

Abstract
The fetus is consistently exposed to repeated periods of impaired oxygen (hypoxaemia) and nutrient supply in labour. This is balanced by the healthy fetus's remarkable anaerobic tolerance and impressive ability to mount protective adaptations to hypoxaemia. The most important mediator of fetal adaptations to brief repeated hypoxaemia is the peripheral chemoreflex, a rapid reflex response to acute falls in arterial oxygen tension. The overwhelming majority of fetuses are able to respond to repeated uterine contractions without developing hypotension or hypoxic–ischaemic injury. In contrast, fetuses who are either exposed to severe hypoxaemia, for example during uterine hyperstimulation, or enter labour with reduced anaerobic reserve (e.g. as shown by severe fetal growth restriction) are at increased risk of developing intermittent hypotension and cerebral hypoperfusion. It is remarkable to note that when fetuses develop hypotension during such repeated severe hypoxaemia, it is not mediated by impaired reflex adaptation, but by failure to maintain combined ventricular output, likely due to a combination of exhaustion of myocardial glycogen and evolving myocardial injury. The chemoreflex is suppressed by relatively long periods of severe hypoxaemia of 1.5–2 min, longer than the typical contraction. Even in this setting, the peripheral chemoreflex is consistently reactivated between contractions. These findings demonstrate that the peripheral chemoreflex is an indefatigable guardian of fetal adaptation to labour.
Keywords: labour, intrapartum deceleration, peripheral chemoreflex, fetal heart rate
Introduction
Labour is inherently associated with intermittent ‘asphyxia’ as each intrapartum uterine contraction impairs gaseous exchange, with transient fetal hypoxaemia, hypercapnia and an obligatory shift to anaerobic metabolism, leading to metabolic acidaemia (Bax & Nelson, 1993). Uterine contractions reduce uteroplacental perfusion, as shown by reduced uterine artery blood flow velocity (Fleischer et al. 1987; Janbu & Nesheim, 1987; Brar et al. 1988; Li et al. 2003) and reduced placental and intervillous perfusion (Ramsey, 1968; Sato et al. 2016; Sinding et al. 2016). Potentially, fetoplacental circulation may also be interrupted, secondary to compression of the umbilical cord. This is unlikely to occur due to increased amniotic pressure per se as the hydraulic pressure will be transmitted equally to the intra‐ and extravascular space, preventing any net compressive effect on the umbilical cord. Cord compression can occur during cord entanglement, knots or physical compression of the cord between the fetus and uterus. Fetoplacental perfusion may additionally be impaired due to upstream compression of the placental vasculature by uterine contractions. Although it is difficult to determine the cause of impaired fetoplacental perfusion in any individual labour, reduced perfusion appears to occur most commonly during contractions associated with fetal heart rate (FHR) decelerations (Murakami et al. 1985; Fairlie et al. 1989; Weiss et al. 1991; Sakai et al. 1997; Tadmor et al. 1999; Li et al. 2003).
Regardless of the upstream mechanism, studies using near‐infrared spectroscopy and pulse oximetry confirm that both first and second stage uterine contractions are associated with reduced fetal oxygenation, which recovers after the end of the contraction (Peebles et al. 1992; McNamara & Johnson, 1995). In the vast majority of labours, these brief periods of impaired gaseous exchange are well tolerated by the healthy fetus. This is partly due to the restoration of placental function between contractions, which is normally sufficient to largely reverse hypoxaemia, hypercapnia and metabolic acidaemia. Nevertheless, this repeated impairment of gaseous exchange leads to a small, but consistent reduction in pH, and an increase in lactate and in normal, uncomplicated labour (Modanlou et al. 1974; Huch et al. 1977; Kro et al. 2010). In contrast, both spontaneous and oxytocin‐induced uterine hyperstimulation are associated with increased risk of fetal acidaemia (Bakker et al. 2007) and a persistent reduction in fetal cerebral oxygenation (Klink et al. 1981; Johnson et al. 1994; Peebles et al. 1994; Simpson & James, 2008).
The defining characteristic of labour is therefore brief, but repeated, periods of fetal hypoxaemia. It is important to appreciate that this intermittent contraction‐related hypoxaemia can be superimposed on pre‐existing mild–moderate hypoxaemia associated with antenatal placental insufficiency (for example in the setting of fetal growth restriction) (Morrison, 2008; Brain et al. 2015; Chauhan et al. 2017). Acute severe hypoxaemia can also occur during labour due to complications such as placental abruption, uterine rupture or cord prolapse. Although such sentinel events are associated with a high risk of hypoxic–ischaemic injury in their own right (Westgate et al. 1999b; Martinez‐Biarge et al. 2012), they contribute about 25% of cases of hypoxic–ischaemic encephalopathy (Westgate et al. 1999b; West et al. 2005; Jonsson et al. 2014). Thus we must appreciate that even in the absence of sentinel events, all labours are still associated with intermittent fetal hypoxaemia.
The fetus has impressive adaptations to defend itself against all of these patterns of hypoxaemia (Giussani, 2016; Bennet et al. 2017). One of the earliest advances was the realisation that the fetus is adapted to live with exceptionally low partial pressures of oxygen, leading to the phrase ‘Everest in utero’ (Barcroft, 1946). This is in part made possible due to the structure of fetal haemoglobin, high basal blood flow and unique vascular shunts which promote preferential streaming of highly oxygenated blood towards critical organs including the heart and brain (Rudolph, 1985; Kiserud et al. 2000; Harding & Bocking, 2001). Critically, the healthy fetus has an impressive anaerobic reserve. This is primarily determined by the exceptionally high level of myocardial glycogen, which is tightly linked to the ability to maintain cardiovascular function during severe hypoxaemia (Dawes et al. 1959; Shelley, 1961). Other organs, including the brain, are inherently resistant to hypoxic injury, partly by suppressing metabolism (Drury et al. 2012). These basal adaptations are coupled with active defences to further prioritise blood supply and reduce oxygen consumption during acute hypoxaemia. Classical studies used fetal sheep to finely dissect the physiology of fetal adaptations to both prolonged mild–moderate hypoxaemia and acute severe hypoxaemia, as reviewed in Giussani (2016) and Bennet (2017). These influential studies showed that the fetus primarily adapts to sustained periods of hypoxaemia with an initial neural reflex but this is quickly augmented and replaced by non‐neural, humoral mechanisms (Giussani, 2016; Bennet, 2017).
It is important to appreciate that such prolonged periods of hypoxaemia are not consistent with the typical pattern of intermittent hypoxaemia associated with labour, and may have inadvertently contributed to a misunderstanding of how the fetus successfully adapts to labour. The impairment of gaseous exchange related to uterine contractions is well appreciated clinically (Ayres‐de‐Campos & Arulkumaran, 2015). Unfortunately a long‐standing belief that the majority of FHR decelerations during labour are triggered by non‐hypoxic events (Hon & Quilligan, 1967; Ayres‐de‐Campos et al. 2015) has de‐emphasised the importance of repeated fetal hypoxaemia. In contrast, modern evidence supports that the vast majority of FHR decelerations are triggered by hypoxaemia and represent the fetus's protective adaptation against hypoxaemia (the peripheral chemoreflex) (Lear et al. 2016b). Understanding the fetal adaptations to brief but repeated hypoxaemia is therefore critical to understanding how the great majority of fetuses survive labour without compromise, in what situations these adaptations are insufficient and, critically, how these adaptations are reflected on the intrapartum FHR trace – the gold standard for assessing fetal wellbeing during labour. This review will therefore focus on the fetal cardiovascular adaptations to intermittent hypoxaemia. In contrast to a reliance on humoral factors during prolonged hypoxaemia, the fetal adaptation to intermittent hypoxaemia is predominantly mediated by repeated activation of the peripheral chemoreflex leading to repeated, intense activation of both arms of the autonomic nervous system.
The peripheral chemoreflex: the rapid adaptation to hypoxaemia
The fetus responds to hypoxaemia with coordinated cardiovascular, neurophysiological and behavioural adaptations (Giussani, 2016; Bennet, 2017). The peripheral chemoreflex mediates the immediate, rapid responses to hypoxaemia. It triggers an increase in parasympathetic activity causing a rapid FHR deceleration (Itskovitz et al. 1983; Giussani et al. 1993), reducing combined ventricular output (CVO). This reduction in CVO is primarily related to the reduction in FHR as indices of preload have consistently been shown to be stable, or even increased, during graded reductions in umbilical blood flow (Edelstone et al. 1980; Itskovitz et al. 1987). This presumptively reduces myocardial oxygen consumption. The peripheral chemoreflex actively accommodates the fall in CVO by simultaneously increasing sympathetic nervous activity (Jensen & Lang, 1992; Giussani et al. 1993). This promotes rapid, intense peripheral vasoconstriction (Fig. 1) resulting in hypertension and centralisation of (the now reduced) CVO to key organs such as the brain, heart and adrenal glands (Jensen et al. 1987; Jensen & Lang, 1992; Giussani et al. 1993). The vasoconstrictor effects of sympathetic neural activation are supplemented by release of humoral factors including adrenal catecholamines, cortisol, angiotensin, vasopressin and neuropeptide Y (Broughton‐Pipkin et al. 1974; Jones & Robinson, 1975; Perez et al. 1989; Giussani et al. 1993, 1994a, b; Fletcher et al. 2000; Galinsky et al. 2014).
Figure 1. Peripheral chemoreflex activation during labour.
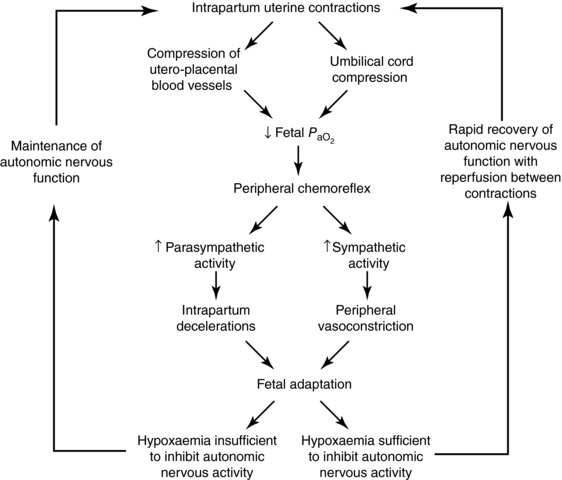
The peripheral chemoreflex is the protective fetal response to acute hypoxaemia caused by intrapartum uterine contractions. Its activation results in a reflex increase in parasympathetic activity, to trigger intrapartum decelerations, and an increase in sympathetic nervous activity, to promote peripheral vasoconstriction and centralisation of blood flow to the heart, brain and adrenal glands. The severity/duration of hypoxaemia during labour is typically brief, meaning that autonomic activity is sustained throughout contractions. However, even if autonomic activity is lost during individual contractions, it rapidly recovers with reperfusion. The peripheral chemoreflex can therefore be continually activated throughout labour, even in the face of evolving fetal compromise. , arterial partial pressure of oxygen.
The peripheral chemoreflex is highly tailored. Greater reductions in oxygen tensions (e.g. due to greater impairment of placental gaseous exchange) are associated with more intense activation and in turn deeper falls in fetal heart rate (decelerations). In healthy near‐term fetal sheep, a 50% reduction in uteroplacental flow is needed to trigger a deceleration (Itskovitz et al. 1983; Ross et al. 2013). Maximal activation of the peripheral chemoreflex is observed when uteroplacental blood flow is reduced to below 90% (Itskovitz et al. 1983). Although it is currently not possible to continuously monitor peripheral vasoconstriction during labour, evidence of centralisation of blood flow has been observed in growth‐restricted fetuses responding positively to an oxytocin challenge test (Li et al. 2006) and during active labour in association with brief periods of fetal deoxygenation as assessed by pulse oximetry (Siristatidis et al. 2004). The parasympathetic component of the peripheral chemoreflex is shown by repetitive FHR decelerations occurring with intrapartum uterine contractions (Lear et al. 2016b). Thus, intrapartum decelerations simply indicate reflex responses to reduced uteroplacental gaseous exchange, while repeated deep decelerations indicate near abolition of gaseous exchange.
The peripheral chemoreflex is capable of repeated activation throughout labour
Most fetuses enter labour with a large reserve of placental capacity and myocardial glycogen stores that help to accommodate the repeated brief reductions in oxygen supply during contractions. When this is coupled with the impressive ability of the healthy fetus to mount protective adaptations against hypoxaemia, the vast majority of fetuses tolerate labour exceptionally well. This is understandably finite, and fetal compromise results when this tolerance is exceeded. In turn, uterine contraction strength, frequency and duration are the key factors that determine whether and how quickly fetuses will develop hypotension. The presence and severity of hypotension is critically associated with both reduced cerebral blood flow and ultimately with intrapartum hypoxic–ischaemic injury across different settings and experimental protocols(Gunn et al. 1992; Mallard et al. 1994; de Haan et al. 1997; Ikeda et al. 1998; Fujii et al. 2003). Thus, we may consider the development of hypotension as an empirical marker of fetal compromise.
Ultimately, the proportion of time the uterus spends at resting tone compared with contracting tone will determine the extent to which fetal gas exchange can be restored between contractions. Consistent with this, reduced relaxation time or increased contraction frequency is associated with persistent reductions in fetal oxygenation (Klink et al. 1981; Johnson et al. 1994; Peebles et al. 1994; Simpson & James, 2008), an increased risk of fetal acidaemia (Bakker et al. 2007) and in turn an increased risk of intrapartum decelerations (Stewart et al. 2012). Thus, interventions, which increase the frequency and/or duration of uterine contractions, place the fetus at a higher risk of compromise.
These clinical findings are strongly paralleled by studies in near‐term fetal sheep, which provide insight into the mechanisms through which the fetus continues to adapt to repeated brief hypoxaemia. In these studies severe hypoxaemia was induced by brief (1 min) complete umbilical cord occlusions repeated every 5 or 2.5 min (consistent with early and active labour, respectively) (Westgate et al. 1999a) or by 2 min complete umbilical cord occlusions repeated every 5 min (Westgate et al. 2001; Galinsky et al. 2014). As explored next, these studies have collectively revealed that the peripheral chemoreflex is exceptionally robust, and able to be repetitively activated over many hours without becoming attenuated. Critically, even with limited time for reperfusion between complete umbilical cord occlusions with progressive hypotension and acidaemia, the peripheral chemoreflex continues to be intensely activated with every episode of hypoxaemia. The peripheral chemoreflex therefore remains the guardian of fetal wellbeing in both cases of successful and unsuccessful adaptation to labour.
Cardiovascular changes in fetuses tolerating labour‐like hypoxaemia
Studies in near‐term fetal sheep have shown that the healthy fetus can tolerate even severe intermittent periods of hypoxaemia essentially indefinitely, providing that a sufficient period of reperfusion is allowed between periods of hypoxaemia. Fetal sheep effectively tolerated 1 min complete umbilical cord occlusions repeated every 5 min (consistent with the contraction frequency during early labour) for 4 h (a total of 49 occlusions) without any hypotension. These fetuses responded to all occlusions with an intense peripheral chemoreflex, including a rapid deep deceleration and hypertension (Fig. 2) and only minimal acidaemia was observed at the end of the experiment (pH 7.34 ± 0.03, base deficit 1.1 ± 1.4 mmol L−1, lactate 4.2 ± 1.5 mmol L−1) (Bennet et al. 2005).
Figure 2. Cardiovascular changes in fetuses tolerating labour‐like hypoxaemia.
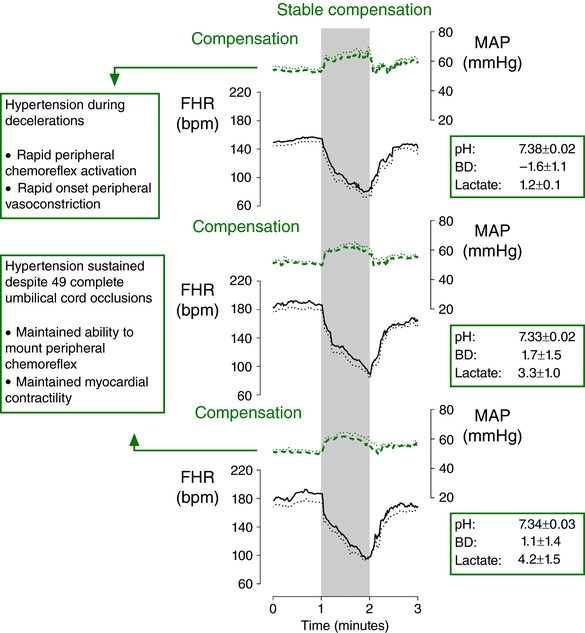
Fetal heart rate (FHR, continuous lines) and mean arterial pressure (MAP, dashed lines) in near‐term fetal sheep during 1 min complete umbilical cord occlusion (UCO) repeated every 5 min (n = 8) for 4 h (total of 49 UCOs). The top panel shows the second UCO, the middle panel shows the middle UCO and the bottom panel shows the penultimate UCO. Fetuses effectively adapted to this severity and frequency of UCO. Each occlusion was associated with a rapid deceleration and sustained hypertension, mediated by peripheral chemoreflex activation. Hypotension was never observed in these fetuses. Data are 1 s means ± SEM (shown as dotted lines). The periods of umbilical cord occlusion are shown in grey. Base deficit (BD) and lactate values are given in mmol L−1. Figure modified from Bennet et al. (2005).
These findings illustrate two important points. Firstly, the peripheral chemoreflex is able to be reliably re‐activated during repeated contractions over prolonged periods of time to effectively promote centralisation of blood flow and hypertension. In turn, this helps explain why the majority of babies are at no risk of hypotension and do not develop marked acidaemia despite showing repeated decelerations during labour: the healthy fetus given sufficient reperfusion between decelerations is well able to adapt to repeated hypoxaemia. The finding that fetuses showed only mild acidaemia is additionally important considering that they were subjected to a cumulative duration of 49 min of complete umbilical cord occlusion over 4 h (Bennet et al. 2005). This illustrates that the healthy placenta is able to rapidly reverse the effects of temporary impaired gaseous exchange, and that the fetus can rapidly metabolise lactate (directly or by conversion to glucose; Carter et al. 1995; Bartelds et al. 2000), and therefore that the effect of uterine contractions on gaseous exchange and lactate will often not be apparent on umbilical blood gases.
Cardiovascular changes in fetuses failing to tolerate labour‐like hypoxaemia
In contrast, fetuses exposed to repeated hypoxaemia with insufficient periods of reperfusion progressively develop hypotension during occlusions, severe acidaemia and eventually neural injury (de Haan et al. 1997). Near‐term fetal sheep exposed to 1 min complete umbilical occlusions repeated every 2.5 min (consistent with active labour) until severe hypotension developed (76.5 ± 21.2 occlusions, or an average 3.2 h of occlusions) responded to the first three cord occlusions with deep decelerations and sustained hypertension during occlusions. From the fourth occlusion onwards, fetuses showed a biphasic arterial pressure response consisting of initial hypertension followed by a fall in blood pressure during the occlusion, before recovering to or above baseline between occlusions (Westgate et al. 1999a; Bennet et al. 2005). The fall in arterial pressure became more rapid and severe with continued occlusions, progressively leading to severe hypotension during occlusions (nadir of 15.5 ± 3.0 mmHg during the final occlusion). Severe acidaemia progressively developed with successive occlusions (pH 6.92 ± 0.04, base deficit 19.7 ± 1.8 mmol L−1, lactate 14.8 ± 1.2 mmol L−1 after the final occlusion) (Bennet et al. 2005).
Mechanisms of evolving hypotension
There is now good evidence that the development of intermittent hypotension during brief repeated umbilical cord occlusions is not due to failure to mount an effective peripheral chemoreflex. Fetal sheep exposed to 2 min complete umbilical cord occlusions repeated every 5 min until severe hypotension developed (18.7 ± 2.3 occlusions) responded to all occlusions with rapid peripheral vasoconstriction intense enough to reduce femoral blood flow essentially to zero (Galinsky et al. 2014). Strikingly, this intense peripheral vasoconstriction was observed even in the final occlusions of the experiment when all fetuses were simultaneously developing severe, rapid hypotension and despite severe acidaemia (pH 6.99 ± 0.03, lactate 12.5 ± 0.8 mmol L−1) (Galinsky et al. 2014). In contrast, chemically sympathectomised fetuses showed impaired and delayed peripheral vasoconstriction during repeated umbilical cord occlusions leading to more rapid onset hypotension (Galinsky et al. 2014). This finding illustrates that the sympathetic nervous system continues to be intensely activated during severe repeated hypoxaemia, even in the face of severe fetal hypotension.
The parasympathetic arm of the peripheral chemoreflex appears to be equally robust. In near‐term fetal sheep exposed to 1 min complete umbilical cord occlusion repeated every 2.5 min until severe hypotension developed, the initial parasympathetic‐mediated deceleration became more rapid as fetal acidaemia and hypotension developed during occlusions (Bennet et al. 2005). The parasympathetic response to repeated hypoxaemia is therefore not only sustained, but augmented during progressive fetal compromise. Metabolic acidosis may have contributed to this, as previous evidence in near‐term fetal sheep has shown that infusion of acidified saline to induce mild acidosis was associated with a greater fall in FHR and increased peripheral vasoconstriction during moderate hypoxaemia (Thakor & Giussani, 2009). Once the initial fall in FHR is achieved, FHR is regulated by a balance between continued parasympathetic activity and the positive chronotropic effects of adrenal catecholamines, acting through β‐adrenergic receptors (Galinsky et al. 2016). This can result in a partial recovery in FHR despite continuing cord occlusion, especially in fetuses tolerating hypoxaemia well (Giussani et al. 1993; Galinsky et al. 2016). In contrast, the nadir of decelerations progressively deepens with evolving fetal compromise and hypotension (Bennet et al. 2005). Given that adrenaline and noradrenaline levels remain extremely high during fetal compromise (Galinsky et al. 2014), deepening decelerations presumptively represent impaired myocardial responsiveness and contractility, potentially due to a combination of intracellular acidosis, depletion of myocardial glycogen and evolving myocardial injury (Dawes et al. 1959; Shelley, 1961; Gunn et al. 2000).
Although these studies demonstrate that the peripheral chemoreflex can be continually activated at the start of labour‐like hypoxaemia for many hours, we know less about how long autonomic tone is maintained during each individual period of deep hypoxaemia. During studies of single periods of prolonged hypoxaemia, the peripheral chemoreflex becomes attenuated after approximately 90 s (Barcroft, 1946). The finding that intense peripheral vasoconstriction was maintained during brief 2 min complete umbilical cord occlusions despite severe hypotension suggests that overall combined sympathetic tone (i.e. neural and adrenal) is sustained even longer (Galinsky et al. 2014). However, there is indirect evidence that parasympathetic tone during repeated labour‐like hypoxaemia becomes shorter‐lived with progressive fetal compromise.
Systematic studies in fetal sheep have shown that overshoot tachycardia occurring immediately after a deceleration is mediated by the combination of loss of parasympathetic tone and β‐adrenergic stimulation (Galinsky et al. 2016). Overshoot tachycardia is always seen after 2 min complete umbilical cord occlusions, confirming that parasympathetic tone is lost by 2 min of severe hypoxaemia (Westgate et al. 2001; Galinsky et al. 2016; Lear et al. 2016a), consistent with studies of parasympathetic blockade (Barcroft, 1946). In contrast, in fetal sheep, overshoot did not occur after 1 min complete umbilical cord occlusions repeated every 2.5 min, until hypotension began to develop (Westgate et al. 2001). This infers that the parasympathetic tone became attenuated earlier during hypoxaemia after the onset of hypotension. The mechanism is unknown, but speculatively may reflect direct neural inhibition due to impaired perfusion of the brainstem (Jensen et al. 1999).
Nevertheless, it is important to appreciate that even if autonomic tone is lost during individual periods of hypoxaemia, both arms of the autonomic nervous system rapidly recover with even brief reperfusion, allowing the peripheral chemoreflex to be reactivated (Figs 1 and 3) (Bennet et al. 2005; Galinsky et al. 2014). This may be partly related to the consistent finding in the above studies that arterial pressure rapidly recovers to above baseline levels between occlusions, even after the onset of severe hypotension during occlusions (Bennet et al. 2005; Westgate et al. 2005; Wassink et al. 2013; Galinsky et al. 2014). This likely allows rapid restoration of brainstem perfusion and substrate supply, and recovery of the autonomic centres.
Figure 3. Cardiovascular changes in fetuses failing to tolerate labour‐like hypoxaemia.
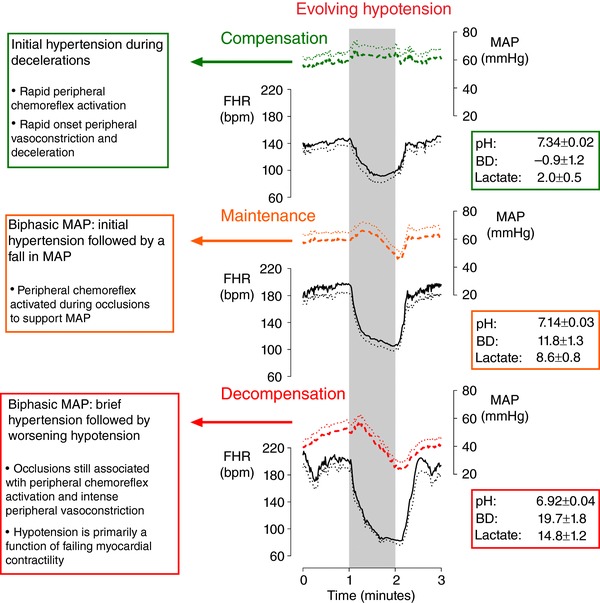
Fetal heart rate (FHR, continuous lines) and mean arterial pressure (MAP, dashed lines) in near‐term fetal sheep during 1 min complete umbilical cord occlusion (UCO) repeated every 2.5 min (n = 8) until severe hypotension developed (76.5 ± 21.2 UCOs). The top panel shows the second UCO, the middle panel shows the middle UCO and the bottom panel shows the penultimate UCO. Fetuses initially tolerated this frequency and severity of UCOs, displaying hypertension and rapid decelerations during UCOs. The arterial pressure response progressively became biphasic as fetal tolerance was overcome, with initial hypertension followed by a fall in arterial pressure during occlusion. Fetal decompensation was associated with short lived hypertension and marked hypotension during occlusion. The associated decelerations became progressively deeper with the onset of hypotension. Data are 1 s means ± SEM (shown as dotted lines). The periods of umbilical cord occlusion are shown in grey. Base deficit (BD) and lactate values are given in mmol L−1. Figure modified from Bennet et al. (2005).
Overall, these studies confirm that the healthy fetus has an impressive tolerance to repeated hypoxaemia. However, even a healthy fetus with normal placental function may be unable to prevent hypotension during tonic contractions or uterine hyperstimulation, as may occur, for example, with oxytocin and prostaglandin for induction or augmentation of labour (Winkler & Rath, 1999). When progressive fetal decompensation during labour occurs, it is not related to the failure of autonomic adaptation, but to an inability to maintain CVO during hypoxaemia. Given that adrenaline and noradrenaline levels remain extremely high during fetal compromise (Galinsky et al. 2014), this strongly denotes that responsiveness to β‐adrenergic stimulation becomes impaired (Bennet et al. 2005). This is likely due to a combination of exhaustion of myocardial glycogen and evolving reversible cardiac injury leading to impaired contractility (Dawes et al. 1959; Shelley, 1961; Gunn et al. 2000). Consistent with a role for cardiac injury in humans, neonatal hypoxic–ischaemic encephalopathy is highly associated with increased plasma troponin‐T levels (Jones et al. 2017).
Does prior hypoxaemia attenuate the peripheral chemoreflex?
There is some evidence in fetal sheep that the peripheral chemoreflex may become attenuated by repeated moderate hypoxaemia (Giussani et al. 1997; Green et al. 2001). Giussani and colleagues found that during a series of partial umbilical cord occlusions for 5 min repeated 12 times with 15 min of reperfusion between occlusions, that FHR fell more slowly (‘was attenuated’) and that peripheral vasoconstriction during the final occlusion was reduced, after 4 h of repeated hypoxaemia (Giussani et al. 1997). These studies could be taken to suggest that exposure to mild hypoxaemia either antenatally, or during early labour, could impair the fetal adaptation to more intense hypoxaemia during active labour. However, it is important to appreciate that this protocol is much more prolonged, less frequent and milder than typical hypoxaemia during labour, and critically did not result in acidaemia or hypotension. It is therefore reasonable to suggest that these findings likely reflect effective adaptation through humoral mechanisms to a non‐life‐threatening insult. More recent studies suggest that peripheral chemoreflex activation becomes progressively more intense and even more rapid during labour‐like hypoxaemia and progressive acidosis/hypotension (Bennet et al. 2005; Ross et al. 2013). The finding of attenuated responses in the studies of mild hypoxaemia reinforces the concept that the peripheral chemoreflex is a tailored response, with reduced responses observed when fetal survival is not threatened but augmented during greater homeostatic challenges. This therefore fulfils a need to balance conservation of oxygen against the need to allow peripheral perfusion and oxygen‐consuming behaviour/development.
The importance of prelabour placental function and anaerobic reserve
The above studies were performed in healthy near‐term fetal sheep. It is well known that fetal growth restriction is associated with high rates of perinatal complications (Chauhan et al. 2017; Temming et al. 2017). Near‐term fetal sheep with spontaneous pre‐existing hypoxaemia showed early onset of hypotension even during 1 min complete umbilical cord occlusions repeated every 5 min (consistent with early labour) whereas normoxic fetal sheep were able to remain normotensive (Westgate et al. 2005). The blood pressure response was likewise characterised by initial hypertension during occlusions at the start of the experiment and shifted to a biphasic pattern of initial hypertension followed by progressive hypotension during occlusions (Wassink et al. 2013). The progressive development of severe acidaemia (pH 7.07 ± 0.05, base deficit 14.5 ± 1.7 mmol L−1, lactate 9.3 ± 2.2 mmol L−1 after the final occlusion) in these fetuses shows that despite 4 min of reperfusion, placental function was insufficient to reverse even a low frequency of impaired gaseous exchange (Westgate et al. 2005; Wassink et al. 2013). The specific mechanisms of the failure of these fetuses to tolerate low frequency occlusions compared to healthy fetal sheep are unclear, but likely reflect reduced reserves of myocardial glycogen (Shelley, 1961).
Perspectives and the search for effective biomarkers
It is often implied that hypoxaemia is only a significant factor in the minority of births associated with severe acidaemia and fetal compromise. This likely originates in the historical belief that most intrapartum decelerations do not reflect hypoxaemia (Lear et al. 2016b). As reviewed here, hypoxaemia, or more correctly the fetal adaptation to repeated brief hypoxaemia, is the key feature regulating intrapartum FHR changes, including the vast majority of intrapartum decelerations. The ubiquitous nature of hypoxaemia during labour is balanced by the remarkable fetal tolerance and ability to mount coordinated adaptations against hypoxaemia. The recent studies highlighted in this review reveal that the peripheral chemoreflex remains an integral part of the fetal adaptation to labour‐like hypoxaemia, even when fetuses are developing severe metabolic acidaemia and hypotension with evolving neural injury (de Haan et al. 1997; Wassink et al. 2013). Because of these robust fetal adaptations, and the fact that injury only occurs in a narrow window between intact survival and death, identification of fetal hypoxaemia per se is not very useful in clinical practice.
There is therefore a critical need to identify new biomarkers to identify fetuses at risk of failing to tolerate labour. The simplest and most straightforward approach to improving the usefulness of current fetal heart rate monitoring is to appreciate the physiological significance of intrapartum decelerations. The focus of clinical training should be redirected to the depth, duration and frequency of intrapartum decelerations, rather than their timing. Supporting this, recent evidence from a large prospective cohort has highlighted that total deceleration area (i.e. taking into account the depth, duration and frequency of all decelerations) shows the best association with fetal acidaemia (Cahill et al. 2018). Impressively, this study suggested that the sole use of total deceleration area would be associated with five Caesareans needing to be performed to prevent one case of acidaemia (Cahill et al. 2018). This and similar studies have highlighted the superior clinical utility of assessing decelerations in a physiologically relevant manner, in contrast to the historical focus on timing (Cahill et al. 2012b; Triebwasser et al. 2016; Martí Gamboa et al. 2017; Georgieva et al. 2017). Future work should establish population‐based thresholds of the burden or area of decelerations that the average healthy human fetus can tolerate during labour. This in turn must be balanced by the understanding that the tolerance of individual fetuses is highly variable and influenced by fetal anaerobic reserve and overall pre‐labour health (Westgate et al. 2005; Wassink et al. 2013; Amaya et al. 2016). There is additional need to better understand the implications of further factors, including antenatal glucocorticoid treatment and gestational age. Fetal sheep experiments have shown that dexamethasone and advancing gestational age affect the pattern and magnitude of FHR responses to moderate isocapnic hypoxaemia (Fletcher et al. 2003, 2006; Jellyman et al. 2005). Further studies are needed to clarify the implications of these factors for severe repetitive labour‐like hypoxaemia.
Clinical monitoring using total deceleration area will likely only allow stratification of risk. We propose that ideally deceleration area should be used in conjunction with additional biomarkers of fetal compromise. The ‘Holy Grail’ would of course be a continuous measure of fetal arterial blood pressure or cerebral perfusion, but this is currently not feasible. Changes in fetal heart rate variability, baseline FHR and additional measures such as ST segment changes can offer information on fetal wellbeing. However, decades of clinical and preclinical work leads to the conclusion that these are only partially effective as biomarkers of fetal compromise (Westgate et al. 2007; Cahill et al. 2012a, 2018; Lear et al. 2016b). Considering the evidence discussed in this review that progressive hypotension and eventual cerebral hypoperfusion are primarily associated with impaired myocardial contractility rather than autonomic impairment, we propose that future searches for biomarkers should be specifically tailored towards markers of failing contractility, assessed during intrapartum decelerations in particular. Given that the current system of intrapartum FHR interpretation is heavily based on outdated physiological concepts, we propose that the first step is the physiologically correct understanding of the big picture – that intrapartum decelerations are a direct index of the fetal reflex responses to fetal hypoxaemia.
Additional information
Competing interests
None declared.
Author contributions
C.A.L. and A.J.G. conceptualised this review. C.A.L. drafted the manuscript. C.A.L., G.W., J.A.W., J.G.N., A.U., R.G., L.B. and A.J.G. were involved in the critical review and revision of this manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This review and the authors’ studies were funded by grants from the Health Research Council of New Zealand (grant numbers 14/216 and 17/601).
Biographies
Christopher Lear is a Research Fellow with the Fetal Physiology and Neuroscience Group, University of Auckland, New Zealand. His interests include the physiological adaptation to labour, and how understanding this fundamental physiology can improve the identification of fetuses at risk of hypoxic–ischaemic brain injury.

Alistair Jan Gunn is a paediatrician–scientist who has conducted ground breaking basic research into ways of identifying compromised fetuses in labour, the mechanisms and treatment of asphyxial brain injury and the mechanisms of life threatening events in infancy. He has helped to develop a range of new, clinically relevant chronically instrumented fetal sheep paradigms to support translation of the team's findings to clinical practice. His research helped to establish mild cooling as the first ever technique to reduce brain injury due to low oxygen levels at birth.
Edited by: Ole Petersen & Dino Giussani
References
- Amaya KE, Matushewski B, Durosier LD, Frasch MG, Richardson BS & Ross MG (2016). Accelerated acidosis in response to variable fetal heart rate decelerations in chronically hypoxic ovine fetuses. Am J Obstet Gynecol 214, 270.e1–270.e8. [DOI] [PubMed] [Google Scholar]
- Ayres‐de‐Campos D & Arulkumaran S (2015). FIGO consensus guidelines on intrapartum fetal monitoring: Physiology of fetal oxygenation and the main goals of intrapartum fetal monitoring. Int J Gynaecol Obstet 131, 5–8. [DOI] [PubMed] [Google Scholar]
- Ayres‐de‐Campos D, Spong CY & Chandraharan E (2015). FIGO consensus guidelines on intrapartum fetal monitoring: Cardiotocography. Int J Gynaecol Obstet 131, 13–24. [DOI] [PubMed] [Google Scholar]
- Bakker PC, Kurver PH, Kuik DJ & Van Geijn HP (2007). Elevated uterine activity increases the risk of fetal acidosis at birth. Am J Obstet Gynecol 196, 313.e1–313.e6. [DOI] [PubMed] [Google Scholar]
- Barcroft J (1946). Researches in Prenatal Life. Blackwell Scientific Publications Ltd, London and Oxford. [Google Scholar]
- Bartelds B, Knoester H, Smid GB, Takens J, Visser GH, Penninga L, van der Leij FR, Beaufort‐Krol GC, Zijlstra WG, Heymans HS & Kuipers JR (2000). Perinatal changes in myocardial metabolism in lambs. Circulation 102, 926–931. [DOI] [PubMed] [Google Scholar]
- Bax M & Nelson KB (1993). Birth asphyxia: a statement. World Federation of Neurology Group. Dev Med Child Neurol 35, 1022–1024. [PubMed] [Google Scholar]
- Bennet L (2017). Sex, drugs and rock and roll: tales from preterm fetal life. J Physiol 595, 1865–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennet L, Wassink G & Gunn AJ. (2017). Fetal responses to asphyxia In Fetal and Neonatal Brain Injury, 5th edn, ed. Stevenson D, Benitz W, Sunshine P, Hintz S. & Druzin M, pp. 187–211. Cambridge University Press, Cambridge. [Google Scholar]
- Bennet L, Westgate JA, Lui YC, Wassink G & Gunn AJ (2005). Fetal acidosis and hypotension during repeated umbilical cord occlusions are associated with enhanced chemoreflex responses in near‐term fetal sheep. J Appl Physiol (1985) 99, 1477–1482. [DOI] [PubMed] [Google Scholar]
- Brain KL, Allison BJ, Niu Y, Cross CM, Itani N, Kane AD, Herrera EA & Giussani DA (2015). Induction of controlled hypoxic pregnancy in large mammalian species. Physiol Rep 3, e12614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar HS, Platt LD, DeVore GR, Horenstein J & Medearis AL (1988). Qualitative assessment of maternal uterine and fetal umbilical artery blood flow and resistance in laboring patients by Doppler velocimetry. Am J Obstet Gynecol 158, 952–956. [DOI] [PubMed] [Google Scholar]
- Broughton‐Pipkin F, Lumbers ER & Mott JC (1974). Factors influencing plasma renin and angiotensin II in the conscious pregnant ewe and its foetus. J Physiol 243, 619–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill AG, Roehl KA, Odibo AO & Macones GA (2012a). Association and prediction of neonatal acidemia. Am J Obstet Gynecol 207, 206.e1–206.e8. [DOI] [PubMed] [Google Scholar]
- Cahill AG, Roehl KA, Odibo AO & Macones GA (2012b). Association of atypical decelerations with acidemia. Obstet Gynecol 120, 1387–1393. [DOI] [PubMed] [Google Scholar]
- Cahill AG, Tuuli MG, Stout MJ, Lopez JD & Macones GA (2018). A prospective cohort study of fetal heart rate monitoring: Deceleration area is predictive of fetal acidemia. Am J Obstet Gynecol (in press; 10.1016/j.ajog.2018.01.026). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter BS, Moores RR Jr, Teng C, Meschia G & Battaglia FC (1995). Main routes of plasma lactate carbon disposal in the midgestation fetal lamb. Biol Neonate 67, 295–300. [DOI] [PubMed] [Google Scholar]
- Chauhan SP, Rice MM, Grobman WA, Bailit J, Reddy UM, Wapner RJ, Varner MW, Thorp JM Jr, Leveno KJ, Caritis SN, Prasad M, Tita ATN, Saade G, Sorokin Y, Rouse DJ & Tolosa JE (2017). Neonatal morbidity of small‐ and large‐for‐gestational‐age neonates born at term in uncomplicated pregnancies. Obstet Gynecol 130, 511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawes GS, Mott JC & Shelley HJ (1959). The importance of cardiac glycogen for the maintenance of life in foetal lambs and newborn animals during anoxia. J Physiol 146, 516–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan HH, Gunn AJ, Williams CE & Gluckman PD (1997). Brief repeated umbilical cord occlusions cause sustained cytotoxic cerebral edema and focal infarcts in near‐term fetal lambs. Pediatr Res 41, 96–104. [DOI] [PubMed] [Google Scholar]
- Drury PP, Bennet L, Booth LC, Davidson JO, Wassink G & Gunn AJ (2012). Maturation of the mitochondrial redox response to profound asphyxia in fetal sheep. PLoS One 7, e39273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstone DI, Rudolph AM & Heymann MA (1980). Effects of hypoxemia and decreasing umbilical flow on liver and ductus venosus blood flows in fetal lambs. Am J Physiol 238, H656–H663. [DOI] [PubMed] [Google Scholar]
- Fairlie FM, Lang GD & Sheldon CD (1989). Umbilical artery flow velocity waveforms in labour. Br J Obstet Gynaecol 96, 151–157. [DOI] [PubMed] [Google Scholar]
- Fleischer A, Anyaegbunam AA, Schulman H, Farmakides G & Randolph G (1987). Uterine and umbilical artery velocimetry during normal labor. Am J Obstet Gynecol 157, 40–43. [DOI] [PubMed] [Google Scholar]
- Fletcher AJ, Edwards CM, Gardner DS, Fowden AL & Giussani DA (2000). Neuropeptide Y in the sheep fetus: effects of acute hypoxemia and dexamethasone during late gestation. Endocrinology 141, 3976–3982. [DOI] [PubMed] [Google Scholar]
- Fletcher AJ, Gardner DS, Edwards CM, Fowden AL & Giussani DA (2003). Cardiovascular and endocrine responses to acute hypoxaemia during and following dexamethasone infusion in the ovine fetus. J Physiol 549, 271–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher AJ, Gardner DS, Edwards CM, Fowden AL & Giussani DA (2006). Development of the ovine fetal cardiovascular defense to hypoxemia towards full term. Am J Physiol Heart Circ Physiol 291, H3023–H3034. [DOI] [PubMed] [Google Scholar]
- Fujii EY, Takahashi N, Kodama Y, Roman C, Ferriero DM & Parer JT (2003). Hemodynamic changes during complete umbilical cord occlusion in fetal sheep related to hippocampal neuronal damage. Am J Obstet Gynecol 188, 413–418. [DOI] [PubMed] [Google Scholar]
- Galinsky R, Jensen EC, Bennet L, Mitchell CJ, Gunn ER, Wassink G, Fraser M, Westgate JA & Gunn AJ (2014). Sustained sympathetic nervous system support of arterial blood pressure during repeated brief umbilical cord occlusions in near‐term fetal sheep. Am J Physiol Regul Integr Comp Physiol 306, R787–R795. [DOI] [PubMed] [Google Scholar]
- Galinsky R, Lear CA, Yamaguchi K, Wassink G, Westgate JA, Bennet L & Gunn AJ (2016). Cholinergic and β‐adrenergic control of cardiovascular reflex responses to brief repeated asphyxia in term‐equivalent fetal sheep. Am J Physiol Regul Integr Comp Physiol 311, R949–R956. [DOI] [PubMed] [Google Scholar]
- Georgieva A, Redman CW & Papageorghiou AT (2017). Computerized data‐driven interpretation of the intrapartum cardiotocogram: a cohort study. Acta Obstet Gynecol Scand 96, 883–891. [DOI] [PubMed] [Google Scholar]
- Giussani DA (2016). The fetal brain sparing response to hypoxia: physiological mechanisms. J Physiol 594, 1215–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giussani DA, McGarrigle HH, Moore PJ, Bennet L, Spencer JA & Hanson MA (1994a). Carotid sinus nerve section and the increase in plasma cortisol during acute hypoxia in fetal sheep. J Physiol 477, 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giussani DA, McGarrigle HH, Spencer JA, Moore PJ, Bennet L & Hanson MA (1994b). Effect of carotid denervation on plasma vasopressin levels during acute hypoxia in the late‐gestation sheep fetus. J Physiol 477, 81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giussani DA, Spencer JA, Moore PJ, Bennet L & Hanson MA (1993). Afferent and efferent components of the cardiovascular reflex responses to acute hypoxia in term fetal sheep. J Physiol 461, 431–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giussani DA, Unno N, Jenkins SL, Wentworth RA, Derks JB, Collins JH & Nathanielsz PW (1997). Dynamics of cardiovascular responses to repeated partial umbilical cord compression in late‐gestation sheep fetus. Am J Physiol 273, H2351–H2360. [DOI] [PubMed] [Google Scholar]
- Green LR, Kawagoe Y, Homan J, White SE & Richardson BS (2001). Adaptation of cardiovascular responses to repetitive umbilical cord occlusion in the late gestation ovine fetus. J Physiol 535, 879–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn AJ, Maxwell L, de Haan HH, Bennet L, Williams CE, Gluckman PD & Gunn TR (2000). Delayed hypotension and subendocardial injury after repeated umbilical cord occlusion in near‐term fetal lambs. Am J Obstet Gynecol 183, 1564–1572. [DOI] [PubMed] [Google Scholar]
- Gunn AJ, Parer JT, Mallard EC, Williams CE & Gluckman PD (1992). Cerebral histologic and electrocorticographic changes after asphyxia in fetal sheep. Pediatr Res 31, 486–491. [DOI] [PubMed] [Google Scholar]
- Harding R & Bocking A (2001). Fetal Growth and Development. Cambridge University Press, Cambridge. [Google Scholar]
- Hon EH & Quilligan EJ (1967). The classification of fetal heart rate. II. A revised working classification. Conn Med 31, 779–784. [PubMed] [Google Scholar]
- Huch A, Huch R, Schneider H & Rooth G (1977). Continuous transcutaneous monitoring of fetal oxygen tension during labour. Br J Obstet Gynaecol 84, 1–39. [DOI] [PubMed] [Google Scholar]
- Ikeda T, Murata Y, Quilligan EJ, Choi BH, Parer JT, Doi S & Park SD (1998). Physiologic and histologic changes in near‐term fetal lambs exposed to asphyxia by partial umbilical cord occlusion. Am J Obstet Gynecol 178, 24–32. [DOI] [PubMed] [Google Scholar]
- Itskovitz J, LaGamma EF & Rudolph AM (1983). Heart rate and blood pressure responses to umbilical cord compression in fetal lambs with special reference to the mechanism of variable deceleration. Am J Obstet Gynecol 147, 451–457. [DOI] [PubMed] [Google Scholar]
- Itskovitz J, LaGamma EF & Rudolph AM (1987). Effects of cord compression on fetal blood flow distribution and O2 delivery. Am J Physiol 252, H100–H109. [DOI] [PubMed] [Google Scholar]
- Janbu T & Nesheim BI (1987). Uterine artery blood velocities during contractions in pregnancy and labour related to intrauterine pressure. Br J Obstet Gynaecol 94, 1150–1155. [DOI] [PubMed] [Google Scholar]
- Jellyman JK, Gardner DS, Edwards CM, Fowden AL & Giussani DA (2005). Fetal cardiovascular, metabolic and endocrine responses to acute hypoxaemia during and following maternal treatment with dexamethasone in sheep. J Physiol 567, 673–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen A, Garnier Y & Berger R (1999). Dynamics of fetal circulatory responses to hypoxia and asphyxia. Eur J Obstet Gynecol Reprod Biol 84, 155–172. [DOI] [PubMed] [Google Scholar]
- Jensen A, Hohmann M & Kunzel W (1987). Dynamic changes in organ blood flow and oxygen consumption during acute asphyxia in fetal sheep. J Dev Physiol 9, 543–559. [PubMed] [Google Scholar]
- Jensen A & Lang U (1992). Foetal circulatory responses to arrest of uterine blood flow in sheep: effects of chemical sympathectomy. J Dev Physiol 17, 75–86. [PubMed] [Google Scholar]
- Johnson N, van Oudgaarden E, Montague I & McNamara H (1994). The effect of oxytocin‐induced hyperstimulation on fetal oxygen. Br J Obstet Gynaecol 101, 805–807. [DOI] [PubMed] [Google Scholar]
- Jones CT & Robinson RO (1975). Plasma catecholamines in foetal and adult sheep. J Physiol 248, 15–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones R, Heep A & Odd D (2017). Biochemical and clinical predictors of hypoxic‐ischemic encephalopathy after perinatal asphyxia. J Matern Fetal Neonatal Med 31, 791–796. [DOI] [PubMed] [Google Scholar]
- Jonsson M, Agren J, Norden‐Lindeberg S, Ohlin A & Hanson U (2014). Neonatal encephalopathy and the association to asphyxia in labor. Am J Obstet Gynecol 211, 667.e1–667.e8. [DOI] [PubMed] [Google Scholar]
- Kiserud T, Rasmussen S & Skulstad S (2000). Blood flow and the degree of shunting through the ductus venosus in the human fetus. Am J Obstet Gynecol 182, 147–153. [DOI] [PubMed] [Google Scholar]
- Klink F, Grosspietzsch R, Klitzing LV & Oberheuser F (1981). Uterine contraction intervals and transcutaneous levels of fetal oxygen pressure. Obstet Gynecol 57, 437–440. [PubMed] [Google Scholar]
- Kro GA, Yli BM, Rasmussen S, Noren H, Amer‐Wahlin I, Saugstad OD, Stray‐Pedersen B & Rosen KG (2010). A new tool for the validation of umbilical cord acid–base data. BJOG 117, 1544–1552. [DOI] [PubMed] [Google Scholar]
- Lear CA, Galinsky R, Wassink G, Mitchell CJ, Davidson JO, Westgate JA, Bennet L & Gunn AJ (2016a). Sympathetic neural activation does not mediate heart rate variability during repeated brief umbilical cord occlusions in near‐term fetal sheep. J Physiol 594, 1265–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lear CA, Galinsky R, Wassink G, Yamaguchi K, Davidson JO, Westgate JA, Bennet L & Gunn AJ (2016b). The myths and physiology surrounding intrapartum decelerations: the critical role of the peripheral chemoreflex. J Physiol 594, 4711–4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Gudmundsson S & Olofsson P (2003). Uterine artery blood flow velocity waveforms during uterine contractions. Ultrasound Obstet Gynecol 22, 578–585. [DOI] [PubMed] [Google Scholar]
- Li H, Gudmundsson S & Olofsson P (2006). Acute centralization of blood flow in compromised human fetuses evoked by uterine contractions. Early Hum Dev 82, 747–752. [DOI] [PubMed] [Google Scholar]
- McNamara H & Johnson N (1995). The effect of uterine contractions on fetal oxygen saturation. Br J Obstet Gynaecol 102, 644–647. [DOI] [PubMed] [Google Scholar]
- Mallard EC, Williams CE, Johnston BM & Gluckman PD (1994). Increased vulnerability to neuronal damage after umbilical cord occlusion in fetal sheep with advancing gestation. Am J Obstet Gynecol 170, 206–214. [DOI] [PubMed] [Google Scholar]
- Martí Gamboa S, Lapresta Moros M, Pascual Mancho J, Lapresta Moros C & Castán Mateo S (2017). Deceleration area and fetal acidemia. J Matern Fetal Neonatal Med 30, 2578–2584. [DOI] [PubMed] [Google Scholar]
- Martinez‐Biarge M, Madero R, Gonzalez A, Quero J & Garcia‐Alix A (2012). Perinatal morbidity and risk of hypoxic‐ischemic encephalopathy associated with intrapartum sentinel events. Am J Obstet Gynecol 206, 148.e1–148.e7. [DOI] [PubMed] [Google Scholar]
- Modanlou H, Yeh SY & Hon EH (1974). Fetal and neonatal acid‐base balance in normal and high‐risk pregnancies: during labor and the first hour of life. Obstet Gynecol 43, 347–353. [PubMed] [Google Scholar]
- Morrison JL (2008). Sheep models of intrauterine growth restriction: fetal adaptations and consequences. Clin Exp Pharmacol Physiol 35, 730–743. [DOI] [PubMed] [Google Scholar]
- Murakami M, Kanzaki T, Utsu M & Chiba Y (1985). Changes in the umbilical venous blood flow of human fetus in labor. Nihon Sanka Fujinka Gakkai Zasshi 37, 776–782. [PubMed] [Google Scholar]
- Peebles DM, Edwards AD, Wyatt JS, Bishop AP, Cope M, Delpy DT & Reynolds EO (1992). Changes in human fetal cerebral hemoglobin concentration and oxygenation during labor measured by near‐infrared spectroscopy. Am J Obstet Gynecol 166, 1369–1373. [DOI] [PubMed] [Google Scholar]
- Peebles DM, Spencer JA, Edwards AD, Wyatt JS, Reynolds EO, Cope M & Delpy DT (1994). Relation between frequency of uterine contractions and human fetal cerebral oxygen saturation studied during labour by near infrared spectroscopy. Br J Obstet Gynaecol 101, 44–48. [DOI] [PubMed] [Google Scholar]
- Perez R, Espinoza M, Riquelme R, Parer JT & Llanos AJ (1989). Arginine vasopressin mediates cardiovascular responses to hypoxemia in fetal sheep. Am J Physiol 256, R1011–R1018. [DOI] [PubMed] [Google Scholar]
- Ramsey EM (1968). Uteroplacental circulation during labor. Clin Obstet Gynecol 11, 78–95. [DOI] [PubMed] [Google Scholar]
- Ross MG, Jessie M, Amaya K, Matushewski B, Durosier LD, Frasch MG & Richardson BS (2013). Correlation of arterial fetal base deficit and lactate changes with severity of variable heart rate decelerations in the near‐term ovine fetus. Am J Obstet Gynecol 208, 285.e1–285.e6. [DOI] [PubMed] [Google Scholar]
- Rudolph AM (1985). Distribution and regulation of blood flow in the fetal and neonatal lamb. Circ Res 57, 811–821. [DOI] [PubMed] [Google Scholar]
- Sakai M, Kozuma S, Okai T, Kagawa H, Ryo E & Taketani Y (1997). Doppler blood flow velocity waveforms of the umbilical artery during variable decelerations in labor. Int J Gynaecol Obstet 59, 207–211. [DOI] [PubMed] [Google Scholar]
- Sato M, Noguchi J, Mashima M, Tanaka H & Hata T (2016). 3D power Doppler ultrasound assessment of placental perfusion during uterine contraction in labor. Placenta 45, 32–36. [DOI] [PubMed] [Google Scholar]
- Shelley HJ (1961). Glycogen reserves and their changes at birth and in anoxia. Br Med Bull 17, 137–143. [Google Scholar]
- Simpson KR & James DC (2008). Effects of oxytocin‐induced uterine hyperstimulation during labor on fetal oxygen status and fetal heart rate patterns. Am J Obstet Gynecol 199, 34.e1–34.e5. [DOI] [PubMed] [Google Scholar]
- Sinding M, Peters DA, Frokjaer JB, Christiansen OB, Uldbjerg N & Sorensen A (2016). Reduced placental oxygenation during subclinical uterine contractions as assessed by BOLD MRI. Placenta 39, 16–20. [DOI] [PubMed] [Google Scholar]
- Siristatidis C, Salamalekis E, Kassanos D, Loghis C & Creatsas G (2004). Evaluation of fetal intrapartum hypoxia by middle cerebral and umbilical artery Doppler velocimetry with simultaneous cardiotocography and pulse oximetry. Arch Gynecol Obstet 270, 265–270. [DOI] [PubMed] [Google Scholar]
- Stewart RD, Bleich AT, Lo JY, Alexander JM, McIntire DD & Leveno KJ (2012). Defining uterine tachysystole: how much is too much? Am J Obstet Gynecol 207, 290.e1–290.e6. [DOI] [PubMed] [Google Scholar]
- Tadmor O, Bocker Y, Rabinowitz R, Aboulafia Y, Yagel S, Stark M, Diamant YZ & Nitzan M (1999). Analysis of umbilical artery flow parameters during fetal variable decelerations using computerized Doppler waveforms. Fetal Diagn Ther 14, 2–10. [DOI] [PubMed] [Google Scholar]
- Temming LA, Dicke JM, Stout MJ, Rampersad RM, Macones GA, Tuuli MG & Cahill AG (2017). Early second‐trimester fetal growth restriction and adverse perinatal outcomes. Obstet Gynecol 130, 865–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakor AS & Giussani DA (2009). Effects of acute acidemia on the fetal cardiovascular defense to acute hypoxemia. Am J Physiol Regul Integr Comp Physiol 296, R90–R99. [DOI] [PubMed] [Google Scholar]
- Triebwasser JE, Colvin R, Macones GA & Cahill AG (2016). Nonreassuring fetal status in the second stage of labor: Fetal monitoring features and association with neonatal outcomes. Am J Perinatol 33, 665–670. [DOI] [PubMed] [Google Scholar]
- Wassink G, Bennet L, Davidson JO, Westgate JA & Gunn AJ (2013). Pre‐existing hypoxia is associated with greater EEG suppression and early onset of evolving seizure activity during brief repeated asphyxia in near‐term fetal sheep. PLoS One 8, e73895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss E, Hitschold T & Berle P (1991). Umbilical artery blood flow velocity waveforms during variable deceleration of the fetal heart rate. Am J Obstet Gynecol 164, 534–540. [DOI] [PubMed] [Google Scholar]
- West CR, Curr L, Battin MR, Harding JE, McCowan LM, Belgrave S, Knight DB & Westgate JA (2005). Antenatal antecedents of moderate or severe neonatal encephalopathy in term infants – a regional review. Aust N Z J Obstet Gynaecol 45, 207–210. [DOI] [PubMed] [Google Scholar]
- Westgate JA, Bennet L, de Haan HH & Gunn AJ (2001). Fetal heart rate overshoot during repeated umbilical cord occlusion in sheep. Obstet Gynecol 97, 454–459. [DOI] [PubMed] [Google Scholar]
- Westgate JA, Bennet L & Gunn AJ (1999a). Fetal heart rate variability changes during brief repeated umbilical cord occlusion in near term fetal sheep. Br J Obstet Gynaecol 106, 664–671. [DOI] [PubMed] [Google Scholar]
- Westgate JA, Gunn AJ & Gunn TR (1999b). Antecedents of neonatal encephalopathy with fetal acidaemia at term. Br J Obstet Gynaecol 106, 774–782. [DOI] [PubMed] [Google Scholar]
- Westgate JA, Wassink G, Bennet L & Gunn AJ (2005). Spontaneous hypoxia in multiple pregnancy is associated with early fetal decompensation and greater T wave elevation during brief repeated cord occlusion in near‐term fetal sheep. Am J Obstet Gynecol 193, 1526–1533. [DOI] [PubMed] [Google Scholar]
- Westgate JA, Wibbens B, Bennet L, Wassink G, Parer JT & Gunn AJ (2007). The intrapartum deceleration in center stage: a physiological approach to interpretation of fetal heart rate changes in labor. Am J Obstet Gynecol 197, 236.e1–236.e11. [DOI] [PubMed] [Google Scholar]
- Winkler M & Rath W (1999). A risk‐benefit assessment of oxytocics in obstetric practice. Drug Saf 20, 323–345. [DOI] [PubMed] [Google Scholar]