

Abstract
Key points
Low weight at birth increases the risk of developing chronic diseases in adulthood
A diet that is high in salt is known to elevate blood pressure, which is a major risk factor for cardiovascular and kidney diseases
The present study demonstrates that growth restricted male rats have a heightened sensitivity to high dietary salt, in the context of raised systolic blood pressure, reduced urinary sodium excretion and stiffer mesenteric resistance vessels
Other salt‐induced effects, such as kidney hyperfiltration, albuminuria and glomerular damage, were not exacerbated by being born small
The present study demonstrates that male offspring born small have an increased cardiovascular susceptibility to high dietary salt, such that that minimizing salt intake is probably of particular benefit to this at‐risk population
Abstract
Intrauterine growth restriction increases the risk of developing chronic diseases in adulthood. Lifestyle factors, such as poor dietary choices, may elevate this risk. We determined whether being born small increases the sensitivity to a dietary salt challenge, in the context of hypertension, kidney disease and arterial stiffness. Bilateral uterine vessel ligation or sham surgery (offspring termed Restricted and Control, respectively) was performed on 18‐day pregnant Wistar Kyoto rats. Male offspring were allocated to receive a diet high in salt (8% sodium chloride) or remain on standard rat chow (0.52% sodium chloride) from 20 to 26 weeks of age for 6 weeks. Systolic blood pressure (tail‐cuff), renal function (24 h urine excretions) and vascular stiffness (pressure myography) were assessed. Restricted males were born 15% lighter than Controls and remained smaller throughout the study. Salt‐induced hypertension was exacerbated in Restricted offspring, reaching a peak systolic pressure of ∼175 mmHg earlier than normal weight counterparts. The natriuretic response to high dietary salt in Restricted animals was less than in Controls and may explain the early rise in arterial pressure. Growth restricted males allocated to a high salt diet also had increased passive arterial stiffness of mesenteric resistance arteries. Other aspects of renal function, including salt‐induced hyperfiltration, albuminuria and glomerular damage, were not exacerbated by uteroplacental insufficiency. The present study demonstrates that male offspring exposed to uteroplacental insufficiency and born small have an increased sensitivity to salt‐induced hypertension and arterial remodelling.
Keywords: developmental programming, growth restriction, blood pressure
Key points
Low weight at birth increases the risk of developing chronic diseases in adulthood
A diet that is high in salt is known to elevate blood pressure, which is a major risk factor for cardiovascular and kidney diseases
The present study demonstrates that growth restricted male rats have a heightened sensitivity to high dietary salt, in the context of raised systolic blood pressure, reduced urinary sodium excretion and stiffer mesenteric resistance vessels
Other salt‐induced effects, such as kidney hyperfiltration, albuminuria and glomerular damage, were not exacerbated by being born small
The present study demonstrates that male offspring born small have an increased cardiovascular susceptibility to high dietary salt, such that that minimizing salt intake is probably of particular benefit to this at‐risk population
Introduction
Hypertension affects 30% of the adult population and is a major risk factor for cardiovascular and kidney diseases (Mills et al. 2016). Studies in both humans and laboratory animals have provided clear evidence that high dietary salt contributes to elevated arterial pressure and that a reduction in salt intake is associated with low average pressures (He & MacGregor, 2003; Appel et al. 2006; Elliott et al. 2007). Blood pressure responses to dietary salt, however, are heterogeneous, with some individuals having greater sensitivity than others. In individuals with hypertension, 30–50% are considered to have salt‐sensitive blood pressure, which is associated with a range of risk factors, including African American ethnicity, older age, obesity and diabetes (Weinberger et al. 1986; Kotchen et al. 2013).
Many animal studies have reported a genetic susceptibility to salt sensitivity, particularly in the context of raised blood pressure. The Dahl rat is the most studied model in the field, in which salt‐induced hypertension can be attenuated and albuminuria prevented upon replacement with specific genetic material from a normotensive rat (Mattson et al. 2008). Furthermore, transgenic mice with various gene deletions, including atrial natriuretic peptide and its receptor, have increased salt‐sensitive blood pressure (John et al. 1995). Increased sensitivity to dietary salt, however, may occur irrespective of the genetic background. In the Dahl rat, the impact of maternal diet on salt‐sensitive hypertension and kidney injury was recently assessed (Geurts et al. 2015). Rats that had been embryo‐transferred from casein‐fed donors to grain‐fed surrogates had attenuated salt‐induced hypertension and kidney damage. The converse was true for rats conceived by grain‐fed parents and transferred to a casein‐fed surrogate. These findings implicate the gestational environment in determining salt sensitivity in adulthood.
Low weight at birth is a surrogate of developmental perturbations and, in such cases, organ structure and function are compromised, including that of the heart, blood vessels, kidneys and pancreas, and the risk of developing chronic diseases increases (Barker et al. 1989, 1990; White et al. 2009; Clough, 2015). Uteroplacental insufficiency is the most common cause of fetal growth restriction in Western nations and affects around one in ten pregnancies (Henriksen & Clausen, 2002). The delivery of nutrients and oxygen to the fetus is compromised because of poor placental function. An established rat model has been used to mimic uteroplacental insufficiency, in which the uterine vessels are bilaterally ligated during late gestation (Simmons et al. 2001; Schreuder et al. 2007; Wlodek et al. 2007). This surgical procedure induces a 10–15% reduction in offspring birth weight along with life‐long deficits in nephron and cardiomyocyte endowment, glomerular hypertrophy, and mesenteric and femoral arterial dysfunction in males (Wlodek et al. 2008; Black et al. 2012; Tare et al. 2012). These can (Wlodek et al. 2008), although not always (Wadley et al. 2016), result in arterial hypertension at 6 months of age. In general, growth restricted males present with more prominent physiological changes but similar structural deficits compared to female counterparts.
We investigated the effects of a short‐term dietary salt challenge on blood pressure, arterial stiffness and kidney function in adult male rats that had been exposed to uteroplacental insufficiency during gestation. We hypothesized that low birth weight rats would have an increased sensitivity to high dietary salt, such that arterial blood pressure, vascular stiffness and renal function would be more adversely affected compared to male rats born of a normal weight.
Methods
Animal procedures
All experiments were approved by The University of Melbourne Animal Ethics Committee prior to commencement. The experiments were carried out in accordance with the Australian Code for the care and use of animals for scientific purposes set out by the National Health and Medical Research Council (2013) and conform to the principles and regulations described by Grundy (2015). Wistar Kyoto (WKY) rats, housed in an environmentally controlled room, had access to food and tap water ad libitum. Rats were mated and uteroplacental insufficiency (offspring termed Restricted) or sham (offspring termed Control) surgery performed at embryonic day (E) 18, as described previously, under anaesthesia (i.p. injection of a mixed solution containing ketamine (50 mg kg−1 body weight) and ilium xylazil‐20 (10 mg kg−1 body weight)) (n = 8–11 per group) (Wlodek et al. 2007). Rats delivered at term (E22). At postnatal day 35 (P35), offspring were weaned and provided with ad libitum access to standard rat chow and water. At 20 weeks of age, male offspring were allocated to one of two diets (Specialty Feeds, Glenn Forrest, WA, Australia): High salt (SF01‐004, 8% sodium chloride) or Normal salt (SF01‐002, 0.52% sodium chloride) with access to water ad libitum (one male offspring per litter per diet, n = 12 per group). Treatment duration was for 6 weeks and rats were killed at 26 weeks of age. Male offspring were used because we have previously reported their increased susceptibility to the developmental programming of high blood pressure (Wlodek et al. 2008).
Blood pressure and blood glucose monitoring
Blood pressure was measured by non‐invasive tail‐cuff plethysmography (ADInstruments Pty Ltd, Castle Hill, NSW, Australia) in rats that were acclimatized to the restraint procedure at 8, 12, 17, 23 and 26 weeks of age (Wlodek et al. 2007; Moritz et al. 2009; Gallo et al. 2012a, b). The final five of 10 acquired traces were recorded and averaged to determine systolic blood pressure. Blood glucose (non‐fasted; tail vein sample) was monitored at 8, 12, 17, 23 and 25 weeks of age using a glucometer (Accu‐Chek Performa, Roche, Mannheim, Germany) during morning daylight hours.
Food and water intake, urinary excretions and biochemistry
Rats were weighed and placed individually into metabolic cages for 24 h measurements of food and water intake and urine production at 8 weeks (pre‐treatment) and 26 weeks (after 6 weeks of normal or high salt diet) of age (Moritz et al. 2009; Gallo et al. 2012a, b). Rats were acclimatized to metabolic cages by placing them in for short daylight periods of 3 h and 8 h on two separate occasions to minimize stress and associated behavioural changes. Measurements of urinary albumin, total protein, sodium, potassium and creatinine were made using commercially available kits in accordance with the manufacturer's instructions (Cobas Integra 400; Roche Diagnostics, Burgess Hill, UK). Plasma samples collected immediately upon removal from metabolic cages, which was at post mortem, were analysed for sodium, potassium, chloride and creatinine. Creatinine clearance (mL min−1) was calculated using: [urinary creatinine (μmol l−1) × 24 h urine production (mL)]/[plasma creatinine (μmol l−1) × 1440 (min)]; predicted sodium filtered (mmol 24 h−1) was calculated using: plasma sodium (mmol l−1) × creatinine clearance (l 24 h−1); and fractional sodium excretion (%) was calculated using: [urinary sodium (mmol l−1) × plasma creatinine (mmol l−1)]/[plasma sodium (mmol l−1) × urinary creatinine (mmol l−1)] × 100.
Post mortem tissue collection
At post mortem, at 26 weeks of age, rats were anaesthetized with an i.p. injection of a mixed solution containing ketamine (100 mg kg−1 body weight) and ilium xylazil‐20 (30 mg kg−1 body weight). Heart, kidneys, liver, pancreas and dorsal fat were excised and weighed. Kidneys were immersion fixed in 10% neutral buffered formalin and processed to paraffin for histological analyses. Small renal and mesenteric arteries were isolated and used for functional studies.
Arterial stiffness
Passive mechanical wall properties were assessed in mesenteric (350–380 μm) and renal (440–460 μm) resistance arteries ∼3‐4 mm long mounted on a pressure myograph (Living Systems Instrumentation, St Albans City, VT, USA), as described previously (Mazzuca et al. 2010; Mazzuca et al. 2012). Briefly, arteries were continuously superfused at ∼15 mL.min−1 with Ca2+‐free, 1 mm EGTA physiological saline solution containing (in mm): 120 NaCl, 5 KCl, 25 NaHCO3, 11 glucose, 1.2 MgSO4 and 1 KH2PO4, gassed with 95% O2 and 5% CO2 at ∼36°C. Each artery was pressurized from 0 to 110 mmHg. Arterial dimensions (length, outer diameter (OD), internal diameter (ID) and wall thickness (WT)) were measured at each 10 mmHg increment in intraluminal pressure. Wall stress and strain were derived as: wall stress (kPa) = (intraluminal pressure × ID)/(2 × WT); wall strain = (ID – ID extrapolated to 5 mmHg pressure) (ID extrapolated to 5 mmHg pressure) (Wigg et al. 2001; Mazzuca et al. 2010). The media‐to‐lumen ratio was calculated as WT/ID.
Kidney morphometry
Representative 5 μm transverse, midline sections from the kidney were collected. In sections stained with Masson's trichrome, perivascular fibrosis and interstitial fibrosis were assessed (Walton et al. 2017). For perivascular fibrosis, nine arterioles per animal were randomly selected from three separate, non‐sequential slides, and the area of adventitial collagen was normalized to vessel lumen area and averaged for each animal. Interstitial fibrosis was quantified in five random cortical/outer medullary fields per animal using a point‐counting technique. In each field, 121 points were counted with 11 equidistant grid lines. Points falling on interstitial fibrosis were expressed as a percentage of the total number of grid points and averaged for each animal. In sections stained with periodic acid‐Schiff (PAS), 20 glomeruli were randomly selected to evaluate glomerulosclerosis using a semi‐quantitative method (Gallo et al. 2016).
Statistical analysis
The results were analysed with Prism, version 6 and above (GraphPad Software, La Jolla, CA, USA). Data were tested for normality and a two‐way ANOVA was performed to determine main effects of diet (normal salt 0.52% and high salt 8%) and uteroplacental insufficiency (Control and Restricted). When significant interactions were observed, individual group means were compared using the Fisher's least significant difference (LSD) test. Stress–strain relationships were analysed using repeated measures two‐way ANOVA followed by Tukey's post hoc multiple comparison testing. P < 0.05 was considered statistically significant and n represents the number of animals per group from different litters.
Results
Uteroplacental insufficiency did not impact salt‐induced alterations in body and organ weights
Uteroplacental insufficiency reduced total litter size (Restricted: 6 ± 1 vs. Control: 10 ± 1, P < 0.05) and litter average male body weight (−15%, Restricted: 3.62 ± 0.12 vs. Control: 4.24 ± 0.09, P < 0.05) compared to Controls at P1. Restricted males remained ∼12 % lighter than Controls throughout the study (Table 1). A diet high in salt (8% sodium chloride) reduced body weight during weeks 3, 5 and 6 of treatment, such that total weight gained over the 6 week treatment period was only 13–15 g in high salt fed rats vs. 34–41 g in normal salt (0.52% sodium chloride) fed rats (Table 1). This reduction in weight gain was not exacerbated by uteroplacental insufficiency (Table 1).
Table 1.
Body weight, food and energy intake and organ weights
| Normal salt (0.52%) | High salt (8%) | Two‐way ANOVA | ||||
|---|---|---|---|---|---|---|
| Control | Restricted | Control | Restricted | Diet | Restricted | |
| Body weight (g) | ||||||
| Before high salt diet – age | ||||||
| P6 | 9.1 ± 0.3 | 7.5 ± 0.5 | 8.5 ± 0.4 | 7.1 ± 0.3 | NS | P < 0.05 |
| P14 | 22.3 ± 0.5 | 18.9 ± 1.1 | 20.9 ± 0.7 | 18.1 ± 1.6 | NS | P < 0.05 |
| P35 | 82 ± 2 | 72 ± 3 | 80 ± 2 | 72 ± 4 | NS | P < 0.05 |
| 8 weeks | 162 ± 4 | 135 ± 8 | 165 ± 5 | 133 ± 7 | NS | P < 0.05 |
| 12 weeks | 293 ± 6 | 261 ± 9 | 288 ± 4 | 251 ± 9 | NS | P < 0.05 |
| 16 weeks | 358 ± 6 | 323 ± 9 | 352 ± 4 | 305 ± 12 | NS | P < 0.05 |
| 20 weeks | 365 ± 7 | 336 ± 10 | 365 ± 5 | 314 ± 13 | NS | P < 0.05 |
| During high salt diet – age | ||||||
| 23 weeks | 388 ± 5 | 348 ± 9 | 365 ± 6 | 318 ± 14 | P < 0.05 | P < 0.05 |
| 25 weeks | 399 ± 5 | 368 ± 10 | 377 ± 6 | 325 ± 13 | P < 0.05 | P < 0.05 |
| 26 weeks (i.e. study end) | 405 ± 7 | 370 ± 12 | 380 ± 6 | 327 ± 13 | P < 0.05 | P < 0.05 |
| Total weight gained | 40.6 ± 2.8 | 34.0 ± 4.4 | 15.1 ± 2.5 | 13.1 ± 2.8 | P < 0.05 | NS |
| Food intake (g 24 h−1) | ||||||
| Before high salt diet (i.e. 8 weeks of age) | 19.9 ± 0.9 | 16.4 ± 0.9 | 19.4 ± 1.0 | 17.2 ± 1.6 | NS | P < 0.05 |
| At 6 weeks of high salt diet (i.e. 26 weeks of age) | 17.6 ± 1.3 | 19.1 ± 1.9 | 19.2 ± 1.0 | 17.4 ± 1.2 | NS | NS |
| Energy intake (kJ 24h−1) | ||||||
| At 6 weeks of high salt diet (i.e. 26 weeks of age) | 246 ± 18 | 268 ± 26 | 248 ± 12 | 224 ± 15 | NS | NS |
| Salt intake (g 24h−1) | ||||||
| At 6 weeks of high salt diet (i.e. 26 weeks of age) | 0.092 ± 0.007 | 0.100 ± 0.010 | 1.537 ± 0.077 | 1.388 ± 0.092 | P < 0.05 | NS |
| Organ weight (mg mm leg length−1) | ||||||
| At 6 weeks of high salt diet (i.e. 26 weeks of age) | ||||||
| Heart | 26.2 ± 0.4 | 24.2 ± 0.6 | 28.1 ± 0.7 | 25.7 ± 1.0 | P < 0.05 | P < 0.05 |
| Left ventricle | 18.8 ± 0.6 | 18.4 ± 0.4 | 20.3 ± 0.9 | 19.3 ± 0.9 | NS | NS |
| Left kidney | 25.4 ± 1.0 | 23.2 ± 0.6 | 28.5 ± 0.7 | 25.2 ± 1.2 | P < 0.05 | P < 0.05 |
| Liver | 222 ± 6 | 204 ± 5 | 207 ± 5 | 192 ± 7 | P < 0.05 | P < 0.05 |
| Pancreas | 12.5 ± 0.9 | 13.2 ± 0.9 | 13.9 ± 1.3 | 12.7 ± 1.3 | NS | NS |
| Dorsal fat | 87.3 ± 11.2 | 64.0 ± 6.2 | 57.6 ± 4.3 | 40.3 ± 6.9 | P < 0.05 | P < 0.05 |
| Leg length (mm) | 53.2 ± 0.4 | 51.4 ± 0.5 | 53.0 ± 0.5 | 50.6 ± 0.7 | NS | P < 0.05 |
Control and Restricted rats were randomized to normal salt or high salt diet for 6 weeks at 20–26 weeks of age. No significant interactions between diet and uteroplacental insufficiency surgery by two‐way ANOVA; n = 7–11 per group. NS, not significant.
At 8 weeks of age, Restricted offspring consumed less food, although this did not persist at 26 weeks of age (Table 1). High dietary salt did not affect the amount of food or energy consumed over a single 24 h period (Table 1) but, over the 6 weeks, total energy intake was less in rats allocated to high vs. normal salt (P < 0.01; high salt Control: 12.17 ± 0.23, high salt Restricted: 12.13 ± 0.67 vs. normal salt Control: 14.39 ± 0.74, normal salt Restricted: 14.40 ± 0.77 MJ/6 weeks). The amount of sodium chloride consumed per day was ∼15‐fold greater in rats allocated to high salt vs. normal salt (P < 0.0001) and not different between Control and Restricted rats (Table 1). The same fold difference was observed for sodium consumption independent of chloride (P < 0.0001; high salt Control: 0.596 ± 0.030, high salt Restricted: 0.538 ± 0.036 vs. normal salt Control: 0.035 ± 0.003, normal salt Restricted: 0.038 ± 0.004 g/24 h).
A high salt diet increased relative heart and kidney weights, although liver and dorsal fat weights were lower at post mortem (Table 1). These changes were not exacerbated by uteroplacental insufficiency (Table 1). Uteroplacental insufficiency, however, independently reduced heart, kidney, liver and dorsal fat weights compared to Control (Table 1). Left ventricular and pancreas weights were not affected by salt intervention or uteroplacental insufficiency (Table 1). The reported organ weights were corrected for leg length which was ∼2 mm shorter in Restricted males (Table 1).
Uteroplacental insufficiency hastened the development of salt‐induced hypertension
Systolic blood pressure was not different between groups at diet baseline (i.e. at 20 weeks of age) (Fig. 1 A and B). Compared to baseline, rats allocated to high dietary salt developed elevated blood pressure at 3 weeks into the diet, which was exacerbated in Restricted offspring (Restricted: +40 mmHg and Control: +27 mmHg vs. baseline) (Fig. 1 B). Between 3 and 6 weeks of high salt feeding, systolic blood pressure continued to rise in Control rats but plateaued in Restricted (Fig. 1 B). Compared to normal salt, 3 weeks of a high salt diet increased systolic blood pressure by 27 mmHg in Restricted and 12 mmHg in Control (Fig. 1 C). Following 6 weeks of high salt feeding (i.e. study end), blood pressure was elevated by a similar degree regardless of birth weight (high salt Control: 176 ± 4 mmHg, high salt Restricted: 171 ± 4 mmHg vs. normal salt Control: 144 ± 4 mmHg, normal salt Restricted: 148 ± 3 mmHg) (Fig. 1 D). Over the 6 week treatment period (i.e. 20–26 weeks of age), all rats that remained on normal chow (both Control and Restricted) did not display changes in blood pressure, highlighting no effect of ageing (Fig. 1 A).
Figure 1. Systolic blood pressure during 6 weeks of a high salt diet.
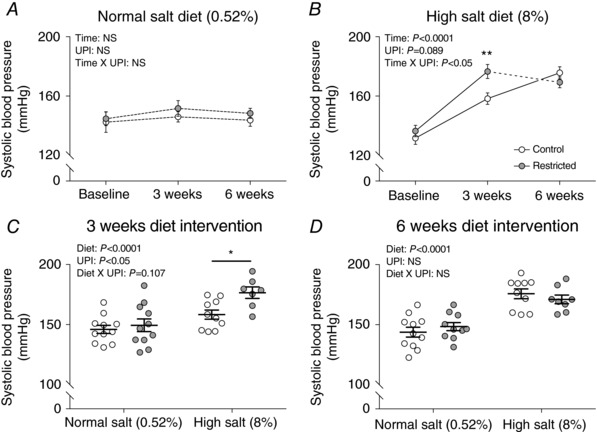
Systolic blood pressure in (A) normal salt diet (0.52%) and (B) high salt diet (8%) groups at baseline (20 weeks of age), 3 weeks (23 weeks of age) and 6 weeks (26 weeks of age) of diet intervention and (C) all groups at 3 weeks (23 weeks of age) and (D) 6 weeks (26 weeks of age) of diet intervention. Data in (A) and (B) are the mean ± SEM; n = 7–11 per group or individual values (C and D) with the mean ± SEM. * P < 0.05, ** P < 0.01 Restricted vs. Control within diet group by Fisher's LSD test. A and B, continuous lines indicate P < 0.05 between connecting time‐points and dotted lines indicate no significance between connecting time‐points. UPI, uteroplacental insufficiency; NS, not significant.
Uteroplacental insufficiency reduced the natriuretic response to high dietary salt
The dietary salt challenge from 20–26 weeks of age induced a 4‐ to 4.5‐fold increase in urine output, which was partially balanced by increased water consumption (+2.9‐fold in Control and +2.1‐fold in Restricted) (Fig. 2 A and B). A positive correlation between urinary output and water intake was observed for Control (r 2 = 0.529) but not Restricted (r 2 = 0.028) offspring on the high salt diet (Fig. 2 A). The 24 h thirst response to high dietary salt (vs. normal salt) was significantly less in Restricted rats (i.e. ∼35 mL of extra water vs. ∼58 mL in Control) (Fig. 2 A). Over the 6 week treatment period, however, there was no statistical difference in the amount of water consumed between Control and Restricted rats and > 2.5 L of extra water was consumed in rats allocated to high vs. normal salt diet (P < 0.05, L/6 weeks; high salt Control: 4.14 ± 0.12, high salt Restricted: 3.96 ± 0.17 vs. normal Control: 1.43 ± 0.07, normal salt Restricted: 1.55 ± 0.09).
Figure 2. Fluid and electrolyte balance following 6 weeks of a high salt diet.
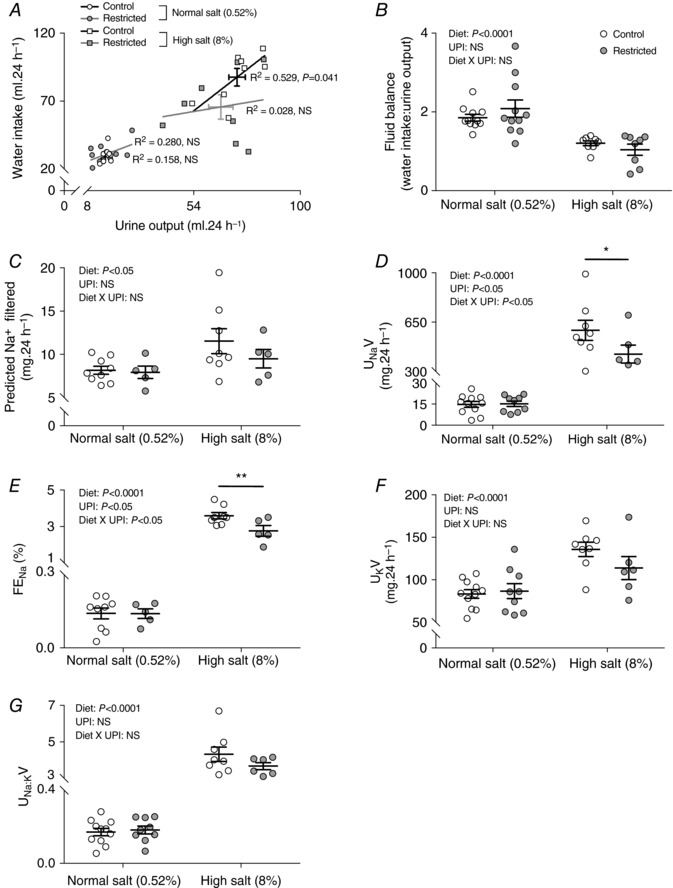
Water intake in relation to urinary ouput (A and B), predicted filtered sodium (C), urinary sodium excretion (UNaV) (D), fractional sodium excretion (FENa) (E), urinary potassium excretion (UKV) (F) and sodium to potassium ratio (UNa:KV) (G) following 6 weeks of diet intervention (26 weeks of age). Data are individual values with the mean ± SEM. Pearson's correlation performed for (A). * P < 0.05, ** P < 0.01 Restricted vs. Control within diet group by Fisher's LSD test. UPI, uteroplacental insufficiency; NS, not significant.
As expected, high dietary salt for 6 weeks increased the filtered load and urinary excretion (total and fractional) of sodium, regardless of birth weight (Fig. 2 C–E). However, in Restricted offspring, total and fractional sodium excretion increased by a lesser magnitude than in Controls (Fig. 2 D and E), suggesting enhanced sodium reabsorption. Urinary excretions of potassium and sodium to potassium ratio were increased in response to high salt feeding and there were no differences between Control and Restricted (Fig. 2 F and G). Plasma concentrations of sodium, potassium, and chloride were not different between Control and Restricted, nor were they affected by high salt consumption (Table 2). Non‐fasted blood glucose levels were also not different between groups throughout the study (Table 2).
Table 2.
Blood and plasma biochemistry
| Normal salt (0.52%) | High salt (8%) | Two‐way ANOVA | ||||
|---|---|---|---|---|---|---|
| Control | Restricted | Control | Restricted | Diet | Restricted | |
| Non‐fasted blood glucose (mmol L−1) | ||||||
| Before high salt diet – age | ||||||
| 8 weeks | 9.2 ± 0.5 | 9.3 ± 0.3 | 8.7 ± 0.3 | 9.0 ± 0.3 | NS | NS |
| 12 weeks | 9.8 ± 0.5 | 10.1 ± 0.6 | 9.6 ± 0.3 | 9.9 ± 0.6 | NS | NS |
| 16 weeks | 9.7 ± 0.3 | 10.3 ± 0.5 | 9.8 ± 0.4 | 8.8 ± 0.4 | NS | NS |
| During high salt diet – age | ||||||
| 23 weeks | 11.1 ± 0.8 | 10.4 ± 0.3 | 9.7 ± 0.4 | 9.8 ± 0.4 | NS | NS |
| 25 weeks | 10.6 ± 0.8 | 11.1 ± 0.3 | 10.6 ± 0.8 | 10.2 ± 0.6 | NS | NS |
| Plasma electrolytes (mmol L−1) | ||||||
| At 6 weeks of high salt diet (i.e. 26 weeks of age) | ||||||
| Na+ | 141.6 ± 3.9 | 138.2 ± 1.5 | 143.6 ± 4.3 | 140.8 ± 1.4 | NS | NS |
| K+ | 6.2 ± 0.3 | 5.9 ± 0.2 | 5.7 ± 0.3 | 5.7 ± 0.2 | NS | NS |
| Cl− | 114.7 ± 2.9 | 112.1 ± 0.7 | 115.7 ± 2.8 | 113.9 ± 1.2 | NS | NS |
Control and Restricted rats were randomized to normal salt or high salt diet for 6 weeks at 20–26 weeks of age. No significant interactions between diet and uteroplacental insufficiency surgery by two‐way ANOVA; n = 6–11 per group. NS, not significant.
Uteroplacental insufficiency modestly attenuated high salt‐induced albuminuria
Creatinine clearance, a surrogate of glomerular filtration rate, was increased by 20–38% following 6 weeks of high dietary salt, although there was no effect of uteroplacental insufficiency (Fig. 3 A). High salt also induced albuminuria and proteinuria in all rats, albeit the degree of albuminuria was ∼30% less in Restricted vs. Control (Fig. 3 B–D).
Figure 3. Kidney function and histopathology following 6 weeks of a high salt diet.
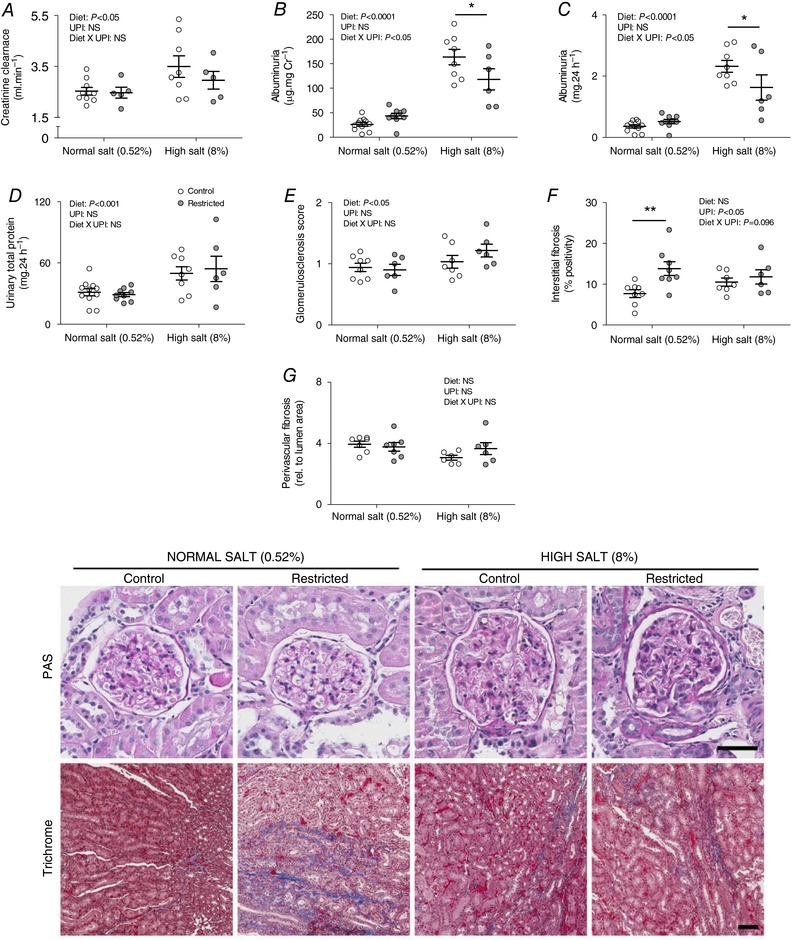
Creatinine clearance (A), albuminuria (B and C), proteinuria (D), glomerulosclerosis (E), tubulointerstitial fibrosis (F) and perivascular fibrosis (G) following 6 weeks of diet intervention (26 weeks of age). Data are individual values with the mean ± SEM. * P < 0.05, ** P < 0.01 Restricted vs. Control within diet group by Fisher's LSD test. UPI, uteroplacental insufficiency; NS, not significant. Representative kidney sections stained with PAS and Masson's trichrome. Scale bars = 50 μm.
High dietary salt and uteroplacental insufficiency induced mild glomerular and tubulointerstitial fibrosis, respectively
Rats allocated to the high salt diet had increased glomerulosclerosis by study end (i.e. 26 weeks of age), regardless of birth weight (Fig. 3 E). Tubulointerstitial and perivascular fibrosis, however, were not affected by the salt challenge (Fig. 3 F and G). Restricted rats had increased tubulointerstitial, but not perivascular, fibrosis compared to Controls (Fig. 3 F and G). Representative sections of kidneys stained with Masson's trichrome and PAS are shown in Fig. 3.
High dietary salt increased mesenteric arterial wall stiffness in Restricted but not Control offspring
For Restricted vs. Control rats on the normal salt diet, there was no significant difference in the passive stress–strain relationship (indicative of arterial stiffness) in mesenteric arteries at study end (i.e. 26 weeks of age) (Fig. 4 A). The mesenteric artery media‐to‐lumen ratio was also not different between Control and Restricted groups (Fig. 4 B and Table 3).
Figure 4. Passive mechanical wall properties of mesenteric and renal arteries following 6 weeks of a high salt diet.
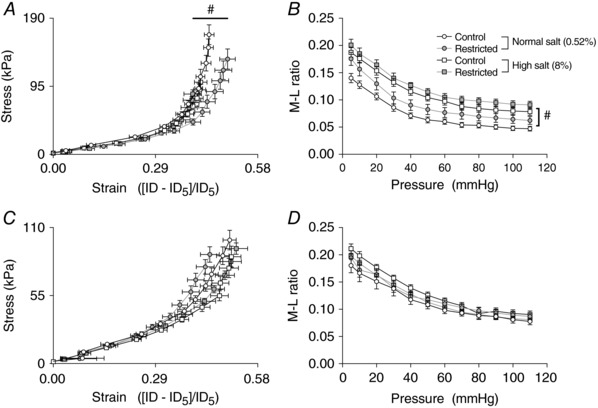
Stress–strain relationships (A and C) and media‐to‐lumen ratio (B and D) for mesenteric (top) and renal (bottom) arteries following 6 weeks of diet intervention (26 weeks of age). Data are the mean ± SEM; n = 10–11 per group. #P < 0.05 High vs. Normal salt within uteroplacental insufficiency group by Tukey's post hoc test.
Table 3.
Passive wall properties in arteries pressurized to 100 mmHg following 6 weeks of a high salt diet
| Normal salt (0.52%) | High salt (8%) | Two‐way ANOVA | ||||
|---|---|---|---|---|---|---|
| Control | Restricted | Control | Restricted | Diet | Restricted | |
| Mesenteric artery | ||||||
| OD (μm) | 462 ± 18 | 438 ± 14 | 457 ± 10 | 427 ± 15 | NS | P = 0.0699 |
| ID (μm) | 422 ± 17 | 389 ± 14 | 395 ± 11 | 362 ± 15 | P = 0.0730 | P < 0.05 |
| WT (μm) | 19.9 ± 1.5 | 24.5 ± 2.4 | 30.9 ± 2.1 | 32.7 ± 1.8 | P < 0.0001 | NS |
| M:L ratio | 0.048 ± 0.004 | 0.065 ± 0.008 | 0.080 ± 0.007 | 0.092 ± 0.007 | P < 0.0001 | P < 0.05 |
| Renal artery | ||||||
| OD (μm) | 575 ± 27 | 540 ± 17 | 552 ± 21 | 573 ± 14 | NS | NS |
| ID (μm) | 496 ± 27 | 459 ± 17 | 466 ± 18 | 492 ± 14 | NS | NS |
| WT (μm) | 39.0 ± 1.9 | 40.5 ± 2.2 | 42.7 ± 3.0 | 40.3 ± 1.6 | NS | NS |
| M:L ratio | 0.081 ± 0.006 | 0.089 ± 0.005 | 0.092 ± 0.007 | 0.083 ± 0.004 | NS | NS |
Control and Restricted rats were randomized to normal salt or high salt diet for 6 weeks at 20–26 weeks of age. No significant interactions between diet and uteroplacental insufficiency surgery by two‐way ANOVA; n = 10–11 per group. NS, not significant; OD, outside diameter; ID, internal diameter; WT, wall thickness; M:L, media‐to‐lumen.
High salt diet was without effect on the stress–strain relationship in mesenteric arteries for Control rats (Fig. 4 A). However, in Restricted rats, the stress–strain curve was shifted to the left, reflecting significantly increased arterial wall stiffness in response to high dietary salt (Fig. 4 A). The media‐to‐lumen ratio of mesenteric arteries from both Control and Restricted rats, however, was significantly increased after 6 weeks of high dietary salt (Fig. 4 B). This salt‐induced change in media‐to‐lumen ratio in mesenteric arteries of Control and Restricted rats was underpinned by a significant increase in wall thickness and a tendency (P = 0.073) for a reduced internal diameter (Table 3).
For renal arteries, there was no effect of uteroplacental insufficiency or high salt diet on the passive mechanical wall properties or arterial stiffness (Fig. 4 C and D and Table 3).
Discussion
Cardiovascular disease is the major cause of death globally and hypertension is considered as a leading preventable risk factor. High dietary salt plays a prominent role in the development of hypertension, particularly in a subset of individuals (Weinberger et al. 1986; Kotchen et al. 2013). Being born small increases the risk of a range of chronic diseases in adulthood and, in general, males are considered to be more at risk than female counterparts. In the present study, we investigated the impact of high dietary salt on adult male rats born small as a result of uteroplacental insufficiency. Salt‐induced hypertension developed earlier in Restricted offspring than in normal weight counterparts and the natriuretic response to high dietary salt was significantly less, which may explain their early rise in arterial pressure. Restricted rats were also more salt‐sensitive in terms of passive arterial wall properties, exhibiting increased arterial stiffness in mesenteric resistance vessels. Other aspects of renal function, however, including salt‐induced hyperfiltration, albuminuria and glomerular damage, were not exacerbated by uteroplacental insufficiency. These findings suggest that males born small as a result of poor development in utero are more susceptible to lifestyle challenges that lead to adverse cardio‐renal health outcomes. Although female offspring were not examined in the present study, future work should ascertain whether a dietary salt challenge unmasks physiological deficits or, indeed, what renders them protected from the long‐term effects of uteroplacental insufficiency.
Our growth restricted males exhibited a greater increase in blood pressure at 3 weeks into the salt challenge, suggesting that they were more salt sensitive than normal weight controls. By 6 weeks, Controls had achieved the same blood pressure level. A chronic salt challenge, even in healthy individuals, will ultimately increase blood pressure (Ha, 2014). The earlier rise in systolic pressure in growth restricted offspring, however, prolongs the exposure time to the negative effects of arterial hypertension which promotes end‐organ damage and cardiovascular disease.
Several mechanisms have been proposed to explain the effects of high dietary salt on blood pressure. Ultimately, a ‘natriuretic handicap’, comprising the limited capacity of the kidneys to excrete sodium, is the common denominator (Kotchen et al. 2013). Indeed, our Restricted offspring displayed an earlier rise in arterial pressure compared to Controls, and this was associated with a ∼25–30% reduction in total and fractional sodium excretion. Impaired natriuresis may occur as a result of an inherent problem with the kidneys and/or factors that increase tubular reabsorption of sodium. Previously, we have reported that Restricted males have a reduction in the number of nephrons (Wlodek et al. 2008) and this is assumed to exacerbate salt‐sensitivity in the context of raised blood pressure (Kotchen et al. 2013). In rat offspring with reduced nephron endowment as a result of a maternal low protein diet during pregnancy, blood pressure was elevated and this was exacerbated by high dietary salt (Woods et al. 2004). This was associated with reduced glomerular filtration rate (GFR), although this was only modest compared to the reduction in nephron number. In the present study, high salt‐induced creatinine clearance and urine production increased equally in Restricted and Control rats, suggesting that whole‐kidney GFR was sufficient despite a known nephron loss (Wlodek et al. 2008).
Hyperfiltration of existing nephrons is considered to contribute to progressive kidney disease (Brenner, 1983). Restricted rats in the present study had increased tubulointerstitial fibrosis which, when superimposed on salt‐induced glomerulosclerosis and increased kidney mass, may have compromised tubular‐glomerular signalling and sodium excretion. In diabetes, proximal tubular growth is considered to contribute, in part, to increased sodium reabsorption and subsequent increases in single nephron GFR (Vallon, 2011). Indeed, in rat offspring prenatally exposed to dexamethasone, proximal tubule NHE3 activity and volume absorption were increased and assumed to contribute to prenatal programming of hypertension (Dagan et al. 2007). Increased expression of renal sodium channels has also been demonstrated in sheep offspring that have a low nephron endowment and develop high blood pressure after exposure to prenatal glucocorticoids (Moritz et al. 2011). In our model, a detailed analysis of renal tubule segment lengths and proximal tubule transport activity is required to determine the mechanisms underlying impaired salt‐induced natriuresis. It is worth noting that, although high dietary salt increased albuminuria, this was surprisingly less in Restricted vs. Control rats. The relevance of this finding requires further study but may have occurred as a consequence of the mild, non‐significant reduction in 24 h urinary output (Restricted: 66 mL vs. Control: 73 mL).
Excessive salt intake stimulates several endocrine, neural and paracrine mechanisms that target the kidneys and blood vessels to help achieve a net sodium balance. In response to high salt intake, endogenous cardiotonic steroids that bind and inhibit the Na+/K+‐ATPase, such as marinobufagenin, are secreted by the brain and adrenal glands (Bagrov et al. 2009). In normotensive women, 6 days of high salt intake increased marinobufagenin excretion that directly correlated with total‐ and fractional‐ sodium excretion and inversely correlated with systolic blood pressure (Anderson et al. 2008). A lack of marinobufagenin‐induced inhibition of Na+/K+‐ATPase in the kidneys and/or blood vessels may therefore render an individual as salt sensitive (Kotchen et al. 2013). Increased activity of the sympathetic nervous system may also enhance sensitivity to dietary salt. Indeed, fetal activation of the sympathetic nervous system is considered to play a role in the developmental programming of sensitivity to cardiovascular stressors (Harris & Seckl, 2011) and we recently reported enhanced respiratory‐related thoracic sympathetic nerve activity in our growth restricted male offspring (Menuet et al. 2016). Experimental studies assessing the development of salt sensitive hypertension also implicate the renin angiotensin system (RAS), formation of reactive oxygen species and reduced production of nitric oxide, which are all similarly implicated in the developmental programming of cardiovascular disease (Alexander et al. 2015). Previous programming studies have reported an upregulation of renal RAS in association with hypertension (Vehaskari et al. 2004; Grigore et al. 2007; Singh et al. 2007; Walton et al. 2017), although this has not been observed in our model (Wlodek et al. 2008) and therefore probably does not contribute to enhanced salt‐sensitivity seen in this study.
The impact of the gestational environment vs. genetic polymorphisms on adulthood blood pressure has been demonstrated previously through embryo transfer approaches in the spontaneously hypertensive rat (SHR) and Dahl salt sensitive rat. Salt sensitive rat embryos transferred to salt resistant dams had blood pressures that matched salt resistant rats (Kubisch et al. 1998) and SHR rats exposed to a WKY uterine environment had a remarkable reduction in blood pressure (Lee & Azar, 2010). In these studies, susceptibility to postnatal dietary salt, however, was either not altered (Kubisch et al. 1998) or not assessed (Lee & Azar, 2010). In the SHR, we have previously shown that the maternal environment does not mediate salt preference (Di Nicolantonio et al. 2005) or blood pressure in adulthood (Di Nicolantonio et al. 2006) and, instead, may be genetically determined. Although embryo transfer, in theory, allows for the separation of genetic and environmental influences, it should be noted that this technique is fraught with inherent confounders that probably interact with the different experimental groups (Tran et al. 2014). This limits our interpretation of the results obtained in such studies. Nevertheless, by using a single rat strain and manipulating the gestational environment in a randomized manner, it has been demonstrated that suboptimal exposures during early development can impact blood pressure responses, independently of genetic inheritance.
The vasculature is sensitive to perturbations in the early‐life environment and adaptations made in this system can predispose to later cardiovascular disease (Clough, 2015). In the present study, passive stiffness of mesenteric and renal arteries was not different between Control and Restricted male offspring. This finding contrasts with earlier studies in male growth restricted offspring by our group (Tare et al. 2012) and most probably reflects differences in early‐life exposure to stress; that is, in our earlier study, male offspring were cross‐fostered to a different dam at birth. However, mesenteric resistance vessels from Restricted males exhibited increased stiffness following a high salt diet. Excess sodium intake leads to progressive deterioration of microvascular and renal function (Weinberger & Fineberg, 1991; Sacks et al. 2001; Yu et al. 2004), including increased vessel stiffness and media thickening, which is independent of age and blood pressure and is mediated, in part, through alterations in the RAS and bradykinin system (Safar et al. 2000). The wall thickness of mesenteric arteries of both Control and Restricted offspring was increased with the high salt diet, probably reflecting alterations in the deposition of extracellular matrix and proliferation and hypertrophy of smooth muscle cells, resulting in inward remodelling (Safar et al. 2000). Despite increased wall thickness in arteries from both Control and Restricted offspring, only the arteries from Restricted males exhibited increased stiffness following a high salt diet. These findings are similar to a previous study in the mouse showing that high salt‐induced mesenteric stiffness was exacerbated by prenatal hypoxia (Walton et al. 2016). Whether this reflects altered deposition or arrangement of extracellular matrix (Jalil et al. 1989; Arribas et al. 2008) requires further investigation. Interestingly, renal arteries from both Control and Restricted offspring were resistant to salt‐induced changes observed in the mesenteric arteries, probably reflecting differences in local regulatory mechanisms between the arteries.
Rats allocated to high salt consumed ∼15‐fold more sodium chloride (or sodium alone) than normal salt rats. This is greater than the extreme differences in sodium intake reported in humans (∼5‐fold), with most adult populations falling within a 2‐fold range (Brown et al. 2009). In the present study, an 8% high salt diet was used to determine whether hypertension was salt‐sensitive. Future studies should determine whether high dietary salt that is more consistent with the range reported in humans yields similar effects.
Finally, we have demonstrated that multiple organ systems controlling blood pressure, namely, the kidneys and microvasculature, are affected differentially by uteroplacental insufficiency, a postnatal high salt diet or a combination of the two. Dietary sodium intake for 6 weeks led to hypertension and signs of glomerular injury in all offspring, irrespective of growth restriction. Growth restricted males, however, exhibited mild renal fibrosis and, when challenged with a high salt diet, developed an earlier peak in arterial pressure, a reduced natriuretic response and stiffening of the mesenteric microvasculature. These findings are pertinent given that most adults consume, on average, five to ten times the daily amount of salt that is physiologically required. The identification of individuals at cardiovascular risk following even short‐term salt loading may warrant targeted dietary recommendations based on birth weight. Furthermore, the long‐term effects of these salt‐induced changes and the effects of chronic high salt intake from childhood are worthy of follow‐up.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
Animal experiments were performed in the Department of Physiology, The University of Melbourne and histological experiments were performed in the School of Biomedical Sciences, The University of Queensland. KMM, MEW and HCP conceived the experiments and obtained funding. LAG, SLW, MQM and MT conducted the experimental work. LAG drafted the manuscript and all authors revised it critically for important intellectual content. All authors approved the final version of the manuscript submitted for publication, agree to be accountable for all aspects of the work, and qualify for authorship.
Funding
This work was funded by the National Health and Medical Research Council and Heart Foundation (Australia). LAG was supported by an Early Career Fellowship from the National Health and Medical Research Council and Heart Foundation (Australia). MQM was supported by a Kidney Health Australia Biomedical Scholarship and The University of Melbourne Fee Remission Scholarship. KMM was supported by a Senior Research Fellowship from the National Health and Medical Research Council (Australia).
Acknowledgements
The authors acknowledge the technical assistance of Kerryn T. Westcott and Andrew J. Jefferies.
Biographies
Linda Gallo completed her PhD at The University of Melbourne in Australia. Her research focused on developmental programming of hypertension and kidney disease associated with uteroplacental insufficiency. She subsequently undertook postdoctoral training at Mater Research in Brisbane with a primary focus on novel anti‐diabetic therapies for diabetic nephropathy. Her current research priorities are to identify therapeutic and/or dietary interventions against heart and kidney complications of diabetes mellitus and to mitigate the transgenerational effects of gestational diabetes mellitus.

Sarah Walton studied biomedical science before obtaining a PhD with Professor Karen Moritz, Professor Melissa Little and Doctor Joan Li at The University of Queensland in Australia. She is presently a postdoctoral research fellow at Monash University. Her research focuses on understanding the long‐term health outcomes of infants following pregnancy complications, particularly with regard to prenatal hypoxia and its association with cardiovascular and renal disease.
Edited by: Laura Bennet & Dino Giussani
References
- Alexander BT, Dasinger JH & Intapad S (2015). Fetal programming and cardiovascular pathology. Compr Physiol 5, 997–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DE, Fedorova OV, Morrell CH, Longo DL, Kashkin VA, Metzler JD, Bagrov AY & Lakatta EG (2008). Endogenous sodium pump inhibitors and age‐associated increases in salt sensitivity of blood pressure in normotensives. Am J Physiol Regul Integr Comp Physiol 294, R1248–R1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel LJ, Brands MW, Daniels SR, Karanja N, Elmer PJ, Sacks FM & American Heart A (2006). Dietary approaches to prevent and treat hypertension: a scientific statement from the American Heart Association. Hypertension 47, 296–308. [DOI] [PubMed] [Google Scholar]
- Arribas SM, Briones AM, Bellingham C, Gonzalez MC, Salaices M, Liu K, Wang Y & Hinek A (2008). Heightened aberrant deposition of hard‐wearing elastin in conduit arteries of prehypertensive SHR is associated with increased stiffness and inward remodeling. Am J Physiol Heart Circ Physiol 295, H2299–H2307. [DOI] [PubMed] [Google Scholar]
- Bagrov AY, Shapiro JI & Fedorova OV (2009). Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Pharmacol Rev 61, 9–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ, Bull AR, Osmond C & Simmonds SJ (1990). Fetal and placental size and risk of hypertension in adult life. BMJ 301, 259–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ, Winter PD, Osmond C, Margetts B & Simmonds SJ (1989). Weight in infancy and death from ischaemic heart disease. Lancet 2, 577–580. [DOI] [PubMed] [Google Scholar]
- Black MJ, Siebel AL, Gezmish O, Moritz KM & Wlodek ME (2012). Normal lactational environment restores cardiomyocyte number after uteroplacental insufficiency: implications for the preterm neonate. Am J Physiol Regul Integr Comp Physiol 302, R1101–R1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner BM (1983). Hemodynamically mediated glomerular injury and the progressive nature of kidney disease. Kidney Int 23, 647–655. [DOI] [PubMed] [Google Scholar]
- Brown IJ, Tzoulaki I, Candeias V & Elliott P (2009). Salt intakes around the world: implications for public health. Int J Epidemiol 38, 791–813. [DOI] [PubMed] [Google Scholar]
- Clough GF (2015). Developmental conditioning of the vasculature. Compr Physiol 5, 397–438. [DOI] [PubMed] [Google Scholar]
- Dagan A, Gattineni J, Cook V & Baum M (2007). Prenatal programming of rat proximal tubule Na+/H+ exchanger by dexamethasone. Am J Physiol Regul Integr Comp Physiol 292, R1230–R1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nicolantonio R, Koutsis K, Westcott KT & Wlodek ME (2006). Relative contribution of the prenatal versus postnatal period on development of hypertension and growth rate of the spontaneously hypertensive rat. Clin Exp Pharmacol Physiol 33, 9–16. [DOI] [PubMed] [Google Scholar]
- Di Nicolantonio R, Westcott KT, Koutsis K & Wlodek ME (2005). Lack of evidence for a role for either the in utero or suckling periods in the exaggerated salt preference of the spontaneously hypertensive rat. Physiol Behav 86, 500–507. [DOI] [PubMed] [Google Scholar]
- Elliott P, Walker LL, Little MP, Blair‐West JR, Shade RE, Lee DR, Rouquet P, Leroy E, Jeunemaitre X, Ardaillou R, Paillard F, Meneton P & Denton DA (2007). Change in salt intake affects blood pressure of chimpanzees: implications for human populations. Circulation 116, 1563–1568. [DOI] [PubMed] [Google Scholar]
- Gallo LA, Denton KM, Moritz KM, Tare M, Parkington HC, Davies M, Tran M, Jefferies AJ & Wlodek ME (2012a). Long‐term alteration in maternal blood pressure and renal function after pregnancy in normal and growth‐restricted rats. Hypertension 60, 206–213. [DOI] [PubMed] [Google Scholar]
- Gallo LA, Tran M, Moritz KM, Jefferies AJ & Wlodek ME (2012b). Pregnancy in aged rats that were born small: cardiorenal and metabolic adaptations and second‐generation fetal growth. FASEB J 26, 4337–4347. [DOI] [PubMed] [Google Scholar]
- Gallo LA, Ward MS, Fotheringham AK, Zhuang A, Borg DJ, Flemming NB, Harvie BM, Kinneally TL, Yeh SM, McCarthy DA, Koepsell H, Vallon V, Pollock C, Panchapakesan U & Forbes JM (2016). Once daily administration of the SGLT2 inhibitor, empagliflozin, attenuates markers of renal fibrosis without improving albuminuria in diabetic db/db mice. Sci Rep 6, 26428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurts AM, Mattson DL, Liu P, Cabacungan E, Skelton MM, Kurth TM, Yang C, Endres BT, Klotz J, Liang M & Cowley AW Jr (2015). Maternal diet during gestation and lactation modifies the severity of salt‐induced hypertension and renal injury in Dahl salt‐sensitive rats. Hypertension 65, 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigore D, Ojeda NB, Robertson EB, Dawson AS, Huffman CA, Bourassa EA, Speth RC, Brosnihan KB & Alexander BT (2007). Placental insufficiency results in temporal alterations in the renin angiotensin system in male hypertensive growth restricted offspring. Am J Physiol Regul Integr Comp Physiol 293, R804–R811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha SK (2014). Dietary salt intake and hypertension. Electrolytes & Blood Pressure 12, 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A & Seckl J (2011). Glucocorticoids, prenatal stress and the programming of disease. Horm Behav 59, 279–289. [DOI] [PubMed] [Google Scholar]
- He FJ & MacGregor GA (2003). How far should salt intake be reduced? Hypertension 42, 1093–1099. [DOI] [PubMed] [Google Scholar]
- Henriksen T & Clausen T (2002). The fetal origins hypothesis: placental insufficiency and inheritance versus maternal malnutrition in well‐nourished populations. Acta Obstet Gynecol Scand 81, 112–114. [DOI] [PubMed] [Google Scholar]
- Jalil JE, Doering CW, Janicki JS, Pick R, Shroff SG & Weber KT (1989). Fibrillar collagen and myocardial stiffness in the intact hypertrophied rat left ventricle. Circ Res 64, 1041–1050. [DOI] [PubMed] [Google Scholar]
- John SW, Krege JH, Oliver PM, Hagaman JR, Hodgin JB, Pang SC, Flynn TG & Smithies O (1995). Genetic decreases in atrial natriuretic peptide and salt‐sensitive hypertension. Science 267, 679–681. [DOI] [PubMed] [Google Scholar]
- Kotchen TA, Cowley AW Jr & Frohlich ED (2013). Salt in health and disease – a delicate balance. N Engl J Med 368, 2531–2532. [DOI] [PubMed] [Google Scholar]
- Kubisch HM, Mathialagan S & Gomez‐Sanchez EP (1998). Modulation of blood pressure in the Dahl SS/jr rat by embryo transfer. Hypertension 31, 540–545. [DOI] [PubMed] [Google Scholar]
- Lee JY & Azar SH (2010). Wistar‐Kyoto and spontaneously hypertensive rat blood pressure after embryo transfer into different wombs and cross‐suckling. Exp Biol Med (Maywood) 235, 1375–1384. [DOI] [PubMed] [Google Scholar]
- Mattson DL, Dwinell MR, Greene AS, Kwitek AE, Roman RJ, Jacob HJ & Cowley AW Jr (2008). Chromosome substitution reveals the genetic basis of Dahl salt‐sensitive hypertension and renal disease. Am J Physiol Renal Physiol 295, F837–F842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzuca MQ, Tare M, Parkington HC, Dragomir NM, Parry LJ & Wlodek ME (2012). Uteroplacental insufficiency programmes vascular dysfunction in non‐pregnant rats: compensatory adaptations in pregnancy. J Physiol 590, 3375–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzuca MQ, Wlodek ME, Dragomir NM, Parkington HC & Tare M (2010). Uteroplacental insufficiency programs regional vascular dysfunction and alters arterial stiffness in female offspring. J Physiol 588, 1997–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menuet C, Wlodek ME, Fong AY & Allen AM (2016). Respiratory modulation of sympathetic nerve activity is enhanced in male rat offspring following uteroplacental insufficiency. Respir Physiol Neurobiol 226, 147–151. [DOI] [PubMed] [Google Scholar]
- Mills KT, Bundy JD, Kelly TN, Reed JE, Kearney PM, Reynolds K, Chen J & He J (2016). Global Disparities of Hypertension Prevalence and Control: A Systematic Analysis of Population‐Based Studies From 90 Countries. Circulation 134, 441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moritz KM, De Matteo R, Dodic M , Jefferies AJ, Arena D, Wintour EM, Probyn ME, Bertram JF, Singh RR, Zanini S & Evans RG (2011). Prenatal glucocorticoid exposure in the sheep alters renal development in utero: implications for adult renal function and blood pressure control. Am J Physiol Regul Integr Comp Physiol 301, R500–R509. [DOI] [PubMed] [Google Scholar]
- Moritz KM, Mazzuca MQ, Siebel AL, Mibus A, Arena D, Tare M, Owens JA & Wlodek ME (2009). Uteroplacental insufficiency causes a nephron deficit, modest renal insufficiency but no hypertension with ageing in female rats. J Physiol 587, 2635–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacks FM, Svetkey LP, Vollmer WM, Appel LJ, Bray GA, Harsha D, Obarzanek E, Conlin PR, Miller ER 3rd, Simons‐Morton DG, Karanja N, Lin PH & Group DA‐SCR (2001). Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH‐Sodium Collaborative Research Group. N Engl J Med 344, 3–10. [DOI] [PubMed] [Google Scholar]
- Safar ME, Thuilliez C, Richard V & Benetos A (2000). Pressure‐independent contribution of sodium to large artery structure and function in hypertension. Cardiovasc Res 46, 269–276. [DOI] [PubMed] [Google Scholar]
- Schreuder MF, Van Wijk JA, Fodor M & Delemarre‐van de Waal HA (2007). Influence of intrauterine growth restriction on renal function in the adult rat. J Physiol Biochem 63, 213–219. [DOI] [PubMed] [Google Scholar]
- Simmons RA, Templeton LJ & Gertz SJ (2001). Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes 50, 2279–2286. [DOI] [PubMed] [Google Scholar]
- Singh RR, Cullen‐McEwen LA, Kett MM, Boon WM, Dowling J, Bertram JF & Moritz KM (2007). Prenatal corticosterone exposure results in altered AT1/AT2, nephron deficit and hypertension in the rat offspring. J Physiol 579, 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tare M, Parkington HC, Bubb KJ & Wlodek ME (2012). Uteroplacental insufficiency and lactational environment separately influence arterial stiffness and vascular function in adult male rats. Hypertension 60, 378–386. [DOI] [PubMed] [Google Scholar]
- Tran M, Gallo LA, Hanvey AN, Jefferies AJ, Westcott KT, Cullen‐McEwen LA, Gardner DK, Moritz KM & Wlodek ME (2014). Embryo transfer cannot delineate between the maternal pregnancy environment and germ line effects in the transgenerational transmission of disease in rats. Am J Physiol Regul Integr Comp Physiol 306, R607–R618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallon V (2011). The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol 300, R1009‐1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vehaskari VM, Stewart T, Lafont D, Soyez C, Seth D & Manning J (2004). Kidney angiotensin and angiotensin receptor expression in prenatally programmed hypertension. Am J Physiol Renal Physiol 287, F262–F267. [DOI] [PubMed] [Google Scholar]
- Wadley GD, Laker RC, McConell GK & Wlodek ME (2016). Endurance training in early life results in long‐term programming of heart mass in rats. Physiol Rep 4, e12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton SL, Bielefeldt‐Ohmann H, Singh RR, Li J, Paravicini TM, Little MH & Moritz KM (2017). Prenatal hypoxia leads to hypertension, renal renin‐angiotensin system activation and exacerbates salt‐induced pathology in a sex‐specific manner. Sci Rep 7, 8241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton SL, Singh RR, Tan T, Paravicini TM & Moritz KM (2016). Late gestational hypoxia and a postnatal high salt diet programs endothelial dysfunction and arterial stiffness in adult mouse offspring. J Physiol 594, 1451–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger MH & Fineberg NS (1991). Sodium and volume sensitivity of blood pressure. Age and pressure change over time. Hypertension 18, 67–71. [DOI] [PubMed] [Google Scholar]
- Weinberger MH, Miller JZ, Luft FC, Grim CE & Fineberg NS (1986). Definitions and characteristics of sodium sensitivity and blood pressure resistance. Hypertension 8, II127‐134. [DOI] [PubMed] [Google Scholar]
- White SL, Perkovic V, Cass A, Chang CL, Poulter NR, Spector T, Haysom L, Craig JC, Salmi IA, Chadban SJ & Huxley RR (2009). Is low birth weight an antecedent of CKD in later life? A systematic review of observational studies. Am J Kidney Dis 54, 248–261. [DOI] [PubMed] [Google Scholar]
- Wigg SJ, Tare M, Tonta MA, O'Brien RC, Meredith IT & Parkington HC (2001). Comparison of effects of diabetes mellitus on an EDHF‐dependent and an EDHF‐independent artery. Am J Physiol Heart Circ Physiol 281, H232–H240. [DOI] [PubMed] [Google Scholar]
- Wlodek ME, Mibus A, Tan A, Siebel AL, Owens JA & Moritz KM (2007). Normal lactational environment restores nephron endowment and prevents hypertension after placental restriction in the rat. J Am Soc Nephrol 18, 1688–1696. [DOI] [PubMed] [Google Scholar]
- Wlodek ME, Westcott K, Siebel AL, Owens JA & Moritz KM (2008). Growth restriction before or after birth reduces nephron number and increases blood pressure in male rats. Kidney Int 74, 187–195. [DOI] [PubMed] [Google Scholar]
- Woods LL, Weeks DA & Rasch R (2004). Programming of adult blood pressure by maternal protein restriction: role of nephrogenesis. Kidney Int 65, 1339–1348. [DOI] [PubMed] [Google Scholar]
- Yu Q, Larson DF, Slayback D, Lundeen TF, Baxter JH & Watson RR (2004). Characterization of high‐salt and high‐fat diets on cardiac and vascular function in mice. Cardiovasc Toxicol 4, 37–46. [DOI] [PubMed] [Google Scholar]