

Abstract
Key points
Sudden infant death syndrome (SIDS) is one of the leading causes of death during the first year of life and abnormalities linked to serotonin (5‐HT) have been identified in many SIDS cases.
Cigarette smoking and associated exogenous stressors, e.g. developmental nicotine exposure, may compound these serotonergic defects and any associated defects in cardiorespiratory function.
Using neonatal rodent pups subjected to medullary 5‐HT deficiency and perinatal nicotine exposure, we examined the impact of this interplay of factors on the neonates’ ability to autoresuscitate at specific ages.
In perinatal nicotine‐exposed 5‐HT deficient pups, impaired autoresuscitation along with significantly delayed post‐anoxic recovery of normal breathing and heart rate was observed at postnatal day 10 (P10).
We found that the interaction between 5‐HT deficiency and perinatal nicotine exposure can significantly increase pups’ vulnerability to environmental stressors and exacerbate defects in cardiorespiratory protective reflexes to repetitive anoxia during the development period.
Abstract
Cigarette smoking during pregnancy increases the risk of sudden infant death syndrome (SIDS), and nicotine replacements, a key ingredient of cigarettes, have been recently prescribed to women who wish to quit smoking during their pregnancy. Serotonin (5‐HT) abnormalities have been consistently identified in many SIDS cases. Here we investigated the effects of perinatal nicotine exposure in mild 5‐HT deficiency rat neonates on autoresuscitation, a protective cardiorespiratory reflex. The mild 5‐HT deficiency was induced by a maternal tryptophan‐deficient diet, and nicotine was delivered from embryonic day (E) 4 to postnatal day (P) 10 at 6 mg kg−1 day−1 through an osmotic pump. In P10 rats, nicotine exposure exacerbates autoresuscitation failure (mortality) in mildly 5‐HT‐deficient rats to a greater extent than in controls (P = 0.029). The recovery of eupnoea and heart rate to baseline values following repetitive anoxic events (which elicit an apnoea accompanied by a bradycardia) is significantly delayed in 5‐HT‐deficient rats treated with nicotine, making them more susceptible to failure of autoresuscitation (eupnoea recovery: P = 0.0053; heart rate recovery: P = < 0.0001). Neither 5‐HT deficiency nor nicotine exposure alone appears to affect the ability to autoresuscitate significantly when compared among the four treatments. The increased vulnerability to environmental stressors, e.g. severe hypoxia, asphyxia, or anoxia, in these nicotine‐exposed 5‐HT‐deficient neonates during postnatal developmental period is evident.
Keywords: Triple Risk hypothesis, Developmental nicotine exposure, Serotonin deficiency
Key points
Sudden infant death syndrome (SIDS) is one of the leading causes of death during the first year of life and abnormalities linked to serotonin (5‐HT) have been identified in many SIDS cases.
Cigarette smoking and associated exogenous stressors, e.g. developmental nicotine exposure, may compound these serotonergic defects and any associated defects in cardiorespiratory function.
Using neonatal rodent pups subjected to medullary 5‐HT deficiency and perinatal nicotine exposure, we examined the impact of this interplay of factors on the neonates’ ability to autoresuscitate at specific ages.
In perinatal nicotine‐exposed 5‐HT deficient pups, impaired autoresuscitation along with significantly delayed post‐anoxic recovery of normal breathing and heart rate was observed at postnatal day 10 (P10).
We found that the interaction between 5‐HT deficiency and perinatal nicotine exposure can significantly increase pups’ vulnerability to environmental stressors and exacerbate defects in cardiorespiratory protective reflexes to repetitive anoxia during the development period.
Introduction
Sudden infant death syndrome (SIDS) is the sudden and unexpected death of an infant under 12 months of age that occurs typically during the sleep period, the cause of which remains unexplained following an autopsy and a death scene investigation (Krous et al. 2004). An increasing number of studies suggest that SIDS infants have deficits in respiratory and autonomic function that increase their vulnerability to severely hypoxic conditions (Franco et al. 1999; Poets et al. 1999; Harper, 2001; Sridhar et al. 2003; Poets, 2004; Kinney & Thach, 2009). Failure to autoresuscitate from events of asphyxia may result from the lack of protective responses against potentially fatal circumstances that may occur during re‐breathing while in the prone position, or in other airway‐obstructing conditions during sleep periods (Kemp et al. 1993; Mitchell et al. 1999). These deficits in respiratory and autonomic control have previously been associated with abnormalities in the medullary serotonin (5‐hydroxytryptamine or 5‐HT) system that have been documented in a majority of SIDS cases studied, including abnormal 5‐HT receptor and transporter binding (Panigrahy et al. 2000; Paterson et al. 2006), reduced tissue 5‐HT and tryptophan hydroxylase 2 (TPH2) levels (Duncan et al. 2010), and increased cell counts of immature and possibly dysfunctional 5‐HT neurons (Panigrahy et al. 2000; Paterson et al. 2006; Machaalani et al. 2009). It is apparent these infants are more vulnerable to stressors that may trigger a life threatening apnoeic event leading to the failure of arousal and autoresuscitation. Neonatal mice and rats expressing a range of medullary 5‐HT deficiencies (Hodges & Richerson, 2008; Hodges et al. 2008; Cummings et al. 2009, 2010, 2011; Penatti et al. 2011) demonstrate homeostatic deficits and exaggerated apnoeas and bradycardias, again implicating the medullary 5‐HT system in maintaining cardiorespiratory homeostasis. Medullary 5‐HT deficiencies in rodent models range from a milder 40–50% 5‐HT deficit in tryptophan‐deficient rat neonates (Penatti et al. 2011) to more severe ∼90% 5‐HT deficit in transgenic Pet‐1−/− mice lacking 60–70% of brainstem 5‐HT cells from early embryogenesis (Cummings et al. 2010, 2011). Pharmacological lesion of 5‐HT neurons via 5,7‐DHT injections during early postnatal period (P2–3) can also induce a severe ∼80% 5‐HT deficit in the brainstem of neonatal rats (Cummings et al. 2009).
Cigarette smoking during pregnancy increases the risk of pre‐term birth and SIDS (MacDorman et al. 1997; Nachmanoff et al. 1998; Waters et al. 1999; Matturri et al. 2006; Duncan et al. 2008; Hakeem et al. 2015). In the last decade, the overall use of cigarettes has declined significantly; however, over 10% of pregnant women still smoke during pregnancy, and there is an increased trend towards using non‐combusted tobacco products (snuff, dissolvable, and electronic nicotine delivery systems) that can deliver nicotine during pregnancy (England et al. 2016, 2017). Nicotine replacement therapies, e.g. nicotine patches or electronic cigarettes, have been prescribed to women who wish to quit smoking during their pregnancy in recent years (Coleman et al. 2012; Oncken, 2012), even though nicotine replacement therapies may not protect neonates from SIDS and other adverse effects of smoking tobacco (England et al. 2016, 2017). Perinatal nicotine exposure can directly or indirectly affect many neurotransmitter systems in the central nervous system, e.g. the serotonergic (Xu et al. 2001; Duncan et al. 2009; Kamendi et al. 2009; Blood‐Siegfried & Rende, 2010; Slotkin et al. 2015) and the cholinergic systems (Duncan et al. 2008), both of which are critically involved in cardiorespiratory function and autoresuscitation. Cigarette smoke can increase the likelihood of inadequate placental perfusion, chronic intra‐uterine hypoxia and intra‐uterine growth restriction (Lambers & Clark, 1996; Andres & Day, 2000) and nicotine itself, via interactions with the brain's endogenous nicotinic receptors, can adversely impair neural development and function (Slikker et al. 2005). Fetal nicotine exposure can impair cardiac and respiratory responses to hypoxia (Slotkin et al. 1995; St‐John & Leiter, 1999; Fewell et al. 2001; Neff et al. 2004; Feng et al. 2010), disrupt autonomic regulation (Zeskind & Gingras, 2006) and increase the incidence of obstructive sleep apnoea (Kahn et al. 1994) in neonates. Any alterations in the course of development of nAChRs and the neurons expressing them, particularly medullary 5‐HT neurons, will inevitably affect how they in turn function and govern cardiorespiratory homeostasis.
Medullary 5‐HT neurons play a critical role in maintaining cardiorespiratory homeostasis and therefore, a dysfunctional 5‐HT network will disrupt this line of defense against perturbations, such as severe hypoxia or asphyxia. During sleep, loss of appropriate responses to hypoxia exposure, e.g. arousal from sleep or gasping, becomes detrimental and makes an infant increasingly vulnerable to the risk of SIDS (Kinney et al. 2009).
The goal of this study was to investigate whether a mild brainstem 5‐HT deficiency induced by maternal tryptophan‐deficient diet would interact with perinatal nicotine exposure to increase neonates’ vulnerability to environmental stressors, e.g. severe hypoxia, anoxia, or asphyxia, during postnatal developmental period. The pre–perinatal nicotine‐exposed mild 5‐HT‐deficient neonates were challenged with repeated bouts of anoxia similar to those experienced during episodes of sleep apnoea in infants at different postnatal ages. We hypothesize that interaction between the mild medullary 5‐HT and developmental nicotine exposure will exacerbate autoresuscitation failure in rat neonates when challenged with environmental anoxia at a specific age.
Methods
Ethical approval
All experimental protocols were approved by the Institutional Animal Care and Use Committee of Dartmouth College and followed the guidelines of the National Institute of Health of animal use and care. The authors have read and complied with guidelines for research in rodents outlined in the Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology (Grundy, 2015).
Tryptophan‐deficient diet treatment
Adult female Sprague‐Dawley rats (Charles River Laboratories, Inc.) were housed with a 12 h light‐dark cycle at an ambient temperature of 21–23°C. They were fed ad libitum isocaloric, amino acid‐based diets containing either the required level of tryptophan (TD.99366; amino acid control diet) or those with levels reduced to approximately 55% of required (TD.140218; tryptophan‐deficient diet diet; Envigo, formerly known as Harlan Teklad). They also had unlimited access to water. The amino acid control (Ctr) diet was composed of the following (in g kg−1): 3.5 L‐alanine, 12.1 L‐arginine HCl, 6.0 L‐asparagine, 3.5 L‐aspartic acid, 3.5 L‐cystine, 40.0 L‐glutamic acid, 23.3 glycine, 4.5 L‐histidine HCl, monohydrate, 8.2 L‐isoleucine, 11.1 L‐leucine, 18.0 L‐lysine HCl, 8.2 L‐methionine, 7.5 L‐phenylalanine, 3.5 L‐proline, 3.5 L‐serine, 8.2 L‐threonine, 1.8 L‐tryptophan, 5.0 L‐tyrosine, 8.2 L‐valine, 351.68 sucrose, 150.0 corn starch, 150.0 maltodextrin, 80.0 soybean oil, 30.0 cellulose, 35.0 mineral mix, 8.2 calcium phosphate, monobasic, monohydrate, 13.0 vitamin mix, 2.5 choline bitartrate, and 0.02 tertiary butylhydroquinone, an antioxidant. The tryptophan‐deficient (TD) diet was identical except that the level of tryptophan was reduced to 1 g kg−1 (approximately 55% of control diet), while L‐glutamic acid was increased to 41.18 g kg−1 in order to maintain the tryptophan‐deficient (TD) diet isonitrogenous and isocaloric to the control diet (Ctr diet). Adult female Sprague‐Dawley rats received the new diets for at least a week prior to mating and continued to receive the diets throughout the gestation period and post birth up to postnatal day 25.
Nicotine treatment and surgery
Pregnant dams on either Ctr or TD diet were surgically implanted subcutaneously with osmotic pumps (model 2ML4; 2.5 μl h−1, 2 ml total; ALZET Osmotic Pumps, DURECT Corporation) filled with a nicotine tartrate solution (6 mg kg−1 day−1) or vehicle (saline) on gestational day 4. The nicotine‐receiving dams were control matched with dams receiving a saline vehicle infusion via the osmotic pumps. Dams were anaesthetized with 4% isoflurane for initial induction; 1–2% isoflurane was used to maintain a surgical level of anaesthesia. Each dam received a subcutaneous injection of ketoprofen (5 mg kg−1) as an analgesic at this stage. While in the prone position, an area approximately 3 cm by 3 cm (fur removed) was cleaned and sterilized with 70% ethanol and topical iodine (Medline Prep Solution; topical antiseptic microbicide) in preparation for the procedure. An incision slightly wider than the width of the pump was made in the back of the rat and a pocket to house the pump was created subcutaneously by breaking apart the connective tissue below the surface of the skin using blunt dissection. Once the pump was in place, the skin was sutured and isoflurane was removed to allow the dam to recover from anaesthesia. Each dam post surgery continued to receive a subcutaneous injection of ketoprofen (5 mg kg−1) as an analgesic for 3 days following pump implantation; dams were also weighed daily during this period to ensure there was no weight loss. Pump contents were released at the rate of 60 μl day−1 and absorbed continuously during the remainder of the gestational and early postnatal period.
HPLC analysis of monoamine medullary content
A separate group of Ctr and TD diet litters that received either vehicle (saline) or nicotine treatments were used for the HPLC analysis of monoamine medullary content. The measured monoamines are as follows: 5‐HT and its metabolite 5‐HIAA (5‐hydroxy‐indole‐acetic acid), noradrenaline (NAd), adrenaline (Adr), dopamine (DA) and their respective major metabolites, HVA (homovanillic acid) and DOP (dihydroxyphenylacetic acid or DOPAC) (Fig. 1 D). Since nicotine alone did not affect any of the monoamine levels measured, nicotine‐ and vehicle‐treated pups were consolidated into their respective diet groups. Ctr diet: P10 (n = 20, 6 litters), P15 (n = 20, 5 litters), and P25 (n = 20, 6 litters); TD diet: P10 (n = 20, 9 litters), P15 (n = 20, 10 litters), and P25 (n = 20, 9 litters). Pups were killed by pentobarbital overdose (>75 mg kg−1, Euthasol, Virbac Inc. Fort Worth, TX, USA) and their brainstems were collected immediately after killing. The method of extracting medullary tissue was similar to those previously reported (Penatti et al. 2011); in short, a tissue wedge was extracted from the ventral midsection of the medulla to include the regions heavily populated by 5‐HT neurons (raphe obscurus, magnus and pallidus), and was stored at −80°C. For the HPLC analysis, the tissues were isolated and homogenized, and the supernatants were harvest. HPLC was performed utilizing an Antec Decade II electrochemical detector. Twenty microlitres of supernatant were injected onto a 100 × 4.60 mm HPLC column (Phenomenex, Torrance, CA, USA). Biogenic amines are eluted with a mobile phase. HPLC data acquisition and analysis are managed by Empower software (Waters Co.) and performed by Neurochemistry Core at Vanderbilt University School of Medicine (Nashville, TN, USA).
Figure 1. The effect of tryptophan‐deficient (TD) on medullary monoamine levels.
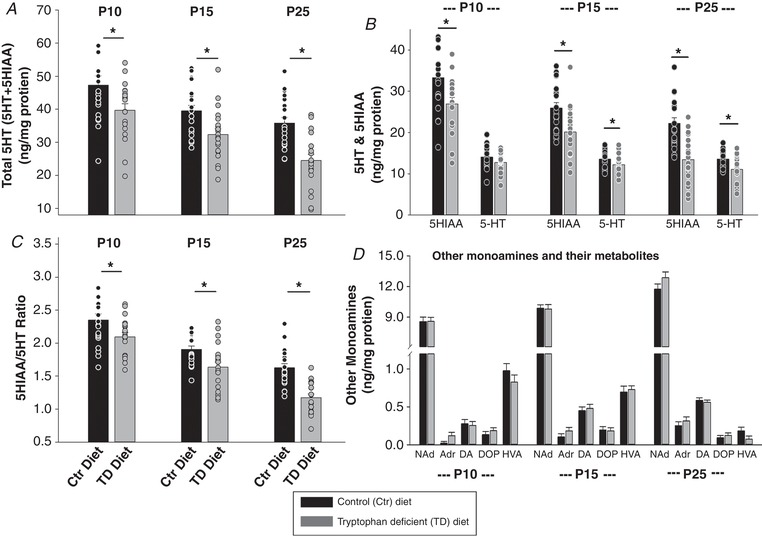
A–C, the total 5‐HT (5‐HT + 5‐HIAA) (A), ratio of 5‐HIAA to 5‐HT (5‐HIAA/5‐HT) (C), and 5‐HT and 5‐HT metabolite (5‐HIAA) levels (B) in the medulla were significantly lower in TD diet pups than control (Ctr) diet pups at P10, P15 and P25 (* P < 0.05). Although 5‐HT levels were lower in TD diet pups than Ctr diet pups at all three ages, they were only significant at P15 and P25 (* P < 0.05). D, there were no significant differences with other monoamines and their metabolite levels between Ctr and TD diet groups at all three ages (P > 0.05). Data are presented as means ± SEM, and each circle represents an individual animal. Two‐way ANOVA followed by the Holm‐Sidak post hoc test were used for all comparisons. Black bar: Ctr diet; grey bar: TD diet. NAd, noradrenaline; Adr adrenaline; DA, dopamine; DOP, DOPAC or dihydroxyphenylacetic acid; HVA, homovanillic acid.
The values obtained are reported in nanograms per gram of tissue protein. The 5‐HT + 5‐HIAA data were used as a measure of total 5‐HT content and the 5‐HIAA/5‐HT ratio was used as an index of serotonergic system activity.
ELISA analysis of serum cotinine level
To confirm nicotine exposure in pups, cotinine (a metabolite of nicotine) levels were quantified using an ELISA kit (Abnova) per the manufacturer's user manual using serum samples from pups treated with nicotine and vehicle on either Ctr or TD diet at P5, P8, P10, and P12. For serum preparation, whole blood samples were collected from the pups following the autoresuscitation experiment. Once pups were killed post experiment, whole blood samples were immediately collected from the heart by making an incision through the cardiac muscle. Blood extraction via cardiac puncture caused haemolysis and was deemed unsuitable for this analysis. Samples collected in a safe‐lock tube were left to clot undisturbed at room temperature for approximately 20 min. The clot was separated from the supernatant (serum) by centrifuging at 2000 × g for 10 min in a refrigerated centrifuge. Serum was transferred immediately into a clean tube, being maintained at 4°C during handling. Samples with visible signs of haemolysis were discarded and those not analysed immediately for cotinine levels were stored at −80°C.
Respiratory and heart rate measurements
The experimental set‐up was similar to those described in a previous study (Cummings et al. 2009, 2011); pups were placed in a water‐jacketed glass chamber with body temperature held at 36 ±0.5°C throughout the experiment by controlling the temperature of the water perfused through the chamber. Water temperature was adjusted frequently in order to maintain a stable body temperature.
Respiratory parameters (tidal volume (V T), breathing frequency (f R), ventilation () and oxygen consumption ()) were measured using head‐out plethysmography; the mask separating the head from the rest of the animal was made by placing a piece of nitrile over a syringe tube (volume approximately 3 ml) and holding it in place with a fitted rubber ring (Terumo Medical Corporation, Japan). A small hole placed in the nitrile acted as an opening for the snout into the mask; the mask was sealed onto the face using Impregum F Impression Material (3M ESPE). A pneumotachograph was mounted onto the other end of the mask, through which the pups breathed in the surrounding air. Inspiratory and expiratory airflows were detected by a differential pressure transducer (Validyne Engineering Corp., Northridge, CA, USA) connected to the side‐arms of the pneumotachograph. Air was drawn through the pneumotachograph and into the mask by a downstream pump to maintain a constant supply of fresh air inside the mask. The expired air was drawn through a desiccant column (Drierite Co. Ltd, Xenia, OH, USA) and then through an O2 analyser (AEI Technologies, Pittsburgh, PA, USA). Body temperature and heart rate were measured using a rectal temperature probe and surface ECG electrodes, respectively. All data were recorded continuously using LabChart (ADinstruments, Colorado Springs, USA).
Experimental design and protocol
For the physiology experiment, animals of either sex were equally divided into four treatment groups based on the diet (Ctr or TD diet) and treatment (nicotine or vehicle). Ctr diet/vehicle group dams were treated with control diet and saline vehicle; TD diet/vehicle group dams were treated with the tryptophan‐deficient diet and saline vehicle; Ctr diet/nicotine group dams were treated with control diet and nicotine; TD diet/nicotine group dams were treated with tryptophan‐deficient diet and nicotine. Within each treatment group, animals were further divided into four age groups (P5, P8, P10 and P12) and each animal was tested only once at one age for the experiment. Table 1 lists the experimental groups at the four age groups including the sample size and the total number of litters used. Large litters were culled to 10 pups at P2 and any litters with fewer than eight pups were excluded from the study in order to eliminate any confounding factors that may arise from differences in litter size.
Table 1.
Number of animals and litters used at each age in each treatment group
| Control vehicle | TD vehicle | Control nicotine | TD nicotine | |||||
|---|---|---|---|---|---|---|---|---|
| Age | n | No. of litters | n | No. of litters | n | No. of litters | n | No. of litters |
| P5 | 10 | 6 | 10 | 5 | 10 | 5 | 10 | 5 |
| P8 | 13 | 6 | 15 | 7 | 13 | 7 | 13 | 7 |
| P10 | 16 | 3 | 16 | 4 | 15 | 4 | 15 | 3 |
| P12 | 14 | 8 | 15 | 9 | 13 | 6 | 13 | 6 |
On the day of the experiment, the animal was instrumented with a temperature probe and ECG electrodes first prior to sealing the mask onto the face. The mask was fitted onto the pre‐heated water‐jacketed body chamber (completing the head‐out plethysmography set‐up) using the same rubber ring used to fix the nitrile onto the syringe tube portion of the mask. The animal could adjust to the experimental setup for 30 min to allow the body temperature to warm to 36°C in addition to stabilizing . Following 10 min of baseline respiration recording, the animal underwent the autoresuscitation protocol, which consists of repeated bouts of anoxia (up to 15 bouts) separated by 5‐min recovery periods of normoxia when respiratory and heart rates returned to ≥ 63% of baseline values. The duration of a typical experiment for the pups that survived all 15 bouts of anoxia was approximately two hours, including the experiment set‐up. For the anoxic bout, gas (97% N2/3% CO2) was delivered directly from the tank to the open end of the pneumotachograph and the downstream pump drew this gas into the mask for a rapid anoxia exposure. Gas was applied until the animal became apnoeic and motionless with lack of muscle tone for 5 s (this period of apnoea will be referred to as ‘primary apnoea’), at which point the gas was immediately removed to flush the mask with room air. Litters from Ctr or TD diet dams treated with either saline vehicle or nicotine underwent the autoresuscitation protocol at four ages (P5, P8, P10 and P12) to assess their ability to survive from events of acute asphyxia. Each pup was tested once at one age only. Following the autoresuscitation experiment, the pups were killed by pentobarbital overdose (>75 mg kg−1, Euthasol, Virbac Inc. USA) and their medullas collected, frozen and stored at −80°C for future analysis.
Data and statistical analysis
Baseline respiratory parameters (, V T, and f R), HR, and metabolic rate () of all four treatment groups are summarized in Table 2 (means ± SD). To evaluate the effects of treatments on baseline parameters at different ages, a three‐way ANOVA (analysis of variance) was employed, with factor 1 being tryptophan deficiency; factor 2 developmental nicotine exposure, and factor 3 age (P5, P8, P10 and P12). Data were collected from both male and female pups of each treatment group at the four experimental ages, and for all statistical analyses an α level of P < 0.05 was used.
Table 2.
Baseline physiological parameters at each age in each group
| HR (min−1) | (ml min−1 kg−1) | V T (ml kg−1) | f R (min−1) | (ml min−1 kg−1) | |
|---|---|---|---|---|---|
| P5 | |||||
| Ctr diet/vehicle | 363.4 ± 29.6 | 705.7 ± 123.7 | 5.8 ± 0.9 | 128.9 ± 21.1 | 24.9 ± 3.8 |
| TD diet/vehicle | 370.7 ± 27.5D | 674.9 ± 68.7 | 5.9 ± 1.4 | 122.2 ± 16.9 | 26.2 ± 3.1 |
| Ctr diet/nicotine | 367.1 ± 28.0 | 748.2 ± 103.8 | 5.7 ± 0.8 | 137.0 ± 22.3 | 27.4 ± 4.7 |
| TD diet/nicotine | 385.7 ± 20.2D | 673.2 ± 89.6 | 6.0 ± 0.9 | 120.5 ± 12.6 | 26.7 ± 2.6 |
| P8 | |||||
| Ctr diet/vehicle | 399.9 ± 25.3 | 757.0 ± 134.3 | 5.2 ± 0.9 | 150.2 ± 13.4 | 25.6 ± 2.7 |
| TD diet/vehicle | 390.2 ± 31.1 | 720.8 ± 108.5 | 5.8 ± 1.3 | 129.1 ± 16.6D | 25.3 ± 3.3 |
| Ctr diet/nicotine | 391.4 ± 11.9T | 704.6 ± 98.2 | 5.3 ± 0.9 | 139.5 ± 21.9 | 28.0 ± 3.7 |
| TD diet/nicotine | 380.3 ± 12.1T | 771.9 ± 100.8 | 5.8 ± 0.7 | 136.8 ± 12.3D | 27.3 ± 3.2 |
| P10 | |||||
| Ctr diet/vehicle | 379.8 ± 21.4 | 782.9 ± 252.6 | 6.4 ± 3.0 | 131.6 ± 19.1 | 27.2 ± 2.6 |
| TD diet/vehicle | 374.1 ± 23.5D | 802.2 ± 213.4 | 6.3 ± 1.4 | 130.5 ± 19.9D | 26.0 ± 1.8 |
| Ctr diet/nicotine | 394.2 ± 31.2T | 845.1 ± 529.2 | 6.0 ± 1.7 | 133.3 ± 27.0 | 26.7 ± 3.6 |
| TD diet/nicotine | 401.3 ± 24.1D,T | 894.8 ± 372.8 | 6.3 ± 1.1 | 138.0 ± 20.7D | 28.2 ± 3.1 |
| P12 | |||||
| Ctr diet/vehicle | 415.5 ± 18.2 | 708.7 ± 198.6 | 5.7 ± 1.6 | 124.6 ± 19.0 | 28.5 ± 2.6 |
| TD diet/vehicle | 409.9 ± 26.3D | 788.8 ± 191.0 | 6.6 ± 1.0 | 119.5 ± 14.0D | 30.1 ± 3.8 |
| Ctr diet/nicotine | 436.4 ± 33.8T | 721.0 ± 148.9 | 5.8 ± 1.5 | 125.9 ± 11.7 | 27.9 ± 2.0 |
| TD diet/nicotine | 408.9 ± 27.8D,T | 743.4 ± 208.8 | 6.2 ± 1.1 | 120.1 ± 20.8D | 27.5 ± 4.6 |
There was a significant difference in HR between TD diet pups and age‐matched Ctr diet pups at P5, P10, and P12 (P < 0.001; ‘D’ denotes diet effect), whereas there was a significance difference in HR between nicotine‐treated and non‐treated pups at P8, P10, and P12 (P < 0.05; ‘T’ denotes nicotine treatment effect). During baseline, HR was significantly affected by age (P < 0.001) and treatment (P = 0.03), and significant interactions were found between age and diet (P = 0.028), as well as between age and treatment (P = 0.018). Three‐way ANOVA with Holm‐Sidak post hoc analysis. Data presented as means ± SD.
Mortality or survival rate was analysed using the Kaplan‐Meier survival analysis (Gehan‐Breslow method) and the Cox Regression–Proportional Hazards Model among the four treatment groups of all ages (P5, P8, P10 and P12). Additionally, at each age, percentage survival was assessed across the 15 anoxic episodes for each treatment group to determine and compare their survival rates. There were no significant differences in survival rates between the treatment groups at P5, P8 and P12; therefore, further detailed analyses and data plots are presented only for P10 groups (Figs 3 and 4).
Figure 3. Percentage survival across 15 bouts of anoxia in P10 neonates.
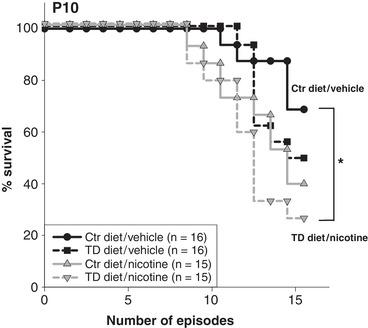
At P10, there is a statistically significant difference between survival curves (P = 0.029, Kaplan‐Meier survival (Gehan‐Breslow) analysis), with 69% of Ctr diet/vehicle‐treated pups (11 of 16), 50% of TD diet/vehicle pups (8 of 16), 40% of Ctr diet/nicotine pups (6 of 15), and only 27% of the TD diet/nicotine‐exposed pups (4 of 15) surviving all 15 episodes. All pairwise comparisons showed that TD diet/nicotine‐treated pups had significant lower surviving rates than Ctr diet and saline (Ctr diet/vehicle)‐treated pups (* P = 0.0425, Survival Gehan‐Breslow–Holm‐Sidak all pairwise analysis).
Figure 4. Mean number of anoxic bouts survived in P10 neonates.
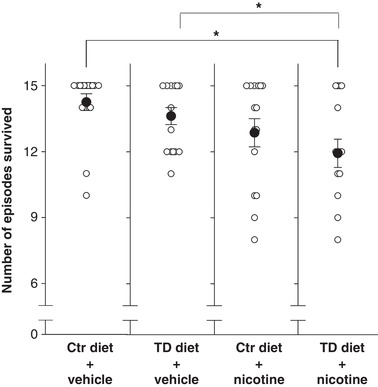
The mean number of anoxic episodes survived (filled circles) was significantly less in TD diet/nicotine‐exposed P10 rats (12 episodes; far right column) than saline vehicle‐treated Ctr and TD diet pups (* P = 0.025). Neither tryptophan deficiency nor nicotine exposure alone significantly altered the mean number of anoxic episodes survived. Data presented as means ± SEM and each circle represents an individual animal.
Breathing and heart rate (HR) indices were defined as gasp latency (duration of primary apnoea), eupnoea recovery (time taken to restore eupnoeic breathing from the primary apnoea elicited by the anoxia to 63% of baseline values), and HR recovery (time taken to restore HR from the bradycardia during primary apnoea elicited by the anoxia to 63% of baseline values). To evaluate whether repetitive anoxia would progressively impair the breathing and HR indices at P10, gasp latency, eupnoea and HR recovery following anoxia episode 1, 5, 10, 15 or last survived anoxic bout were determined and compared among all experimental groups using a repeated‐measures (RM) ANOVA followed by Holm‐Sidak post hoc test (repeat with episode). Animals that died before episode 10 were not included in this analysis, and numbers of animals in each group used for this analysis are shown in Fig. 5. A value of 63% was used since that is the definition of the time constant, tau (Fewell et al. 2000).
Figure 5. Gasp latency, eupnoea and HR recovery at anoxic episodes 1, 5, 10 and the last survived episode in P10 neonates.
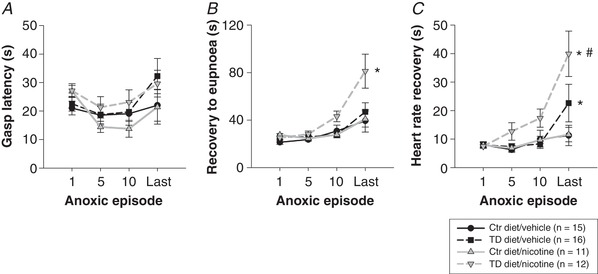
A, no significant differences in gasp latency (duration of primary apnoea) were observed amongst the four treatment groups (no treatment effect) across the repeated bouts of anoxia (no episode effect). B, recovery to eupnoea was delayed with progressive anoxic bouts (episode effect: P < 0.0001,) in all treatment groups, with the longest recovery period observed during the last survived anoxic episode. Eupnoea recovery was significantly delayed in TD diet/nicotine‐treated pups at P10 (grey inverted triangles, dashed line; treatment effect: * P = 0.0053) compared to all other treatment groups during the last survived anoxic episode. C, HR recovery was delayed with progressive anoxic bouts in all treatment groups. The pups treated with TD diet had longer HR recovery periods during the last anoxia episode than the pups treated with Ctr diet (* P ≤ 0.05). TD diet/nicotine pups had the slowest HR recovery compared to all other treatment groups (grey inverted triangles, dashed line; # P < 0.0001). Two‐way RM ANOVA with Holm‐Sidak post hoc analysis. Data presented as means ± SEM.
The effects of tryptophan‐deficient diet and developmental nicotine exposure on the medullary tissue monoamine levels were analysed using a two‐way ANOVA at each age, followed by Holm‐Sidak method post hoc test. Since nicotine alone has no effect on the levels of medullary monoamines, the results from nicotine‐ and saline vehicle‐treated pups were combined based on their diet treatments (Fig. 1).
Results
Effect of nicotine on serum cotinine level
Cotinine (a metabolite of nicotine) levels were undetectable in the offspring of saline vehicle‐treated dams on either Ctr or TD diet at any age. The offspring of nicotine‐treated dams had serum cotinine levels between 70 and 80 ng ml−1 (Ctr diet/nicotine pups: 80.4 ± 26.3; TD diet/nicotine: 76.2 ± 22.5; data are means ± SD) between P8 and P12, with no significant difference between the two diet groups. These levels are comparable to those seen in other studies of developmental nicotine exposure in rat pups (Benowitz et al. 1982; Lichtensteiger et al. 1988; Slotkin et al. 1995; Milerad et al. 1998; Hafstrom et al. 2005).
Effect of tryptophan‐deficient diet on 5‐HT and 5‐HIAA content in medulla
The levels of medullary monoamines, including 5‐HT, noradrenaline (NAd), adrenaline (Adr), dopamine (DA) and their major metabolites (5‐HIAA, homovanillic acid (HVA), dihydroxyphenylacetic acid (DOPAC or DOP)), were measured in the offspring from Ctr and TD diet dams. There was no significant difference in medullary monoamine levels between nicotine‐ and vehicle‐treated pups in either Ctr or TD diet groups; therefore, the pups were regrouped into two groups based only on their diet, Ctr or TD diet (Fig. 1).
Nicotine treatment alone did not significantly affect levels of catecholamine, dopamine, 5‐HT and their metabolites in the medulla (data not shown). TD diet did not significantly affected levels of catecholamines, dopamine and their metabolites in the medulla (Fig. 1 D, P > 0.05) but significantly decreased medullary tissue total 5‐HT (5‐HT + 5‐HIAA) content and 5‐HT activity index (ratio of 5‐HIAA/5‐HT) (Fig. 1 A–C). The levels of 5‐HIAA and 5‐HIAA/5‐HT are highly correlated with 5‐HT level in the brain, and therefore the changes of 5‐HT, 5‐HT and its metabolite 5‐HIAA, and the 5‐HIAA/5‐HT ratio were further analysed to evaluate the total 5‐HT activity in the brain in this study. At P10, the total 5‐HT (5‐HT + 5‐HIAA) content, 5‐HIAA, and 5‐HIAA/5‐HT ratio were all significantly lower in TD diet pups than Ctr diet pups (diet effect: P = 0.035; 0.03 and 0.04, respectively; two‐way ANOVA) (Fig. 1 A–C). At P10, even though the medullary 5‐HT only decreased mildly (∼10%) and was not statistical significant (P = 0.116, two‐way ANOVA) between TD diet and Ctr diet pups, the level of 5‐HT metabolite, 5‐HIAA, decreased significantly (∼19%; P = 0.03, two‐way ANOVA), and both 5‐HT and 5‐HIAA contribute to a significant decrease in total 5‐HT (∼16%; P = 0.035, two‐way ANOVA). At P15 and P25, all 5‐HT product levels in the medulla, including 5‐HT, 5‐HIAA, total 5‐HT, and 5‐HIAA/5‐HT, were significantly lower in TD diet pups than in Ctr diet pups (diet effect: P ≤ 0.05, two‐way ANOVA) (Fig. 1 A–C). An age difference effect on the severity of 5‐HT deficiency (total 5‐HT content) in TD diet pups was also observed (P < 0.001, two‐way ANOVA) and this was expected as the pups started to consume more TD diet food themselves as they were ageing.
Effects of tryptophan‐deficient diet and nicotine on baseline body weight, respiratory and heart rate parameters
Baseline values, including respiratory parameters, HR, and metabolic rates of all four treatment groups at P5, P8, P10 and P12, have been summarized in Table 2.
A three‐way ANOVA analysis showed that body weight was significantly affected by age (P ≤ 0.001), diet (TD or Ctr diet; P ≤ 0.001) and treatment (nicotine or vehicle control; P = 0.005), and the significant interactions were found between age and diet (P ≤ 0.001), diet and treatment (P = 0.003) (three‐way ANOVA with Holm‐Sidak post hoc test). TD diet pups had significantly lower body weights than age‐matched Ctr diet pups at P8, P10 and P12 (P < 0.001, three‐way ANOVA), and when treated with nicotine, the TD diet/nicotine pups exhibited significantly lower body weights than the other three treatment groups at P8, P10 and P12 ((P < 0.001, three‐way ANOVA; Fig. 2). Body weight changes induced by the TD diet are similar to those observed in our previous study (Penatti et al. 2011).
Figure 2. The effects of diet and nicotine on body weights in P5, 8, 10 and 12 neonates.
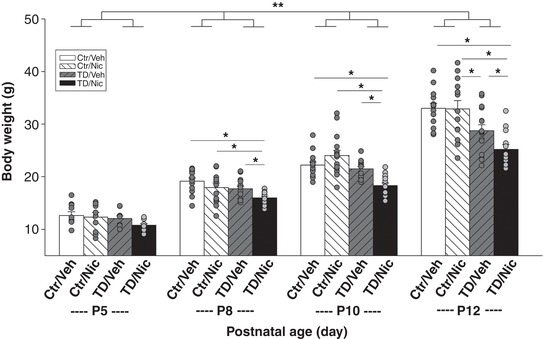
Body weight was significantly affected by age (P ≤ 0.001), diet (P ≤ 0.001) and treatment (P = 0.005), and the significant interactions were found between age and diet (P ≤ 0.001), diet and treatment (P = 0.003) (three‐way ANOVA with Holm‐Sidak post hoc test; **significant interaction between age and diet). At P8, P10 and P12, the TD diet/nicotine (TD/Nic)‐treated pups (black bars) were significantly smaller than pups from other treatment groups at P8, P10 and P12 (*statistical significance; P ≤ 005). At P12, TD diet/vehicle (TD/Veh) pups (grey hatched bars) were significantly smaller than the Ctr/Veh (white bars) and Ctr/Nic (white hatched bar) pups (P = 0.011). Data presented as means ± SEM and each circle represents an individual animal.
Baseline HR was significantly affected by age (P < 0.001) and treatment (P = 0.03), and significant interactions were found between age and diet (P = 0.028), as well as between age and treatment (P = 0.018) using a three‐way ANOVA. In particular, there was a significant difference in HR between TD diet pups and age‐matched Ctr diet pups at P5, P10 and P12 (P < 0.001), whereas there was a significance difference in HR between nicotine‐treated and non‐treated pups at P8, P10 and P12 (P < 0.05) (three‐way ANOVA, Holm‐Sidak post hoc test; Fig. 2).
Baseline respiratory parameters, f R , V T and , were significantly affected by age (P < 0.001, P < 0.001, and P = 0.027, respectively, three‐way ANOVA). The treatment and diet had no effect on baseline V T and , whereas diet did significantly affect f R (P = 0.01) at P8, P10 and P12 (three‐way ANOVA, Holm‐Sidak post hoc test; Fig. 2).
Effect of tryptophan‐deficient diet and nicotine on autoresuscitation
The ability to autoresuscitate from repetitive episodic anoxia was assessed in pups of all treatment groups at each age, P5, P8, P10 and P12. The percentage of pups that survived all 15 anoxic episodes was not significantly different between the treatment groups at P5, P8 and P12 (a Kaplan‐Meier survival analysis, Gehan‐Breslow method; data not shown), whereas at P10, the pups from the dams treated with both maternal TD diet and nicotine (TD diet/nicotine) exhibited the highest mortality rate when challenged with repeated bouts of anoxia (Fig. 3). The percentage of pups that survived all 15 bouts of anoxia was significantly different among the treatment groups (P = 0.029, Gehan‐Breslow analysis, Fig. 3). The subsequent all pairwise multiple comparison analysis showed that, compared to the Ctr diet/vehicle pups, tryptophan deficiency or nicotine exposure alone did not significantly change the survival rate; however, when the two factors, tryptophan deficiency and nicotine exposure, were combined, percentage survival decreased significantly (P = 0.045, Survival Gehan‐Breslow and Holm‐Sidak method). P10 TD diet/nicotine‐exposed pups also survived fewer anoxic episodes than non‐nicotine (Ctr diet/vehicle and TD diet/vehicle; P = 0.03 and 0.025 respectively, one‐way ANOVA)‐treated pups (Fig. 4). In addition, a Cox regression proportional hazards survival model further showed that TD diet/nicotine pups at P10 had a significantly lower survival rate than those at P5 and P8 (P < 0.001 and P = 0.002, respectively). These data suggest that a mild 5‐HT deficiency (induced by a maternal tryptophan‐deficient diet) together with pre‐ and early postnatal nicotine exposure can exacerbate autoresuscitation failure at a specific age.
Effect of tryptophan‐deficient diet and nicotine on cardiorespiratory recovery from apnoea during autoresuscitation
Breathing and heart rate indices (gasp latency, eupnoea recovery and HR recovery) were quantified as possible indicators of successful vs. failed autoresuscitation in this study. There were no differences in both breathing and HR indices between treatment groups at P5, P8 and P12 (data not shown).
At P10, gasp latency (the time between the last breath prior to the primary apnoea and the first gasp post apnoea) did not differ among the four treatment groups at any anoxic episode (Fig. 5 A). Neither maternal tryptophan deficiency diet nor developmental nicotine exposure on their own or combined altered the offspring's gasp latency.
Recovery to eupnoea (the time from the first gasp post apnoea to when breathing frequency reaches 63% of baseline values) was delayed with progressive anoxic bouts in all treatment groups with the longest recovery period observed during the last survived anoxic episode (episode effect: P = < 0.0001, two‐way RM ANOVA) (Fig. 5 B). In the TD diet/nicotine‐treated P10 neonates, eupnoea recovery was significantly delayed (2‐fold longer) compared to all other treatment groups (treatment effect: P = 0.0053) during the last survived anoxic episode. Eupnoea recovery was prolonged by 4‐fold during the last survived episode vs. the first in the TD diet/nicotine‐exposed pups. In contrast, eupnoea recovery was only 2‐fold longer by the last survived anoxic episode in other treatment groups.
HR recovery (the time from the first gasp post apnoea to when HR reaches 63% of baseline) was also delayed with increasing number of repeated anoxic episodes in all treatment groups, particularly in the TD diet/nicotine‐exposed P10 animals (Fig. 5 C). When compared to the other three treatment groups, HR recovery in TD diet/nicotine pups was notably delayed from episode 5 onward and was significantly longer during the last survived anoxic episode (P = < 0.0001, two‐way RM ANOVA). HR recovery in TD diet/nicotine‐treated pups was 4‐fold longer during the last survived episode compared to the first anoxic episode. HR recovery was also significantly delayed in TD diet/vehicle‐treated pups (P ≤ 0.05) during the last survived anoxic episode compared to both vehicle‐ and nicotine‐treated Ctr diet groups. These data suggest that the 5‐HT deficiency induced by the maternal TD diet may exacerbate the delay of HR recovery from repetitive anoxia, which can be even further exacerbated by pre‐ and perinatal nicotine exposure in neonates at P10.
Discussion
The main findings include the following: (1) maternal tryptophan‐deficient diet resulted in a mild brainstem 5‐HT deficiency in the developing offspring, (2) autoresuscitation was significantly compromised when perinatal nicotine exposure interacted with the mild 5‐HT deficiency at P10, even though the mild 5‐HT deficiency or developmental nicotine exposure alone did not significantly alter the cardiorespiratory response to repetitive anoxia, (3) the impaired autoresuscitation was characterized as a progressively delayed restoration of eupnoea and HR over subsequent episodes of anoxia, despite a normal initiation of gasping, and (4) the combination of perinatal nicotine exposure and mild 5‐HT deficiency increased resting HR in P10 neonates. These key findings related to the gestational nicotine exposure and 5‐HT deficiency with altered cardiorespiratory responses to repetitive anoxia at a specific developmental period are relevant to malnutrition, smoking/nicotine exposure and homeostasis in early life, including SIDS.
Maternal tryptophan‐deficient diet and nicotine administration on the medullary 5‐HT system in the offspring
Serotonin synthesis in the brain depends on levels of tryptophan (the primary precursor), which in turn depends on blood tryptophan concentrations that are highly affected by diet. Elimination of dietary tryptophan can profoundly lower the brain serotonin levels (Culley et al. 1963; Gessa et al. 1975; Franklin et al. 1995; Gonzalez et al. 2008), while increased tryptophan administration can increase plasma tryptophan and whole brain 5‐HT and 5‐HIAA (Haider et al. 2006; Khaliq et al. 2006). 5‐HIAA is the primary metabolite of 5‐HT, and the 5‐HIAA/5‐HT ratio is an index that measures brain serotonin turnover; the levels of 5‐HIAA and 5‐HIAA/5‐HT are highly correlated with 5‐HT levels in the brain. In this study, we showed that treating pregnant dams with a tryptophan‐deficient diet (55% of control level) until postnatal day 25 can result in a mild brainstem 5‐HT deficiency in neonatal offspring with the total 5‐HT (5‐HT + 5‐HIAA) content in the medulla reduced 16%, 18% and 32% at P10, P15 and P25, respectively, compared to age‐matched Ctr diet pups (Fig. 1). The changes of total 5‐HT in the medulla are associated with significantly decreased 5‐HT turnover (5‐HIAA/5‐HT) in TD diet pups. In TD diet pups at P10, there was a strong trend towards a significant decrease in medullary 5‐HT content alone (−10%), in addition to significant decreases in 5‐HIAA, total 5‐HT, and 5‐HIAA/5‐HT ratio (−19%, 16%, and 11%, respectively), indicating a measureable 5‐HT deficiency in the medulla from the age of P10. At P15 and P25, all 5‐HT activity, i.e. 5‐HT, 5‐HIAA and 5‐HIAA/5‐HT levels, were significantly lower in TD diet pups. The lower levels of all 5‐HT parameters in the older TD diet pups may reflect longer exposure to the tryptophan‐deficient diet through the dam's milk and the food itself as the pups start to consume solid foods from 2 weeks of age onward. The degradation of monoamines, e.g. catecholamine, serotonin and dopamine, requires the monoamine oxidase enzyme (MAO), and the normal levels of catecholamines and dopamine and their metabolites support the interpretation that the lower medullary 5‐HIAA in TD diet pups is due to lower 5‐HT content and not abnormal MAO. Unlike other transgenic 5‐HT‐deficient mice, e.g. pet1, Lmxb or TPH2 knockout, the reduction in the medullary 5‐HT levels in these dietary‐induced 5‐HT‐deficient pups was only partial, and appeared to be more closely aligned to those documented in SIDS cases (Duncan et al. 2010).
The degree of 5‐HT deficiency in the offspring at P25 seen here is less severe than those reported previously (Penatti et al. 2011); unfortunately, we could not verify whether there is any difference between the two sets of data in P10 and P15 animals as these data were not available from the earlier study by Penatti et al. The discrepancy in the resultant brainstem 5‐HT levels from the two studies at P25 is very likely due to the variable sizes and the precise location of the tissue samples used for the HPLC analysis in the two studies. Both studies collected tissue wedge sections enriched with 5‐HT neurons from the brainstem for the HPLC analysis; however, the precise region and size of the tissue wedges were heavily dependent on the judgement of the individual investigator at the time. A larger tissue wedge could by chance include nearby regions less concentrated in 5‐HT and dilute the 5‐HT levels detectable by HPLC. In addition, there is potential variability between the two batches of samples used for the HPLC analysis which were performed 6 years apart. Nevertheless, both studies were internally consistent in terms of who performed the tissue dissection, the diet batch used, and the timing of HPLC. Both studies showed that maternal dietary tryptophan deficiency can lead to 5‐HT deficiency in offspring. Here we further demonstrated that the brainstem 5‐HT deficiency induced by maternal dietary tryptophan deficiency is measurable as early as P10 in these pups.
Perinatal nicotine exposure can directly or indirectly affect the 5‐HT system (Xu et al. 2001; Kamendi et al. 2009; Blood‐Siegfried & Rende, 2010; Slotkin et al. 2015), e.g. increased serotonin 5‐HT1AR binding in raphe obscurus, and nicotinic receptor binding in the raphe obscurus and vagal complex of baboons (Duncan et al. 2009), as well as both decreased and increased 5‐HT transporter expression in the cortex and the midbrain/brainstem, respectively, with the midbrain/brainstem being the region that contains the 5‐HT cell bodies that project to the cerebral cortex (Xu et al. 2001). Cerpa et al. (2015) recently further showed that the increased 5‐HT1AR expression in raphe 5‐HT neurons is accompanied by reduced spontaneous firing frequency and reduced hypercapnia activated c‐Fos‐positive 5‐HT neuron numbers in the raphe obscurus of P3–5 mice. Taken together, results of these studies suggest that perinatal nicotine exposure can lead to a dysfunctional 5‐HT system in the neonate offspring, and the phenotypes observed in our current study may reflect the results of the combination of nicotine exposure and 5‐HT deficiency.
Impaired autoresuscitation in perinatal nicotine‐exposed 5‐HT‐deficient neonates at a critical age
Autoresuscitation is considered as the last protective mechanism to severe hypoxia, anoxia and asphyxia in newborn mammals and successful autoresuscitation from hypoxia‐induced apnoea requires integration of several physiological systems including the central nervous, respiratory and cardiovascular systems (Sridhar et al. 2003; Fewell, 2005). A typical autoresuscitation from anoxia or asphyxia includes a sequence of events, hyperpnoea, apnoea, gasping, which is then followed by either successful termination of apnoea and restoration of eupnoea and HR or unsuccessful termination of apnoea or HR and death (Fewell et al. 2000; Sridhar et al. 2003). Autoresuscitation failure has been reported in some SIDS cases (Poets et al. 1999; Sridhar et al. 2003) and in several severe 5‐HT deficiency animal models at specific ages (Cummings et al. 2009, 2010; Erickson & Sposato, 2009; Chen et al. 2013; Barrett et al. 2016), with defects including prolonged apnoea or delayed onset of gasping and delayed restoration of eupnoea and heart rate. The reported deficiency of brainstem 5‐HT includes ∼26% in SIDS cases (Duncan et al. 2010), 35% in Pet1::Flpe‐silenced pups (Barrett et al. 2016), and over 80% in Pet‐1 −/− knockout mouse (Cummings et al. 2009, 2011) and serotonin lesioned rat pups (Cummings et al. 2009). In contrast to these severely 5‐HT‐deficient animal models, a mild 5‐HT deficiency induced by the maternal tryptophan‐deficient diet alone did not alter autoresuscitation significantly; however, when such mild 5‐HT deficiency interacted with developmental nicotine exposure, the ability to autoresuscitate from repetitive anoxia was significantly compromised and mortality rate was exacerbated at P10 (Fig. 3). The impaired autoresuscitation is characterized by a relatively normal gasp latency, delayed cardiorespiratory (eupnoea and HR) recovery from primary apnoea, and fewer number of hypoxic exposures tolerated in P10 TD diet/nicotine‐treated pups. These phenotypes are similar to those previously reported in perinatal nicotine‐exposed developing rat pups, piglets and lambs (Fewell & Smith, 1998; Froen et al. 2000; Fewell et al. 2001; Hafstrom et al. 2005). However, there are a few notable differences, e.g. the age and the effect of nicotine alone vs. combined nicotine and 5‐HT deficiency. Fewell et al. showed a similar phenotype of impaired autoresuscitation in P5–6 but not P10–11 perinatal nicotine‐exposed rat pups, whereas in the current study, the impaired phenotype was only observed at P10. In our study, the autoresuscitation failure rate is ranked as TD diet/nicotine > Ctr diet/nicotine > TD diet/vehicle > Ctr diet/ vehicle, with the highest mortality rate observed in the perinatal nicotine‐treated 5‐HT‐deficient (TD diet/nicotine) pups (Fig. 3). The differences between our study and Fewell et al. (2001) can be attributed to the diet‐induced 5‐HT deficiency in the pups. Reduced concentrations of central 5‐HT are related to failure of autoresuscitation and spontaneous cardiorespiratory recovery from episodes of anoxia and apnoea during the first few weeks of life, especially between the ages of P8 and 13 (Fewell & Smith, 1998; Fewell et al. 2001; Cummings et al. 2009, 2010, 2011; Erickson & Sposato, 2009; Chen et al. 2013; Barrett et al. 2016). It is noteworthy that the TD diet/nicotine pups have a relatively normal autoresuscitation at an older age (P12 or older) even when their brainstem 5‐HT deficiency persists, and this time‐specific effect could be the result of developmental compensatory changes with age. In this study, we have demonstrated that the interaction between a mild 5‐HT deficiency and developmental nicotine exposure can significantly increase pups’ vulnerability to environmental stress, e.g. repetitive anoxia, and exacerbate defects in cardiorespiratory protective reflexes at a specific age, in this case P10. The age‐specific phenotype probably exists due to the changes in neurochemical development and in cardiorespiratory responses that occur within a matter of days or even less.
Altered resting HR and cardiorespiratory recovery from anoxia in perinatal nicotine‐exposed 5‐HT deficiency neonates
Perinatal nicotine‐exposed 5‐HT‐deficient (TD diet/nicotine) pups have significantly elevated HR at rest (Table 2) at P10, and yet a significantly delayed or reduced HR recovery or response to repetitive anoxia. HR recovery in TD diet/nicotine pups was notably delayed from episode 5 onwards and was significantly longer during the last survived anoxic episode, which was 4‐fold longer than the first anoxic episode (Fig. 5). The results are consistent with reports that nicotine exposure results in autonomic imbalance in developing offspring (Slotkin et al. 1995; Hafstrom et al. 2002b; Fewell, 2005; Hafstrom et al. 2005; Zeskind & Gingras, 2006; Slotkin et al. 2015). In contrast to the elevated HR during rest, nicotine‐exposed 5‐day‐old lambs did not increase HR in response to hypoxia to the same extent as unexposed controls (Hafstrom et al. 2002a). Perinatal nicotine exposure can delay the development of β‐adrenergic receptors in the heart (Slotkin et al. 1997) and abolish serotonergic neurotransmission to cardiac vagal neurons (Kamendi et al. 2009), both of which can lead to an altered neuronal input in cardiac control, imbalanced autonomic function, and reduced HR responsiveness to severe hypoxia and anoxia.
Inability to maintain adequate cardiac output, blood pressure and its associated poor reoxygenation process during autoresuscitation may also contribute to the delayed cardiorespiratory recovery and autoresuscitation failure (Gershan et al. 1992; Fewell et al. 2000; Fewell, 2005; Yang & Cummings, 2013). In acute 5‐HT depleted rat pups, Yang et al. showed that a progressive and premature deterioration of blood pressure is associated with delayed HR recovery with successive episodes of anoxia despite normal gasping (Yang & Cummings, 2013), suggesting 5‐HT deficiency can also lead to an autonomic imbalance and impair autoresuscitation process.
Developmental nicotine exposure can also functionally reduce the ventilatory response to hypercapnia and hypoxia in mice, rats and lambs (see review by Hafstrom et al. 2005), impair autoresuscitation from severe hypoxia and anoxia, and anatomically alter multiple neurotransmitter systems, e.g. catecholamines and serotonin. When nicotine exposure interacts with a mild 5‐HT deficiency in developing neonates, autoresuscitation failure is exacerbated.
The underlying mechanism for eupnoea and HR recovery and the involvement of 5‐HT neurons and nicotine exposure is still unclear at the present time. Regardless of the origin of the autonomic dysregulation, the autonomic imbalance is amplified in perinatal nicotine‐exposed 5‐HT‐deficient pups at P10.
Perspective and concluding remarks
SIDS persists as a major cause of death in infants under 1 year of age (Kochanek et al. 2017). Maternal cigarette smoking remains a major preventable risk factor for SIDS. With increasing numbers of nicotine patch and electronic cigarette users during pregnancy, there is an increasing urgency to better understand the impact of developmental nicotine exposure on the health of neonates, especially those who are more vulnerable with an intrinsic medullary 5‐HT defect. SIDS is a complex disorder of homeostasis, and while abnormalities in the brainstem 5‐HT system have been consistently found in most SIDS cases studied, it is likely that SIDS involves the interactions of multiple neurotransmitters and neuromodulators, multiple stressors acting simultaneously, and multiple genetic and environmental factors (Kinney & Thach, 2009; Kinney et al. 2009; Duncan et al. 2010). In this study, we further investigated the effect of a specific interaction between developmental nicotine exposure and mild brainstem 5‐HT deficiency on autoresuscitation in developing animals. Our data suggest that the interaction of developmental nicotine exposure and a mild brainstem 5‐HT deficiency can significantly compromise autoresuscitation in response to environmental stressors, e.g. severe hypoxia, anoxia, or asphyxia, at a specific developmental age. The age‐specific effect probably exists due to either changes in neurochemical development or changes in cardiorespiratory responses that occur within a matter of days during the development. We speculate that maternal nicotine exposure places infants who have other vulnerabilities, e.g. mild 5‐HT deficiency, at high risk for an impairment of protective response to severe hypoxia, anoxia and asphyxia.
Additional information
Competing interests
There are no competing interests for the authors of this paper.
Author contributions
S.Y.L., E.N., and A.L. conceived of and designed the work. S.Y.L. and A.L. wrote the manuscript, performed data analysis and interpretation. S.Y.L. performed the whole animal experiments and the tissue sampling. C.M.S. acquired data for the cotinine analysis. All authors contributed to the editing of the manuscript, gave approval for the final version, and agreed to be accountable for all content.
Funding
The research was supported by the NIH Program Project Grant HD036379.
Biography
Stella Y. Lee completed her PhD studies in the Department of Physiology and Neurobiology at the Geisel School of Medicine at Dartmouth in Hanover, New Hampshire. Her primary research focus was on the physiological consequences of neurochemical abnormalities in a rodent model of SIDS. She is currently working as a teaching faculty at the University of British Columbia in the Department of Zoology in Vancouver, British Columbia. She integrates her research background into her teaching, specifically animal physiology and developmental neurobiology.

Edited by: Harold Schultz & Gregory Funk
References
- Andres RL & Day MC (2000). Perinatal complications associated with maternal tobacco use. Semin Neonatol 5, 231–241. [DOI] [PubMed] [Google Scholar]
- Barrett KT, Dosumu‐Johnson RT, Daubenspeck JA, Brust RD, Kreouzis V, Kim JC, Li A, Dymecki SM & Nattie EE (2016). Partial raphe dysfunction in neurotransmission is sufficient to increase mortality after anoxic exposures in mice at a critical period in postnatal development. J Neurosci 36, 3943–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz NL, Kuyt F & Jacob P 3rd (1982). Circadian blood nicotine concentrations during cigarette smoking. Clin Pharmacol Ther 32, 758–764. [DOI] [PubMed] [Google Scholar]
- Blood‐Siegfried J & Rende EK (2010). The long‐term effects of prenatal nicotine exposure on neurologic development. J Midwifery Womens Health 55, 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerpa VJ, Aylwin Mde L, Beltran‐Castillo S, Bravo EU, Llona IR, Richerson GB & Eugenin JL (2015). The alteration of neonatal raphe neurons by prenatal‐perinatal nicotine. meaning for sudden infant death syndrome. Am J Respir Cell Mol Biol 53, 489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Magnusson J, Karsenty G & Cummings KJ (2013). Time‐ and age‐dependent effects of serotonin on gasping and autoresuscitation in neonatal mice. J Appl Physiol (1985) 114, 1668–1676. [DOI] [PubMed] [Google Scholar]
- Coleman T, Cooper S, Thornton JG, Grainge MJ, Watts K, Britton J & Lewis S (2012). A randomized trial of nicotine‐replacement therapy patches in pregnancy. N Engl J Med 366, 808–818. [DOI] [PubMed] [Google Scholar]
- Culley WJ, Saunders RN, Mertz ET & Jolly DH (1963). Effect of a tryptophan deficient diet on brain serotonin and plasma tryptophan level. Proc Soc Exp Biol Med 113, 645–648. [DOI] [PubMed] [Google Scholar]
- Cummings KJ, Commons KG, Fan KC, Li A & Nattie EE (2009). Severe spontaneous bradycardia associated with respiratory disruptions in rat pups with fewer brain stem 5‐HT neurons. Am J Physiol Regul Integr Comp Physiol 296, R1783–R1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings KJ, Commons KG, Hewitt JC, Daubenspeck JA, Li A, Kinney HC & Nattie EE (2011). Failed heart rate recovery at a critical age in 5‐HT‐deficient mice exposed to episodic anoxia: implications for SIDS. J Appl Physiol 111, 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings KJ, Li A, Deneris ES & Nattie EE (2010). Bradycardia in serotonin‐deficient Pet‐1‐/‐ mice: influence of respiratory dysfunction and hyperthermia over the first 2 postnatal weeks. Am J Physiol Regul Integr Comp Physiol 298, R1333–R1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JR, Garland M, Myers MM, Fifer WP, Yang M, Kinney HC & Stark RI (2009). Prenatal nicotine‐exposure alters fetal autonomic activity and medullary neurotransmitter receptors: implications for sudden infant death syndrome. J Appl Physiol 107, 1579–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JR, Paterson DS, Hoffman JM, Mokler DJ, Borenstein NS, Belliveau RA, Krous HF, Haas EA, Stanley C, Nattie EE, Trachtenberg FL & Kinney HC (2010). Brainstem serotonergic deficiency in sudden infant death syndrome. JAMA 303, 430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JR, Randall LL, Belliveau RA, Trachtenberg FL, Randall B, Habbe D, Mandell F, Welty TK, Iyasu S & Kinney HC (2008). The effect of maternal smoking and drinking during pregnancy upon 3H‐nicotine receptor brainstem binding in infants dying of the sudden infant death syndrome: initial observations in a high risk population. Brain Pathol 18, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England LJ, Aagaard K, Bloch M, Conway K, Cosgrove K, Grana R, Gould TJ, Hatsukami D, Jensen F, Kandel D, Lanphear B, Leslie F, Pauly JR, Neiderhiser J, Rubinstein M, Slotkin TA, Spindel E, Stroud L & Wakschlag L (2017). Developmental toxicity of nicotine: A transdisciplinary synthesis and implications for emerging tobacco products. Neurosci Biobehav Rev 72, 176–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England LJ, Tong VT, Koblitz A, Kish‐Doto J, Lynch MM & Southwell BG (2016). Perceptions of emerging tobacco products and nicotine replacement therapy among pregnant women and women planning a pregnancy. Prev Med Rep 4, 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JT & Sposato BC (2009). Autoresuscitation responses to hypoxia‐induced apnea are delayed in newborn 5‐HT‐deficient Pet‐1 homozygous mice. J Appl Physiol (1985) 106, 1785–1792. [DOI] [PubMed] [Google Scholar]
- Feng Y, Caiping M, Li C, Can R, Feichao X, Li Z & Zhice X (2010). Fetal and offspring arrhythmia following exposure to nicotine during pregnancy. J Appl Toxicol 30, 53–58. [DOI] [PubMed] [Google Scholar]
- Fewell JE (2005). Protective responses of the newborn to hypoxia. Respir Physiol Neurobiol 149, 243–255. [DOI] [PubMed] [Google Scholar]
- Fewell JE & Smith FG (1998). Perinatal nicotine exposure impairs ability of newborn rats to autoresuscitate from apnea during hypoxia. J Appl Physiol 85, 2066–2074. [DOI] [PubMed] [Google Scholar]
- Fewell JE, Smith FG & Ng VK (2001). Prenatal exposure to nicotine impairs protective responses of rat pups to hypoxia in an age‐dependent manner. Respir Physiol 127, 61–73. [DOI] [PubMed] [Google Scholar]
- Fewell JE, Smith FG, Ng VK, Wong VH & Wang Y (2000). Postnatal age influences the ability of rats to autoresuscitate from hypoxic‐induced apnea. Am J Physiol Regul Integr Comp Physiol 279, R39–46. [DOI] [PubMed] [Google Scholar]
- Franco P, Szliwowski H, Dramaix M & Kahn A (1999). Decreased autonomic responses to obstructive sleep events in future victims of sudden infant death syndrome. Pediatr Res 46, 33–39. [DOI] [PubMed] [Google Scholar]
- Franklin M, Cowen PJ & Craven RD (1995). The effects of a low tryptophan diet on brain 5‐HT metabolism and 5‐HT‐mediated neuroendocrine responses in the male rat. J Psychopharmacol 9, 336–341. [DOI] [PubMed] [Google Scholar]
- Froen JF, Akre H, Stray‐Pedersen B & Saugstad OD (2000). Adverse effects of nicotine and interleukin‐1beta on autoresuscitation after apnea in piglets: implications for sudden infant death syndrome. Pediatrics 105, E52. [DOI] [PubMed] [Google Scholar]
- Gershan WM, Jacobi MS & Thach BT (1992). Mechanisms underlying induced autoresuscitation failure in BALB/c and SWR mice. J Appl Physiol (1985) 72, 677–685. [DOI] [PubMed] [Google Scholar]
- Gessa GL, Biggio G, Fadda F, Corsini GU & Tagliamonte A (1975). Tryptophan‐free diet: a new means for rapidly decreasing brain tryptophan content and serotonin synthesis. Acta Vitaminol Enzymol 29, 72–78. [PubMed] [Google Scholar]
- Gonzalez S, Huerta JM, Fernandez S, Patterson AM & Lasheras C (2008). Differences in overall mortality in the elderly may be explained by diet. Gerontology 54, 232–237. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafstrom O, Milerad J, Sandberg KL & Sundell HW (2005). Cardiorespiratory effects of nicotine exposure during development. Respir Physiol Neurobiol 149, 325–341. [DOI] [PubMed] [Google Scholar]
- Hafstrom O, Milerad J & Sundell HW (2002a). Altered breathing pattern after prenatal nicotine exposure in the young lamb. Am J Respir Crit Care Med 166, 92–97. [DOI] [PubMed] [Google Scholar]
- Hafstrom O, Milerad J & Sundell HW (2002b). Prenatal nicotine exposure blunts the cardiorespiratory response to hypoxia in lambs. Am J Respir Crit Care Med 166, 1544–1549. [DOI] [PubMed] [Google Scholar]
- Haider S, Khaliq S, Ahmed SP & Haleem DJ (2006). Long‐term tryptophan administration enhances cognitive performance and increases 5HT metabolism in the hippocampus of female rats. Amino Acids 31, 421–425. [DOI] [PubMed] [Google Scholar]
- Hakeem GF, Oddy L, Holcroft CA & Abenhaim HA (2015). Incidence and determinants of sudden infant death syndrome: a population‐based study on 37 million births. World J Pediatr 11, 41–47. [DOI] [PubMed] [Google Scholar]
- Harper RM (2001). Autonomic control during sleep and risk for sudden death in infancy. Arch Ital Biol 139, 185–194. [PubMed] [Google Scholar]
- Hodges MR & Richerson GB (2008). Interaction between defects in ventilatory and thermoregulatory control in mice lacking 5‐HT neurons. Respir Physiol Neurobiol 164, 350–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges MR, Tattersall GJ, Harris MB, McEvoy SD, Richerson DN, Deneris ES, Johnson RL, Chen ZF & Richerson GB (2008). Defects in breathing and thermoregulation in mice with near‐complete absence of central serotonin neurons. J Neurosci 28, 2495–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn A, Groswasser J, Sottiaux M, Kelmanson I, Rebuffat E, Franco P, Dramaix M & Wayenberg JL (1994). Prenatal exposure to cigarettes in infants with obstructive sleep apneas. Pediatrics 93, 778–783. [PubMed] [Google Scholar]
- Kamendi HW, Cheng Q, Dergacheva O, Gorini C, Jameson HS, Wang X, McIntosh JM & Mendelowitz D (2009). Abolishment of serotonergic neurotransmission to cardiac vagal neurons during and after hypoxia and hypercapnia with prenatal nicotine exposure. J Neurophysiol 101, 1141–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp JS, Kowalski RM, Burch PM, Graham MA & Thach BT (1993). Unintentional suffocation by rebreathing: a death scene and physiologic investigation of a possible cause of sudden infant death. J Pediatr 122, 874–880. [DOI] [PubMed] [Google Scholar]
- Khaliq S, Haider S, Ahmed SP, Perveen T & Haleem DJ (2006). Relationship of brain tryptophan and serotonin in improving cognitive performance in rats. Pak J Pharm Sci 19, 11–15. [PubMed] [Google Scholar]
- Kinney HC, Richerson GB, Dymecki SM, Darnall RA & Nattie EE (2009). The brainstem and serotonin in the sudden infant death syndrome. Annu Rev Pathol 4, 517–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney HC & Thach BT (2009). The sudden infant death syndrome. N Engl J Med 361, 795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochanek KD, Murphy SL, Xu J & Arias E (2017). Mortality in the United States, 2016. NCHS Data Brief 293, 1–8. [PubMed] [Google Scholar]
- Krous HF, Beckwith JB, Byard RW, Rognum TO, Bajanowski T, Corey T, Cutz E, Hanzlick R, Keens TG & Mitchell EA (2004). Sudden infant death syndrome and unclassified sudden infant deaths: a definitional and diagnostic approach. Pediatrics 114, 234–238. [DOI] [PubMed] [Google Scholar]
- Lambers DS & Clark KE (1996). The maternal and fetal physiologic effects of nicotine. Semin Perinatol 20, 115–126. [DOI] [PubMed] [Google Scholar]
- Lichtensteiger W, Ribary U, Schlumpf M, Odermatt B & Widmer HR (1988). Prenatal adverse effects of nicotine on the developing brain. Prog Brain Res 73, 137–157. [DOI] [PubMed] [Google Scholar]
- MacDorman MF, Cnattingius S, Hoffman HJ, Kramer MS & Haglund B (1997). Sudden infant death syndrome and smoking in the United States and Sweden. Am J Epidemiol 146, 249–257. [DOI] [PubMed] [Google Scholar]
- Machaalani R, Say M & Waters KA (2009). Serotoninergic receptor 1A in the sudden infant death syndrome brainstem medulla and associations with clinical risk factors. Acta Neuropathol 117, 257–265. [DOI] [PubMed] [Google Scholar]
- Matturri L, Ottaviani G & Lavezzi AM (2006). Maternal smoking and sudden infant death syndrome: epidemiological study related to pathology. Virchows Arch 449, 697–706. [DOI] [PubMed] [Google Scholar]
- Milerad J, Vege A, Opdal SH & Rognum TO (1998). Objective measurements of nicotine exposure in victims of sudden infant death syndrome and in other unexpected child deaths. J Pediatr 133, 232–236. [DOI] [PubMed] [Google Scholar]
- Mitchell EA, Thach BT, Thompson JM & Williams S (1999). Changing infants' sleep position increases risk of sudden infant death syndrome. New Zealand Cot Death Study. Arch Pediatr Adolesc Med 153, 1136–1141. [DOI] [PubMed] [Google Scholar]
- Nachmanoff DB, Panigrahy A, Filiano JJ, Mandell F, Sleeper LA, Valdes‐Dapena M, Krous HF, White WF & Kinney HC (1998). Brainstem 3H‐nicotine receptor binding in the sudden infant death syndrome. J Neuropathol Exp Neurol 57, 1018–1025. [DOI] [PubMed] [Google Scholar]
- Neff RA, Simmens SJ, Evans C & Mendelowitz D (2004). Prenatal nicotine exposure alters central cardiorespiratory responses to hypoxia in rats: implications for sudden infant death syndrome. J Neurosci 24, 9261–9268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oncken C (2012). Nicotine replacement for smoking cessation during pregnancy. N Engl J Med 366, 846–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahy A, Filiano J, Sleeper LA, Mandell F, Valdes‐Dapena M, Krous HF, Rava LA, Foley E, White WF & Kinney HC (2000). Decreased serotonergic receptor binding in rhombic lip‐derived regions of the medulla oblongata in the sudden infant death syndrome. J Neuropathol Exp Neurol 59, 377–384. [DOI] [PubMed] [Google Scholar]
- Paterson DS, Trachtenberg FL, Thompson EG, Belliveau RA, Beggs AH, Darnall R, Chadwick AE, Krous HF & Kinney HC (2006). Multiple serotonergic brainstem abnormalities in sudden infant death syndrome. JAMA 296, 2124–2132. [DOI] [PubMed] [Google Scholar]
- Penatti EM, Barina AE, Raju S, Li A, Kinney HC, Commons KG & Nattie EE (2011). Maternal dietary tryptophan deficiency alters cardiorespiratory control in rat pups. J Appl Physiol 110, 318–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poets CF (2004). Apparent life‐threatening events and sudden infant death on a monitor. Paediatr Respir Rev 5 (Suppl. A), S383–386. [DOI] [PubMed] [Google Scholar]
- Poets CF, Meny RG, Chobanian MR & Bonofiglo RE (1999). Gasping and other cardiorespiratory patterns during sudden infant deaths. Pediatr Res 45, 350–354. [DOI] [PubMed] [Google Scholar]
- Slikker W Jr, Xu ZA, Levin ED & Slotkin TA (2005). Mode of action: disruption of brain cell replication, second messenger, and neurotransmitter systems during development leading to cognitive dysfunction–developmental neurotoxicity of nicotine. Crit Rev Toxicol 35, 703–711. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Lappi SE, McCook EC, Lorber BA & Seidler FJ (1995). Loss of neonatal hypoxia tolerance after prenatal nicotine exposure: implications for sudden infant death syndrome. Brain Res Bull 38, 69–75. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Saleh JL, McCook EC & Seidler FJ (1997). Impaired cardiac function during postnatal hypoxia in rats exposed to nicotine prenatally: implications for perinatal morbidity and mortality, and for sudden infant death syndrome. Teratology 55, 177–184. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Skavicus S, Card J, Stadler A, Levin ED & Seidler FJ (2015). Developmental neurotoxicity of tobacco smoke directed toward cholinergic and serotonergic systems: more than just nicotine. Toxicol Sci 147, 178–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar R, Thach BT, Kelly DH & Henslee JA (2003). Characterization of successful and failed autoresuscitation in human infants, including those dying of SIDS. Pediatr Pulmonol 36, 113–122. [DOI] [PubMed] [Google Scholar]
- St‐John WM & Leiter JC (1999). Maternal nicotine depresses eupneic ventilation of neonatal rats. Neurosci Lett 267, 206–208. [DOI] [PubMed] [Google Scholar]
- Waters KA, Meehan B, Huang JQ, Gravel RA, Michaud J & Cote A (1999). Neuronal apoptosis in sudden infant death syndrome. Pediatr Res 45, 166–172. [DOI] [PubMed] [Google Scholar]
- Xu Z, Seidler FJ, Ali SF, Slikker W Jr & Slotkin TA (2001). Fetal and adolescent nicotine administration: effects on CNS serotonergic systems. Brain Res 914, 166–178. [DOI] [PubMed] [Google Scholar]
- Yang HT & Cummings KJ (2013). Brain stem serotonin protects blood pressure in neonatal rats exposed to episodic anoxia. J Appl Physiol (1985) 115, 1733–1741. [DOI] [PubMed] [Google Scholar]
- Zeskind PS & Gingras JL (2006). Maternal cigarette‐smoking during pregnancy disrupts rhythms in fetal heart rate. J Pediatr Psychol 31, 5–14. [DOI] [PubMed] [Google Scholar]