


Abstract
High-throughput sequencing of RNAs isolated by crosslinking immunoprecipitation (HITS-CLIP, also called CLIP-Seq) has been used to map global RNA–protein interactions. However, a critical caveat of HITS-CLIP results is that they contain non-linear background noise—different extent of non-specific interactions caused by individual transcript abundance—that has been inconsiderately normalized, resulting in sacrifice of sensitivity. To properly deconvolute RNA–protein interactions, we have implemented CLIPick, a flexible peak calling pipeline for analyzing HITS-CLIP data, which statistically determines the signal-to-noise ratio for each transcript based on the expression-dependent background simulation. Comprising of streamlined Python modules with an easy-to-use standalone graphical user interface, CLIPick robustly identifies significant peaks and quantitatively defines footprint regions within which RNA–protein interactions were occurred. CLIPick outperforms other peak callers in accuracy and sensitivity, selecting the largest number of peaks particularly in lowly expressed transcripts where such marginal signals are hard to discriminate. Specifically, the application of CLIPick to Argonaute (Ago) HITS-CLIP data were sensitive enough to uncover extended features of microRNA target sites, and these sites were experimentally validated. CLIPick enables to resolve critical interactions in a wide spectrum of transcript levels and extends the scope of HITS-CLIP analysis. CLIPick is available at: http://clip.korea.ac.kr/clipick/
INTRODUCTION
Complexity of RNAs in sequences and structures overwhelms the limited number of primary transcripts, conferring diverse functions through sophisticated regulatory mechanisms (1). The roles of RNA-binding proteins (RBPs) underscore this with their relatedness to phenotypic complexity because they directly interact numbers of mRNAs to regulate their splicing, stability, translation and/or cellular localization (2). Depending on the target RNAs with which a specific RBP interacts, the biological mechanisms and consequences of RBP regulation can be determined; thus, it is important to understand RNA–protein interactions in biology and pathophysiology (1,2).
RNA–protein interactions was initially attempted to be isolated by RNA immunoprecipitation (RIP) with a cognate RBP antibody (3), but the technique raised the concern of non-specific interactions introduced by in vitro rearrangement (4). To overcome this, the crosslinking and immunoprecipitation (CLIP) method has been developed to secure RNA–protein complexes in living cells by irradiating ultraviolet (UV) to induce covalent bonds (5). This method allows extremely stringent conditions for immunoprecipitation, minimizing non-specific interactions while specifically purifying RBP complexes. In combination with high-throughput sequencing (HITS-CLIP, also called CLIP-Seq), the recovered fragments of target RNAs have been comprehensively identified, mapped, and compiled into clusters (overlapping of reads) as read-counts on genome sequences (6).
HITS-CLIP has been successfully applied to various RBPs (7), including Argonaute (Ago), for the identification of microRNA (miRNA) target sites (8) and even to the antibody recognizing N6-methyladenosine (m6A) for mapping the RNA modification sites (9). However, not all these regions covered by CLIP fragments were analyzed to represent canonical RBP binding sites (10), especially ambiguous when non-canonical RBP interactions were marginally mediated in lowly expressed transcripts (11). Increasing evidences, especially for non-canonical miRNA binding sites, showed the biological importance of the marginally effective non-canonical RBP interactions (11). Therefore, HITS-CLIP analysis required to devise experimental and analytical procedures for refining binding regions with higher resolution and dealing with the remaining background noise.
In lieu of this, sequences of CLIP reads were further investigated to detect crosslinking-induced mutation sites (CIMS) (12). During the preparation of CLIP sequencing libraries, reverse transcriptase (RT) often skipped crosslinked residues and thus resulted in predominant deletion in CIMS. By performing permutation and reclustering of deleted sites, the statistical significance of the CIMS has been estimated, enabling the mapping of specific RBP interactions at a higher resolution (13). Alternatively, by circularly cloning truncated RT products caused by crosslinking, the repertoire of informative CLIP reads has also been expanded, achieving the individual nucleotide resolution of UV crosslinking and immunoprecipitation (iCLIP) based on the analysis of the crosslink-induced truncation sites (14,15). Later, positioning of the truncated iCLIP fragments was improved by accounting for fragment length-dependent distribution changes (16). Moreover, the method was also experimentally devised to specifically purify cDNA products using an antibody that recognizes BrdU, wherein BrdU was introduced to cDNA during RT reaction (17).
The CLIP experiment was also modified to increase the rate of CIMS by using the uracil (U) analog, 4-thiouridine, in cultured cells (photoactivatable-ribonucleoside-enhanced crosslinking and immunoprecipitation; PAR-CLIP), resulting in the enhancement of crosslinking efficiency and the subsequent production of U>C transitions in CLIP reads (18). However, the usage of 4-thiouridine is concerned in causing artificial side effects due to nucleoside cytotoxicity (19). Furthermore, all of these CIMS in CLIP reads were generally rare—the most of CLIP clusters (∼80–92%) could not be analyzed by these features (12). Therefore, to utilize the majority of CLIP reads, it is also important to systematically analyze the distribution of CLIP reads regardless of CIMS, eventually resolving binding sites based only on the statistical observation of read-counts and overlaps.
By examining the shapes and frequencies of aligned reads, RBP binding sites were tried to be specifically determined in the CLIP cluster via ‘peak calling’ (20,21). The peak calling comprises three central steps: (i) selecting CLIP clusters with significant height over the background noise, (ii) refining the selected CLIP clusters into peaks with defined positions and heights, and (iii) resolving regions within which RNA–protein interactions have occurred. Initially, background clusters were filtered out based on the iterative simulation of random CLIP experiments with matched transcript profiles (8). Then, the shapes of reads in the CLIP clusters were interpolated by cubic splines, enabling to pinpoint the locations and heights of the peaks. Similarly, the modified false discovery rate algorithm (later implemented in the ‘Pyicoclip’ program (22)) determined the significance of CLIP clusters by enumerating the random rearrangement of extended CLIP reads within specific exploratory regions based on the transcript annotation (23). Probabilistic models of Poisson distributions were also applied to filter out the background peaks globally by examining all other reads on the length of the whole transcriptome and also locally by calculating the gene-specific frequency, the total number of overlapping reads divided by the length of the corresponding transcript (24). Taken together, a peak calling tool, named ‘CLIPper’ (HITS-CLIP peak enrichment recognition), was implemented by interrogating the modified false discovery rate algorithm, the probabilistic models of Poisson distribution, and the cubic spline interpolation methods (25).
Alternatively, based on the observation that CLIP peak heights (PHs) globally fit a zero-truncated version of negative binomial distribution (ZTNB), peak callers such as Piranha (26) and PIPE-CLIP (27) used ZTNB for estimating background distribution by dividing genomic portions into bins with fixed sizes. To separate CLIP peaks in abundant transcripts, the CLIP tool kit (CTK) used the ‘valley seeking’ algorithm, which calls peaks if valleys of certain depths are identified on both sides, evaluating the statistical significance of PH using different background models and scan statistics (28). Different from conventional peak calling, the MiClip program refined the binding sites of CLIP clusters by using Hidden Markov Models (HMMs) and assessed the spatial dependency of CLIP clusters, which were divided into bins of small lengths (29). Analogously, dCLIP performed comparative analysis of CLIP clusters by applying modified Bland–Altman (MA) normalization and HMMs, enabling to identify differential binding regions without calling peaks (30).
To accurately and sensitively resolve CLIP peaks, the proper estimation of background is important. Indeed, CLIP peak signals were found to be differentially affected by non-specific interactions due to the variability in gene expression (8). Non-linear noise was imposed in peak calling analysis, initially attempted to be estimated based on microarray data (8). Transcript abundances were also accounted for by another peak caller, Piranha, as an external covariate by employing zero-truncated negative binomial regression (26). Additionally, recently optimized enhanced CLIP (eCLIP) procedures also used sequencing results of a size-matched input (SM-Input) to consider such non-linear noises (31).
Despite the improved sensitivity brought by considering expression levels, which successfully discovered non-canonical target sites of miRNAs in Ago HITS-CLIP data (10), the expression-dependent simulation method for background noise was not generally implemented in a peak calling analysis. Here, we devised and optimized the expression-dependent background simulation of random CLIP, implemented together with the cubic spline interpolation and the peak width refining method, and ultimately offered CLIPick, an expression-based deconvolution pipeline for HITS-CLIP analysis. CLIPick outperformed other pre-existing peak calling programs in sensitivity. By applying CLIPick to Ago HITS-CLIP data, we even discovered the additional new features of miRNA target sites, extended AU-rich motifs of seed sites, particularly enriched in lowly expressed transcripts. CLIPick includes several devised features that improve sensitivity, accuracy and usability of the peak calling process, significantly expanding the range of HITS-CLIP analysis. CLIPick offers both an easy-to-use standalone software with graphical user interface (GUI) and a flexible streamlined Python modules, of which installation packages can be downloaded from: http://clip.korea.ac.kr/clipick/.
MATERIALS AND METHODS
Implementation
CLIPick was implemented by Python 2.7 or 3.5 using BedTools (http://bedtools.readthedocs.io), matplotlib (https://matplotlib.org/), Numpy (http://www.numpy.org/), Scipy (https://www.scipy.org/) and Pandas (https://pandas.pydata.org/), Matplotlib (https://matplotlib.org/) and PyQt5 (https://pypi.org/project/PyQt5/). Both implemented Python modules and a standalone program with a GUI are provided with their pre-compiled versions (http://clip.korea.ac.kr/clipick/) and detailed documents (https://naturale0.github.io/CLIPick/). Source codes of the programs are accommodated by GitLab (https://gitlab.com/CLIPick/), where Integrity of the pipeline has been tested (passed with 95% coverage). Especially for CLIPick with GUI, instructions are also provided (Supplementary Figure S1) together with video tutorial (Supplementary Video S1, http://clip.korea.ac.kr/clipick/gui-tutorial.html).
Defining peaks of CLIP clusters
Based on the positions of aligned CLIP reads on the genome (given as a BED file), CLIP clusters were defined by using BedTools, which calculated compiled read-counts at each nucleotide position (BEDGraph). In the case of single-end reads, experimentally determined insert size during the preparation of CLIP sequencing libraries was utilized by defining only start position of mapped reads with extension of the given insert size. When the inserted size cannot be definitely determined within intended read length, 50-nt is used as a default value, as it was generally reported in the standard CLIP experiments (5,6,8). Thus, any continuous overlaps of the read lengths was condensed into one CLIP cluster, behaving that read-count should be reach zero between clusters. Based on the distribution of CLIP reads on each cluster, positions of peaks were defined by cubic spline interpolation, enabling to select multiple peaks in a given cluster depending on the extent of smoothness. The cubic spline interpolation method was applied by using Scipy (scipy.interpolate.splrep) as described previously (8). The amount of smoothness is adjusted by defining s parameter in splrep as 0 (no smoothing), m-sqrt(2*m) (weak), m (moderate) or m+sqrt(2*m) (strong), where m is the number of datapoints in the BEDGraph. s = m-sqrt(2*m) is recommended as a default smoothing value for CLIPick, because increasing the smoothness more than weak did not improve or change the performance in general (Supplementary Figure S5E). By determining the derivative of the function at each point of the interpolation and locating the point where the derivative became zero, the location and height of peaks per cluster were calculated.
Expression-dependent background simulation
The CLIP cluster could be derived from transient non-specific RNA–protein interactions, which are known to correlate with transcript abundance (8). Therefore, expression-dependent background noise was estimated by iteratively simulating random CLIP for each transcript. For transcript abundance, mRNA expression data were provided for each transcript  (annotated in RefSeq) either with normalized probe intensity from microarrays (
(annotated in RefSeq) either with normalized probe intensity from microarrays ( ) or with the
) or with the  (Reads Per Kilobase Million) value from RNA-Seq experiments. Then, expected read-counts for transcript
(Reads Per Kilobase Million) value from RNA-Seq experiments. Then, expected read-counts for transcript  (
( were calculated by multiplying the total read count from actual CLIP experiments (
were calculated by multiplying the total read count from actual CLIP experiments ( ) with the fraction of the expected fragment number of transcript
) with the fraction of the expected fragment number of transcript 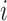 in the transcriptome (
in the transcriptome ( as follows:
as follows:
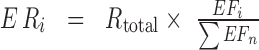 |
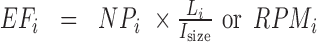 |
The expected fragment number of transcript  ), which accounts for its length and abundance, was estimated by assigning
), which accounts for its length and abundance, was estimated by assigning 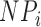 as the number for each transcript and multiplying it by the number of fragments per transcript, estimated by dividing the length of transcript
as the number for each transcript and multiplying it by the number of fragments per transcript, estimated by dividing the length of transcript 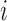 (
( ) with the observed average size of fragments generated in RNase treatment (
) with the observed average size of fragments generated in RNase treatment (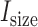 , 50 nt is used as a default value, generally reported in CLIP experiments (5,6,8)). In the case of using RNA-Seq data,
, 50 nt is used as a default value, generally reported in CLIP experiments (5,6,8)). In the case of using RNA-Seq data, 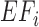 was simply calculated from
was simply calculated from 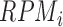 (reads per million mapped reads), derived from
(reads per million mapped reads), derived from  value in a baseline expression profile. As mRNA was fragmented in CLIP experiments, simulated fragments of transcript
value in a baseline expression profile. As mRNA was fragmented in CLIP experiments, simulated fragments of transcript 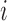 were generated based on a Gaussian distribution (mean =
were generated based on a Gaussian distribution (mean = 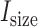 , standard deviation = 20% of
, standard deviation = 20% of 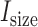 , based on observations in the RNase treatment of CLIP experiments) until it reached the number of
, based on observations in the RNase treatment of CLIP experiments) until it reached the number of 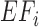 .
.
To estimate background distribution, the immunoprecipitation process of CLIP was iteratively simulated until the number of trials reached a user-selected number of times (e.g. n = 1000, default value). The fragments of transcript  were randomly chosen as many as
were randomly chosen as many as  , only if the size of the fragments was within an user defined range (between lower and upper bound). After trimming the selected fragments into the size of a given read length (except in cases where the size of the fragment was shorter than the given read length), they were aligned with their position in the transcript
, only if the size of the fragments was within an user defined range (between lower and upper bound). After trimming the selected fragments into the size of a given read length (except in cases where the size of the fragment was shorter than the given read length), they were aligned with their position in the transcript 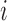 . Then, the maximum background cluster height (maximum number of overlapping reads in each running) was counted from the random alignment. By repeating this procedure for every transcript, the P-values of corresponding background PHs were differentially calculated. Based on a user-defined P-value threshold (e.g.P < 0.05 as a default value), significant CLIP clusters, of which assigned P-values for their heights were less than the threshold, were ultimately deconvoluted.
. Then, the maximum background cluster height (maximum number of overlapping reads in each running) was counted from the random alignment. By repeating this procedure for every transcript, the P-values of corresponding background PHs were differentially calculated. Based on a user-defined P-value threshold (e.g.P < 0.05 as a default value), significant CLIP clusters, of which assigned P-values for their heights were less than the threshold, were ultimately deconvoluted.
Resolving width of deconvoluted CLIP peaks
As analyzed and validated previously (8), the relative distances of the deconvoluted CLIP clusters to peak positions were compiled to refine putative regions of RBP interactions. Adjustment of peak width was attempted by examining the performance of CLIPick and PH with variable window size (200, 100, 50 and 20 nt) (Supplementary Figure S4). By narrowing down the size of the window to span more than 95% of all the significant CLIP clusters, where optimal tradeoff between precision and recall was achieved (proportion of true sites >60%, ratio of observed versus expected target sites >1.5, Supplementary Figure S4), RBP footprints were resolved relative to peak positions, finally offering the regions to be searched for RBP binding sites. In other words, all CLIP clusters were compiled depending on relative positions from the selected peaks, then peak width was determined by examining the fraction of overlaps in each nucleotide relative to peak positions, at which 95% of clusters have larger size than the defined range relative to peak positions (as illustrated in outputs of CLIPick, Figure 2E and F). Of note, all peak positions, deconvoluted by CLIPick, were retained but only their widths were adjusted by this newly defined peak width, of which size was statistically supported by 95% of significant clusters.
Figure 2.

CLIPick outputs. (A) A screen shot of the CLIPick GUI for choosing output options (texts and figures). (B) Output text describing selected significant peaks, comprising genomic coordinates of the peak width (resolved footprints), position and height together with estimated P-values. Moreover, names (RefSeq) and mRNA abundance are also provided. Partial results from Ago HITS-CLIP data are shown. (C) Cumulative distribution of the number of peaks was plotted depending on P-values assigned by the background simulation. As an example, the result from the robust Ago HITS-CLIP peaks (n = 61 554, derived from single-end reads of ∼50 nt insert size) is represented with P-value cutoffs (indicated as a red dotted line, P < 0.05, n = 23 876). (D) Graphical output of selected peaks. Significantly called peaks (P < 0.05, colored in red) are plotted on a genomic coordination together with all peaks (colored in black). Partial results from Ago HITS-CLIP data are shown as an example. (E and F) Figures indicating footprint analyses were provided to refine width of the significantly selected peaks, which covered 95% of the selected clusters (0.95 in fraction), shown separately for upstream (E) and downstream (F) regions from the peak positions. As a result, regions spanning −48 to +40 (relative position from the peaks) were resolved as peak widths indicating Ago footprint regions.
Datasets
For the validation of the CLIPick program, previously verified CLIP results were used. Robust Ago HITS-CLIP reads (8), which were reproducibly derived from experiments using two different Ago antibodies [biological complexity (BC) ≥ 2], were retrieved (http://ago.rockefeller.edu/rawdata.php or http://ago.korea.ac.kr/Ago_Clip_data) by downloading mapped results on the mouse genome (mm8) within transcribed regions (RefSeq) or processing raw sequencing results (FASTQ files). To assess the performance of CLIPick in parallel with CIMS analysis, raw sequencing data were processed by following the previously used criteria (12,13). In brief, FASTQ files were initially filtered based on quality scores (fastq_filter.pl –f mean:0–24:20) and collapsed (fastq2collapse.pl) (28). Then, the pre-processed reads, of which length was stretched to the average size of inserts (50 nt), were mapped to the mouse genome (mm10) using the NovoAlign program (http://www.novocraft.com) (Supplementary Table S1A) with the same parameters as described previously (13). After selecting reads on the reproducible clusters (BC ≥ 2) and annotated transcripts (RefSeq), aligned Ago CLIP reads were finally generated as BED or BAM files using BedTools and SAMtools (http://samtools.sourceforge.net/) and used for the rest of the analyses. The matched expression profile of the P13 mouse brain, measured by microarray, was obtained from the GEO database (GSE16338).
Ago HITS-CLIP reads performed in the frontal cortex (BA4 region, n = 5) of the human brain were downloaded from the GEO database (GSE52084) (32), mapped using Bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/) with the local alignment option, allowing one mismatch in each seed (–local -N1) (Supplementary Table S1B) and then polymerase chain reaction (PCR) duplicates in each experiment were removed. After examining variability of a result from each replicate (GSM1259105, GSM1259107, GSM259109, GSM1259112 or GSM1259114) (Supplementary Figure S7B–F), all reads were compiled for the following analysis (Supplementary Figure S7A). For CLIPick, a matched expression profile of the normal BA4 region was derived as the median value from 16 replicates of microarray data (GSM86957–GSM86972) in the GEO database (GSE3790) (33). Ago HITS-CLIP reads from human cardiac tissues (GSE83410) were also mapped (Supplementary Table S2A) and analyzed by using the same methods, utilizing the reported abundance of miRNA families (relative normalized seed abundance, Supplementary Table S2B) (34). Transcript abundance of human heart was derived from RNA-Seq Atlas (35), pre-built in CLIPick. Of note, all peak calling programs used the CLIP reads derived from expressed transcripts, at which the transcript level of target genes was substantially measured.
eCLIP results (31) performed in HepG2 (n = 42) and K562 (n = 60) were retrieved from ENCODE project site (https://www.encodeproject.org/) with their normalized outputs from CLIPper using size-matched input control (SM-Input). RNA-Seq results from HepG2 (ENCSR329MHM) and K562 (ENCSR000AEL) were also obtained by calculating average FPKM (Fragments Per Kilobase Milliion) for background simulation of CLIPick. Intron enrichment of CLIP reads relative to SM-Input reads (intron enrichment) was calculated to identify eCLIP data for cytoplasmic RBPs as previously reported (31).
Analysis of miRNA seed sites in Ago CLIP peaks
For the analysis of miRNA target sites in Ago CLIP peaks, 6mer (positions 2–7) or 7mer (positions 2–8) matches to seed sequences were searched in Ago CLIP cluster regions or derived from the miRTCat database (http://ago.korea.ac.kr/mirtcat/) (36). Ago CLIP peaks and miRNA target sites were visualized as a heat map indicating gene expression or PH using the UCSC genome browser (http://genome.ucsc.edu/) or Treeview (http://rana.lbl.gov/EisenSoftware.htm), as described previously (8). Top 20 expressed miRNA families were used as in Supplementary Table S1C. But for Ago HITS-CLIP data from human brain, top 30 expressed miRNA families (∼80% of miRNAs) were used according to the previous results (32). Ago HITS-CLIP results from human cardiac tissues were also analyzed with top 30 expressed miRNA families (Supplementary Table S2B) (34). miRNA sequences were obtained from miRBase (http://www.mirbase.org/). To examine the effects of sequencing coverage on the peak analysis, given percentage of random sampling (25, 50 and 75%) was iterated (n = 10) and analyzed by CLIPick and PH method (Supplementary Figure S3). To visualize peaks, compiled reads, and putative miRNA target sites on genome, Integrative Genomics Viewer (https://software.broadinstitute.org/software/igv/) was applied. For the analysis of adjacent sequence motifs, WebLogo 3 (http://weblogo.threeplusone.com) was used.
Comparison of CLIP peak calling programs
To compare the performance of different peak detection methods, CLIP peak callers were run under different stringency of P-values. For analyzing overlaps of CLIP peaks, we used P < 0.01 with default parameters unless otherwise indicated: CLIPick, default parameters in GUI (P < 0.01) except for the robust Ago HITS-CLIP data (8), where no smoothing (based on Supplementary Figures S5 and 6) and 50 nt stretches of reads (insert size of 50 nt, single-end reads) were used; CIMS analysis (https://zhanglab.c2b2.columbia.edu/index.php/CIMS), the same procedures and parameters as described previously (P < 0.01) (13); CLIPper (https://github.com/YeoLab/clipper/wiki/CLIPper-Home), –FDR 0.01 –binomial 0.01 –poisson-cutoff 0.01; Piranha (http://smithlabresearch.org/software/piranha/), -s –b 20 –p 0.01; CTK (http://zhanglab.c2b2.columbia.edu/index.php/CTK), default parameters with valley depth 0.9 as described previously (P < 0.01) (28). When a specific version of genome assembly is required, LiftOver (https://genome.ucsc.edu/cgi-bin/hgLiftOver) was used to convert genomic coordinates of mapped reads. In the case of using expression profiles as covariates for the Piranha program, a baseline expression profile corresponding to each binned position was generated and used (derived from GSE16338 for Ago HITS-CLIP data, mouse brain). When peaks were linearly filtered, PH > 6 was used as a default cutoff based on ZTNB analysis unless otherwise indicated. Positional comparison of CLIP peaks was performed for the results from different programs, wherein overlaps in genomic coordination were considered as sharing at least 1 nt between defined peak widths. To properly compare the result from CIMS analysis, realigned reads from the NovoAlign program were used as recommended (12,13). In the case of examining precision versus recall for peak calling programs, Ago HITS-CLIP data from moue brain were used (8). Top 20 miRNA target sites (6mer seed sites) were searched within the same window sizes (the peak width determined by CLIPick) and their proportion of peaks was calculated by averaging ratios in 100 nt window, plotted with the number of significant peaks for different programs with varying P-value thresholds (derived from the results running with P < 0.1).
For Ago HITS-CLIP from human brain (32) and heart (34), 6mer sites of top 30 expressed miRNAs were examined within peak widths determined by each program otherwise indicated. For the comparison between CLIPick and CLIPper (with SM-Input), eCLIP data for cytoplasmic RBPs were retrieved from ENCODE based on intron enrichment values (Supplementary Figure S8A), selecting FXR1 (ENCSR774RFN), HNRNPA1 (ENCSR769UEW) and HNRNPU (ENCSR520BZQ) (intron enrichment = 0.262, 0.563 and 0.662, respectively). For the application of CLIPick, aligned reads from the eCLIPs were used from BAM files (FXR1; ENCFF070XXP, HNRNPA1; ENCFF963VIU, HNRNPU; ENCFF881JZG) and further selected to be mapped on expressed transcripts (average FPKM>0) in the corresponding cell line, HepG2 or K562. Because of the similarity (median = 1 749 167 ± 50 000) in total counts of mapped reads (Rtotal), additional 24 eCLIP data (n = 25, in case of including eCLIP data for HNRNPA1) from HepG2 (n = 80, including all replicates) were attempted to be analyzed by CLIPick (Supplementary Figure S8C and D). After narrowing down to 10 eCLIP data for cytoplasmic RBPs (intron enrichment < 0.84), analysis of relative abundance between CLIPick (P < 0.01) and CLIPper with SM-Input (enrichment relative to SM-Input > 0, P < 0.01) was performed. For analyzing accuracy of the selected eCLIP data (BED files) for cytoplasmic RBPs (FXR1; ENCFF963VIU, HNRNPA1; ENCFF070XXP, HNRNPU; ENCFF881JZG), a known binding site of each RBPs (ACUK or WGGA; FXR1 (37), UAGG; HNRNPA1 (38,39), GUGUG; HNRNPU (40)) was used. Distribution of transcript abundance within which eCLIP peaks were selected by CLIPper with SM-Input (enrichment relative to SM-Input > 0, P < 0.05) or CLIPick (P < 0.05) was examined for every available eCLIPs in ENCODE (n = 102), which included ones analyzed by CLIPick (n = 27; hepG2, n = 2; K562), and depicted as a box plot (Supplementary Figure S9).
Meta-analysis of miRNA-mediated global target repression
Meta-analysis was performed by obtaining published compiled data, comprising normalized microarray data from 74 different miRNA or siRNA transfections as described previously (41). Based on nucleotides at position 9 or 10, different sets of transcripts with corresponding miRNA seed sites (7mer, positions 2–8) were separately examined by cumulative fraction analyses depending on fold change (log2 ratio). To delineate the compounding effects of multiple sites on target repression, transcripts with only one site of interest were analyzed in the cumulative distribution. Kolmogorov–Smirnov tests (KS tests) were performed using Scipy [scipy.stats.ks_2samp].
Construction of luciferase reporters
To measure the efficiency of miRNA-mediated gene silencing, the psiCheck-2 vector (Promega) was used to construct luciferase reporters as described previously (42). In the 3′-UTR of synthetic Renilla luciferase, following sites were inserted. In general, synthetic duplex oligos (Bioneer, Korea) containing various target sites for miR-124 (8mer-AA, forward: 5′-TCGAGAAGTGCCTTAAAGTGCCTTAGC-3′, reverse: 5′-GGCCGCTAAGGCACTTTAAGGCACTTC-3′; 9merC-C, forward: 5′-TCGAGCCGTGCCTTACCGTGCCTTAGC-3′, reverse: 5′-GGCCGCTAAGGCACGGTAAGGCACGGC-3′; 10merCG, forward: 5′-TCGAGGCGTGCCTTAGCGTGCCTTAGC-3′, reverse: 5′-GGCCGCTAAGGCACGCTAAGGCACGCC-3′; 8mer-GC, forward: 5′-TCGAGCGGTGCCTTACGGTGCCTTAGC-3′, reverse: 5′-GGCCGCTAAGGCACCGTAAGGCACCGC-3′) and for miR-9 (8mer-AA, forward: 5′-TCGAGAAACCAAAGAAAACCAAAGAGC-3′, reverse: 5′-GGCCGCTCTTTGGTTTTCTTTGGTTTC-3′; 8mer-CC, forward: 5′-TCGAGCCACCAAAGACCACCAAAGAGC-3′, reverse: 5′-GGCCGCTCTTTGGTGGTCTTTGGTGGC-3′; 10merUA, forward: 5′-TCGAGTAACCAAAGATAACCAAAGAGC-3′, reverse: 5′-GGCCGCTCTTTGGTTATCTTTGGTTAC-3′; 8mer-AU, forward: 5′-TCGAGATACCAAAGAATACCAAAGAGC-3′, reverse: 5′-GGCCGCTCTTTGGTATTCTTTGGTATC-3′) were cloned into the psiCheck-2 plasmid through XhoI and NotI sites.
Luciferase reporter assays
Luciferase reporter assays were performed as described previously (42). In brief, psiCheck-2 plasmids (Promega) were co-transfected with duplexed miRNAs into HeLa cells (ATCC CCL-2) using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol. HeLa cells were maintained in Dulbecco’s modified Eagle's medium (Gibco) supplemented with 10% fetal bovine serum (Gibco), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin at 37°C with 5% CO2 incubation. Custom RNA synthesis services of Bioneer (Korea) were used for constructing miRNAs (mmu-miR-124, mmu-miR-9) according to the sequences in miRBase. miRNA duplexes were produced in vitro by the following reaction: 90°C for 2 min, 30°C for 1 h and 4°C for 5 min. Twenty-four hours after the transfection, relative activity (Renilla luciferase activity normalized to firefly luciferase) was measured by the Dual-Luciferase Reporter Assay System (Promega) with the GloMax-Multi Detection System (Promega) with replicates (n = 6) according to the manufacturer’s protocol. Ultimately, half inhibitory concentration (IC50) was calculated by performing non-linear least squares fitting for the sigmoid function using Scipy [scipy.optimize.curve_fit].
RESULTS
Overview of CLIPick
In HITS-CLIP analysis, clusters (overlapping of aligned sequencing reads) were interpreted as signatures of consistent RNA–protein interactions in vivo (7). In contrast to cluster signals derived from analogous ChIP-Seq experiments, where the interactions occurred in even amounts of DNA, read-counts of CLIP clusters and their backgrounds were differentially generated depending on the level of transcripts (e.g. high expression versus low expression, Figure 1A).
Figure 1.

CLIPick overview. (A) Different extent of non-specific CLIP peaks caused by varying transcript abundance [CLIP reads on highly expressed mRNA (upper panel) versus lowly expressed mRNA (lower panel)] is schematically represented to determine the different stringencies of cutoffs. (B) Summary of three central peak calling steps. Positions of peaks, defined by cubic-spline interpolation, are displayed with both arrows and dotted lines. Cutoffs used for deconvolution are represented as dotted arrows. Compilation of selected significant clusters relative to peak positions is indicated and used to refine the peak widths of CLIP footprints (denoted as a double-headed arrow in the filled box, covering ∼95% of selected clusters). (C) Diagrams illustrate the required inputs for CLIPick comprising HITS-CLIP data (BED format of aligned reads, upper panel) and a matched expression profile (RNA-Seq or microarray, lower panel). (D) Illustration of expression-based background simulation, referred to as ‘in silico random CLIP,’ shows iterative processes of random fragmentation, random selection as many as expected read-count (ERi) and calculation of maximum height distribution (n = 1000, P < 0.05, used as default parameters). Details in ‘Materials and Methods’ section. (E) Python modules implemented and provided in CLIPick. (F) Adjustable parameters shown in the CLIPick GUI for in silico random CLIP simulation.
For this reason, a peak calling program, named ‘CLIPick,’ was developed to evaluate the significance of peak signals based on expression-dependent background simulation. CLIPick is organized into three main steps (as schematically represented in Figure 1B): (i) refining CLIP clusters (overlapping reads) into peaks with defined positions and heights, (ii) selecting CLIP peaks with significant height over the estimated background noise, (iii) resolving RBP footprint regions as peak widths within which RNA–protein interactions were mediated. First, from aligned CLIP reads (Figure 1C, upper panel), CLIPick interpolates the distribution of CLIP clusters with cubic splines, thus enabling to pinpoint peak positions and heights of the clusters based on read-counts (Figure 1B, left panel). Second, CLIPick uses matched expression profiles either derived from RNA-Seq or microarray experiments (Figure 1C, lower panel) and iteratively performs in silico random CLIP (Figure 1D) to evaluate the probability of PHs that can be observed significantly by chance (e.g. P < 0.05, n = 1000). Therefore, CLIPick can determine the significant threshold of PHs differentially by considering the abundance and length of each transcript (Figure 1B, middle panel). Third, with selected peaks, the relative distribution of CLIP clusters from peak positions is examined to resolve the regions of RBP footprints, narrowing down the relative size of the window from peak positions (peak width) where it covers the majority of CLIP clusters (95%; Figure 1B, right panel). Details of algorithms were described in the ‘Materials and Methods’ section.
As a standalone program with a GUI, CLIPick software takes inputs with aligned CLIP reads, transcript annotation and matched expression profiles (Figure 1C and Supplementary Figure S1A–C). Separately, streamlined Python modules used in CLIPick were also offered for the advanced utility (Figure 1E, details are described in the CLIPick project website; http://clip.korea.ac.kr/CLIPick/). Users should provide aligned CLIP reads either in single-end or paired-end reads (BED format) with notice of the mapped reference genome (RefSeq annotation, Supplementary Figure S1A). Transcript annotation can be either uploaded or selected in CLIPick, where the several annotations of the human and mouse genomes (e.g. RefSeq for mm10 or hg38) were available. PCR duplicates also could be processed either by looking at both start and end positions (generally for paired-end reads) or start position only (generally for single-end reads). Users could adjust the amount of smoothness for the interpolation (e.g. ‘moderate’, default option in CLIPick), controlling tradeoff between closeness and smoothness of fit to find optimal peaks. If the length of mapped read was not the same as the actual fragmented RNA in the case of single-end reads, CLIPick offered an option to extend its mapped reads up to the given size of stretch (e.g. 50 nt was used for the robust Ago HITS-CLIP dataset (8)), recommended to use the average fragment size estimated during the preparation of HITS-CLIP experiments. Types of matched expression data (microarray or RNA-Seq) should be instructed to CLIPick (Supplementary Figure S1B). In the current version of CLIPick, expression data for eleven human tissues were already available (prebuilt from RNA-Seq Atlas (35)) or could be uploaded by users.
For the accurate simulation of CLIP experiments, conditions of preparing HITS-CLIP libraries, such as the average size of inserts (which can also be further defined as lower and upper bound, mimicking size selection in CLIP experiments), should be provided with the read length (Figure 1F and Supplementary Figure S1C). Furthermore, for the flexibility of stringency in the deconvoluting and resolving process, default parameters, such as the P-value cutoff (0.05), number of iterations (n = 1000), and coverage used for refining peak width (95%), are adjustable. CLIPick offers options to select several types of outputs either in text or figures (Figure 2A; Supplementary Figure S1D and E). In addition to summary as a text, detailed information about identified significant peaks is provided, containing all genomic locations of resolved RBP footprints (as refined peak width), peak positions, PHs and P-values, together with the abundance of mRNA transcripts at which the corresponding peaks resided (Figure 2B). As a report of expression-based background simulation, the number of significant CLIP peaks depending on the determined P-value threshold is represented as a cumulative distribution (Figure 2C).
The application of CLIPick to the robust Ago HITS-CLIP data (8), which were derived in single-end reads of ∼50 nt insert size, selected 23 876 peaks (P < 0.05) from 61 554 raw peaks (Figure 2C). Furthermore, the selected peaks were also graphically plotted on genomic coordination in comparison with all peaks (Figure 2D). Finally, output figures that indicated RBP footprints were offered as a part of the process to refine the width of the deconvoluted peaks, which cover 95% of the clusters. As a result, regions spanning −48 to +40 (relative position from the peaks) were resolved as Ago footprint regions (Figure 2E and F). Similar results with 18 100 peaks (P < 0.05) and 59 nt peak width (−32 to +26) were also obtained when the size of insert was not stretched to 50 nt (Supplementary Figure S2A–C).
CLIPick sensitively and accurately defines RBP footprints in a broad range of expression
To evaluate the results from CLIPick, the selected peaks (n = 18 100, P < 0.05) from the robust Ago HITS-CLIP data (8) were examined for the prevailing binding sites of miR-124, relative to peak positions (Figure 3A). There, 7mer seed sites (positions 2–8) were enriched near the selected peaks (excess kurtosis [k] = 1.45), more leptokurtic than the distribution of width of all CLIP clusters (k = −1.39 versus −1.2 in uniform distribution). Within a 100-nt distance from the peak position (from −100 to +100), miRNA binding sites were observed (n = 1120, true positives) ∼5 times more than expected (n = ∼221, false positives). However, the resolved footprints by CLIPick (from −32 to +26, n = 779) showed ∼12 times more binding sites than expected (n = 64, calculated as explained in Figure 3A legend). The results indicated that the refined width by CLIPick outperformed with 92.4% precision (improved from 83.5% in ±100 nt windows) while preserving sensitivity (69.6%) and specificity (71.0%) (details are described in Figure 3A legend). Moreover, transcripts containing the deconvoluted peaks with binding sites showed a wide range of expression levels, often observed in low abundance (Figure 3A).
Figure 3.

CLIPick enables the deconvolution of peaks in lowly expressed transcripts. (A) Deconvoluted peaks (CLIPick, 18 100 peaks, P < 0.05) from robust Ago HITS-CLIP reads (66 266 peaks), where the size of insert was not stretched to 50 nt, were examined for miR-124 binding sites (7mer seed sites, −100 to +100, n = 1120) within refined footprint regions (−32 to +26, indicated by the blue double arrow, n = 779) relative to peak positions. Transcript expression levels are represented by different intensities of gray coloring as indicated (0–3000, normalized probe intensity, microarray). Of note, the expected number of 7mer sites in footprint regions (−32 to +26) is ∼64 = [18 100 (total peaks) × 58 (window size)]/47, but 779 sites were observed. Since the expected number of sites in the given interval (±100 nt) is ∼221 = (18 100 × 200)/47, the resolved peak width (−32 to +26) was estimated to perform 92.4% precision [779/(779 + 64)], 69.6% sensitivity (779/1120) and 71.0% specificity [(221 − 64)/221]. (B and C) Genome-wide mapping of all Ago CLIP peaks (n = 66 266) with defined heights (B) and matched mRNA expression level (C) are displayed as intensity as in (A). Chromosome numbers are indicated. (D) The same analysis as (B) except for Ago CLIP peaks after the deconvolution (n = 18 100, P < 0.05). Representative examples of peaks are shown in the zoomed inset (B–D). (E) Number of peaks depending on the level of located mRNA transcripts [log10(normalized probe intensity)] is plotted before (all peaks, n = 61 554, considering ∼50 nt insert size in single-end reads) and after the deconvolution (CLIPick, n = 23 876), compared with applying static threshold of PH (PH > 6, n = 14 790, determined by ZTNB as equivalent to P < 0.05). (F) The same analysis as (E) but represented as a cumulative fraction.
When Ago CLIP peaks were inspected across the genome-wide landscape (Figure 3B), PH had the propensity to be affected by gene expression levels (Figure 3C). However, CLIPick properly deconvoluted peak signals considering non-linear noise from transcript abundance (Figure 3D), which was especially evident for the selected peaks in lowly expressed (Figure 3B–D, lower panel of zoomed inset) versus highly expressed regions (Figure 3B–D, upper panel of zoomed inset). Indeed, the distribution of CLIP peaks was skewed toward the high mRNA level, implicating the existence of bias compounded by mRNA abundance (Figure 3E). Such bias became more distinct when the global threshold of PH (PH > 6, determined by ZTNB of all CLIP peaks as equivalent to P < 0.05) was applied. In comparison, CLIPick (23 876 selected out of 61 554 peaks, P < 0.05) found more peaks in lowly expressed transcripts as a consequence of normalizing the bias toward high expression, but still enable to select large number of peaks in highly expressed transcripts (Log10(mRNA level) = ∼4–4.5) comparable to the PH method (Figure 3E). The relative enrichment of peaks in low expression was also significantly observed in CLIPick by cumulative distribution analyses (Figure 3F, P < 0.01, KS test, CLIPick versus all peaks). All these results demonstrate that CLIPick can specifically resolve RBP binding regions with increasing accuracy and sensitivity. This is presumably due to its ability to normalize expression-dependent bias, particularly useful in identifying CLIP peaks for low-level transcripts.
CLIPick robustly outperforms other peak callers in both accuracy and sensitivity
To prove the improvement of CLIPick over other peak callers, corresponding peaks from different programs were tested using the same CLIP dataset (realigned Ago HITS-CLIP reads, Supplementary Table S1A) under the same cutoff stringency (P < 0.01; Figure 4A). In the pairwise comparison of peak positions, CLIPick covered most of the peaks identified by other programs (∼63–97%). Despite the fact that only few peaks overlapped between CIMS (∼27%) and Piranha (∼8%), CLIPick also shared ≥94% of both peaks, representing its superior sensitivity which could cover results from different peak callers. Of note, CIMS has generally been reported to be accurate but called fewer peaks because of the examination of rare events, crosslinking-induced mutations (12,43). Thus, albeit precise, CIMS identified only a limited number of peaks (n = 249), raising concerns about its performance in recall. The same problem in sensitivity was observed for Piranha (n = 532), of which peaks were still restricted regardless of using expression profiles as covariates (+cov, n = 497). Although CLIPper and CTK also exhibited superior performance, its number of peaks (CLIPper; n = 20 780, CTK; n = 16 883) was average ∼1.7 times (CLIPper; 1.51 times, CTK; 1.87 times) less than that of CLIPick (n = 31 561) with more than 65% overlap, indicating insufficient coverage of true positives. The similar results were also observed for different Ago CLIP datasets derived from human brain (left panel, Figure 4B) (32)— CLIPick selected ∼1.5 times more peaks (n = 39 432 versus n = 26 714) than CLIPper, which shared ∼34% more peaks with Piranha (77 versus 43%) than CLIPper, and ∼1.3 times more peaks than CTK, which shared ∼9% more peaks with Piranha than CTK—that supports the comprehensiveness of the peak calling process in CLIPick (n = 39 432, P < 0.01). In addition, CLIPick was also applied to Ago CLIP data from human heart (34), where CLIPick (n = 88 265, P < 0.01) showed comparable sensitivity to CLIPper (n = 98 323, P < 0.01) (right panel, Figure 4B).
Figure 4.

Comparative evaluation of CLIPick and other peak calling programs for precision and sensitivity. w(A) Ago CLIP peaks identified by the indicated programs (P < 0.01) were analyzed for positional overlaps, and the pairwise comparison results are shown. Each panel indicates the ratio and number of total peaks from one program (Y column) that are shared with another program (X row). From realigned Ago CLIP reads by NovoAlign (Supplementary Table S1A), peak widths were refined by each program and used in the analysis as follows: CLIPick (74 nt; −40/+33), CLIPper (46 ± 21 nt), CTK (74 nt was used as CLIPick, since CTK could not define size of peak width), CIMS (42 nt), Piranha (24 ± 15 nt) and Piranha+cov (24 ± 15 nt, ‘+cov’ denotes running with covariate of transcript abundance). Of note, realignment of the reads by NovoAlign gave CLIPick to have different peak width from the robust Ago HITS-CLIP results. (B) The same comparison analyses as performed in (A) except for the Ago HITS-CLIP from human brain (left panel, details in Supplementary Table S1B) using CLIPick (79 nt; −41/+37), CLIPper (56 ± 30 nt), CTK (79 nt as in CLIPIck) and Piranha (22 ± 8 nt). The same analyses were conducted for the data from human heart (right panel, details in Supplementary Table S2A) using CLIPick (47nt, −26/+21), CLIPper (28 ± 18 nt), CTK (47 nt as in CLIPIck) and Piranha (26 ± 27 nt). The ratio is also denoted by a color code (upper panel) for (A) and (B). (C) The fraction of peaks with miRNA seed sites (top 20 expressed miRNAs, Supplementary Table S1C; 6mers) in robust Ago CLIP reads (http://ago.korea.ac.kr/Ago_Clip_data) is shown for different programs depending on varying thresholds up to P < 0.1. PH denotes ‘peak height,’ which was used as a global cutoff in peak selection. To avoid bias from varying peak widths, 89 nt windows (−48/+40) determined by CLIPick were also used for other programs. (D) The same analyses as performed in (C) except applied to the realigned Ago CLIP reads to include CIMS results. An inset figure represented only up to 1000 peaks because numbers of significant peaks from using Piranha (P < 0.05), CIMS (P < 0.05), Piranha+cov (P < 0.05) were relatively low. Although Piranha, CIMS and Piranha+cov were run with P < 0.05 as threshold, their top margins of P-values were often less than the threshold. A total of 74 nt peak width (−40/+33), defined by CLIPick, was used for this analysis. (E) Similar analysis in (C) and (D) applied to the human brain Ago HITS-CLIP datasets (Supplementary Table S1B) for top 30 expressed miRNAs (P < 0.1). For this analysis, peak widths determined by each program were used as in (B). Piranha results were also examined after using peak widths of CLIPick (Piranha+CLIPick width). (F) The same analyses as in (E) except for human heart (Supplementary Table S2). Details were described in ‘Materials and Methods’ section.
Next, the accuracy of CLIPick was investigated for the robust Ago CLIP dataset (8) by taking advantage of miRNA binding sites (6mer seed sites), which could serve as an objective estimation of precision (Figure 4C). As shown in the ratio of seed sites of top 20 miRNAs (Supplementary Table S1C) within a refined peak width of CLIPick (89 nt, −48/+40), CTK outperformed in accuracy but selected smaller number of peaks, which was ∼1.6 times less than CLIPper and ∼2.4 times less than CLIPick (P < 0.1, the end of the line, Figure 4C). Compared with CLIPIck, CLIPper also showed limited potency in expanding sensitivity—both CTK and CLIPper performed more accurately for a limited number of peaks with low P-values, but the number of significant peaks negatively correlated with accuracy. Including PH methods (with global threshold), every pre-existing peak calling program showed a decrease in accuracy that was proportional to the increasing number of peaks, resulted from lowering stringency of thresholds. However, CLIPick consistently achieved high precision (0.63 ± 0.02) throughout all acceptable P-value thresholds (all values ≤0.1). Even with the least stringent P-value cutoff (P < 0.1, the end of the line), the accuracy shown in CLIPick was maintained to the extent that is only observed with higher P-value stringency in CLIPper (0.61, P < 0.0005). Moreover, under default P-value cutoff (P < 0.05), CLIPick sensitively deconvoluted ∼1.4 times more peaks than CLIPper and ∼2.4 times more peaks than CTK (Figure 4C).
Such improved accuracy and sensitivity of CLIPick was hypothesized to be achieved by deconvoluting signals from expression-dependent background noises, supported by observing enhanced precision relative to PH method—especially evident when analyzed from the lowly expressed transcripts (Supplementary Figure S2D and E). CLIPick also exerted robust performance, consistently behaving accurate and sensitive peak calling rather than the PH method regardless of sequencing coverage (Supplementary Figure S3), size of peak width (Supplementary Figure S4) or the amount of smoothness in the interpolation (Supplementary Figure S5). Of note, sequencing depth correlated number of peaks with sustained accuracy in CLIPick (∼0.61, P < 0.1, Supplementary Figure S3) but increasing peak width did sacrifice specificity to enhance precision (with consistent sensitivity, n = 26 012, Supplementary Figure S4). Thus, resolving the peak width by determining the window size that covered 95% of all the significant CLIP clusters, was supported in CLIPick due to appropriate tradeoff between precision and specificity (Supplementary Figure S4). The extent of smoothness did not affect the accuracy of CLIPick, but impacting sensitivity—resolution of the fitting determined the number of peaks in one cluster, where increasing the smoothness could miss true positives (Supplementary Figure S6). Therefore, no smoothing was used for robust Ago HITS-CLIP dataset due to its performance in choosing large number of true positive peaks (Supplementary Figure S5).
After validating the performance and set-up of CLIPick, realigned Ago HITS-CLIP reads from mouse brain were also analyzed to further compare with other peak callers including CIMS, wherein CLIPick performed more accurately than CLIPper throughout all ranges of P-value thresholds (e.g. 0.56 versus 0.49, P < 0.05, Figure 4D). Although only restricted to small number of peaks (<1000 peaks), Piranha showed to perform the most precisely within top ranked peaks and CIMS showed similar accuracy with CLIPick (P < 0.05; inset, Figure 4D). By considering transcript abundance as a covariate (+cov), Piranha was able to improve precision but the number was still much smaller than that in CLIPIck (Piranha+cov, inset, Figure 4D). Furthermore, CLIPick was also validated to show superior sensitivity with sustained accuracy for the Ago CLIP results from human brain (32), where its accuracy within selected peak widths were comparing with those determined by other methods (Figure 4E). Of note, variability of experimental replicates, derived from different patient samples, was shown to confound the analysis of CLIP data from human brain, wherein compiling of replicates could overcome such fluctuation in the results (Supplementary Figure S7). Similar results were also observed for the Ago CLIP data from human heart (34), showing improved accuracy of CLIPick comparing with others. In some cases, CLIPper performed comparable (Figure 4E) to or even better sensitivity (Figure 4D and F) than CLIPick, but the results were always less accurate than CLIPick (Figure 4C–F). Notably, Piranha became ∼2 or ∼3.5 times more accurate when its peak widths were changed to those defined by CLIPick (Piranha+CLIPick width, Figure 4E and F), implicating the importance of resolving the appropriate size of peak width as implemented in CLIPick.
Beside Ago HITS-CLIP, 102 eCLIP results (42 from HepG2 and 60 from K562 cells), processed by CLIPper and further normalized by sequencing results of a size-matched input (SM-Input) (31), were examined for the comparison. To be properly compared with CLIPick, that requires an accurate gene expression profile, eCLIP data were initially selected into only ones for the cytoplasmic RBPs (HNRNPA1, FXR1 and HNRNPU) according to intron enrichment values (Supplementary Figure S8A and B). In the enrichment analyses of known binding sites of the selected RBPs (ACUK or WGGA; FXR1 (37), UAGG; HNRNPA1 (38,39), GUGUG; HNRNPU (40)), CLIPick also outperformed CLIPper (even with SM-Input normalization) significantly in sensitivity and generally in accuracy, selecting at least two times more peaks in all cases (Figure 5A–C) while exhibiting ∼1.3 times more improved precision throughout all ranges of P-value threshold in the cases of HNRNPA1 (Figure 5A) and HNRNPU (Figure 5C). For FXR1 eCLIP, CLIPick outperformed within only top ranked peaks of FXR1 eCLIP data and became slightly less accurate after it reached the same number of peaks that CLIPper with SM-Input called at P < 0.01 (Figure 5B). After that, additional 26 eCLIP data (n = 27, including HNRNPA1 in Figure 5A) were selected based on their similar numbers in total read count (Rtotal) out of all data including replicates (n = 80, HepG2) for CLIPIck (Supplementary Figure S8C), showing that CLIPick tended to select more peaks in lowly expressed transcripts than CLIPper with SM-Input (Figure 5D). Notably, 10 eCLIP data for cytoplasmic RBPs (intron enrichment <0.84) had average 3.48 times (±1.86) more peaks in CLIPick than in CLIPper with SM-Input (Supplementary Figure S8D). Such increased sensitivity of CLIPick could be brought by the capability of selecting peaks in transcript with low abundance, whereas all eCLIP results analyzed by CLIPper (with SM-Input) showed that expression of their target transcripts were relatively high in HepG2 (n = 80, Supplementary Figure S9A) and K562 (n = 108, including FXR1 and HNRNPU in Figure 5B and C; Supplementary Figure S9B). Taken together, we concluded that CLIPick outperformed existing peak callers in sensitivity with high precision, yielding expanded numbers in the peak calling process.
Figure 5.

Comparative analyses of eCLIP results with CLIPick. (A–C) Analysis of precision and sensitivity of eCLIP peaks, previously analyzed and normalized by CLIPper with SM-Input (blue line) (31) for HNRNPA1 (A), FXR1 (B) or HNRNPU (C), compared with results from CLIPick (red line) using reported binding site (UAGG for HNRNPA1, ACUK or WGGA for FXR1 and GUGUG for HNRNPU). CLIPick used RNA-Seq data from the corresponding cell lines (HNRNPA1; HepG2, FXR1; K562, HNRNPU; K562) as analyzed in Figure 4C. The same peak widths defined by CLIPick (HNRNPA1; −38/+36, FXR1; −68/+54, HNRNPU; −46/+43) were used for CLIPper. Of note, these three RBPs were selected due to their preferential interactions with mRNA in cytoplasm based on intron enrichment values (0.262 from HNRNPA1, 0.563 from FXR1 and 0.662 from HNRNPU; Supplementary Figure S8). (D) Twenty-seven eCLIP data were analyzed by CLIPick in HepG2 (right panel, shaded in red), where abundance of transcripts (log2(FPKM)) harboring the selected peaks were compared with results from CLIPper (with SM-Input, left panel, shaded in blue) and represented as box plots. The rest of the analyses were also displayed in Supplementary Figure S9. Details were described in ‘Materials and Methods’ section.
CLIPick uncovers extended AU-rich motifs of miRNA seed sites
As expected from expression-dependent deconvolution, CLIPick was able to identify more true positives in lowly expressed transcripts, which was confirmed by estimating the cumulative enrichment of seed sites in the robust Ago CLIP dataset (especially evident for the transcripts ranked in low 25%, Figure 6A). Such improved performance of CLIPick perpetuated to search any additional sequence feature of miRNA binding sites. Focusing on miR-124, the nucleotide composition in the adjacent positions of seed sites (6mer, positions 2–7) was analyzed according to transcript levels (Figure 6B and C). In low abundant transcripts (ranked in low 25%), miR-124 seed sites (6mer, positions 2–7) seemed to preferentially contain A or U in positions 9 and 10 as well as known features—A in position 1 and a match in position 8 (G for miR-124, the rest of the nucleotide within the seed region)—based on analyses of information content (Figure 6B, lower panel) and probability (Figure 6C, lower panel). The extended AU-rich motifs were also confirmed for 7mer seed sites in the transcripts of which expression ranked in low 50% (Figure 6C, middle panel). Generally, the AU-rich motifs in positions 9 and 10 were frequently observed for other miRNAs (top 20 miRNAs, 8mer seed sites; Figure 6C, upper panel), prevailing as much as A in position 1 in the CLIPick-selected peaks (Figure 6D).
Figure 6.

Identification of the extended AU-rich feature of miRNA target sites by CLIPick. (A) Numbers of identified Ago CLIP peaks by CLIPick (P < 0.05) versus PH threshold (PH > 6), which contain 6mer (upper panel) and 7mer (lower panel) seed sites (top 20 miRNAs), were compared as cumulative distribution depending on the expression level of located mRNA transcripts in the mouse brain. (B) Adjacent sequences of miR-124 6mer sites (positions 2–7) in the deconvoluted CLIPick peaks are represented as bits scores (analyzed by Weblogo 2.0), based on expression levels categorized as high 50% (n = 1510, upper panel), low 50% (n = 230, middle panel) and low 25% (n = 16, lower panel) in (A). (C) The same analysis as in (B) except represented as probability for 7mer sites (positions 2–8) of top 20 expressed miRNAs in low 25% (upper panel), 7mer sites of miR-124 in low 50% (middle panel, n = 121) and 6mer sites of miR-124 in low 25% (lower panel, n = 16). (D) Plotting of AU versus GC rich motifs in positions 1–10 of seed sites, identified in the deconvoluted Ago CLIP peaks. (E and F) Meta-analysis of compiled microarray data showing the fold changes induced by the overexpression of 74 individual small RNAs. (E) Cumulative distributions are represented for transcripts containing 8mer seed sites of corresponding miRNAs with different nucleotides in position 9; 8mer-9A (n = 1491), 8mer-9U (n = 1554), 8mer-9G (n = 952) and 8mer-9C (n = 933). Indicated P-value was from the KS test between the combined distribution of 8mer-9A (A) and 8mer-9U (U) versus 8mer-9G (G) and 8mer-9C (C). (F) The same as (D) except for the analysis of nucleotide composition at position 10. (G) Luciferase reporter assays for estimating the efficiency of suppressing 10mer (8mer-CG), 9mer (8mer-CC), 8mer-GC and 8mer-AA sites by different concentrations of miR-124 (left panel). Sequences of the sites and corresponding half maximal inhibitory concentrations (IC50) are indicated (right panel). (H) Same luciferase assays as performed in (F) except for miR-9 seed sites.
Next, the efficacy of miRNA-mediated target repression was examined for the AU-rich motifs by analyzing compiled microarray data (41), which measured the global alteration of transcript abundance induced by expressing 74 different small RNAs. In the analysis of the cumulative fraction depending on nucleotide composition in positions 9 and 10 (Figure 6E and F), putative miRNA targets (8mer seed sites) with extended A or U seemed to be more susceptible, wherein the AU content significantly enhanced the repression relative to the GC content in both position 9 (P = 6.8 × 10−7, KS test, Figure 6E) and 10 (P = 8.5 × 10−7, KS test, Figure 6F). To further confirm this, luciferase reporter assays were performed for miR-124 seed sites with AA in positions 9 and 10, showing increasing efficiency of inhibitory activity (8mer-AA, IC50 = 0.18 nM) compared with GC (8mer-GC, IC50 = 5.96nM) and CG (10merCG, IC50 = 4.83 nM) regardless of pairing potency (Figure 6G). Furthermore, luciferase reporter assays for miR-9 (Figure 6H) also showed similar results—all extended AU-rich motifs (positions 9 and 10) improved miRNA-mediated silencing (10merUA, IC50 = 0.58nM; 8mer-AU, IC50 = 0.89nM; 8mer-AA, IC50 = 0.36 nM) relative to the other without any A or U (8mer-CC, IC50 = 1.54 nM). Overall, CLIPick was sensitive enough to discover extended AU-rich motifs of seed sites in Ago HITS-CLIP data, supporting the strength and usage of CLIPick in resolving specific interactions in lowly expressed transcripts.
DISCUSSION AND CONCLUSION
Among features that influence peak calling procedures, transcript abundance substantially correlates with non-specific interactions in CLIP, exerting different signal-to-noise ratios depending on the individual transcript level. Since assessing CLIP peaks is crucial in determining precise RBP binding sites (43), such non-linear background noise must be taken into account to delineate their compounding effects. Nevertheless, CLIP peaks have often been analyzed without additional information of gene expression by trying to indirectly infer a different statistical background on a gene-by-gene basis (such as in Pyicoclip (22) and CLIPper (25)). Such inconsiderate normalization could sacrifice sensitivity by using a stringent threshold for precision although they could become specific. Particularly when the background probability model was derived from an underlying distribution of all read-counts (26,27), it was not feasible to infer noise differentially for each transcript. Although Piranha (26) and PIPE-CLIP (27) allow external covariates, such as transcript abundance, by employing zero-truncated negative binomial regression, they rather generally sacrificed too much sensitivity for accuracy—calling much fewer peaks than other programs (21,43).
Particularly in cases where CLIP sequencing reads contain little noise, background estimation from all read counts, used in ZTNB-based methods, could be inappropriate, requiring to separate background reads from the majority of signals in all reads. Of note, Piranha was initially designed to be usable on general cases of RIP, intending to deal with noisy data inherently derived from RIP-Seq. Thus, Piranha may not be well tuned to analyze more specific HITS-CLIP data. Furthermore, the bin size used for fitting a ZTNB-based model is manually selected, which could make the analysis variable depending on its size (21,43). In lieu of this, simple changes of peak widths in Piranha to the size that defined by CLIPick were observed to increase the accuracy of Ago HITS-CLIP results (Figure 4E, Piranha versus Piranha+CLIPick width), suggesting that adjustment of peak width is also important determinant for the performance of peak calling. Although CTK was generally accurate in our benchmark studies, CTK showed limited sensitivity comparing with CLIPick (Figure 4C, E and F) and possibly had the same variability problem caused by manually deciding its peak width (28).
By determining the signal-to-noise ratio for each transcript based on expression-dependent background simulation, CLIPick can properly deconvolute RBP binding sites. Based on the distribution of CLIP clusters, CLIPick also statistically refines peak widths, within which RNA–protein interactions have been occurred. CLIPick offers an easy-to-use standalone program with a GUI and streamlined Python modules as a unified pipeline for peak calling. CLIPick is generalized to accept either single-end or paired-end CLIP reads. For the matched information of transcript abundance, users can select corresponding gene expression profiles listed in CLIPick (currently pre-built for several human tissues from RNA-Seq Atlas (35)) or manually provide their own microarray or RNA-Seq results. Besides the expression profile, matched IgG CLIP results have been used often to estimate background control, but they were unfavorably sparse and artificially over-amplified (31). Although sequencing of a size-matched input control prior to immunoprecipitation is informative to improve the peak calling process, shown in the eCLIP (31) and an HMM-based peak calling method with individual crosslink site detection (PureCLIP) (44), performing such matched experiments with CLIPper is neither always feasible nor so much beneficial in terms of accuracy and sensitivity as show in the comparison (Figure 5 and Supplementary Figure S8). This is likely due to relatively superior performance of CLIPick in refining peak signals from lowly expressed transcripts (Figure 5D and Supplementary Figure S9). In contrast to the limited availability of the matched CLIP controls, gene expression profiles were easily accessible for diverse tissues and cell lines, thus generally applicable to normalize the variety of CLIP results by simply utilizing the CLIPick program.
CLIPick robustly outperformed other peak calling programs in terms of accuracy and sensitivity, regardless of variability in sequencing depth (Supplementary Figure S3), size of peak width (Supplementary Figure S4), or the amount of smoothness in the interpolation (Supplementary Figure S5), implicating that expression-dependent background noise matters the most. CLIPick was proven to be especially good at detecting specific RBP interactions in transcripts at low expression level where the signal-to-noise difference is difficult to discriminate. With sustained accuracy, CLIPick identified at least 1.5 times more significant peaks (P < 0.01) than CLIPper (Figure 4), of which core implementation, Pyicoclip (22), had been reported to outperform in previous benchmark studies (43). CLIPick was also able to determine the resolution of binding sites in CLIP peaks by examining the distribution of CLIP cluster widths (e.g. 95% of overlaps as default), within which RBP binding sites were validated to be sensitively identified (69.6%) with high accuracy (90.8%) and specificity (71.1%; Figure 3A). Notwithstanding, other peak callers generally use a user-defined size of peak widths (∼50–100 nt windows), often neither optimized nor narrow enough to be accurate. Moreover, every peak callers including CLIPick requires to determine smoothness of the interpolation although amount of closeness and smoothness of the fits is important for peak calling process.
With combination of expression-dependent background estimation and statistical determination of peak widths, CLIPick enables to call more peaks with high resolution and accuracy, informative enough to identify sequence features of RBP interactions. Therefore, by applying CLIPick to Ago HITS-CLIP data, we were able to discover and validate extended AU motifs (positions 9 and 10) of miRNA seed sites enriched in lowly expressed target transcripts. Intriguingly, neither the extended AU motifs nor the well-known feature of A in position 1 was observed to be enriched in the highly or moderately expressed transcripts (ranked in high 50% of expression; Figure 6B and C, upper panels). This could imply that lowly expressed miRNA targets might require additional features to be efficiently recognized by Ago–miRNA complex because they are too rare in abundance to be passively bound by Ago–miRNA. As it has been practically validated, CLIPick analysis should be used for revisiting CLIP data or applying forthcoming CLIP results to identify unknown or non-canonical features of RBP binding sites, of which the signals are too marginal to be detected by conventional peak callers, but biologically important to understand their mechanisms of RNA regulation (11).
In summary, we developed an expression-based deconvolution pipeline, named ‘CLIPick’, to sensitively resolve HITS-CLIP peaks with ease, facilitating and expanding usage of HITS-CLIP for studying RBP regulations. CLIPick showed the unprecedented sensitivity with sustained accuracy and usability which have not been offered by other CLIP analysis programs. By applying CLIPick to Ago HITS-CLIP data, we even discovered additional new features of miRNA target sites, extended AU-rich motifs of seed sites, especially enriched in lowly expressed transcripts. CLIPick extends the current scope to a wide range of transcript levels and provides new opportunities to uncover detailed characteristics of RBP binding sites that would otherwise be invisible.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr Heeyoung Seok for helpful discussions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Korean Health Technology R&D Project, Ministry of Health and Welfare, Republic of Korea [HI15C3137]. Korea University Future Research Grant.
Conflict of interest statement. None declared.
REFERENCES
- 1. Licatalosi D.D., Darnell R.B.. RNA processing and its regulation: global insights into biological networks. Nat. Rev. Genet. 2010; 11:75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hentze M.W., Castello A., Schwarzl T., Preiss T.. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018; 19:327–341. [DOI] [PubMed] [Google Scholar]
- 3. McHugh C.A., Russell P., Guttman M.. Methods for comprehensive experimental identification of RNA-protein interactions. Genome Biol. 2014; 15:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mili S., Steitz J.A.. Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA. 2004; 10:1692–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ule J., Jensen K.B., Ruggiu M., Mele A., Ule A., Darnell R.B.. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003; 302:1212–1215. [DOI] [PubMed] [Google Scholar]
- 6. Licatalosi D.D., Mele A., Fak J.J., Ule J., Kayikci M., Chi S.W., Clark T.A., Schweitzer A.C., Blume J.E., Wang X. et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008; 456:464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Darnell R.B. HITS-CLIP: panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip. Rev. RNA. 2010; 1:266–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chi S.W., Zang J.B., Mele A., Darnell R.B.. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009; 460:479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ke S., Alemu E.A., Mertens C., Gantman E.C., Fak J.J., Mele A., Haripal B., Zucker-Scharff I., Moore M.J., Park C.Y. et al. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev. 2015; 29:2037–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chi S.W., Hannon G.J., Darnell R.B.. An alternative mode of microRNA target recognition. Nat. Struct. Mol. Biol. 2012; 19:321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Seok H., Ham J., Jang E.S., Chi S.W.. MicroRNA target recognition: insights from transcriptome-wide non-canonical interactions. Mol. Cells. 2016; 39:375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang C., Darnell R.B.. Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat. Biotechnol. 2011; 29:607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moore M.J., Zhang C., Gantman E.C., Mele A., Darnell J.C., Darnell R.B.. Mapping Argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. Nat. Protoc. 2014; 9:263–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Konig J., Zarnack K., Rot G., Curk T., Kayikci M., Zupan B., Turner D.J., Luscombe N.M., Ule J.. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010; 17:909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haberman N., Huppertz I., Attig J., Konig J., Wang Z., Hauer C., Hentze M.W., Kulozik A.E., Le Hir H., Curk T. et al. Insights into the design and interpretation of iCLIP experiments. Genome Biol. 2017; 18:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hauer C., Curk T., Anders S., Schwarzl T., Alleaume A.M., Sieber J., Hollerer I., Bhuvanagiri M., Huber W., Hentze M.W. et al. Improved binding site assignment by high-resolution mapping of RNA-protein interactions using iCLIP. Nat. Commun. 2015; 6:7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weyn-Vanhentenryck S.M., Mele A., Yan Q., Sun S., Farny N., Zhang Z., Xue C., Herre M., Silver P.A., Zhang M.Q. et al. HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep. 2014; 6:1139–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M. Jr, Jungkamp A.C., Munschauer M. et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010; 141:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burger K., Muhl B., Kellner M., Rohrmoser M., Gruber-Eber A., Windhager L., Friedel C.C., Dolken L., Eick D.. 4-thiouridine inhibits rRNA synthesis and causes a nucleolar stress response. RNA Biol. 2013; 10:1623–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang T., Xiao G., Chu Y., Zhang M.Q., Corey D.R., Xie Y.. Design and bioinformatics analysis of genome-wide CLIP experiments. Nucleic Acids Res. 2015; 43:5263–5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bottini S., Pratella D., Grandjean V., Repetto E., Trabucchi M.. Recent computational developments on CLIP-seq data analysis and microRNA targeting implications. Brief. Bioinform. 2017; 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Althammer S., Gonzalez-Vallinas J., Ballare C., Beato M., Eyras E.. Pyicos: a versatile toolkit for the analysis of high-throughput sequencing data. Bioinformatics. 2011; 27:3333–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yeo G.W., Coufal N.G., Liang T.Y., Peng G.E., Fu X.D., Gage F.H.. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat. Struct. Mol. Biol. 2009; 16:130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zisoulis D.G., Lovci M.T., Wilbert M.L., Hutt K.R., Liang T.Y., Pasquinelli A.E., Yeo G.W.. Comprehensive discovery of endogenous Argonaute binding sites in Caenorhabditis elegans. Nat. Struct. Mol. Biol. 2010; 17:173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lovci M.T., Ghanem D., Marr H., Arnold J., Gee S., Parra M., Liang T.Y., Stark T.J., Gehman L.T., Hoon S. et al. Rbfox proteins regulate alternative mRNA splicing through evolutionarily conserved RNA bridges. Nat. Struct. Mol. Biol. 2013; 20:1434–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Uren P.J., Bahrami-Samani E., Burns S.C., Qiao M., Karginov F.V., Hodges E., Hannon G.J., Sanford J.R., Penalva L.O., Smith A.D.. Site identification in high-throughput RNA-protein interaction data. Bioinformatics. 2012; 28:3013–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen B., Yun J., Kim M.S., Mendell J.T., Xie Y.. PIPE-CLIP: a comprehensive online tool for CLIP-seq data analysis. Genome Biol. 2014; 15:R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shah A., Qian Y., Weyn-Vanhentenryck S.M., Zhang C.. CLIP Tool Kit (CTK): a flexible and robust pipeline to analyze CLIP sequencing data. Bioinformatics. 2017; 33:566–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang T., Chen B., Kim M., Xie Y., Xiao G.. A model-based approach to identify binding sites in CLIP-Seq data. PLoS One. 2014; 9:e93248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang T., Xie Y., Xiao G.. dCLIP: a computational approach for comparative CLIP-seq analyses. Genome Biol. 2014; 15:R11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Van Nostrand E.L., Pratt G.A., Shishkin A.A., Gelboin-Burkhart C., Fang M.Y., Sundararaman B., Blue S.M., Nguyen T.B., Surka C., Elkins K. et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods. 2016; 13:508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boudreau R.L., Jiang P., Gilmore B.L., Spengler R.M., Tirabassi R., Nelson J.A., Ross C.A., Xing Y., Davidson B.L.. Transcriptome-wide discovery of microRNA binding sites in human brain. Neuron. 2014; 81:294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hodges A., Strand A.D., Aragaki A.K., Kuhn A., Sengstag T., Hughes G., Elliston L.A., Hartog C., Goldstein D.R., Thu D. et al. Regional and cellular gene expression changes in human Huntington's disease brain. Hum. Mol. Genet. 2006; 15:965–977. [DOI] [PubMed] [Google Scholar]
- 34. Spengler R.M., Zhang X., Cheng C., McLendon J.M., Skeie J.M., Johnson F.L., Davidson B.L., Boudreau R.L.. Elucidation of transcriptome-wide microRNA binding sites in human cardiac tissues by Ago2 HITS-CLIP. Nucleic Acids Res. 2016; 44:7120–7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krupp M., Marquardt J.U., Sahin U., Galle P.R., Castle J., Teufel A.. RNA-seq Atlas–a reference database for gene expression profiling in normal tissue by next-generation sequencing. Bioinformatics. 2012; 28:1184–1185. [DOI] [PubMed] [Google Scholar]
- 36. Kim K.K., Ham J., Chi S.W.. miRTCat: a comprehensive map of human and mouse microRNA target sites including non-canonical nucleation bulges. Bioinformatics. 2013; 29:1898–1899. [DOI] [PubMed] [Google Scholar]
- 37. Ascano M. Jr, Mukherjee N., Bandaru P., Miller J.B., Nusbaum J.D., Corcoran D.L., Langlois C., Munschauer M., Dewell S., Hafner M. et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012; 492:382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huelga S.C., Vu A.Q., Arnold J.D., Liang T.Y., Liu P.P., Yan B.Y., Donohue J.P., Shiue L., Hoon S., Brenner S. et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 2012; 1:167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bruun G.H., Doktor T.K., Borch-Jensen J., Masuda A., Krainer A.R., Ohno K., Andresen B.S.. Global identification of hnRNP A1 binding sites for SSO-based splicing modulation. BMC Biol. 2016; 14:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xiao R., Tang P., Yang B., Huang J., Zhou Y., Shao C., Li H., Sun H., Zhang Y., Fu X.D.. Nuclear matrix factor hnRNP U/SAF-A exerts a global control of alternative splicing by regulating U2 snRNP maturation. Mol. Cell. 2012; 45:656–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Agarwal V., Bell G.W., Nam J.W., Bartel D.P.. Predicting effective microRNA target sites in mammalian mRNAs. eLife. 2015; 4:e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee H.S., Seok H., Lee D.H., Ham J., Lee W., Youm E.M., Yoo J.S., Lee Y.S., Jang E.S., Chi S.W.. Abasic pivot substitution harnesses target specificity of RNA interference. Nat. Commun. 2015; 6:10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bottini S., Hamouda-Tekaya N., Tanasa B., Zaragosi L.E., Grandjean V., Repetto E., Trabucchi M.. From benchmarking HITS-CLIP peak detection programs to a new method for identification of miRNA-binding sites from Ago2-CLIP data. Nucleic Acids Res. 2017; 45:e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Krakau S., Richard H., Marsico A.. PureCLIP: capturing target-specific protein-RNA interaction footprints from single-nucleotide CLIP-seq data. Genome Biol. 2017; 18:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.