with CBP and p300 antibodies, to compare the interaction efficiency of DYRK1A-WT and DYRK1A-KD with CBP/p300.
DYRK1A exogenous expression causes hyper-phosphorylation of p300 and CBP
Interestingly, overexpressed WT DYRK1A leads to a small upshift of p300 and CBP proteins on the western blot (Figure 1C and D), whereas the catalytically inactive F-DYRK1A-KD did not (Figure 1D). As DYRK1A is known to phosphorylate some of its substrates at multiple residues (21–23), we reasoned that DYRK1A may be phosphorylating p300 and CBP at multiple residues, leading to their reduced migration on SDS denaturing gel. Co-overexpression of DYRK1A with either p300 or CBP resulted in significant upshift of exogenously expressed p300 and CBP proteins in the SDS PAGE gel (Figure 2A and B). To test if the upshifted p300 and CBP signals are indeed phosphorylated p300/CBP and are not other post-translational modifications such as sumoylation or ubiquitination, we treated lysates with λ-phosphatase. The λ-phosphatase treatment of lysates made from cells overexpressing DYRK1A-WT led to increased migration of p300/CBP signals comparable to vector or DYRK1A-KD samples (Figure 2A and B). Moreover, an in vitro kinase assay, indicated that DYRK1A, and not kinase inactive mutant can significantly increase the phosphorylation of CBP, as probed using a pan-phospho antibody (Supplementary Figure S2). To determine the DYRK1A mediated potential phosphorylation sites on these extremely large proteins, we performed mass spectrometry analysis of CBP in DYRK1A overexpression cells, and identified five putative phosphorylation sites—S380, T1004, S2062, S2079 and S2420. However, mutating these sites did not rescue the mobility shift of CBP mediated by overexpressed DYRK1A (data not shown). There are two likely explanations, (i) DYRK1A may phosphorylate CBP at additional sites, which were missed by our MS analysis, and (ii) DYRK1A may promiscuously phosphorylate other sites when its substrate sites are mutated. Further, we analysed if DYRK1A may mediate phosphorylation of p300 at S1834, a site which is phosphorylated by AKT, and is proposed to regulate the activity of p300 (24). However, S1834 phosphorylation level was not altered by DYRK1A overexpression (data not shown). Taken together, the above results indicate that overexpression of DYRK1A leads to hyper-phosphorylation of p300 and CBP.
Figure 2.

Overexpression of DYRK1A leads to hyper-phosphorylation of CBP and p300. (A) Whole cell lysates from 293T cells co-transfected with Myc-p300 and Flag-DYRK1A-WT or Flag-DYRK1A-KD were treated with λ phosphatase, and analysed by western blot. Arrows indicate the slower migrating p300 signal in vector vs DYRK1A overexpression. (B) Whole cell lysates from 293T cells co-transfected with HA-CBP and Flag-DYRK1A-WT or Flag-DYRK1A-KD were treated with λ phosphatase, and analysed by western blot. Arrows indicate the slower migrating CBP signal in vector vs DYRK1A overexpression.
Since in Down Syndrome patients DYRK1A is overexpressed by 1.5-fold and CBP/p300 expression in Down Syndrome is unknown, we next analysed the effect of DYRK1A expression levels on the transcription of CBP or p300 genes and their corresponding protein levels. GAPDH expression was used as reference, as the gene has been shown to be a non-target of DYRK1A (12). Inducing overexpression of DYRK1A in Flag-DYRK1A 293 stable cells did not significantly alter the transcription of CBP and p300 genes (Supplementary Figure S3), suggesting that DYRK1A may not regulate the transcription of CBP or p300. Next, we analysed the effect of downregulation of DYRK1A on endogenous CBP and p300 levels. Reduction in DYRK1A levels by shRNA led to a reduction in p300 and CBP mRNA levels (Figure 3A), however, the levels of CBP and p300 proteins were only slightly affected (Figure 3B). This suggests that though DYRK1A might be involved in transcriptional regulation of CBP and p300 to some extent, protein levels of CBP and p300 may not be significantly affected by DYRK1A. Further, we analysed H3K27acetylation levels in DYRK1A knockdown cells, but found no obvious alteration in bulk acetylation levels (data not shown). Finally, we also analysed if p300 or CBP regulated the expression of DYRK1A, but found no significant changes in DYRK1A expression after knockdown of CBP or p300 (Figure 3C–F).
Figure 3.

Reduction in DYRK1A leads to a weak reduction in expression of p300 and CBP. (A) RT-qPCR analysis of CBP and p300 mRNA levels after knockdown of DYRK1A with shRNA in 293 cells. GAPDH mRNA was used to normalize RNA in RT-qPCR samples. Data represent the mean ± SD (n = 3 biological replicates). (B) Western blot analysis of p300, CBP and DYRK1A proteins levels in whole cell extracts, after knockdown of DYRK1A with shRNA in 293 cells (n = 3 biological replicates). Actin was used as loading control in all western samples. Right graph shows quantification of western signal as the mean ±SD (n = 3 biological replicates). Signals were quantitated using Image J software. (C and E) RT-qPCR analysis of DYRK1A mRNA levels after knockdown of CBP or p300 with shRNAs in 293 cells. GAPDH mRNA was used to normalize RNA in RT-qPCR samples. Data represent the mean ± SD (n = 3 biological replicates). (D and F) Western blot analysis of DYRK1A protein levels in whole cell extracts, after knockdown of CBP or p300 with shRNAs in 293 cells. Actin was used as a loading control in all western samples. (n = 3 biological replicates). Graph on the right shows quantitation as in figure B. Student's t test were done to compare samples. P value = * (P < 0.05).
DYRK1A co-localizes with p300 and CBP at enhancer regions
Various studies have reported the association of DYRK1A with chromatin, including localization at TSS where it carries out phosphorylation of RNA Polymerase II (12) and phosphorylation of H3T45 and H3S57 at promoters of inducible genes (13,25). To better understand the function of genome-wide localization of DYRK1A, we analysed the chromatin occupancy of DYRK1A using previously reported chromatin-immunoprecipitation-sequencing (ChIP-seq) data performed in T98G glioblastoma cell line (12). Our analysis of DYRK1A peak distribution in cycling cells identified 596 peaks using a P value of 10−8, the majority of which are located near the TSS, but ∼ 9% are located within the introns, and up to 29% are located in the intergenic region (not shown). These values are similar to the published values (12). Therefore, up to 40% of the peaks are not in the promoter region. Considering our observation that DYRK1A interacts with the two HATs, CBP and p300, which occupy enhancers either located near or distal to TSS, and in the intergenic/intragenic region, we hypothesized that DYRK1A co-occupies enhancer regions with CBP/p300. We analysed overlap in peaks of DYRK1A, p300 and CBP in T98G cells (12,20) and found that the majority (79.5%) of the DYRK1A peaks co-localized with either p300 alone (226 peaks), or with p300 and CBP (232 peaks) (Figure 4A). A non-significant number of sites (9 peaks) were co-occupied by DYRK1A and CBP. Moreover, heatmap analysis of DYRK1A, p300 and CBP at the DYRK1A-occupied sites showed that almost all sites were co-occupied by all the three factors (Figure 4C and D). We also analysed the localization of DYRK1A in HeLa cells, as previously reported (12), and found that about 64% peaks were located in intergenic regions (not shown). Our observations suggest that DYRK1A almost always localizes with either CBP or p300 on the chromatin. Manual inspection of DYRK1A peaks at intergenic regions located near many genes including DYNC1H1 (Figure 4E), KDM4C, WNT11, DNAJC12, ZNF692, FOXK2, and PLLP genes showed that DYRK1A co-occupies sites together with p300 and CBP, corroborating our observation that DYRK1A localizes to the enhancers of select group of genes. Moreover, two of these enhancer sites were also occupied by DYRK1A in HeLa cells (Figure 4E, bottom track). To further explore if DYRK1A localizes to enhancer regions in another cellular milieu, we performed ChIP in HEK293 cells, and analysed DYRK1A enrichment at the enhancer sites located upstream to DNAJC12, DYNC1H1 and WNT11 genes. DYRK1A indeed localized to these sites (Figure 4F). However, several other enhancer sites bound by DYRK1A in T98G cells were not occupied by HEK293 cells (data not shown). This was expected as enhancers are tissue specific, and show distinct binding patterns of enhancer proteins based on enhancer activity. Further, we also confirmed that the enhancer sites located near DNAJC12, DYNC1H1 and WNT11, bound by DYRK1A in HEK293 cells were also occupied by CBP and marked by H3K27acetylation, indicating that these regions indeed function as enhancers in HEK293 cells (Figure 4F). Taken together, our data suggests that DYRK1A localizes to enhancer regions in multiple cell systems.
Figure 4.

DYRK1A is associated with enhancer regions in T98G cells. (A) Venn diagram analysis of ChIP-seq data of DYRK1A, p300 and CBP in cycling T98G cells. Raw ChIP-seq data were obtained from GEO (GSE63712 and GSE21026) (12,20). (B) Profiles of CBP (black), p300 (red) centered on DYRK1A (yellow) peak. (C) Analysis of DYRK1A, p300 and CBP ChIP-seq data in cycling T98G cells. Occupancy levels of DYRK1A, p300 and CBP are shown. Profiles are centered on DYRK1A-occupied peaks (±10 kb) and sorted in descending order of DYRK1A occupancy. (D) Analysis of DYRK1A, p300 and CBP ChIP-seq data as in (C), but centered at TSS of the nearest gene and extends (±10 kb) on each side. (E) Genome browser track example showing CBP, p300 and DYRK1A ChIP-seq signal in T98G cells. Bottom track shows DYRK1A ChIP-seq signal in HeLa cells. 16 and 42 are arbitrary units depicting enrichment in genome tracks. (F) ChIP analysis shows enrichment of DYRK1A, CBP and H3K27acetylation at enhancer sites upstream to DNAJC12, WNT11, and DYNC1H1 in HEK293 cells. Data represent the mean ± SD (n = 3 biological replicates). Student's t test were done to compare samples. P value = * (P < 0.05). (G) GO term analysis of genes located near or downstream to DYRK1A occupied peaks.
In order to investigate if DYRK1A functions in specific biological pathways through chromatin occupancy, we performed DAVID GO enrichment analysis of genes nearest to DYRK1A peaks. We found that the genes co-occupied by DYRK1A, p300, and CBP were significantly enriched for the GO class of cell cycle function (Figure 4G and Supplementary Table S4). These genes included CYCLIN T1, NCAGP, NCAGP2, CDC20, CDC25B, CHAF1A, MAU2 and SMC4. Various studies have investigated function of DYRK1A in cell cycle, many of which have proposed a cell proliferation promoting function (12,26). Based on our findings, the function of DYRK1A promoting cell proliferation may partially be explained by occupancy of DYRK1A and CBP/p300 at promoters of cell cycle genes.
DYRK1A kinase activity is required for expression of its target genes
A previous study using microarray reported that DYRK1A knockdown by shRNA affected the expression of only 30% of the genes that bound DYRK1A at their promoter region (12). We then asked if localization of DYRK1A to enhancer regions affected the expression of genes that are located upstream or downstream of these enhancers. We chose a select group of genes, which exhibited well-defined DYRK1A peaks in their 5′ upstream regions/enhancer regions, including DNAJC12, ZNF692, WNT11, DYNC1H1, KDM4C, FOXK2, USPL1 and PLLP. We performed knockdown of DYRK1A, CBP or p300 in T98G cells, and interestingly, found that knockdown of DYRK1A resulted in robust down-regulation of half of the tested genes (Figure 5A). Interestingly, these genes also showed strong down-regulation in CBP or p300 knockdown samples. This is in agreement with previously published results that p300 and CBP are required for expression of only some genes, and not all, even if the gene promoters/promoter distal regions are bound by CBP/p300 (27,28). Our results suggest that DYRK1A may regulate the expression of these genes by modulating the CBP/p300 recruitment or function at the enhancers of these genes.
Figure 5.

Knockdown of DYRK1A affects the expression of genes located near its binding sites. (A) Expression analysis of a select group of genes located near the DYRK1A- CBP/p300 occupied regions, in T98G cells infected with lentivirus expressing shRNAs targeting control, DYRK1A, p300, or CBP. (B) Expression analysis of a select group of genes located near DYRK1A-CBP/p300 occupied regions in HEK293 cells, infected with lentivirus expressing shRNA targeting DYRK1A, p300 or CBP. Lentivirus infected cells were selected with 1μg/ml puromycin for 4 days before harvesting (A and B). (C) Expression analysis of genes analysed in (B) in HEK293 cells inducibly expressing Flag-DYRK1A. For induction, cells were treated with 250ng/ml Doxycycline for 40 h. (D) Expression analysis of genes analysed in (B) in HEK293 cells inducibly expressing Flag-DYRK1A-KD. For induction, cells were treated with 250 ng/ml Doxycycline for 40 h. GAPDH mRNA was used to normalize RNA in RT-qPCR samples. Data represent the mean ± SD (n = 3 biological replicates). Student's t test were done to compare samples. P value = * (P < 0.05).
We further investigated whether occupancy of DYRK1A at enhancer sites was also required for expression of downstream genes in another cell system, HEK293 cells, where we have confirmed the localization of DYRK1A at enhancer regions of DNAJC12, DYNC1H1 and WNT11 (Figure 4F). We performed knockdown of DYRK1A, p300 or CBP in HEK293 cells, and found that DYRK1A as well as CBP and p300 are required for proper expression of these genes (Figure 5B), suggesting that DYRK1A regulates expression of its target genes, in different cell systems. However, the requirement of CBP and/or p300 may be context specific.
Next, we tested if overexpression of DYRK1A can up-regulate putative target genes. Using the HEK293-Flag-DYRK1A inducible system, we found that all the three genes tested were upregulated (Figure 5C). We then examined whether the kinase function of DYRK1A is involved in the regulation of its target genes. We over-expressed kinase dead DYRK1A-KD using the inducible system and found that all the genes which were upregulated upon DYRK1A overexpression, were downregulated (Figure 5D). Taken together, our data suggests that the kinase activity of DYRK1A regulates the expression of a select group of genes by binding to their enhancers.
DYRK1A is required for H3K27 acetylation at DYRK1A-p300/CBP co-bound loci
CBP and p300 acetylate H3K27 at their binding sites on the genome and promote transcription (29,30). Considering our observation that DYRK1A co-localizes with p300/CBP at enhancer regions and regulates the expression of downstream genes, we hypothesized that DYRK1A may promote the histone acetyltransferase activity of CBP/p300 at these sites. We performed ChIP for DYRK1A, H3K27ac and CBP after knocking down DYRK1A by shRNA. DYRK1A enrichment at enhancers near DNAJC12, DYNC1H1 and WNT11 was significantly reduced in DYRK1A knockdown samples (Figure 6A). We also analysed the occupancy of DYRK1A, H3K27ac and CBP at promoters of RPS6 and CDK12, which were previously shown to be occupied by DYRK1A (12). Interestingly, H3K27ac enrichment at all these sites was significantly reduced, indicating a role for DYRK1A in activity and/or stability of HATs at these sites (Figure 6B). A reduction in H3K27ac at the promoter proximal regions of RPS6 and CDK12 suggests that DYRK1A may modulate the activity of CBP/p300 and alter the levels of H3K27ac at both enhancers and promoters. However, occupancy of CBP was either not altered at these sites, or was slightly higher in DYRK1A shRNA samples at these sites (Figure 6C). Taken together, above observations suggests that DYRK1A may modulate the histone acetylation activity of CBP, and possibly p300, but not their stability on these chromatin loci. Further, we investigated if knockdown of CBP affected the DYRK1A localization on the chromatin. Knockdown of CBP resulted in a decrease in enrichment of CBP (Figure 6D) and also of H3K27ac (Figure 6E). Interestingly, DYRK1A ChIP exhibited a decrease in enrichment at all but one loci (Figure 6F). These observations indicate that CBP may help stabilize DYRK1A on its target chromatin loci. Taken together, our data suggests that DYRK1A likely phosphorylates CBP on the target chromatin loci and promotes its catalytic and transcription activities.
Figure 6.

DYRK1A is required for p300/CBP mediated H3K27 acetylation at its target enhancers (A–C) ChIP analysis of DYRK1A, H3K27acetylation and CBP in cells treated with control or DYRK1A shRNA. DYRK1A bound enhancer sites (near DNAJC12, DYNC1H1 and WNT11) and at TSS (RPS6 and CDK12) were probed for enrichment. (D–F) ChIP analysis of CBP, H3K27acetylation and DYRK1A in cells treated with control or CBP shRNA. Sites probed as in (A–C). Data represent the mean ± SD (n = 3 biological replicates). Student's t test were done to compare samples. P value = * (P < 0.05).
DYRK1A knockdown affects proliferation of THP1 cells
Previously, multiple reports using overexpression studies have indicated that DYRK1A promotes quiescence in a number of cell types (5,26,31). Moreover, Chen et al. showed that DYRK1A knockdown affects proliferation of a subgroup of cells in a population (32). As our analysis of ChIP-seq data suggests that DYRK1A, CBP, and p300 co-binds cell cycle genes, we wondered if DYRK1A played a role in cell proliferation. We chose to study the effect of DYRK1A in proliferation of immune cells, where CBP and p300 have been previously shown to affect cell proliferation. Moreover, DYRK1A is highly expressed in myeloid cells (Human Protein Atlas Project) and in Down syndrome patients, who carry an extra copy of DYRK1A gene due to trisomy of chromosome 21 and have a 20-fold higher risk of developing leukaemia. We infected two different shRNA viruses each for DYRK1A, p300 and CBP, and performed a cell proliferation assay in THP1 cells. Knockdown of p300 and CBP reduced the rate of cell proliferation (Figure 7). Interestingly, DYRK1A knockdown also significantly reduced the rate of proliferation (Figure 7). Further, a similar requirement of DYRK1A, p300 and CBP for cell proliferation was also observed in HEK293 cells (Supplementary Figure S4). Taken together, reduction in DYRK1A by shRNA reveals its function in promotion of cell growth.
Figure 7.

Knockdown of DYRK1A, p300 and CBP in THP1 cells affects cell proliferation. THP1 cells were infected with shRNA lentivirus targeting DYRK1A, p300 and CBP, and sorted to isolate GFP expressing cells. Sorted cells were seeded in 6cm plates in triplicates. Cell proliferation was measured by counting number of cells using haemocytometer every 48 hours for six days. (A) Cell numbers are plotted. (B) Area under curve (AUC) analysis of data in (A). P values between control and experimental group were calculated using a two-tailed Student's t test. Data represent the mean ± SD (n = 3 biological replicates). P value = * (P < 0.05).
DISCUSSION
DYRK1A localizes within multiple organelles in the cell, however, our knowledge of its nuclear functions remains poorly understood. Here, using proteomics and ChIP-seq analysis, we show that DYRK1A interacts with the two major HATs, CBP and p300, and co-localizes with them on the chromatin. CBP and p300 are transcription coactivators which localize to both enhancers and TSS (20,33) and promote transcription activation. In T98G cells, up to ∼60% of the co-occupied sites are located near the promoters, while about 40% were located further upstream or downstream. Interestingly, nearly all DYRK1A occupied sites were occupied by both CBP and p300, both at TSS and in intergenic regions (Figure 4C and D). Moreover, all these sites also harboured H3K27 acetylation, which is a mark for enhancers and promoters (15).
Co-occupancy of DYRK1A with p300/CBP suggests that DYRK1A promotes gene transcription by modulating the function of CBP or p300. Previously, Di Vona et al. (12), showed that DYRK1A can phosphorylate RNA polymerase II, and promote transcription. It is possible that DYRK1A initially functions in modulating the activity of p300/CBP upon receiving the right stimulus or signaling for activating transcription and subsequently phosphorylates RNA polymerase II. Interestingly, half the genes located near DYRK1A bound peaks we analysed in this study, required DYRK1A for expression (Figure 5A–D). Also, most but not all the analysed genes required p300 or CBP for expression, which supports the current understanding that CBP and p300 may act redundantly at some chromatin loci (Figure 5A). Also, context, such as signaling, temporal, or developmental events, may play a role in determining if CBP or p300 function with DYRK1A at a given enhancer.
Previous studies have shown that CBP can be stimulated by phosphorylation (34,35), and phosphorylation of p300 at S1834 is essential for its HAT and transcriptional activity (24), whereas phosphorylation of p300 at S89 by PKC represses its function (36). Moreover, it was suggested that phosphorylation of p300 at S1038/S2039, destabilizes the protein (37). Interestingly, a previous report suggested that wild type DYRK1A, but not kinase dead DYRK1A, enhances RUNX1 mediated phosphorylation of p300 (38). However, in our hands, DYRK1A overexpression alone consistently lead to slower migration of both CBP and p300 in denaturing PAGE. Our ChIP-seq analysis of DYRK1A, CBP and p300 occupancy suggests that DYRK1A may phosphorylate p300/CBP on chromatin and may stimulate their acetyltransferase activity. However, due to the extreme size of p300/CBP, and the multiple possible phosphorylation sites, our multiple mapping efforts have failed to identify DYRK1A phosphorylation sites on CBP and p300. Using mass spectrometry, we have identified up to five potential phosphorylation sites, however, mutating these sites did not relive the DYRK1A mediated upshift of CBP (data not shown). Future investigations into the phosphorylation sites may provide further insight into the regulation of CBP/p300 activity.
Multiple laboratories have investigated the function of DYRK1A in cell cycle, where it has been proposed to act as an oncogene as well as a tumour suppressor. Studies with mnb, the Drosophila homolog of DYRK1A, have suggested that it is required for organ growth (8,39). In human and mice, altered DYRK1A protein levels have been associated with impaired brain development (10). Moreover, studies with mammalian cellular systems have also reported that DYRK1A promotes cell proliferation and tumour growth (2,40,41). However, a plethora of studies have found that DYRK1A is a negative regulator of cell cycle, and various mechanisms have been proposed to explain the phenomenon. In neuroepithelial cells, DYRK1A promotes expression of p27kip1, leading to a decrease in cell proliferation (42). DYRK1A phosphorylates LIN52 and facilitates the formation of cell cycle exit complex, DREAM complex (31). DYRK1A also prevents cell cycle entry by phosphorylating and degrading cyclin D1 (32). In addition, overexpression of DYRK1A in AML cells inhibit proliferation by downregulation of c-Myc (43). The differences in cell cycle investigation outcomes are surprising, but could arise due to dosage sensitive functions of DYRK1A, and also could be affected by cellular context. The majority of negative cell proliferation effects were observed in overexpression studies, whereas all cell promoting effects were observed in shRNA or knockout studies. In this study, we found that DYRK1A interacts with and modulates the function of two major histone acetyltransferases, CBP and p300, which are also known to regulate the expression of cell cycle genes and proliferation of myeloid cells. We observed that knockdown of DYRK1A significantly affects the proliferation rate of THP1 cells. Taken together with previous findings, it is likely that DYKR1A could regulate cell cycle by multiple mechanisms. Various cellular systems may utilize various aspects of DYRK1A functions to modulate cell cycle in a spatiotemporal manner, not only in a given cell type, but also during development and tumorigenesis.
Overall, our study provides a new mechanism by which DYRK1A regulates gene transcription through enhancer regulation. DYRK1A co-localizes with CBP/300 at enhancers of a select group of genes and regulates their expression. Our findings add new complexity to the DYRK1A biology and provides new direction for investigating the function of DYRK1A in development, and in human pathologies.
DATA AVAILABILITY
Original data underlying this manuscript can be accessed after publication from the Stowers Original Data Repository at http://www.stowers.org/research/publications/libpb-1222. DYRK1A ChIP-seq data is from GEO accession number GSE63712 (12); p300 and CBP ChIP-seq data are from GEO accession number GSE21026 (20).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Ali Shilatifard for providing initial support for starting the project. We also thank Drs Zhaoyuan Hou, Jinke Cheng and Y. Eugene Chin for helpful discussions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31471206] (in part); Shanghai Science and Technology grant [14JC1404000]; Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning, and Overseas introduction of non-Chinese Foreign Teacher Research Incentive Program from Shanghai Jiaotong University grant (to M.M.); Stowers Institute for Medical Research (to Y.Z.H., M.P.W. and L.F.); intramural funding of TIFR (to M.J). Funding for open access charge: Shanghai Science and Technology grant.
Conflict of interest statement. None declared.
REFERENCES
- 1. Becker W., Sippl W.. Activation, regulation, and inhibition of DYRK1A. FEBS J. 2011; 278:246–256. [DOI] [PubMed] [Google Scholar]
- 2. Malinge S., Bliss-Moreau M., Kirsammer G., Diebold L., Chlon T., Gurbuxani S., Crispino J.D.. Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of Down syndrome. J. Clin. Invest. 2012; 122:948–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Courcet J.B., Faivre L., Malzac P., Masurel-Paulet A., Lopez E., Callier P., Lambert L., Lemesle M., Thevenon J., Gigot N. et al. . The DYRK1A gene is a cause of syndromic intellectual disability with severe microcephaly and epilepsy. J. Med. Genet. 2012; 49:731–736. [DOI] [PubMed] [Google Scholar]
- 4. Moller R.S., Kubart S., Hoeltzenbein M., Heye B., Vogel I., Hansen C.P., Menzel C., Ullmann R., Tommerup N., Ropers H.H. et al. . Truncation of the Down syndrome candidate gene DYRK1A in two unrelated patients with microcephaly. Am. J. Hum. Genet. 2008; 82:1165–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kay L.J., Smulders-Srinivasan T.K., Soundararajan M.. Understanding the Multifaceted Role of Human Down Syndrome Kinase DYRK1A. Adv. Protein Chem. Struct. Biol. 2016; 105:127–171. [DOI] [PubMed] [Google Scholar]
- 6. Fotaki V., Dierssen M., Alcantara S., Martinez S., Marti E., Casas C., Visa J., Soriano E., Estivill X., Arbones M.L.. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol. Cell. Biol. 2002; 22:6636–6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fotaki V., Martinez De Lagran M., Estivill X., Arbones M., Dierssen M.. Haploinsufficiency of Dyrk1A in mice leads to specific alterations in the development and regulation of motor activity. Behav. Neurosci. 2004; 118:815–821. [DOI] [PubMed] [Google Scholar]
- 8. Tejedor F., Zhu X.R., Kaltenbach E., Ackermann A., Baumann A., Canal I., Heisenberg M., Fischbach K.F., Pongs O.. minibrain: a new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron. 1995; 14:287–301. [DOI] [PubMed] [Google Scholar]
- 9. Degoutin J.L., Milton C.C., Yu E., Tipping M., Bosveld F., Yang L., Bellaiche Y., Veraksa A., Harvey K.F.. Riquiqui and minibrain are regulators of the hippo pathway downstream of Dachsous. Nat. Cell Biol. 2013; 15:1176–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guedj F., Pereira P.L., Najas S., Barallobre M.J., Chabert C., Souchet B., Sebrie C., Verney C., Herault Y., Arbones M. et al. . DYRK1A: a master regulatory protein controlling brain growth. Neurobiol. Dis. 2012; 46:190–203. [DOI] [PubMed] [Google Scholar]
- 11. Duchon A., Herault Y.. DYRK1A, a Dosage-Sensitive gene involved in neurodevelopmental disorders, is a target for drug development in down syndrome. Front. Behav. Neurosci. 2016; 10:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Di Vona C., Bezdan D., Islam A.B., Salichs E., Lopez-Bigas N., Ossowski S., de la Luna S.. Chromatin-wide profiling of DYRK1A reveals a role as a gene-specific RNA polymerase II CTD kinase. Mol. Cell. 2015; 57:506–520. [DOI] [PubMed] [Google Scholar]
- 13. Jang S.M., Azebi S., Soubigou G., Muchardt C.. DYRK1A phoshorylates histone H3 to differentially regulate the binding of HP1 isoforms and antagonize HP1-mediated transcriptional repression. EMBO Rep. 2014; 15:686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zaret K.S., Carroll J.S.. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011; 25:2227–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heintzman N.D., Hon G.C., Hawkins R.D., Kheradpour P., Stark A., Harp L.F., Ye Z., Lee L.K., Stuart R.K., Ching C.W. et al. . Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009; 459:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kalkhoven E. CBP and p300: HATs for different occasions. Biochem. Pharmacol. 2004; 68:1145–1155. [DOI] [PubMed] [Google Scholar]
- 17. Mohan M., Herz H.M., Takahashi Y.H., Lin C.Q., Lai K.C., Zhang Y., Washburn M.P., Florens L., Shilatifard A.. Linking H3K79 trimethylation to Wnt signaling through a novel Dot1-containing complex (DotCom). Genes Dev. 2010; 24:574–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin C.Q., Smith E.R., Takahashi H., Lai K.C., Martin-Brown S., Florens L., Washburn M.P., Conaway J.W., Conaway R.C., Shilatifard A.. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol. Cell. 2010; 37:429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al. . Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ramos Y.F., Hestand M.S., Verlaan M., Krabbendam E., Ariyurek Y., van Galen M., van Dam H., van Ommen G.J., den Dunnen J.T., Zantema A. et al. . Genome-wide assessment of differential roles for p300 and CBP in transcription regulation. Nucleic Acids Res. 2010; 38:5396–5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Adayev T., Chen-Hwang M.C., Murakami N., Wang R., Hwang Y.W.. MNB/DYRK1A phosphorylation regulates the interactions of synaptojanin 1 with endocytic accessory proteins. Biochem. Biophys. Res. Commun. 2006; 351:1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu H., Wang K., Chen S., Sun Q., Zhang Y., Chen L., Sun X.. NFATc1 phosphorylation by DYRK1A increases its protein stability. PLoS One. 2017; 12:e0172985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Glenewinkel F., Cohen M.J., King C.R., Kaspar S., Bamberg-Lemper S., Mymryk J.S., Becker W.. The adaptor protein DCAF7 mediates the interaction of the adenovirus E1A oncoprotein with the protein kinases DYRK1A and HIPK2. Sci. Rep. 2016; 6:28241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang W.C., Chen C.C.. Akt phosphorylation of p300 at Ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Mol. Cell. Biol. 2005; 25:6592–6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Himpel S., Tegge W., Frank R., Leder S., Joost H.G., Becker W.. Specificity determinants of substrate recognition by the protein kinase DYRK1A. J. Biol. Chem. 2000; 275:2431–2438. [DOI] [PubMed] [Google Scholar]
- 26. Fernandez-Martinez P., Zahonero C., Sanchez-Gomez P.. DYRK1A: the double-edged kinase as a protagonist in cell growth and tumorigenesis. Mol. Cell. Oncol. 2015; 2:e970048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kasper L.H., Thomas M.C., Zambetti G.P., Brindle P.K.. Double null cells reveal that CBP and p300 are dispensable for p53 targets p21 and Mdm2 but variably required for target genes of other signaling pathways. Cell Cycle. 2011; 10:212–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kasper L.H., Qu C., Obenauer J.C., McGoldrick D.J., Brindle P.K.. Genome-wide and single-cell analyses reveal a context dependent relationship between CBP recruitment and gene expression. Nucleic Acids Res. 2014; 42:11363–11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vo N., Goodman R.H.. CREB-binding protein and p300 in transcriptional regulation. J. Biol. Chem. 2001; 276:13505–13508. [DOI] [PubMed] [Google Scholar]
- 30. Chen J., Li Q.. Life and death of transcriptional co-activator p300. Epigenetics. 2011; 6:957–961. [DOI] [PubMed] [Google Scholar]
- 31. Litovchick L., Florens L.A., Swanson S.K., Washburn M.P., DeCaprio J.A.. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 2011; 25:801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen J.Y., Lin J.R., Tsai F.C., Meyer T.. Dosage of Dyrk1a shifts cells within a p21-cyclin D1 signaling map to control the decision to enter the cell cycle. Mol. Cell. 2013; 52:87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang Z., Zang C., Cui K., Schones D.E., Barski A., Peng W., Zhao K.. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009; 138:1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ait-Si-Ali S., Carlisi D., Ramirez S., Upegui-Gonzalez L.C., Duquet A., Robin P., Rudkin B., Harel-Bellan A., Trouche D.. Phosphorylation by p44 MAP Kinase/ERK1 stimulates CBP histone acetyl transferase activity in vitro. Biochem. Biophys. Res. Commun. 1999; 262:157–162. [DOI] [PubMed] [Google Scholar]
- 35. Ait-Si-Ali S., Ramirez S., Barre F.X., Dkhissi F., Magnaghi-Jaulin L., Girault J.A., Robin P., Knibiehler M., Pritchard L.L., Ducommun B. et al. . Histone acetyltransferase activity of CBP is controlled by cycle-dependent kinases and oncoprotein E1A. Nature. 1998; 396:184–186. [DOI] [PubMed] [Google Scholar]
- 36. Yuan L.W., Gambee J.E.. Phosphorylation of p300 at serine 89 by protein kinase C. J. Biol. Chem. 2000; 275:40946–40951. [DOI] [PubMed] [Google Scholar]
- 37. Wang S.A., Hung C.Y., Chuang J.Y., Chang W.C., Hsu T.I., Hung J.J.. Phosphorylation of p300 increases its protein degradation to enhance the lung cancer progression. Biochim. Biophys. Acta. 2014; 1843:1135–1149. [DOI] [PubMed] [Google Scholar]
- 38. Wee H.J., Voon D.C., Bae S.C., Ito Y.. PEBP2-beta/CBF-beta-dependent phosphorylation of RUNX1 and p300 by HIPK2: implications for leukemogenesis. Blood. 2008; 112:3777–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang L., Paul S., Trieu K.G., Dent L.G., Froldi F., Fores M., Webster K., Siegfried K.R., Kondo S., Harvey K. et al. . Minibrain and Wings apart control organ growth and tissue patterning through down-regulation of Capicua. PNAS. 2016; 113:10583–10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rachdi L., Kariyawasam D., Aiello V., Herault Y., Janel N., Delabar J.M., Polak M., Scharfmann R.. Dyrk1A induces pancreatic beta cell mass expansion and improves glucose tolerance. Cell Cycle. 2014; 13:2221–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pozo N., Zahonero C., Fernandez P., Linares J.M., Ayuso A., Hagiwara M., Perez A., Ricoy J.R., Hernandez-Lain A., Sepulveda J.M. et al. . Inhibition of DYRK1A destabilizes EGFR and reduces EGFR-dependent glioblastoma growth. J. Clin. Invest. 2013; 123:2475–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hammerle B., Ulin E., Guimera J., Becker W., Guillemot F., Tejedor F.J.. Transient expression of Mnb/Dyrk1a couples cell cycle exit and differentiation of neuronal precursors by inducing p27KIP1 expression and suppressing NOTCH signaling. Development. 2011; 138:2543–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu Q., Liu N., Zang S., Liu H., Wang P., Ji C., Sun X.. Tumor suppressor DYRK1A effects on proliferation and chemoresistance of AML cells by downregulating c-Myc. PLoS One. 2014; 9:e98853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang Y., Wen Z., Washburn M.P., Florens L.. Refinements to label free proteome quantitation: how to deal with peptides shared by multiple proteins. Anal. Chem. 2010; 82:2272–2281. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original data underlying this manuscript can be accessed after publication from the Stowers Original Data Repository at http://www.stowers.org/research/publications/libpb-1222. DYRK1A ChIP-seq data is from GEO accession number GSE63712 (12); p300 and CBP ChIP-seq data are from GEO accession number GSE21026 (20).