

Abstract
Key points
This study identifies phosphorylated extracellular signal‐regulated kinase (ERK) to be immediately diminished followed by a rapid if transient increase for up to 4 h following hypoxic–ischaemic insult (HI) in the neonatal mouse.
Phosphorylated ERK up‐regulation was prevented with systemic injection of the mitogen‐activated protein kinase kinase (MEK) inhibitor SL327. Treatment with SL327 both pre‐ and post‐HI gave a strong reduction in the number of dying cells and microgliosis.
By utilising transgenic mouse mutations, we observe that neuronal ERK2 significantly contributes to tissue damage, while ERK1 and astrocytic ERK2 are neuroprotective.
Compared to global inactivation, selective cell‐specific interference with ERK activity could result in stronger neuroprotection.
Abstract
Hypoxia–ischaemia (HI) is a major cause of neonatal brain injury resulting in cerebral palsy, epilepsy, cognitive impairment and other neurological disabilities. The role of extracellular signal‐regulated kinase (ERK) isoforms and their mitogen‐activated protein kinase kinase (MEK)‐dependent phosphorylation in HI has previously been explored but remains unresolved at cellular level. This is pertinent given the growing awareness of the role of non‐neuronal cells in neuroprotection. Using a modified Rice–Vannucci model of HI in the neonatal mouse we observed time‐ and cell‐dependent ERK phosphorylation (pERK), with strongly up‐regulated pERK immunoreactivity first in periventricular white matter axons within 15–45 min of HI, followed by forebrain astrocytes and neurons (1–4 h post‐HI), and return to baseline by 16 h. We explored the effects of pharmacological ERK blockade through the MEK inhibitor SL327 on neonatal HI‐brain damage following HI alone (30 or 60 min) or lipopolysaccharide (LPS)‐sensitised HI insult (30 min). Global inhibition of ERK phosphorylation with systemically applied SL327 abolished forebrain pERK immunoreactivity, and significantly reduced cell death and associated microglial activation at 48 h post‐HI. We then explored the effects of cell‐specific ERK2 deletion alone or in combination with global ERK1 knockout under the same conditions of HI insult. Neuronal ERK2 deletion strongly decreased infarct size, neuronal cell death and microglial activation in grey matter following both HI alone or LPS‐sensitised HI. ERK1 deletion attenuated the protective effect of neuronal ERK2 deletion. Removal of astroglial ERK2 produced a reverse response, with a 3‐ to 4‐fold increase in microglial activation and cell death. Our data suggest a cell‐specific and time‐dependent role of ERK in neonatal HI, with a predominant, neurotoxic effect of neuronal ERK2, which is counteracted by neuroprotection by ERK1 and astrocytic ERK2. Overall, global pharmacological inhibition of ERK phosphorylation is strongly neuroprotective.
Keywords: Neonate, Hypoxia, ischemia, Brain, extracellular signal‐related kinase, Neuron, Astrocyte
Key points
This study identifies phosphorylated extracellular signal‐regulated kinase (ERK) to be immediately diminished followed by a rapid if transient increase for up to 4 h following hypoxic–ischaemic insult (HI) in the neonatal mouse.
Phosphorylated ERK up‐regulation was prevented with systemic injection of the mitogen‐activated protein kinase kinase (MEK) inhibitor SL327. Treatment with SL327 both pre‐ and post‐HI gave a strong reduction in the number of dying cells and microgliosis.
By utilising transgenic mouse mutations, we observe that neuronal ERK2 significantly contributes to tissue damage, while ERK1 and astrocytic ERK2 are neuroprotective.
Compared to global inactivation, selective cell‐specific interference with ERK activity could result in stronger neuroprotection.
Introduction
Cerebral hypoxia–ischaemia (HI) is a leading cause of neurological deficits in neonates. It affects 1–5 per 1000 live births worldwide (Vannucci, 1990; Vannucci & Hagberg, 2004). The pattern of damage depends on the severity, gestational age, as well as antenatal/perinatal factors such as maternal/fetal infection (Vincer et al. 2006; Higgins & Shankaran, 2009).
The immature brain is particularly vulnerable to HI due to insufficient anti‐oxidant and scavenging systems preventing the elimination of endogenous free radicals (Ferriero et al. 1996; Volpe, 2001; Vannucci & Hagberg, 2004). The subcortical white matter of the immature brain is unmyelinated and predominantly populated with oligodendrocyte (ODC) precursors susceptible to an imbalance between pro‐ and anti‐inflammatory cytokines (Skoff et al. 2001; Dewar et al. 2003; Bain et al. 2010). Alteration in that cytokine balance shifts the differentiation of ODC precursors towards astrocytes instead of oligodendrocytes, thus affecting subsequent myelination (Bain et al. 2010). Additionally, inflammatory changes in the white matter also activate resident microglia causing release of neurotoxic substances including nitric oxide (NO), reactive oxygen species and pro‐inflammatory cytokines (Dommergues et al. 2003; Chock & Giffard, 2005; Polazzi & Monti, 2010; Kendall et al. 2011; Hagberg et al. 2012).
During neonatal HI multiple chemical stimuli, including growth factors, cytokines, glutamate and free radicals, interact with corresponding target cell membrane receptors (Irving & Bamford, 2002). Activation of their receptor‐linked tyrosine kinases stimulates signal transduction via the Ras/Raf/MEK1&2 pathway causing up‐regulation of phosphorylated extracellular signal‐regulated kinase 1 and 2 (pERK1&2), and affecting a range of transcription factors, protein kinases, cytoskeletal elements and regulators of apoptosis (Lu & Xu, 2006).
Previous studies of neonatal HI in rodents revealed phosphorylated ERK (pERK) positive neurons with signs of DNA damage at the core of infarct and the border zones to undamaged tissue (Wang et al. 2004). Pharmacological inhibitors of MEK/ERK (PD98059, U0126) reduced NO‐induced neuronal cell death following glutathione depletion in vitro (Canals et al. 2003; de Bernardo et al. 2004). In an adult mouse model of middle cerebral artery occlusion (MCAO), PD98059 administered prior to insult reduced infarct volume by 40–50%, accompanied by reduction in neurobehavioural defects (Alessandrini et al. 1999). A similar effect was observed in an adult gerbil model of cerebral ischaemia using the more selective MEK1/2 inhibitor UO126, with significant reduction in cerebral infarct and protection against hippocampal CA1 pyramidal neuron loss (Namura et al. 2001). On the other hand, administration of neuroprotective brain‐derived neurotrophic factor (BDNF) to postnatal day 7 (P7) HI mice resulted in a rapid increase in pERK and in phosphatidylinositol 3‐kinase (PI3K)/AKT. When ERK but not PI3K/AKT was inhibited, the neuroprotective effect of BDNF was abolished (Han & Holtzman, 2000).
Thus, despite the association of ERK activation to regions of neurodegeneration, the precise role of ERK1/2 in neonatal HI brain damage remains unclear. We hypothesise that ERK1 and ERK2 have cell‐ and time‐dependent effects following neonatal HI. The aim of our study was to explore the outcomes of cell‐specific ERK2 removal and global ERK1 deletion, as well as ERK1/2 inactivation in neonatal HI brain damage using a modified P7 Rice–Vannucci mouse model of HI insult.
Methods
Animals
All animal experiments and techniques were approved by the Ethics Committee of the University College London and were carried out by licensed personnel in accordance with the UK Home Office Guidelines (Animals (Scientific Procedures) Act, 1986). C57/Bl6 (Charles River, UK), ERK1KO, ERK2ΔSyn, ERK2ΔGFAP and ERK1KOERK2ΔSyn mice were bred in‐house with a 12 h light/dark cycle and had free access to water and food. The ARRIVE guidelines (Kilkenny et al. 2010) were followed in all animal experiments. Animals were killed at different time points (see Fig. 1) post‐HI by intraperitoneal injection of sodium pentobarbital (2.5 μg/g) and confirmed by exsanguination.
Figure 1. Schedule of experimental procedures.
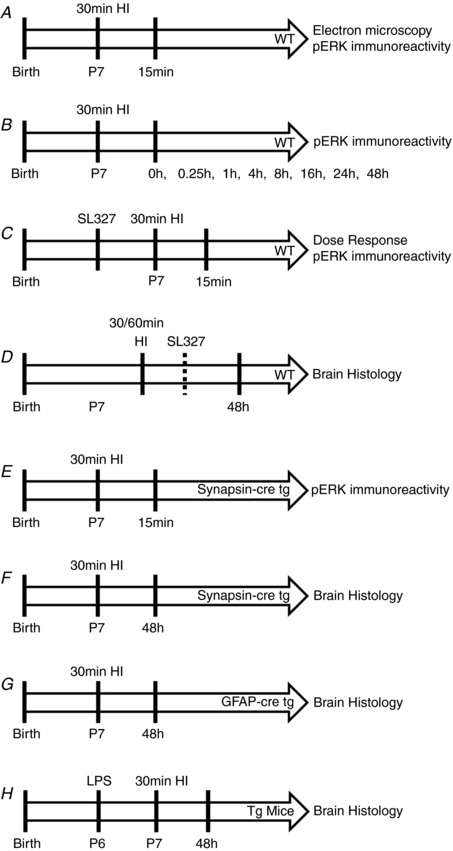
A, WT (C57/Bl6) mice underwent 30 min hypoxic–ischaemic (HI) insult and were then killed at 15 min post‐hypoxia for pERK immunoreactivity evaluation. B, pERK immunoreactivity was assessed at multiple time points up to 48 h post‐insult to P7 WT mice. C, a dose response of SL327, controlled to vehicle alone, was administered 20 min prior to 30 min HI and pERK immunoreactivity was assessed at 15 min post‐insult. D, WT mice were subject to either 30 min or 60 min HI, with 133 μg/g SL327 or EtOH (vehicle) administered either 20 min prior to or 60 min post‐insult. Brain histology was assessed at 48 h. E, inhibition of neuronal pERK immunoreactivity was confirmed at 15 min post‐HI in synapsin‐cre driven ERK tg mutant mice compared to littermate WT controls. F, brain histology was assessed at 48 h after 30 min HI in synapsin‐cre driven ERK tg mutant mice and littermate WT controls. G, brain histology was assessed at 48 h after 30 min HI in GFAP‐cre driven ERK tg mutant mice and littermate WT controls. H, saline or LPS was injected at 12 h prior to 30 min HI in both synapsin‐cre and GFAP‐cre driven ERK tg mutant mice and littermate WT controls. Brain histology was assessed at 48 h.
Global ERK1 deletion was described by Nekrasova et al. (2005). Heterozygous ERK1 mice were bred together to produce both homozygous null mutants (ERK1KO) and wild‐type controls (ERK1WT). To ablate ERK2 expression in the CNS, and to overcome embryonic lethality of global ERK2 deletion, we utilised a cell‐specific approach with Cre recombinase driven transgenic mutation of LoxP site flanked ERK2 under the control of either the synapsin (ERK2ΔSyn) or glial fibrillary acidic protein (GFAP) (ERK2ΔGFAP) promoters in order to remove ERK2 expression from neurons or astrocytes, respectively. Syn‐Cre mice were provided by Dr Axel Behrens from the Mammalian Genetics Laboratory, Cancer Research UK, and animals expressing Cre recombinase under the control of GFAP promoter (GFAP‐Cre) were from Jackson Labs (USA, http://jaxmice.jax.org/strain/004600.html). Mice were bred heterozygous with C57/Bl6 to produce both homozygous null mutants (ERK2ΔSyn or ERK2ΔGFAP) and wild‐type littermate controls (ERK2WT). Deletion of both ERK1 and 2 in neurons or astrocytes was achieved by breeding heterozygous ERK1 mutants with ERK2ΔSyn or ERK2ΔGFAP, respectively.
DNA extraction and genotyping
DNA extraction was performed with the ‘Wizard’ Genomic DNA purification system according to the manufacturer's instructions (Promega, Southampton, UK), using mouse tail tips taken before the perfusion. Specific oligonucleotide primers (Invitrogen, Loughborough, UK) were used for genotyping against erk1, erk2, synapsin‐cre, GFAP‐cre.
Hypoxia–ischaemia surgery
Surgeries were performed on C57/Bl6 and transgenic ERK mutant mice at P7, the age equivalent of late preterm human brain maturation. Animals were anaesthetised with isoflurane (5% induction, 1.5% maintenance), the left common carotid artery permanently occluded and the wound closed with tissue glue. The mice were returned to the dam for 2 h of recovery before being placed in a hypoxia chamber and exposed to continuous humidified 8% oxygen–92% nitrogen at 36°C for 30 or 60 min. The mice were left to recover at 36°C and returned to the dam for 2 h until being killed at 48 h post‐HI for analysis of neuropathological markers.
We have previously shown that lipopolysaccharide (LPS) bacterial endotoxin administered 12 h before the start of HI surgery confers a sensitising effect, as seen through a significant increase in neuropathological markers, when compared to saline‐injected controls (Rocha‐Ferreira et al. 2015). This is in concordance with other studies that show this sensitisation is mediated by the binding of LPS to toll‐like receptor 4 (TLR4) and recruitment of MyD88 adaptor protein (Wang et al. 2009). In the current study, P6 mouse pups were injected intraperitoneally (i.p.) with a single dose of LPS (serotype 055:B5, Fluka, Loughborough, UK; 0.6 μg/g), or saline as control (10 μl/g body weight (BW)). Twelve hours following injection, animals were exposed to carotid occlusion and 30 min hypoxia as above.
All animals were operated blindly with genotypes established after killing.
See Fig. 1 for all experimental outlines.
Pharmacological manipulation
SL327 toxicity
SL327 (Tocris, Bristol, UK), a MEK1/2 inhibitor (Atkins et al. 1998) proved to be toxic when dissolved in DMSO (data not shown), and therefore, was dissolved in 100% EtOH instead. P7 C57/Bl6 mice were treated with a single i.p. injection of SL327 (Dommergues et al. 2003) at 0, 15, 30, 65 or 133 μg/g BW (n = 5 per group) 20 min prior to the start of 30 min hypoxia. Fifteen minutes after HI, brains were assessed for intensity of pERK staining.
Time window for pharmacological ERK inhibition
In order to investigate the effect of MEK1/2 inhibition on the brain regions of interest – isocortex, pyriform cortex, hippocampus, striatum, thalamus and external capsule – P7 C57/Bl6 animals were injected with a single i.p. dose of SL327 (133 μg/g BW) either 20 min preceding or 1 h following both 30 and 60 min hypoxia (n = 10 per group). Control animals received the corresponding volume of 100% EtOH alone (10 μl/g BW). Following 48 h survival, animals were killed and brains collected for histopathological analysis of tissue infarction (Nissl), cell fragmentation (TUNEL), microglial activation (CD11b) and astrogliosis (GFAP).
Tissue sample preparation
For immunohistochemistry (IHC) analysis all animals were perfused with 4% paraformaldehyde (PFA)/PBS. The brains were excised, post‐fixed in 4% PFA/PBS for 1 h at 4°C, followed by cryoprotection for 24 h in 30% sucrose/PBS at 4°C and snap frozen on dry ice. Brains were sectioned on a cryostat into 40 μm sequential coronal sections starting from the point of fusion of the corpus callosum, in a total of 50 serial sections terminating in the hippocampus.
For electron microscopy, tissue was cut and stained as previously described (Hristova et al. 2010). In brief: glutaraldehyde‐fixed forebrain vibrotome sections of 100 μm were rinsed overnight in 2% sodium acetate solution and then pre‐treated for 6 h in 10% thioglycolic acid. Sections were developed for 25–30 min under visual control in a physical developer suspension containing 0.1% AgNO3, 0.1% NH4NO3, 0.5% H4SiW12O40 (silicotungstic acid) and 0.9% paraformaldehyde in distilled water, to which concentrated, aqueous Na2CO3 solution was added under vigorous stirring, to a final concentration of 2.5% Na2CO3. Following 2 min fixation and wash with 1% acetic acid, silver deposits were replaced with gold by immersing sections in 0.02% AuCl3 for 10 min. Sections were washed again for 10 min in two changes of 1% acetic acid, and 10 min in two changes of 2% sodium acetate, before transfer back into glutaraldehyde fixative until further use.
Immunohistochemistry: frozen
Tissue IHC staining was performed as previously described (Hristova et al. 2010). In brief, sections were fixed in 4% formaldehyde/0.1 M phosphate buffer for 5 min before acetone treatment for antigen retrieval (50, 100, 50%: 2 min each). Sections were incubated with CD11b (αMβ2) (1:5000, Serotec, Kidlington, UK), glial fibrillary acidic protein (GFAP, 1:6000, Dako, Santa Clara, CA, US), pERK (1:100, Cell Signalling Systems, Hitchen, UK) and pC‐Jun (1:200, Santa Cruz Biotechnology, Heidleberg, UK) primary antibodies overnight at 4°C. Sections were then incubated with biotinylated secondary antibody (1:100 anti‐rabbit or anti‐rat IgG, Vector Laboratories, Inc., Burlingame, CA, USA) and visualised with avidin‐biotinylated peroxidase complex (ABC, Vector Laboratories, Peterborough, UK) and diaminobenzidine/H2O2 stain. Lastly, sections were air‐dried, immersed in xylene and covered using DEPEX (Fluka).
Immunohistochemistry: free floating
This technique was utilised to stain for phosphorylated ERK due to the polyclonal primary antibody's sensitivity to dehydration. Sections were washed in PBS and endogenous peroxidases were blocked using 3% H2O2 in dH2O for 10 min at room temperature (RT). Antigen retrieval was achieved using 0.3% Triton (Triton X‐100, Bio‐Rad, Kidlington, UK) in a 5% goat serum/PBS solution for 30 min. Sections were incubated in wells containing polyclonal pERK antibody (1:100 in 5% goat serum/PBS) overnight at 4°C. Samples were washed in PBS and incubated for 1 h with secondary goat‐anti‐rabbit antibody (1:6000 dilution), before ABC incubation for 1 h at RT. The stain was visualised with 3,3'‐diaminobenzidine (DAB)/H2O2 and the reaction was stopped in dH2O and covered as described above.
Electron microscopy silver–gold intensification
Brain sections were osmicated in reduced osmium tetroxide, dehydrated in serial dilutions of ethanol and embedded in Epon 208 (Taab Laboratories, Aldermaston, UK). Semi‐thin sections were counterstained with toluidine blue for light microscopy, and ultrathin, 80 nm sections were counterstained with uranyl acetate and lead citrate for examination in a JEOL 1010 transmission electron microscope.
Histological analysis
Forty‐eight hours after HI insult, a time point where secondary energy failure has already initiated (Inder & Volpe, 2000), and with the highest level of widespread neuronal caspase‐3 expression within the acute period of injury (Johnston et al. 2011), the differences in microglial activation, astrocyte recruitment, infarct size and cell death were compared between littermate control animals and the corresponding SL327 treated, or mutant animals. All tissue sections were analysed blindly.
CD11b score
Activated microglia scores were assigned as previously described (Kendall et al. 2006; Hristova et al. 2016). In brief, a grade between 0 and 4 was assigned to each section for αMβ2 (0 = no activation, 1 = foci of non‐ramified active microglia, 2 = <50% coverage of active microglia, 3 = widespread active and predominantly phagocytic microglia, 4 = near full coverage of active and predominantly phagocytic microglia in addition to tissue infarct).
TUNEL+ cell death
Brain sections were stained at 400 μm intervals for DNA fragmentation using a TUNEL kit (Roche, Welwyn Garden City, UK) according to the manufacturer's instructions. Cell death was quantified at ×20 magnification by counting the number of TUNEL positive nuclei per brain region (3 microscope fields/region) of experimental and control animals.
Infarct volume measurement
Infarct volume was measured in 10 coronal sections at 400 μm intervals, stained with cresyl violet (Nissl stain). Optimas 6.2 image analysis software (Meyer Instruments Inc.) was utilised to calculate the intact brain tissue of each forebrain region by converting the measured injured and uninjured areas into square millimetres and then converting to a volume by multiplying by 400 μm. The sum of these volumes was converted into a percentage of surviving brain tissue by: injured/uninjured volume × 100 (Kendall et al. 2006).
Optical luminosity
A Sony AVT‐Horn camera was used to capture three 8‐bit RBG images of each of our regions of interest under a ×20 magnification with three eye fields/region. Images were then imported into the Optimas v6.5 Software. The mean and standard deviation (SD) of luminosity was obtained through the regions (Carsten Möller et al. 1996). For each image, the SD was subtracted from the mean, and the resulting value further subtracted from the mean optical luminosity of the empty glass slide. This provides a specific optical luminosity value (OLV).
Statistics
When only two groups, experimental and control, were present, statistical analysis for tissue loss, cell death, CD11b and GFAP immunoreactivity in HI forebrain regions was performed by either a two‐tailed, unpaired Student's t test or two‐way ANOVA, post hoc Sidak. When more than two groups were present, statistical analysis was performed using one‐way ANOVA followed by post hoc Tukey. The confidence interval for all assessments was set at 95%. For a global hemispheric response, statistical significance was assessed through one‐way ANOVA, post hoc Tukey. Animals of different genotypes or treatment were compared in their response to injury using the combined regions ipsilateral to carotid artery occlusion.
Results
Rapid ERK phosphorylation occurs across whole brain in the neonatal mouse brain following HI
ERK activation in response to HI was visualised using immunohistochemistry for pERK immunoreactivity (pERK‐IR) in C57/Bl6 mouse pups at postnatal day 7 (P7). These animals underwent unilateral left carotid artery occlusion (CROC) and were exposed to 8% O2 for 30 min. The distribution of normal pERK‐IR in the forebrain of a sham‐operated animal is shown in Fig. 2 A. OLV for pERK immunoreactivity in animals with CROC (ischaemic insult) only (Fig. 2 B) was unchanged compared to sham operated littermates and were referred to as controls (or CTRL in Fig. 2 D). Intensity of pERK‐IR was comparable in both ipsilateral (occluded, hypoxic–ischaemic insult) and contralateral (non‐occluded, ischaemia alone) hemispheres.
Figure 2. Histological and electron microscopy assesment of pERK expression in P7 mouse forebrain following 30 min HI.
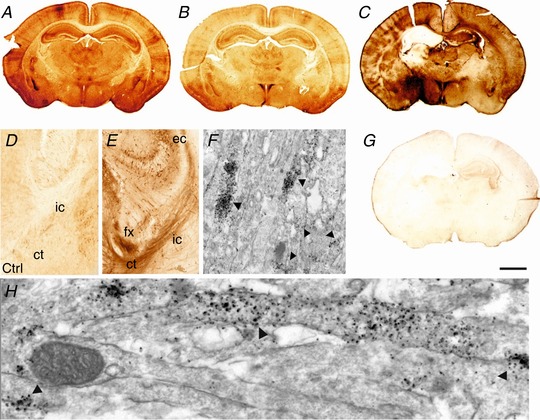
A and B, distribution of normal pERK immunoreactivity in the forebrain of sham animal (A) and an animal with unilateral carotid occlusion (B). C and G, increased pERK immunoreactivity in untreated animals at 15 min following 30 min HI (C). Response was ablated with the application of MEK inhibitor SL327 (133 μg/g BW) (G). D and E, schematic summary of white matter pERK‐IR in naive (D) and after a 30 min HI insult (E). Light microscopy overview at the intersection between hippocampus (top), thalamus (left) and cerebral cortex (right), coronal section at mid‐parietal level (D and E). Note the faint pERK‐IR in control animal with carotid occlusion only (D, Ctrl), and the strong increase of expression in fibre tracts in external capsule (ec), fornix (fx), cortico‐thalamic fibres (ct), and descending tracts of the internal capsule (ic) at 1 h recovery following HI (E). F and H, electron microscopy of the internal capsule, at 15 min recovery following HI. Early pERK reactivity is located to the axons only. Arrows point to pERK positive clusters within adjacent axons. Scale bar (A–C, G): 1.5 mm [Color figure can be viewed at http://wileyonlinelibrary.com]
Compared to controls, the animals exposed to both carotid occlusion and 30 min hypoxia exhibited a rapid increase in white matter pERK‐IR, reaching a maximum at 15 min (Fig. 2 C). Up to a 2‐fold increase in pERK‐IR was observed in cortical and subcortical white matter tracts (Fig. 2 D and E).
White matter pERK expression is isolated to clusters within parallel axonal tracts
To determine the precise ultrastructural pERK localisation within the subcortical white matter, pERK immunoreactivity augmented with silver–gold intensification (Hristova et al. 2010) revealed pERK clusters 200–500 nm in size, occurring in 1–2 μm‐long segments within central axons (Fig. 2 F). The segments of pERK expression within axons run parallel with neighbouring axonal segments within the white matter, with pERK clusters sometimes also in two adjacent axons (Fig. 2 H, black arrows). This fibre tract labelling disappeared within 60 min post‐HI, but was contrasted by a cell body pERK‐IR+ labelling in grey matter forebrain areas with a more delayed response (Fig. 3).
Figure 3. Number of phosphorylated ERK‐positive cells following HI insult per ×20 microscopy field (mean + SEM, n = 4 animals per time point) in forebrain regions.
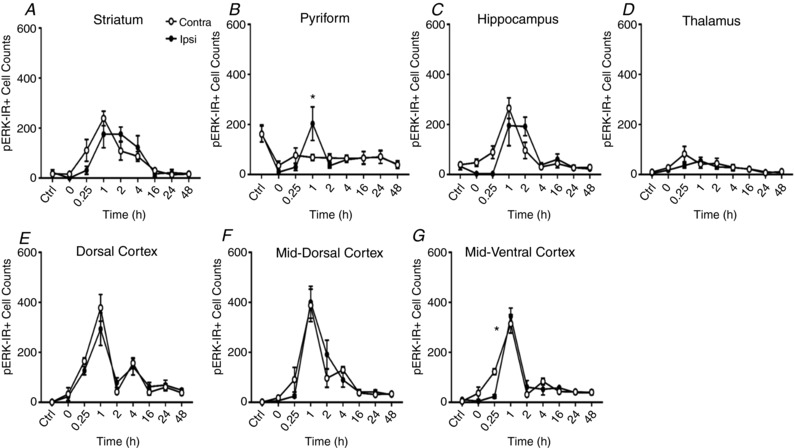
Striatum (A), pyriform (B), hippocampus (C), thalamus (D), dorsal cerebral cortex (E), mid‐dorsal cortex (F) and mid‐ventral cerebral cortex (G). HI insult induces drastic changes in ERK phosphorylation in postnatal mouse forebrain, causing an initial ipsilateral blanking out (0–15 min) and then a bilateral peak at 1–2 h. * P < 0.05 in paired Student's t test for ipsilateral versus contralateral hemispheres.
Grey matter pERK is bilaterally increased following HI, with a common temporal pattern and peak expression at 1 h post‐insult
With the exception of thalamus, there is a strong increase in pERK‐IR with a peak at 1 h following recovery from HI, declining back to approximately baseline levels at 16 h (Fig. 3 A–G). A degree of regional specificity was observed with reference to pERK+ cells. While the striatum, pyriform cortex, hippocampus, dorsal, mid‐dorsal and mid‐ventral cortex all displayed a robust increase in pERK+ cells; the thalamus did not (Fig. 3 D). Of interest, the brain hemisphere contralateral to CROC saw a mildly, but not significantly, higher number of pERK+ cells compared to the ipsilateral. Sub‐regional exceptions were observed in pyriform cortex and mid‐ventral cortex where ipsilateral expression at 1 h was significantly higher (P = 0.00008 and P = 0.0001, respectively). Table 1 illustrates the peak number of pERK+ cells in grey matter regions of both hemispheres plus time of peak occurrence.
Table 1.
pERK positive cell counts in grey matter regions of P7 mouse brains following 30 min HI
| Region | Contralateral peak (mean ± SEM) | Peak time (h) | Ipsilateral peak (mean ± SEM) | Peak time (h) |
|---|---|---|---|---|
| Striatum | 222.00 ± 30.53 | 1 | 202.86 ± 40.63 | 1 |
| Pyriform | 151.81 ± 25.77 | 0.25 | 304.38 ± 35.33 | 1 |
| Hippocampus | 269.69 ± 32.92 | 1 | 201.70 ± 34.33 | 2 |
| Thalamus | 63.93 ± 14.46 | 0.25 | 53.40 ± 11.29 | 1 |
| Dorsal cortex | 376.59 ± 27.95 | 1 | 298.50 ± 40.81 | 1 |
| Mid‐dorsal cortex | 274.32 ± 40.57 | 1 | 403.73 ± 44.47 | 1 |
| Mid‐ventral cortex | 315.91 ± 35.49 | 1 | 345.83 ± 30.68 | 1 |
MEK inhibitor SL327 is efficient in blocking ERK phosphorylation
Previous studies of ERK inhibition in rat neonatal HI injury introduced the MEK‐selective inhibitor UO126 intracerebroventricularly (i.c.v.) in two doses prior to CROC and 2.5 h hypoxia (Han & Holtzman, 2000). Due to the potentially traumatic nature of i.c.v. administration, we wished to examine the efficacy of our inhibitor, SL327, an analogue of UO126, when introduced via the intraperitoneal route. To determine the optimal dose and subsequent dose–response curve for pERK inhibition, five groups of animals (n = 5/group) were pre‐exposed to a step‐wise, 2‐fold dilution of 133 μg/g (i.e. 15, 30, 65, 133 μg/g) or to 100% EtOH (vehicle) alone (dose 0). As shown in Fig. 4 A–C, increasing doses of SL327 led to reduced pERK‐IR across six forebrain regions, with reliable suppression of immunoreactivity at 133 μg/g.
Figure 4. Ipsilateral ERK phosphorylation.
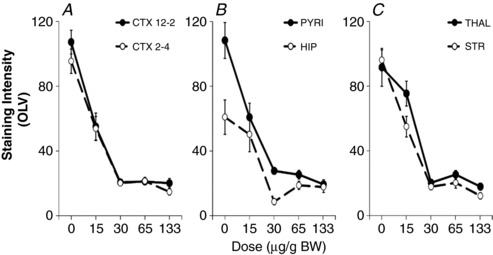
Dose response for SL327 inhibition of pERK immunoreactivity (optical luminosity value (OLV)), applied 20 min before a 30 min HI insult in hemispheric regions ipsilateral to carotid occlusion. A, CTX 12–2 dorsal cerebral cortex (12 to 2 o'clock segment), CTX 2–4 middle cerebral cortex (2 to 4 o'clock segment). B, PYRI: pyriform cortex, HIP: hippocampus. C, THAL: thalamus, STR; striatum. Increasing the dose of SL327 from 15 to 30 μg/g BW correlates to an 80% reduction in immunoreactivity.
Pre‐treatment with SL327 significantly reduces damage after HI insult
To determine the effect of pERK inhibition on neonatal HI brain damage, P7 mouse pups (C57/Bl6) were pre‐exposed to SL327 (133 μg/g BW, n = 10) or EtOH (n = 8), 20 min prior to insult. Here, they were subject to a 30 min HI insult. Results were assessed 48 h post‐HI. Histopathological assessment, shown in Fig. 5, revealed that 20 min pre‐treatment with SL327 significantly reduced levels of microglial activation in all brain regions (apart from thalamus and cortex, Fig. 5 A), neuronal loss, via the presence of Nissl bodies in the cortex (P = 0.001, Fig. 5 E), and overall TUNEL+ cell death in striatum and hippocampus (Fig. 5 G, P = 0.06, P = 0.0001). The levels of reactive astrogliosis assessed through OLV of GFAP‐IR remained unaffected (Fig. 5 B).
Figure 5. Effect of SL327 on αMβ2+ microglial activation, astroglial activation, neuronal tissue loss (Nissl body presence) and TUNEL+ cell death, when applied 20 min before (A, C, E, G) or 1 h post (B, D, F, H, I) 30 or 60 min HI.
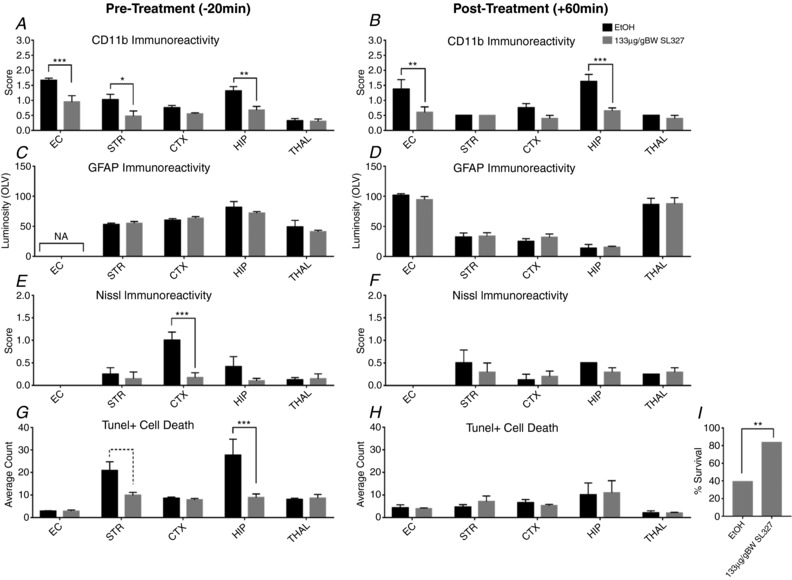
Assessment at ×20 microscopy field magnification (mean + SEM over 3 fields). A and B, the levels of CD11b+ microglia are significantly decreased in the SL327 group in white matter (EC) as well as in most grey matter regions (STR, CTX, HIP). E and F, Nissl score was decreased in pre‐treated animals. Cortex was particularly spared compared to vehicle alone. G and H, this trend to decrease is observed with number of TUNEL+ cells. SL327 treated pups have a reduction in dying cells compared to EtOH treated animals, significantly so in pre‐treated STR and HIP. C and D, extent of gliosis and reactive (GFAP+) astrocyte activation was unaffected by the application of SL327. * P < 0.05, ** P < 0.01, and *** P < 0.001 using ANOVA and post hoc Tukey. I, χ2 analysis showed survival rate was significantly increased in animals treated with SL327 1 h following a more severe 60 min HI.
Post‐treatment with SL327 reduces microglial activation but not HI damage
To examine the time dependent window for pharmacological pERK inhibition, a second set of P7 animals was injected 60 min post‐30 min HI with 133 μg/g BW SL327 or EtOH alone (n = 6 animals per group). Post‐treatment with SL327 caused a significant reduction of CD11b immunoreactivity in external capsule white matter (P = 0.01) and hippocampus (P = 0.0001, Fig. 5 B). Markers for either activated astrocytes, TUNEL+ cell death or neuronal loss remained unaffected (Fig. 5 D, F and H).
A significant number of neonatal mice survived severe HI brain injury with SL327 treatment 1 h after insult
Previous work by our group and others have shown that a severe form of insult, exposure to 60 min hypoxia alone, leads to an almost complete loss of hippocampal neurons and severe tissue damage in cortex, thalamus and basal ganglia, as well as in the subcortical white matter (Lehnardt et al. 2003; Kendall et al. 2006). To test the efficacy of ERK inhibition in a severe HI insult to P7 mice following 60 min of hypoxia, 133 μg/g BW SL327 was injected 60 min after completing the hypoxia.
The control group (EtOH) showed a significantly higher incidence of death in the 16–48 h interval, with survival of only 9 of 23 animals at 48 h (39%, P = 0.002), compared to the SL327 treated group with a survival of 83% (Fig. 5 I). Despite this, cortex, pyriform cortex, hippocampus, striatum, thalamus and external capsule revealed little difference in microglial activation and TUNEL+ cell death in SL327‐treated pups when compared to vehicle‐treated controls when killed at 48 h (data not shown).
Neuronal deletion of ERK2 has no effect on 30 min HI forebrain injury in the neonatal mouse
Due to embryonic lethality of the ERK2 null (−/−) phenotype (Yao et al. 2003; Satoh et al. 2007), the effect of ERK2 on neonatal HI brain injury was examined using cell type‐specific deletions with LoxP‐tagged (floxed) ERK2 genes and assessed alone and in combination with global ERK1 deletion. Mice with global ERK1 deletion (ERK1KO) were crossed with animals carrying the ERK2 gene flanked by LoxP sites on either side (ERK2f/f), and then further crossed with those expressing Cre recombinase under the control of neuronal synapsin promoter (Syn::Cre) (Ruff et al. 2012) or astroglial GFAP promoter (GFAP::Cre). Heterozygous breeding for Syn::Cre and ERK1KO gave rise to four distinct genotypes used in the study: ERK2f/f alone, which were functionally wild‐type (ERK1/2WT); Syn::Cre and ERK2f/f, where neurons lack ERK2 but ERK1 is expressed normally (ERK2ΔSyn); global deletion of ERK1 plus ERK2f/f (ERK1KO); and Syn::Cre plus both ERK1KO and ERK2f/f (ERK1KOERK2ΔSyn). All animals were genotyped after completing the HI experiment; heterozygous mice with the single wild‐type (WT) copy of ERK1 were excluded from assessment of 48 h histopathology.
The effects of ERK2 deletion were first assessed through pERK‐IR at 15 min post 30 min HI insult, as shown in Fig. 6. Control animals showed similar pERK expression compared to WT as displayed in Figs 2 A and 6 A. Homozygous ERK1 deletion led to a visible decrease (Fig. 6 B), and deletion of both ERK2 copies in ERK1KOERK2ΔSyn to an almost complete disappearance of pERK‐IR (Fig. 6 C). At high resolution (Fig. 6 D and G) there was a complete disappearance of the neuronal pERK‐IR (Fig. 6 G) compared to ERK1/2WT littermate controls (Fig. 6 D). In contrast, this homozygous deletion of ERK1 and neuron‐specific deletion of ERK2 did not interfere with the pERK+ astroglial cells (Fig. 6 E–I).
Figure 6. Effects of global ERK1 and neuronal ERK2 deletion on pERK immunoreactivity at 15 min post‐30 min HI insult.
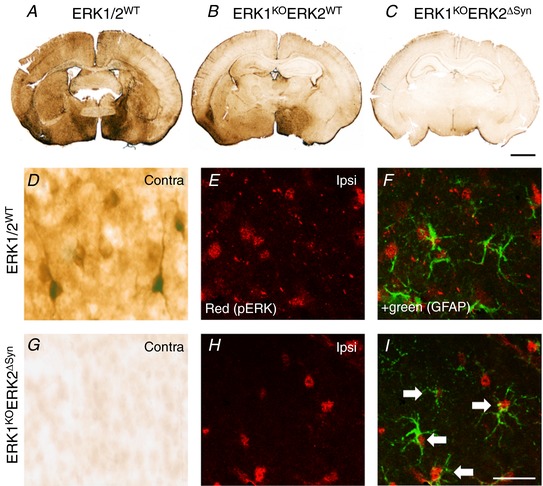
A–C, control (ERK1/2WT) animal (A), global deletion of ERK1 and ERK2WT (ERK1KO) (B), global ERK1 deletion and homozygous neuronal ERK2 deletion (ERK1KOERK2ΔSyn) (C). C, pERK immunoreactivity is almost completely reduced following deletion of both copies of ERK1 and ERK2. D–I, quantification and distribution of pERK immunoreactivity at high magnification. D, pERK staining in the contralateral pyriform cortex of ERK1/2WT with strong neuronal reactivity and prominent dendritic staining which disappears in the presence of global ERK1 deletion and homozygous neuronal ERK2 mutation ERK1KOERK2ΔSyn. E–I, residual immunoreactivity on the ipsilateral side. E and H, pERK alone. F and I, immunofluorescence double labelling with GFAP demonstrating co‐localisation of pERK in astrocytes, particularly pronounced in ERK1WTERK2ΔSyn (white arrows). Scale bar = 25 µm [Color figure can be viewed at http://wileyonlinelibrary.com]
In the mild HI model of 30 min hypoxia following carotid occlusion, homozygous global deletion of ERK1, alone or in combination with homozygous neuronal Syn::Cre‐mediated deletion of ERK2, did not lead to a significant change in histopathology, based on microglial (CD11b) activation score (Fig. 7 A and B), neuronal tissue loss (Fig. 7 C and D) and the number of TUNEL+ dying cells (Fig. 7 E and F). In total 24 pups at P7 were subject to 30 min HI with a survival time of 48 h, with the groups of ERK1/2WT (n = 5 pups), ERK2ΔSyn (n = 5), ERK1KO (n = 6) and ERK1KOERK2ΔSyn (n = 8) at completion of the experiment. Both at a sub‐regional level, and over total hemisphere, changes in neuronal ERK expression gave no significant effect on damage markers.
Figure 7. Forty‐eight hours post‐insult, damage markers were analysed in four neuronal ERK variations: ERK1/2WT, neuronal ERK2 deletion only (ERK1WTERK2ΔSyn), global ERK1 deletion only (ERK1KO) and global ERK1 deletion plus neuronal ERK2 deletion (ERK1KOERK2ΔSyn).
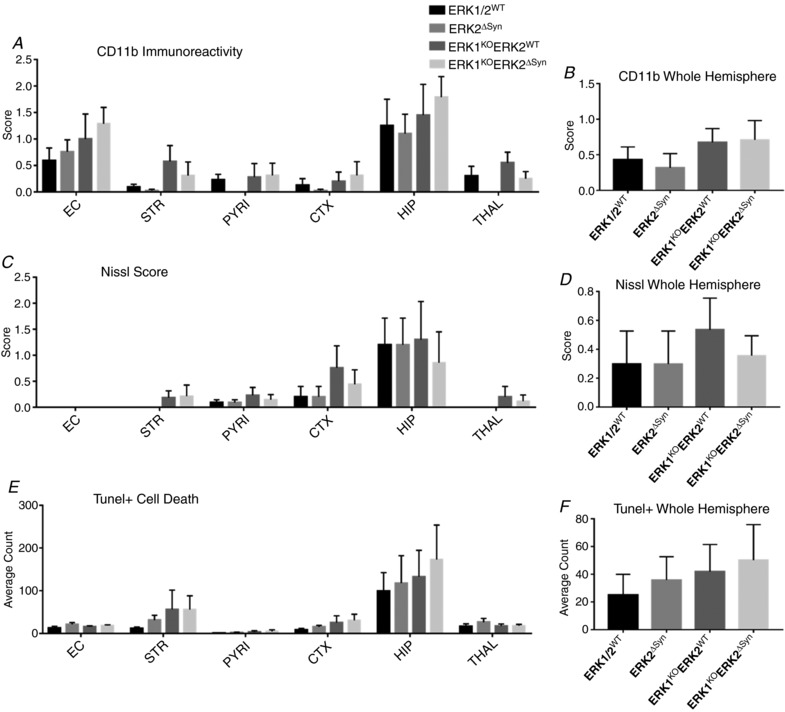
Forebrain sections were analysed for microglial activation (A and B), Nissl score (C and D), and TUNEL+ cell death (E and F). A–F, for neuron‐specific transgenic mutants there was no significant change in damage markers for each sub‐region nor over the whole ipsilateral hemisphere. Despite this, inclusion of ERK1KO gave a trend to increased injury over whole hemisphere (B, D and F). Analysis at ×20 eye field (mean + SEM over 3 fields) using ANOVA and post hoc Tukey.
Astrocytic expression of ERK2 is required for neuroprotection following 30 min HI insult
To further investigate the role of astroglial ERK in HI, mice carrying the GFAP::Cre promoter were crossed with the ERK1KOERK2f/f (without Syn::Cre) for four generations to produce ERK1KOERK2ΔGFAP and ERK1KO mouse pups. Post‐HI genotyping revealed 9 controls (ERK1/2WT) and 10 each for the ERK1KO and ERK1KOERK2ΔGFAP groups.
Global deletion of ERK1 alone was associated with higher averages for activated microglia in cortex (P = 0.01) and a trend to increase in thalamus (P = 0.06) compared to ERK1/2WT littermate controls. However, the number of Nissl bodies and total cell death (TUNEL+) were not statistically changed (Fig. 8 A, C and E).
Figure 8. Three astrocytic mutant groups were assessed for brain damage markers: ERK1/2WT, ERK1KO and ERK1KOERK2ΔGFAP .
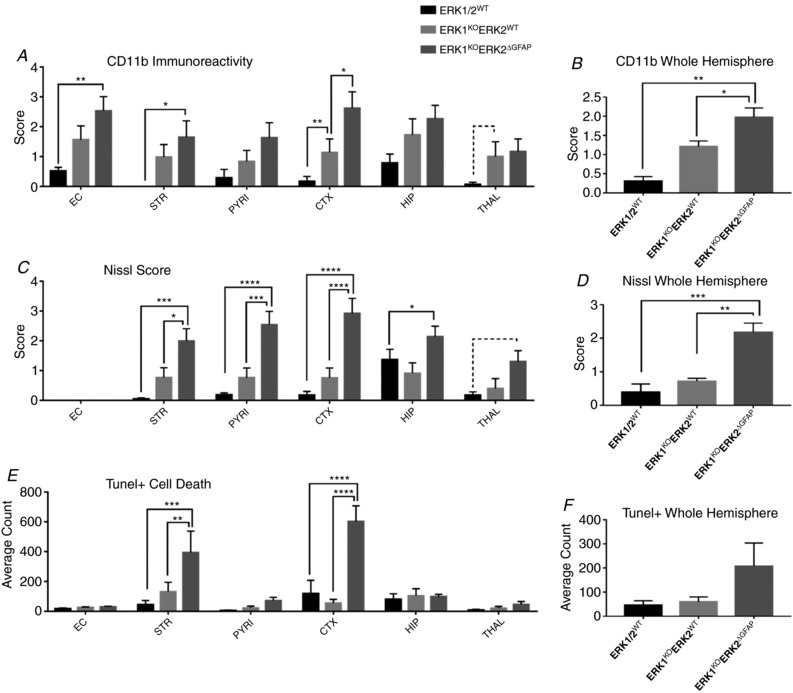
Deletion of ERK1 resulted in a strong increase in microglial activation in the ipsilateral hemispheres of both ERK1KO and ERK1KOERK2ΔGFAP groups compared to controls. A and B, increased ipsilateral microglia activation both in grey matter regions and over total hemisphere. Similarly, an increased level of Nissl bodies was observed (C and D), the exception being in thalamus where P = 0.06. E and F, increased density of TUNEL+ dying cells in striatum and cortex, although this is not reflected statistically in total hemisphere counts. Analysis at ×20 eye field (mean + SEM over 3 fields). * P < 0.05, ** P < 0.01, *** P < 0.001 and **** P < 0.0001 using ANOVA and post hoc Tukey.
Selective deletion of both ERK2 gene copies in the GFAP‐expressing cells (ERK1KOERK2ΔGFAP) resulted in a strong and robust increase in all damage markers, both on a regional basis and over total hemisphere (Fig. 8). All forebrain regions showed an increased presence of activated microglia, particularly in white matter where P = 0.01. More notably both tissue loss and cell death were greatly enhanced in grey matter regions (apart from thalamus), where striatum and cortex were most affected (Fig. 8 C–F). Nissl bodies were observed at a higher abundance in striatum (P = 0.001), pyriform cortex (P < 0.0001), cortex (P < 0.0001) and hippocampus (P = 0.04). TUNEL+ cell death was significantly increased in both striatum (P = 0.001) and cortex (P < 0.0001); however, this was reflected as a trend but with no statistical effect over the whole hemisphere (Fig. 8 F).
Interestingly, this effect was bilateral, with contralateral cortex of ERK1KOERK2ΔGFAP resulting in significant increase of microglial activation in regions contralateral to carotid occlusion with sub‐regional differences in external capsule (P = 0.03), pyriform cortex (P = 0.04), striatum (P = 0.03) and cortex (P = 0.01). In addition there was an increase in the overall number of TUNEL+ cells (P = 0.003) (data not shown).
Neuronal ERK2 is a significant contributor to forebrain response after LPS‐sensitised HI insult in the P7 mouse
Compared to 30 min HI, systemic pre‐exposure to LPS endotoxin from Escherichia coli results in strongly increased tissue loss and neuronal and astroglial cell death (Kendall et al. 2011; Jã et al. 2013), mediated by the LPS/TLR4/MyD88 pathway induction of genes encoding pro‐inflammatory factors, particularly the tumour necrosis factor (TNF) family of cytokines (Wang et al. 2009; Kendall et al. 2011). In peripheral tissues, these effects involve ERK signalling (An et al. 2002; Watts et al. 2011). To determine the role of ERK1/2 in LPS‐sensitisation to cerebral HI insult, P6 mice were injected intraperitoneally with LPS (E. coli 055/B5 serotype) 0.6 μg/g BW 12 h prior to a 30 min insult.
Recruitment of activated microglia over total hemisphere was strongly and significantly decreased in animals lacking neuronal ERK2 expression (P = 0.008, Fig. 9 A). ERK1 KO and double ERK KO were unaffected compared to littermate controls. Looking at individual regions, there is a significant decrease in thalamus (P = 0.01) with a similar trend, but not reaching significance, for pyriform cortex, hippocampus and striatum (not shown). Additional deletion of ERK1 (ERK1KOERK2ΔSyn) completely abolished this ERK2ΔSyn mediated reduction in microglial activation in total hemisphere (P = 0.048, Fig. 9 A).
Figure 9. Neuronal ERK2 is required for non‐neuronal cell activation and recruitment after combined LPS and 30 min HI insult.
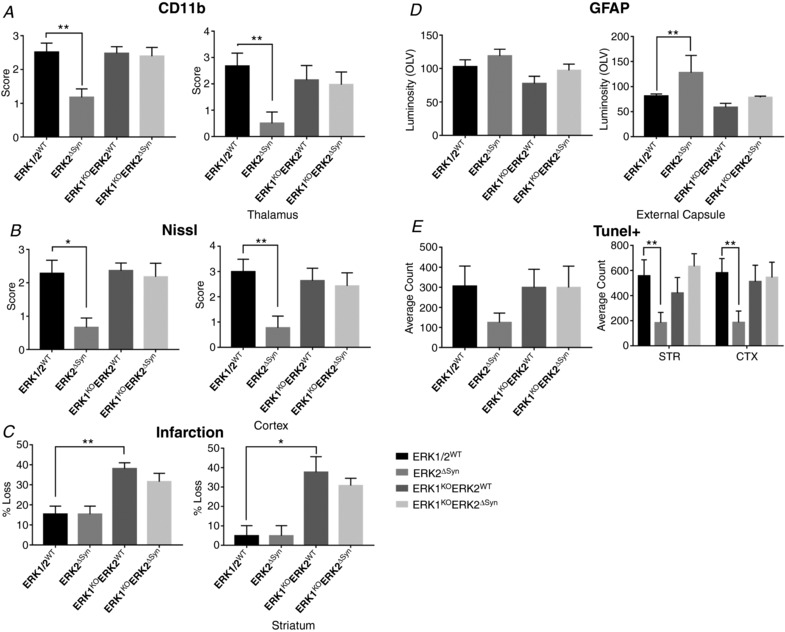
Neuronal ERK2 deletion, in the presence of WT ERK1 expression significantly reduces activated microglia (A), neuronal cell death (B), infarction (C) and astrogliosis (GFAP, D). In ERK2ΔSyn mutants this response is significantly decreased. E, quantification of TUNEL+ dying cells. ERK2ΔSyn exhibited a decrease in the number of dying cells; although not significant over the whole hemisphere, both striatum (STR) and cortex (CTX) saw a highly significant reduction in the number of TUNEL positive cells. Cell death patterns were not reflected in a decrease in lesion size compared to ERK1/2WT controls. However, global deletion of ERK1 or double knockout animals saw a significant increase in lesion size and tissue loss with STR being most affected (C). Regions of significant changes are shown for each corresponding damage marker. Analysis at ×20 eye field (mean + SEM over 3 fields) using ANOVA and post hoc Tukey where * P < 0.05, ** P < 0.01.
Nissl and TUNEL+ cell death assessments (Fig. 9 B and E) in the ERK2ΔSyn subgroup were similar to the ones observed in respect to microglial activation, i.e. reduction of both markers. Similarly to the trend observed in the microglial activation this reduction disappeared in ERK1KOERK2ΔSyn. Using a one‐way ANOVA with post hoc TUKEY the number of Nissl bodies was significantly different in ERK2ΔSyn animals only, with cortex (P = 0.02, Fig. 9 B) as the most affected region and an overall trend to decrease seen in all other grey matter regions (not shown). TUNEL+ cell counts were lower in all regions with single ERK2ΔSyn mutations. Both striatum and cortex saw significant reduction (P = 0.009 and P = 0.008, respectively, Fig. 9 E), although counts over total hemisphere were not statistically significant. Interestingly this cumulatively did not contribute to total hemisphere tissue loss in the ERK2ΔSyn cohort; what was observed, though, was a significant involvement of ERK1 mutations, either alone or in combination with neuronal ERK2 deletion, in infarction and overall loss (P = 0.009, Fig. 9 C) of 72%, particularly in striatum (P = 0.03, Fig. 9 C).
GFAP‐IR remains unaffected between all four mutant groups (Fig. 9 D), the exception being an increase in white matter GFAP expression (P = 0.001) in ERK2ΔSyn animals. As before, this effect was ameliorated by the presence of ERK1KO to baseline expression (Fig. 9 D).
Astrocyte deletion of ERK1/2 is protective in cerebral cortex after LPS‐sensitised HI insult
We explored the effects of LPS‐sensitised 30 min HI insult and GFAP‐specific ERK2 deletion on ERK1 null background (ERK1KO n = 6, ERK1KOERK2ΔGFAP n = 4) against WT littermate controls (n = 8).
CD11b‐IR was reduced in double mutant mice (P = 0.03), which was a cumulative effect with no individual region seeing significant change (Fig. 10 A). A similar trend was observed with Nissl bodies score (Fig. 10 B). TUNEL+ cell death was reduced in ERK1KOERK2ΔGFAP compared to WT littermate controls although significance was not reached. A strong contributor to this was a clear and significant sparing of the cortex (P = 0.03, Fig. 10 E) and this is confirmed by the same pattern of sparing observed by tissue loss, where damage to the cerebral cortex was prevented in the ERK1KOERK2ΔGFAP animals (P = 0.04, Fig. 10 C).
Figure 10. Astrocytic ERK directs astrogliosis and tissue loss after combined LPS and 30 min HI insult.
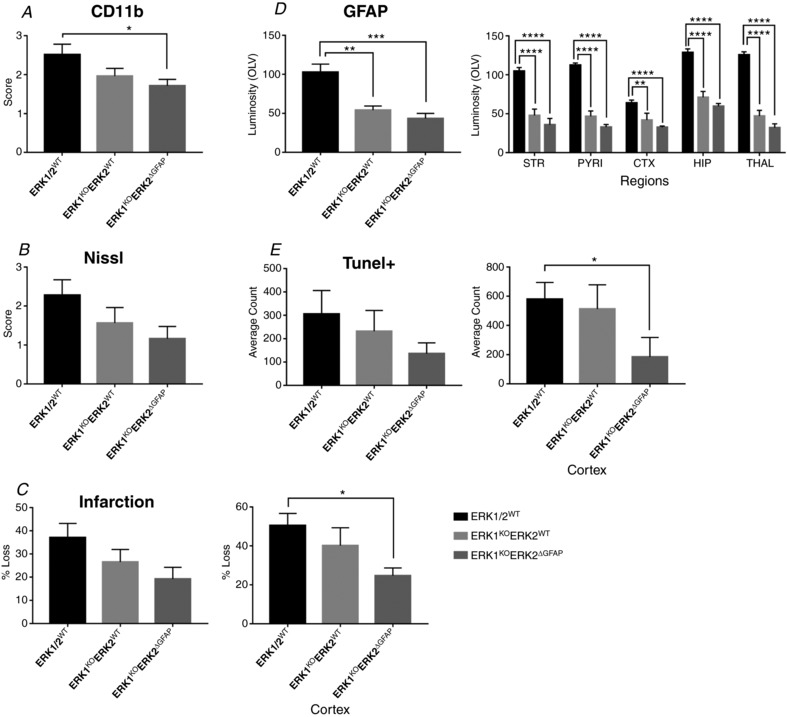
Global deletion of ERK1 had no direct effect on microglial activation (A), neuronal damage (B), tissue loss (C) or overall cell death (E). Double ERK mutations in astrocytes gave a reduction in microglial activation, cell death and tissue loss with cortex being the region most spared (A, C and E). ERK strongly drives astrogliosis in response to HI. Deletion of even one copy results in a dramatic decrease of GFAP, marker for gliosis, with significance seen in all grey matter regions (D). * P < 0.05, ** P < 0.01, *** P < 0.001, and **** P < 0.0001 using ANOVA and post hoc Tukey.
ERK1/2 expression in the brain is a strong contributor to astrocyte activation and astrogliosis after LPS‐sensitised HI insult
In contrast to the mild effect on cell death, GFAP‐IR did show a strong and significant reduction (P < 0.01) in astrogliosis in all grey matter regions. This was true for both single ERK1 and double ERK1 and 2 mutant animals compared to littermate controls (Fig. 10 D). Whereas neuronal ERK2 deletion increases astrocyte reactivity in white matter (Fig. 9 D), astrocyte‐specific deletion of ERK2 has no effect on white matter (not shown). This is a robust indication that ERK phosphorylation is required for astrocyte activation and a master modulator of astrogliosis observed with neonatal brain injury.
Discussion
During neonatal HI insult, once a critical threshold is reached, cellular mechanisms begin to fail due to ATP insufficiency leading to a two‐phase neurotoxic cascade (Sanders et al. 2010). Primary energy failure, resulting in immediate and necrotic cell death, is followed by a period of up to 6 h in which reperfusion can occur. Afterwards, secondary neuronal cell death ensues due to multiple molecular imbalances instigated by excitotoxic oxidative stress and mitochondria failure (Rocha‐Ferreira & Hristova, 2016). Depending on the severity of the insult, tertiary energy failure associated with late cell death, astrogliosis, as well as remodelling and repair, lasting for weeks and months following the initial HI insult may occur (Rocha‐Ferreira & Hristova, 2016). Owing to their extensive control over cellular physiology, the mitogen‐activated protein kinase kinases have a potential role in hypoxia induced cell death.
Our study shows rapid phosphorylation of ERK following mild HI insult. In line with previous studies, ipsilateral expression of pERK is immediately nullified for the first 15 min after insult followed by a rapid bilateral increase in expression that peaked at 1 h and returned to baseline by 4 h (Alessandrini et al. 1999; Wang et al. 2003, 2004; van den Tweel et al. 2006) establishing a consistent temporal pattern of pERK expression in neonatal rodent models of HI.
Initial pERK reduction was equally observed in both white and grey matter. The appearance of axonal pERK was seen from 15 min onwards, and normalised by 1 h (Fig. 2). Expression of pERK was observed in white matter, bound within the cytoplasm beyond 2 h post‐HI. Our data show clustering of pERK positive axons following HI, which has not been reported before (Fig. 2). This effect coincides with other in vivo studies where neurons showing cytoplasmic pERK activity are commonly adjacent to one another at the borders of ischaemia‐induced micro‐lesions in adult mice (Wang et al. 2003).
Pre‐treatment with SL327, a selective inhibitor of MEK, resulted in decreased levels of microglia activation, histological brain injury, and TUNEL+ cell death. These SL327 mediated protective outcomes were retained when administered up to 1 h post‐insult. This is in line with current protocols where pre‐treatment with U0126 to bilateral carotid artery occlusion reduced the loss of hippocampal neurons in addition to an overall decrease of infarct size associated with improved neurological outcome in adult rodents (Namura et al. 2001). Other in vivo studies explored the suppression of cytokine release following HI by application of U0126 both 20 min prior to and immediately following MCAO in the adult mouse (Namura et al. 2001). This suggests a mechanism by which the protective effects are due to restoration of the balance between pro‐ and anti‐inflammatory cytokines thus maintaining subsequent myelination (Bain et al. 2010).
Post‐treatment with SL327 after 60 min of 8% O2 exposure had diminished efficacy, compared to 30 min exposure, in protecting the neonatal mouse brain from ischaemic damage. Whilst markers for damage remained unaffected, the morbidity of this cohort was dramatically improved with survival rate increasing from 30% in EtOH treated controls to 83% in the SL327 group.
Some data suggest that transient suppression of ERK phosphorylation by an intraperitoneal injection of SL327 at P6 without a surgical procedure significantly increases cleaved caspase‐3 expression, and respectively apoptosis (Yufune et al. 2016). Nevertheless, our data are obtained using TUNEL+ cell death as a broader marker of damage (Hristova et al. 2010) having in mind that in neonatal HI apoptosis, necrosis and autophagy take place (Rocha‐Ferreira & Hristova, 2016). Thus SL327 might cause apoptosis, but at the same time prevent necrosis and autophagy thus having an overall neuroprotective function in neonatal HI. However, this requires further investigation.
Use of neuronal specific ERK2 knockout mice resulted in a clear and significant reduction of dying neurons, active microglia and brain injury with cortex, pyriform cortex and striatum being regions of particular sensitivity. The neuroprotection following ERK2 deletion correlates well with the actions of MEK inhibitors in adult MCAO studies (Alessandrini et al. 1999). Alessandrini's group showed that in the adult gerbil model of focal cerebral ischaemia, infarct size was reduced by 55%, suggesting that similarly to our results, ERK must be acting alongside other complementary or parallel pro‐apoptotic pathways in order to induce cell death after HI (Namura et al. 2001).
Global ERK1 deletion is embryonically viable with no phenotypic differences to wild‐type littermates. By deleting ERK1 globally, we observed an effect contrasting the neuroprotection achieved through neuronal ERK2 deletion, where ERK1 mutation increased HI brain damage following 30 min HI compared to littermate controls.
In adult mouse forebrain, expression of ERK1 is significantly lower than that of ERK2 – up to 6‐fold less in the frontal cortex (Ortiz et al. 1995). The complementary and ubiquitous co‐expression of ERK1 and 2 has led to the paradigm that ERK1 regulates ERK2 actions on cell growth and survival (Pouysségur & Lenormand, 2003; Lefloch et al. 2008). However, developmental complications have associated ERK1 with thrombocyte dysfunction (Nekrasova et al. 2005; Lefloch et al. 2008), as well as behavioural studies that link the MAP3K gene on chromosome 16, encoding ERK1, to altered synaptic plasticity and subsequent behavioural abnormalities, including autism (Campbell et al. 2008; Engel et al. 2008; Fernandez et al. 2010; Pucilowska et al. 2012). In addition, under certain circumstances ERK1 attenuates the ERK2 signal; indeed, ERK2 up‐regulation is seen in ERK1KO mice (Selcher et al. 2003; Lefloch et al. 2008; Samuels et al. 2008) which, as shown herein, promotes neuronal death following HI. Despite these studies, to the best of our knowledge there is little information as to how suppression of ERK1 exacerbates injury in the neonatal HI mouse and further work is required to elucidate the role of ERK1 in injury response.
Global deletion of ERK1 and astrocyte‐specific ERK2 deletion resulted in a greater deleterious effect than global ERK1 deletion alone, with significantly higher expression of each damage marker. In vitro studies suggest a protective role of astrocytes to ODC precursor cells (Arai & Lo, 2010). As such they are suggested to be partially responsible for subsequent white matter injury, via H2O2 insult, by the up‐regulation of pERK. Application of U0126 abolishes the protective nature of astrocytes (Arai & Lo, 2010). Other in vivo studies looked at ERK2 excision under the GFAP promoter in the developing mouse and observed that whilst ODC precursor cells develop normally, there is a significant delay in maturation and reduced myelin production. (Fyffe‐Maricich et al. 2011). This suggests a developmental requirement of astrocytic ERK2 expression for normal ODC genesis.
Following oxidative stress, reactive oxygen species activation of ERK results in dysfunction of the mitochondrial outer membrane inducing cytochrome c release and cleavage of caspases 3 and 8 (Nowak, 2002; Nowak et al. 2006; Martin & Pognonec, 2010). ERK can further promote cytochrome c release via the up‐regulation of the pro‐apoptotic proteins Bax, PUMA and Bad (Cagnol & Chambard, 2010). Our study elucidates that ERK can be both protective and detrimental in neonatal HI injury depending on the cellular localisation of ERK.
Our data suggest that ERK2 deletion on its own seems to have ERK1 independent and cell‐specific function with a detrimental effect in neurons and a protective one in astrocytes. Our previous data demonstrate that inhibition of phosphorylated STAT3 (pSTAT3) has a neuroprotective effect in neonatal HI brain damage in a cell‐ and time‐specific manner (Hristova et al. 2016). While the pSTAT3 Tyr705 phosphorylation site, which is Jak2‐dependent, is associated with the transcriptional properties of STAT3, the Ser727 site (downstream of MAPK and ERK1/2) is responsible for recruitment to mitochondria and regulates functions alternative to transcription (Yang & Rincon, 2016). This suggests that inhibition of both ERK1 and 2 could result in mitochondrial dysfunction and might be a possible explanation for the detrimental effects observed when both ERK1 and ERK2 deletions combined were used. Although possible, this mechanism would require further investigation.
In neonatal brain injury studies, systemic injection of LPS leads to an up‐regulation of pro‐inflammatory cytokines and consequently increased neuronal and astroglial cell death (Jã et al. 2013). The synergistic nature of LPS to HI shows the same underlying white matter and grey matter lesion formation as those seen in human babies subject to infection as well as hypoxic ischemic encephalopathy (Wang et al. 2009; Kendall et al. 2011). LPS/TLR4/MyD88 induction of genes encoding pro‐inflammatory cytokines is mediated by the phosphorylation and nuclear translocation of ERK1/2 (An et al. 2002; Watts et al. 2011). MyD88 can form a functional complex directly with ERK1/2 via the recruitment of a scaffold protein MKP3 (tpl2) which prevents ERK dephosphorylation, rendering it constitutively active (Bandow et al. 2012). MKP3 had been previously implicated in the dysregulation of TLR2/MyD88 activation of ERK, resulting in transcription of its nuclear target Elk‐1.
Our data indicate that both single neuronal ERK2 mutation and double ERK1 and neuronal ERK2 mutation result in strong reduction in damage. Single mutants exhibited up to a 90% decrease in microglia and 40% decrease in astrocyte activation that correlate with a reduction in tissue loss and TUNEL+ cell death. Preservation of regional cell loss was validated by measurements of infarct. No reduction of cell death or tissue loss was observed with double mutation of ERK1 and neuronal ERK2.
In vitro dendritic cell cultures from ERK1 null mice show an increased expression of interleukin (IL)‐12p70 and a decrease of anti‐inflammatory IL‐10 secretion in response to TLR stimulation (Bandow et al. 2012). Hippocampal cultures exposed to combined LPS and interferon γ (IFNγ), a pro‐death cytokine, were susceptible to damage due to NO production by co‐cultured microglia (Xiao et al. 1996). Using the MEK inhibitor PD98059, IFNγ‐induced NO production was reduced by 40% (Bandow et al. 2012). In human monocytes, PD98059 reduced LPS induction of TNFα gene expression in a dose‐dependent manner. Indeed, inhibition of ERK decreased the release of several pro‐inflammatory cytokines including IL‐1 and IL‐18 (An et al. 2002; Gorina et al. 2011). To date, this is the first time evidence has been provided to implicate the capacity of ERK to modulate both neuronal and glial damage response following endotoxin‐sensitised ischaemia in the neonate.
Our results would need to be further confirmed in large animals as the rodent Rice–Vannucci model does not mimic the human condition optimally. This includes the level of white matter development, as well as the type of injury caused by the insult. In the rodent it is mostly severe injury with multiple infarctions involving both white matter and grey matter compared to diffuse apoptotic and relatively small necrotic areas in the human infant brain, affecting mostly white matter in most cases of periventricular leukomalacia resulting in cerebral palsy (Rumajogee et al. 2016).
As a conclusion our data confirm that neuronal ERK2 has a pivotal role in the development of neonatal HI brain damage. In contrast ERK1 and astrocytic ERK2 show a clear involvement in the pro‐survival response to insult. There is clear therapeutic potential for ERK2 inhibition after HI with the possibility of combined therapy to contest neurodegeneration by severe insult. Similar patterns of expression suggest that the therapeutic window of 1 h where inhibition could be beneficial is restricted and that pre‐emptive treatment during the developmental stage of infancy could diminish endogenous biochemical survival mechanisms, significantly worsening the outcome.
Additional information
Competing interests
None declared.
Author contributions
L.T.: acquisition, analysis and interpretation of data; drafting and critically analysing the work. E.R.‐F.: acquisition and interpretation of data; drafting the work or revising it critically for important intellectual content. D.P.: conception of the work and revision of content; G.R.: conception of the work and revision of content; M.H.: acquisition, analysis and interpretation of data and revising the work. We confirm that all authors approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed. Work was carried out at the Institute for Womens Health, University, College London.
Funding
This work was funded by SPARKS 07UCL02, and Welcome Trust grants: WT088646MA and WT089624MA.
Acknowledgements
The authors wish to acknowledge Dr Alejo J. Nevado‐Holgado for all his intellectual and technical advice on the statistics.
Biography
Laura Thei currently holds a postdoctoral researcher position at the Department of Pharmacy, University of Reading. She has over 12 years of research background in the fields of immunology, oncology and neuroscience. Current studies are on the role of microglia and downstream signalling cascades to amyloid‐β pathology in Alzheimer's disease. For this she is generating models of how amyloid‐β protofibrils selectively activate ion channels and how disruption to these has a knock‐on effect on metabolism and cell death mechanisms. Prior to this, her work focused on the role of the MAPK signalling cascade in neonatal cerebral brain damage following hypoxia/ischaemia, and endotoxin‐sensitised hypoxia/ischaemia insult.

Edited by: Laura Bennet & Maike Glitsch
This is an Editor's Choice article from the 1 December 2018 issue.
References
- Alessandrini A, Namura S, Moskowitz MA & Bonventre JV (1999). MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci U S A 96, 12866–12869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An H, Yu Y, Zhang M, Xu H, Qi R, Yan X, Liu S, Wang W, Guo Z, Guo J, Qin Z & Cao X (2002). Involvement of ERK, p38 and NF‐κB signal transduction in regulation of TLR2, TLR4 and TLR9 gene expression induced by lipopolysaccharide in mouse dendritic cells. Immunology 106, 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai K & Lo EH (2010). Astrocytes protect oligodendrocyte precursor cells via MEK/ERK and PI3K/Akt signaling. J Neurosci Res 88, 758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trazaskos JM & Sweatt JD (1998). The MAPK cascade is required for mammalian associative learning. Nat Neurosci 1, 602–609. [DOI] [PubMed] [Google Scholar]
- Bain JM, Ziegler A, Yang Z, Levison SW & Sen E (2010). TGFβ1 stimulates the over‐production of white matter astrocytes from precursors of the “brain marrow” in a rodent model of neonatal encephalopathy. PLoS One 5, e9567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandow K, Kusuyama J, Shamoto M, Kakimoto K, Ohnishi T & Matsuguchi T (2012). LPS‐induced chemokine expression in both MyD88‐dependent and ‐independent manners is regulated by Cot/Tpl2‐ERK axis in macrophages. FEBS Lett 586, 1540–1546. [DOI] [PubMed] [Google Scholar]
- Cagnol S & Chambard J‐C (2010). ERK and cell death: mechanisms of ERK‐induced cell death – apoptosis, autophagy and senescence. FEBS J 277, 2–21. [DOI] [PubMed] [Google Scholar]
- Campbell DB, Li C, Sutcliffe JS, Persico AM & Levitt P (2008). Genetic evidence implicating multiple genes in the MET receptor tyrosine kinase pathway in autism spectrum disorder. Autism Res 1, 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals S, Casarejos MJ, de Bernardo S, Solano RM & Mena MA (2003). Selective and persistent activation of extracellular signal-regulated protein kinase by nitric oxide in glial cells induces neuronal degeneration in glutathione-depleted midbrain cultures. Mol Cell Neurosci 24, 1012–1026. [DOI] [PubMed] [Google Scholar]
- Carsten Möller J, Klein MA, Haas S, Jones LL, Kreutzberg GW & Raivich G (1996). Regulation of thrombospondin in the regenerating mouse facial motor nucleus. Glia 17, 121–132. [DOI] [PubMed] [Google Scholar]
- Chock VY & Giffard RG (2005). Development of neonatal murine microglia in vitro: changes in response to lipopolysaccharide and ischemia‐like injury. Pediatr Res 57, 475–480. [DOI] [PubMed] [Google Scholar]
- de Bernardo S, Canals S, Casarejos MJ, Solano RM, Menendez J & Mena MA (2004). Role of extracellular signal-regulated protein kinase in neuronal cell death induced by glutathione depletion in neuron/glia mesencephalic cultures. J Neurochem 91, 667–682. [DOI] [PubMed] [Google Scholar]
- Dewar D, Underhill SM & Goldberg MP (2003). Oligodendrocytes and ischemic brain injury. J Cereb Blood Flow Metab 23, 263–274. [DOI] [PubMed] [Google Scholar]
- Dommergues M‐A, Plaisant F, Verney C & Gressens P (2003). Early microglial activation following neonatal excitotoxic brain damage in mice: a potential target for neuroprotection. Neuroscience 121, 619–628. [DOI] [PubMed] [Google Scholar]
- Engel SR, Creson TK, Hao Y, Shen Y, Maeng S, Nekrasova T, Landreth GE, Manji HK & Chen G (2008). The extracellular signal‐regulated kinase pathway contributes to the control of behavioral excitement. Mol Psychiatry 14, 448–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez BA, Roberts W, Chung B, Weksberg R, Meyn S, Szatmari P, Joseph‐George AM, MacKay S, Whitten K, Noble B, Vardy C, Crosbie V, Luscombe S, Tucker E, Turner L, Marshall CR & Scherer SW (2010). Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. J Med Genet 47, 195–203. [DOI] [PubMed] [Google Scholar]
- Ferriero DM, Holtzman DM, Black SM & Sheldon RA (1996). Neonatal mice lacking neuronal nitric oxide synthase are less vulnerable to hypoxic–ischemic injury. Neurobiol Dis 3, 64–71. [DOI] [PubMed] [Google Scholar]
- Fyffe‐Maricich SL, Karlo JC, Landreth GE & Miller RH (2011). The ERK2 mitogen‐activated protein kinase regulates the timing of oligodendrocyte differentiation. J Neurosci 31, 843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorina R, Font‐Nieves M, Márquez‐Kisinousky L, Santalucia T & Planas AM (2011). Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88‐dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia 59, 242–255. [DOI] [PubMed] [Google Scholar]
- Hagberg H, Gressens P & Mallard C (2012). Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann Neurol 71, 444–457. [DOI] [PubMed] [Google Scholar]
- Han BH & Holtzman DM (2000). BDNF protects the neonatal brain from hypoxic‐ischemic injury in vivo via the ERK pathway. J Neurosci 20, 5775–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins RD & Shakaran S (2009). Hypothermia for hypoxic ischemic encephalopathy in infants ≥ 36 weeks. Early Hum Dev 85, S49–S52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hristova M, Cuthill D, Zbarsky V, Acosta‐Saltos A, Wallace A, Blight K, Buckley SMK, Peebles D, Heuer H, Waddington SN & Raivich G (2010). Activation and deactivation of periventricular white matter phagocytes during postnatal mouse development. Glia 58, 11–28. [DOI] [PubMed] [Google Scholar]
- Hristova M, Rocha‐Ferreira E, Fontana X, Thei L, Buckle R, Christou M, Hompoonsup S, Gostelow N, Raivich G & Peebles D (2016). Inhibition of signal transducer and activator of transcription 3 (STAT3) reduces neonatal hypoxic‐ischaemic brain damage. J Neurochem 136, 981–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inder TE & Volpe JJ (2000). Mechanisms of perinatal brain injury. Semin Neonatol 5, 3–16. [DOI] [PubMed] [Google Scholar]
- Irving EA & Bamford M (2002). Role of mitogen‐ and stress‐activated kinases in ischemic injury. J Cereb Blood Flow Metab 22, 631–647. [DOI] [PubMed] [Google Scholar]
- Jã K, Naylor AS, Dean J, Hagberg H, Mallard C & Järlestedt K (2013). Decreased survival of newborn neurons in the dorsal hippocampus after neonatal LPS exposure in mice. Neuroscience 253, 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston MV, Fatemi A, Wilson MA & Northington F (2011). Treatment advances in neonatal neuroprotection and neurointensive care. Lancet Neurol 10, 372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall GS, Hristova M, Hirstova M, Horn S, Dafou D, Acosta‐Saltos A, Almolda B, Zbarsky V, Rumajogee P, Heuer H, Castellano B, Pfeffer K, Nedospasov SA, Peebles DM & Raivich G (2011). TNF gene cluster deletion abolishes lipopolysaccharide‐mediated sensitization of the neonatal brain to hypoxic ischemic insult. Lab Invest 91, 328–341. [DOI] [PubMed] [Google Scholar]
- Kendall GS, Robertson NJ, Iwata O, Peebles D & Raivich G (2006). N‐Methyl‐isobutyl‐amiloride ameliorates brain injury when commenced before hypoxia ischemia in neonatal mice. Pediatr Res 59, 227–231. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M & Altman DG (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br J Pharmacol 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefloch R, Pouysségur J & Lenormand P (2008). Single and combined silencing of ERK1 and ERK2 reveals their positive contribution to growth signaling depending on their expression levels. Mol Cell Biol 28, 511–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ & Vartanian T (2003). Activation of innate immunity in the CNS triggers neurodegeneration through a Toll‐like receptor 4‐dependent pathway. Proc Natl Acad Sci U S A 100, 8514–8519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z & Xu S (2006). ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life 58, 621–631. [DOI] [PubMed] [Google Scholar]
- Martin P & Pognonec P (2010). ERK and cell death: cadmium toxicity, sustained ERK activation and cell death. FEBS J 277, 39–46. [DOI] [PubMed] [Google Scholar]
- Namura S, Iihara K, Takami S, Nagata I, Kikuchi H, Matsushita K, Moskowitz MA, Bonventre JV & Alessandrini A (2001). Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc Natl Acad Sci U S A 98, 11569–11574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrasova T, Shive C, Gao Y, Kawamura K, Guardia R, Landreth G & Forsthuber TG (2005). ERK1‐deficient mice show normal T cell effector function and are highly susceptible to experimental autoimmune encephalomyelitis. J Immunol 175, 2374–2380. [DOI] [PubMed] [Google Scholar]
- Nowak G (2002). Protein kinase C‐α and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin‐induced apoptosis in renal cells. J Biol Chem 277, 43377–43388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak G, Clifton GL, Godwin ML & Bakajsova D (2006). Activation of ERK1/2 pathway mediates oxidant‐induced decreases in mitochondrial function in renal cells. Am J Physiol Renal Physiol 291, F840–F855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz J, Harris HW, Guitart X, Terwilliger RZ, Haycock JW & Nestler EJ (1995). Extracellular signal‐regulated protein kinases (ERKs) and ERK kinase (MEK) in brain: regional distribution and regulation by chronic morphine. J Neurosci 15, 1285–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polazzi E & Monti B (2010). Microglia and neuroprotection: From in vitro studies to therapeutic applications. Prog Neurobiol 92, 293–315. [DOI] [PubMed] [Google Scholar]
- Pouysségur J & Lenormand P (2003). Fidelity and spatio‐temporal control in MAP kinase (ERKs) signalling. Eur J Biochem 270, 3291–3299. [DOI] [PubMed] [Google Scholar]
- Pucilowska J, Puzerey PA, Karlo JC, Galán RF & Landreth GE (2012). Disrupted ERK signaling during cortical development leads to abnormal progenitor proliferation, neuronal and network excitability and behavior, modeling human neuro‐cardio‐facial‐cutaneous and related syndromes. J Neurosci 32, 8663–8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha‐Ferreira E & Hristova M (2016). Plasticity in the neonatal brain following hypoxic‐ischaemic injury. Neural Plast 2016, 4901014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha‐Ferreira E, Phillips E, Francesch‐Domenech E, Thei L, Peebles DM, Raivich G & Hristova M (2015). The role of different strain backgrounds in bacterial endotoxin‐mediated sensitization to neonatal hypoxic–ischemic brain damage. Neuroscience 311, 292–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff CA, Staak N, Patodia S, Kaswich M, Rocha‐Ferreira E, Da Costa C, Brecht S, Makwana M, Fontana X, Hristova M, Rumajogee P, Galiano M, Bohatschek M, Herdegen T, Behrens A & Raivich G (2012). Neuronal c‐Jun is required for successful axonal regeneration, but the effects of phosphorylation of its N‐terminus are moderate. J Neurochem 121, 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumajogee P, Bregman T, Miller SP, Yager JY & Fehlings MG (2016). Rodent hypoxia–ischemia models for cerebral palsy research: a systematic review. Front Neurol 7, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K, Sweatt JD, Saitta SC & Landreth GE (2008). Deletion of ERK2 mitogen‐activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J Neurosci 28, 6983–6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders RD, Manning HJ, Robertson NJ, Ma D, Edwards AD, Hagberg H & Maze M (2010). Preconditioning and postinsult therapies for perinatal hypoxic-ischemic injury at term. Anesthesiology 113, 233–249. [DOI] [PubMed] [Google Scholar]
- Satoh Y, Endo S, Ikeda T, Yamada K, Ito M, Kuroki M, Hiramoto T, Imamura O, Kobayashi Y, Watanabe Y, Itohara S & Takishima K (2007). Extracellular signal‐regulated kinase 2 (ERK2) knockdown mice show deficits in long‐term memory; ERK2 has a specific function in learning and memory. J Neurosci 27, 10765–10776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selcher JC, Weeber EJ, Christian J, Nekrasova T, Landreth GE & Sweatt JD (2003). A role for ERK MAP kinase in physiologic temporal integration in hippocampal area CA1. Learn Mem 10, 26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoff RP, Bessert DA, Barks JDE, Song D, Cerghet M & Silverstein FS (2001). Hypoxic–ischemic injury results in acute disruption of myelin gene expression and death of oligodendroglial precursors in neonatal mice. Int J Develop Neurosci 19, 197–208. [DOI] [PubMed] [Google Scholar]
- van den Tweel ERW, Kavelaars A, Lombardi MS, Nijboer CHA, Groenendaal F, van Bel F & Heijnen CJ (2006). Bilateral molecular changes in a neonatal rat model of unilateral hypoxic‐ischemic brain damage. Pediatr Res 59, 434–439. [DOI] [PubMed] [Google Scholar]
- Vannucci RC (1990). Experimental biology of cerebral hypoxia‐ischemia: relation to perinatal brain damage. Pediatr Res 27, 317–326. [DOI] [PubMed] [Google Scholar]
- Vannucci SJ & Hagberg H (2004). Hypoxia–ischemia in the immature brain. J Exp Biol 207, 3149–3154. [DOI] [PubMed] [Google Scholar]
- Vincer MJ, Allen AC, Joseph KS, Stinson DA, Scott H & Wood E (2006). Increased prevalence of cerebral palsy amongst very preterm infants: a population-based study. Pediatrics 188, e1621–1626. [DOI] [PubMed] [Google Scholar]
- Volpe JJ (2001). Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res 50, 553–562. [DOI] [PubMed] [Google Scholar]
- Wang X, Stridh L, Li W, Dean J, Elmgren A, Gan L, Eriksson K, Hagberg H & Mallard C (2009). Lipopolysaccharide sensitizes neonatal hypoxic‐ischemic brain injury in a MyD88‐dependent manner. J Immunol 183, 7471–7477. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang H, Xu L, Rozanski DJ, Sugawara T, Chan PH, Trzaskos JM & Feuerstein GZ (2003). Significant neuroprotection against ischemic brain injury by inhibition of the MEK1 protein kinase in mice: exploration of potential mechanism associated with apoptosis. J Pharmacol Exp Ther 304, 172–178. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhu C, Qiu L, Hagberg H, Sandberg M & Blomgren K (2004). Activation of ERK1/2 after neonatal rat cerebral hypoxia‐ischaemia. J Neurochem 86, 351–362. [DOI] [PubMed] [Google Scholar]
- Watts BA, George T, Sherwood ER & Good DW (2011). Basolateral LPS inhibits NHE3 and HCO3 − absorption through TLR4/MyD88‐dependent ERK activation in medullary thick ascending limb. Am J Physiol Cell Physiol 301, C1296–C1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B‐G, Bai X‐F, Zhang G‐X, Höjeberg B & Link H (1996). Shift from anti‐ to proinflammatory cytokine profiles in microglia through LPS‐or IFN‐γ‐mediated pathways. Neuroreport 7, 1893–1898. [DOI] [PubMed] [Google Scholar]
- Yang R & Rincon M (2016). Mitochondrial Stat3, the need for design thinking. Int J Biol Sci 12, 532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Li W, Wu J, Germann UA, Su MSS, Kuida K & Boucher DM (2003). Extracellular signal‐regulated kinase 2 is necessary for mesoderm differentiation. Proc Natl Acad Sci U S A 100, 12759–12764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yufune S, Satoh Y, Akai R, Yoshinaga Y, Kobayashi Y, Endo S & Kazama T (2016). Suppression of ERK phosphorylation through oxidative stress is involved in the mechanism underlying sevoflurane‐induced toxicity in the developing brain. Sci Rep 6, 21859. [DOI] [PMC free article] [PubMed] [Google Scholar]