

Abstract
Properties of the local internal environment of the adult brain are tightly controlled providing a stable milieu essential for its normal function. The mechanisms involved in this complex control are structural, molecular and physiological (influx and efflux transporters) frequently referred to as the ‘blood–brain barrier’. These mechanisms include regulation of ion levels in brain interstitial fluid essential for normal neuronal function, supply of nutrients, removal of metabolic products, and prevention of entry or elimination of toxic agents. A key feature is cerebrospinal fluid secretion and turnover. This is much less during development, allowing greater accumulation of permeating molecules. The overall effect of these mechanisms is to tightly control the exchange of molecules into and out of the brain. This review presents experimental evidence currently available on the status of these mechanisms in developing brain. It has been frequently stated for over nearly a century that the blood–brain barrier is not present or at least is functionally deficient in the embryo, fetus and newborn. We suggest the alternative hypothesis that the barrier mechanisms in developing brain are likely to be appropriately matched to each stage of its development. The contributions of different barrier mechanisms, such as changes in constituents of cerebrospinal fluid in relation to specific features of brain development, for example neurogenesis, are only beginning to be studied. The evidence on this previously neglected aspect of brain barrier function is outlined. We also suggest future directions this field could follow with special emphasis on potential applications in a clinical setting.
Keywords: blood‐brain barrier, cerebrospinal fluid, choroid plexus, embryo, transport, protein, amino acids, electrolytes, ion gradients, gene transcripts, immunohistochemistry, electron microscope, meninges, blood vessels, tight junctions, epithelium, endothelium
Gene abbreviations: (For gene products (proteins), capitals are used.)
- Cacn
calcium voltage‐gated channel
- Clcnka
chloride voltage‐gated channel Ka
- Clic
chloride channel
- Gypa
glycophorin A
- Kcnh8
potassium voltage‐gated channel subfamily H member 8
- Kcnk9
potassium two pore domain channel subfamily K member 9
- Cacn
calcium voltage‐gated channel
- Kcnmb1
potassium calcium‐activated channel subfamily M regulatory beta subunit 1
- Kcnmb2
potassium calcium‐activated channel subfamily M regulatory beta subunit 2
- Scn
voltage gated sodium channels
- Scnn1g
sodium channel, nonvoltage‐gated 1 gamma
- Sparc
Secreted protein acidic and rich in cysteine; vamp, vesicle‐associated membrane proteins
- Tmc5
transmembrane channel‐like 5
- Trpv
transient receptor potential cation channel
- Wnt
wingless‐type MMTV (mouse mammary tumor virus) integration site
Introduction
The internal environment of the adult brain is defined and controlled by a series of mechanisms usually referred to colloquially as the blood–brain barrier. However, these are not unified identities, either in terms of their anatomical situation and structure, or in terms of their functional properties. The multifactorial characteristics of brain–blood interfaces are the main reason why misconceptions and controversies continue in the field (see box). One of the main areas of dispute has been the so‐called ‘immaturity’ of brain barriers, especially the blood–brain barrier proper (cerebral endothelial interface), in the developing brain.
It has been widely believed for nearly a century that these mechanisms in the fetal and newborn brain are absent or poorly developed. This belief appears to stem from misunderstandings and mistranslations of early studies, not helped by some poorly designed experiments (reviewed in detail in Saunders et al. 2014). In spite of extensive evidence to the contrary, this belief still persists (Allen, 2015; Oberdick et al. 2016; Panfoli et al. 2016; Amaraneni et al. 2017) with some new inventive terms appearing that imply some level of dysfunction: ‘inefficient’ (Panfoli et al. 2016), ‘primitive’ (Zhao et al. 2015). In place of this rather un‐illuminating view of blood–brain barrier mechanisms in the developing brain, we propose that the specific barrier mechanisms present at any particular stage of brain development are ones that are appropriate for that stage of its development. They can thus be investigated in relation to specific features of brain maturation, such as neurogenesis (Lehtinen & Walsh, 2011; Lehtinen et al. 2013; Lun et al. 2015).
Misconceptions and controversies discussed below
Continued use of the term ‘immature’ to indicate a functional deficiency in brain barrier mechanisms.
Do the first blood vessels that grow into the brain anlage have functionally effective intercellular tight junctions?
Does increased apparent permeability (leakiness) to dyes and small molecular mass markers reflect ‘breakdown’ of tight junctions?
Does greater accumulation of a molecule in developing brain and cerebrospinal fluid reflect greater permeability of barriers in the developing brain?
What is the role of astrocytes in formation (initiation) and maintenance of blood–brain barrier properties?
Permeability of the paracellular pathway.
In support of this proposition we summarize data that have accumulated over the past few decades from studies using classical physiological approaches and in more recent years from the application of molecular techniques. We also outline some key points in the biology of the field that are still controversial (box above) and indicate where this vital research may take us in the future.
Barrier interfaces between the brain and the rest of the organism
The fundamental structural component of almost all of the barrier interfaces is the tight junctions between the cells forming the interface. There are at least six identifiable barrier interfaces in the brain (including one that is exclusive to the fetal brain). These are illustrated and described in Fig. 1, with morphological details in Fig. 2. In summary these barrier interfaces are:
The blood–brain barrier (Figs 1 A and 2 Aa and b) is situated between the lumen of cerebral blood vessels and brain parenchyma. Tight junctions are present between the endothelial cells restricting permeability of the paracellular cleft (Brightman & Reese, 1969) to an extent that is still controversial (see below); however, the tight junctions in blood vessels of early developing brain appear to be impermeable to even very small molecules (Fig. 2 A). Additional details are in legends to Figs 1 and 2.
The blood‐cerebrospinal fluid barrier (Fig. 1 B) in the choroid plexus within each brain ventricle. The barrier forming cells are the epithelial cells, which have tight junctions at the apices of adjacent epithelial cells forming the structural basis of this barrier; they prevent penetration of even small molecules from blood to cerebrospinal fluid (CSF; Fig. 2 B). The blood vessels in the stroma of the choroid plexus are fenestrated and are thought not to form a barrier, although their endothelial cells appear to contain some key efflux transporters that prevent entry of many lipid‐soluble molecules into the brain and CSF (Møllgård et al. 2017). Extracellular matrix basement membrane may also contribute, but there are no astrocytic endfeet or pericytes.
Circumventricular organs (Fig. 1 C). These comprise the organum vasculosum laminae terminalis (OVLT), subfornical organ, median eminence, subcommissural organ (SCO)–pineal complex and area postrema. Apart from the SCO, which has a normal blood–brain barrier, blood vessels in the circumventricular organs have similar permeability characteristics to vessels elsewhere in the body. This allows feedback penetration of circulating peptide hormones. However, these and other molecules are prevented from entering the CSF by tanycytes, connected by tight junctions between their apices; the perivascular space is separated off by tight junctions from the CSF milieu of the adjacent neuropil, as illustrated in Fig. 2 Ca and b for the subcommissural organ (see Madsen & Møllgård, 1979).
Ependyma in adult brain (Figs 1 D and 2 D. Ependymal cells are linked by gap junctions; there is unrestricted exchange of even large molecules such as proteins between CSF and brain interstitial space (Brightman & Reese, 1969; Fossan et al. 1985).
The embryonic CSF–brain barrier (Fig. 1 E). This is a transient barrier between the CSF and brain parenchyma formed by strap junctions between adjacent neuroepithelial cells (Fig. 2 Ea and b; Møllgård et al. 1987) which restrict all but the smallest lipid insoluble molecules from entering the brain (Fossan et al. 1985; Whish et al. 2015). They disappear progressively during development and are no longer present when this interface becomes ependyma (Fig. 2 D; Møllgård et al. 1987).
The meningeal barrier (Fig. 1 F). This consists of three distinct interfaces: (i) blood–arachnoid–outer CSF, (ii) blood–pia microvessel–outer CSF and (iii) outer CSF–brain surface interfaces (Fig. 2 F). The outer CSF is the fluid‐filled subarachnoid space. Tight junctions are present between the cells of the arachnoid barrier layer restricting permeability between fenestrated blood vessels in the dura and the subarachnoid space. The blood vessels within this space have tight junctions between the endothelial cells, but no astroctyic end feet or pericytes. This is the interface that restricts exchange between the blood and the outer CSF.
Figure 1. Schematic diagram (middle right) of the five main barrier interfaces (A–D and F) in the adult and developing brain and an additional one present only in the embryo (E).
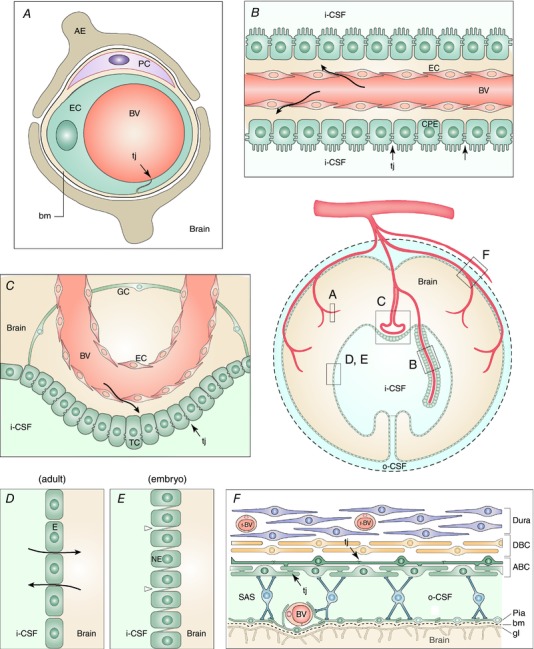
The barrier‐forming cellular layers at each interface are coloured green.
A, the blood–brain barrier is situated at the level of cerebral blood vessels (BV). Tight junctions (tj, arrowhead) are present between the endothelial cells (EC) restricting the paracellular cleft. AE, end feet from astroglial cells; bm, basement membrane; PC, pericytes. Other important components of this interface are a basement membrane of extracellular matrix, within which are embedded pericytes (Engelhardt & Sorokin, 2009) surrounding the endothelial cells (Daneman et al. 2010b, Errede et al. 2014). Astroglial end feet encircle cerebral blood vessels during the first 2–3 weeks of postnatal development in rodents (Caley & Maxwell, 1970) although the encirclement appears to be less complete than previously thought (Korogod et al. 2015). The contribution of astrocytes to development and maintenance of barrier properties is controversial as will be discussed. These cellular structures are known collectively as the neurovascular unit (Neuwelt, 2004). B, the blood–CSF barrier is situated in the choroid plexus within each brain ventricle. Barrier‐forming cells are the epithelial cells (CPE), which have tight junctions at their apical side (CSF facing, arrowheads). Blood vessels (BV) are fenestrated and do not form a barrier (arrows); apical microvilli increase exchange surface of epithelial cells to the internal CSF (i‐CSF) C, circumventricular organs (including median eminence, pineal gland, area postrema, subfornical organ). Blood vessels have permeability characteristics similar to elsewhere in the body and have the functional property of allowing feedback penetration of peptide hormones controlled by the hypothalamic–pituitary axis. These peptides and other molecules are prevented from entering the CSF by tanycytes (TC), the specialized ependymal cells of these brain areas, connected by tight junctions between their apices (arrowhead); entry into the rest of the brain is prevented by tight junctions between astroglial cells (GC). Away from the tanycyte layer, ependymal cells lining the ventricular system are linked by gap junctions that allow free exchange between the CSF and brain interstitial fluid. D, ependyma in adult brain. Apart from areas where there are specialized tanycytes, ependymal cells are linked by gap junctions that do not restrict exchange of even large molecules, such as proteins, between CSF and interstitial space of brain (arrows). E, the embryonic CSF–brain barrier. In early brain development, strap junctions (arrowheads) are present between adjacent neuroepithelial cells (NE); these form a barrier restricting the movement of larger molecules, such as proteins, but not smaller molecules. F, the meningeal barrier is structurally the most complex of all the brain barriers. Barrier‐forming cells are the outer layer of the arachnoid membrane (the arachnoid barrier cells; ABC); these have tight junctions (arrowheads) between adjacent cells forming a barrier between the outer cerebrospinal fluid (o‐CSF) in the subarachnoid space (SAS) and more superficial dural layers (dural border cells (DBC) and the dura mater). Blood vessels (BV) in the SAS have tight junctions with similar barrier characteristics as cerebral blood vessels without surrounding pericytes and astrocytic end‐feet. Blood vessels within the dura mater are fenestrated (f‐BV). bm, basement membrane; gl, glia limitans. Redrawn from Saunders et al. (2016b).
Figure 2. Morphology of the brain barriers illustrated in Fig. 1 .
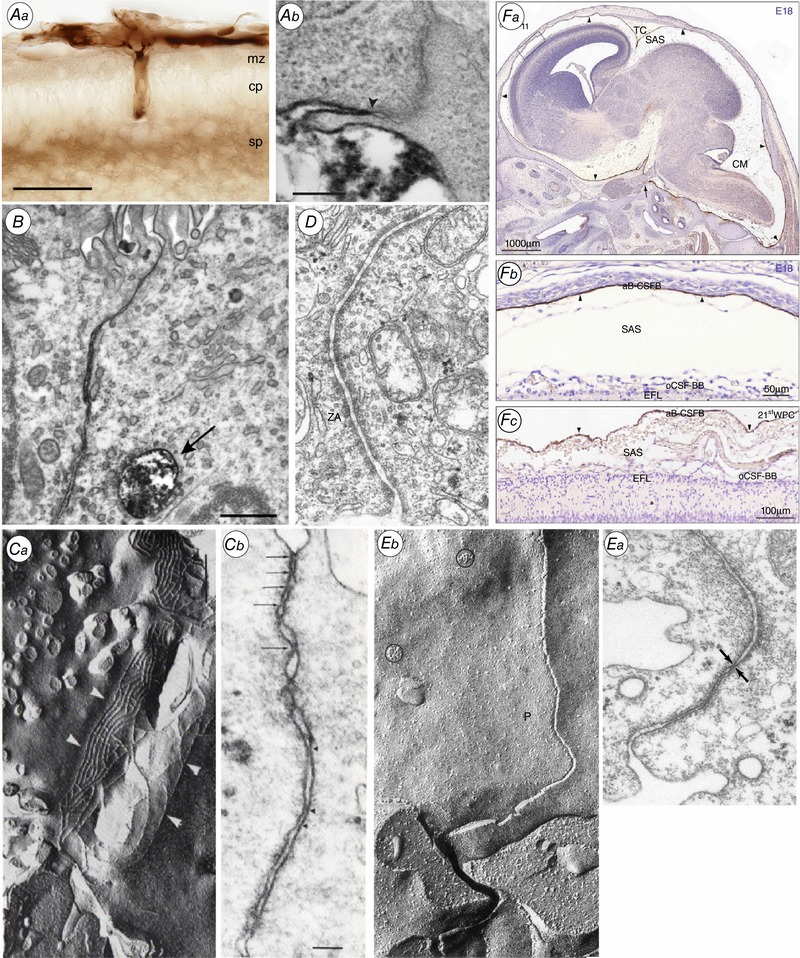
Aa, the blood–brain barrier. Light micrograph showing the localization of biotin ethylenediamine (BED) in the neocortex of opossum at P5 20–25 min after an intraperitoneal injection. Note that the staining for BED is most visible within the vessels, in the marginal and subplate zones. cp, cortical plate; mz, marginal zone; sp, subplate. Ab, localization of biotin–dextran (BDA3000) in the neocortex of P2 opossum 20–25 min after an intraperitoneal injection. Arrowhead points to site of the tight junction. Note that the marker is prevented from passing through the intercellular space by the tight junction. From Ek et al. (2006). B, the blood–CSF barrier. Lateral ventricular choroid plexus (blood–CSF barrier) in P13 Monodelphis. Tight junction excludes entry of BDA3000 into CSF. Labelled dextran present in one large and several smaller endosomes. From Ek et al. (2003). C, Circumventricular organs. a, adjacent tanycytes facing the CSF in circumventricular organs are connected by an extensive network of apical tight junctional strands shown here by freeze fracture of the adult Mongolian gerbil subcommissural organ. b, a thin section electron micrograph of the same region in a neonatal animal shows multiple ‘kissing points’ (arrows) between neighbouring tanycytes indicating a complete occlusion of the paracellular pathway. From Madsen & Møllgård (1979). D, ependyma in adult brain. Junctional configuration in the ependymal layer of 125‐day sheep fetus similar to those found in mature ependyma. ZA, zonulae adherens. Note unobstructed intercellular space. E, the embryonic CSF–brain barrier. a, thin‐section electron micrograph of the neuroepithelial lining of the cerebral vesicle from an E19 sheep. The junctional zone exhibits very narrow intercellular clefts which at places seem to be totally occluded (arrows). This junctional configuration has been named ‘strap’ junction. b, characteristic freeze fracture single strand of strap junction perpendicular to CSF surface. From Møllgård et al. (1987). F, the meningeal barrier. Distribution of claudin‐11 immunoreactivity in sagittal sections of E18 rat (a and b) and 21st wpc human (c) brain. Demonstrates a strong reactivity of the entire arachnoid barrier cell layer = arachnoid blood–CSF barrier (aB‐CSFB, arrowheads). From Brøchner et al. (2015). CM, cisterna magna; EFL, radial glial end feet layer; SAS, subarachnoid space; TC, tentorium cerebelli.
Molecular structure of tight junctions in brain barrier interfaces
The molecular structure of tight junctions in brain barrier interfaces has been extensively studied both in vivo and in vitro. There are numerous proteins that interact to form intercellular junctional structures in many tissues including ZO‐1, ZO‐2, occludin and several claudins. However, there is some degree of specificity in the brain barriers. For example, in the mammalian blood–brain barrier claudin 5 expression is specific to this interface (Nitta et al. 2003; Kratzer et al. 2012) although in zebrafish it is apparently also expressed at the blood–CSF barrier (van Leewen et al. 2018). Angiogensis and blood–brain barrier formation appear to be linked and occur simultaneously; by using genetic mouse models, the effectors of Wnt/b‐catenin signaling, including Lef1, Apcdd1 and tnfrsf19, have been shown to control these processes (reviewed in Daneman & Prat, 2015). Recently it has been shown that Reck (a glycosylphosphatidylinositol‐anchored membrane protein) and Gpr124, an orphan G‐protein‐coupled receptor, promote both CNS angiogenesis and blood–brain barrier formation by activating the canonical Wnt7a/7b pathway (Cho et al. 2017).
Functional implications of the junctional structures of brain barriers
It is important to stress that these interfaces are functionally much more than a structural barrier preventing passive exchange of molecules between the blood, CSF and brain. Nevertheless this seal is important not just because it prevents the intercellular passage of even very small (lipid insoluble) molecules, but also because this restriction allows the numerous cellular exchange mechanisms to act in concert over the full extent of the interface. For example without such a diffusion restriction it would not be possible for the ion exchange mechanisms to tightly control the ionic milieu of the brain's extracellular fluid that is essential for normal neuronal function.
Alternative route into the brain
There have also been suggestions of ‘another route’ of entry into the very early embryonic brain before most of the characteristics of the six barrier interfaces have appeared. Bueno and colleagues have carried out extensive studies of the properties of CSF in chick embryos (eCSF) before the appearance of the choroid plexuses. They have also studied the transfer of proteins and other molecules between the blood and neural tube tissue (Parvas & Bueno, 2010; Bueno et al. 2014). Their evidence suggests that the internal environment of the CNS at this very early stage of development is already well controlled. The route of transfer appears to be in the brainstem lateral to the floor plate, in the ventral mesencephalon and in the most anterior part of the ventral prosencephalon. This location does not correspond to the site at which the choroid plexus later develops. From studies of zebrafish embryos, Lowery & Sive (2009) suggest that eCSF is a secretion of the neuroepithelium forming the neural tube. However, the studies of Bueno and colleagues demonstrate clearly that the composition of eCSF in the chick embryo depends importantly on transfer from the blood into eCSF (Parvas & Bueno, 2010; Bueno et al. 2014).
Lehtinen and colleagues (Zappaterra et al. 2007; Chau et al. 2015) have carried out extensive studies of the composition of amniotic fluid and eCSF in the period after neural tube closure (described below in the section ‘Proteins in fetal and newborn CSF’).
Physiological and molecular evidence for effective barrier mechanisms in the developing brain
Evidence for early establishment of ion gradients between CSF and plasma
The identification of a gradient between CSF and plasma for even a single ion implies that at least two essential features of a barrier mechanism are present: (i) tight junctions between the cells of the interface that are functionally effective in enabling the gradient to be established, and (ii) a cellular pump that sets up the gradient. This is amongst the most convincing evidence for functional barrier mechanisms in the developing brain. The ionic composition of CSF is thought to reflect that of the general internal environment of the developing brain, although there is some controversy about the extent to which CSF composition actually reflects blood–brain barrier properties; this is important for correct interpretation of CSF data in clinical conditions in adult patients (Lange, 2013) but does not seem to be used in neonates (see below). There have been numerous studies in a variety of species showing the presence of ion gradients between CSF and plasma, some from very early in development, as summarized in Table 1 and illustrated for the rat in Fig. 3.
Table 1.
Electrolyte concentration (mmol L−1 or mEq kg−1 H2O) in CSF and plasma of embryos/newborns and adults
| Species | Age | Units | Fluid | Na+ | K+ | Cl− | HCO3 − | Ca2+ | Mg2+ | Ref |
|---|---|---|---|---|---|---|---|---|---|---|
| Monkey, term 168 days | E50–60 | mEq kg−1 H2O | CSF | 3.6 | 3.8 | 1.85 | 1 | |||
| Plasma | 4.1 | 4.9 | 1.5 | 1 | ||||||
| E90 | mEq kg−1 H2O | CSF | 3.2 | 3.5 | 2.3 | 1 | ||||
| Plasma | 4.0 | 5.0 | 1.5 | 1 | ||||||
| E130 | mEq kg−1 H2O | CSF | 2.9 | 2.9 | 2.2 | 1 | ||||
| Plasma | 3.8 | 4.9 | 1.5 | 1 | ||||||
| Adult | mEq kg−1 H2O | CSF | 2.6 | 2.3 | 1.9 | 1 | ||||
| Plasma | 4.5 | 4.6 | 1.2 | 1 | ||||||
| Rabbit, term 31 days | E23 | mEq L−1 | CSF | 142 | 4.1 | 113 | 1.5 | 2 | ||
| Plasma | 143 | 6.5 | 104 | 3.2 | 2 | |||||
| E27 | mEq L−1 | CSF | 140 | 4.0 | 110 | 1.6 | 2 | |||
| Plasma | 147 | 6.0 | 108 | 3.6 | 2 | |||||
| Adult | mEq L−1 | CSF | 150 | 3.0 | 120 | 1.4 | 2 | |||
| Plasma | 154 | 3.2 | 100 | 2.2 | 2 | |||||
| Rat, term 21 days | P0 | mEq L−1 | CSF | 144 | 3.8 | 109 | 2 | |||
| Plasma | 150 | 6.5 | 102 | 2 | ||||||
| P8 | mEq L−1 | CSF | 144 | 3.8 | 107 | 2 | ||||
| Plasma | 166 | 7.1 | 98 | 2 | ||||||
| Adult | mEq L−1 | CSF | 152 | 3.0 | 122 | 2 | ||||
| Plasma | 153 | 5.0 | 106 | 2 | ||||||
| Sheep, term 150 days | E44–50 | mEq L−1 | CSF | 135 | 5.4 | 113 | — | 2.0 | 3 | |
| Plasma | 138 | — | 113 | — | 1.6 | 3 | ||||
| E85–92 | mEq L−1 | CSF | 144 | 3.5 | 123 | 3.4 | 1.9 | 3 | ||
| Plasma | 136 | 3.8 | 104 | 6.05 | 1.8 | 3 | ||||
| Adult | mEq L−1 | CSF | 148 | 3.1 | 128 | 2.45 | 1.8 | 3 | ||
| Plasma | 138 | 3.4 | 115 | 3.4 | 1.4 | 3 | ||||
| Pig, term 115 days | 5 cm CRL | mmol L−1 H2O | CSF | 126 | 115 | 4 | ||||
| Plasma† | 125 | 112 | 4 | |||||||
| 6.5 cm CRL | mmol L−1 H2O | CSF | 120 | 115 | 4 | |||||
| Plasma† | 110 | 98 | 4 | |||||||
| 12.5 cm CRL | mmol L−1 H2O | CSF | 125 | 122 | 4 | |||||
| Plasma† | 105 | 96 | 4 | |||||||
| Pony, term 342 days | E312 | mmol L−1 | CSF | 143 | 3.7 | 109 | 5 | |||
| Serum | 134 | 5.7 | 5 | |||||||
| E342 | mmol L−1 | CSF | 143 | 2.9 | 112 | 5 | ||||
| Serum* | — | — | — | 5 | ||||||
| Adult | mmol L−1 | CSF | 144 | 2.9 | 101 | 5 | ||||
| Serum | 132 | 4.7 | 94 | 5 | ||||||
| Chick, hatch 20–21 days | E13 | mEq L−1 | CSF | 121 | 3.9 | 104 | 6 | |||
| Plasma | 117 | 6.3 | 101 | 6 | ||||||
| E15 | mEq L−1 | CSF | 125 | 3.6 | 103 | 6 | ||||
| Plasma | 120 | 5.8 | 91 | 6 | ||||||
| E19 | mEq L−1 | CSF | 129 | 3.8 | 109 | 6 | ||||
| Plasma | 134 | 7.8 | 93 | 6 | ||||||
| Adult | mmol kg−1 H2O | CSF | 159 | 4.2 | 142 | 7 | ||||
| Plasma | 160 | 4.7 | 115 | 7 | ||||||
| Human, 38 weeks’ post–conception | P0–4 weeks | mmol L−1 | CSF | 138 | 3.2 | 117 | 1.6 | 8 | ||
| Plasma | 136 | 5.4 | 105 | 2.3 | 8 | |||||
| P7–12 months | mmol L−1 | CSF | 142 | 2.5 | 120 | 1.4 | 8 | |||
| Plasma | 136 | 4.9 | 107 | 2.7 | 8 | |||||
| P7–14 years | mm L−1 | CSF | 141 | 2.8 | 122 | 1.3 | 8 | |||
| Plasma | 138 | 4.6 | 107 | 2.7 | 8 | |||||
| Adult | mEq kg−1 H2O | CSF | 147 | 2.9 | 113 | 23 | 2.3 | 2.2 | 9 | |
| Plasma | 150 | 4.6 | 99 | 27 | 4.7 | 1.6 | 9 |
References: (1) Bito & Myers (1970), cisternal CSF; (2) Amtorp & Sorensen (1974), cisternal CSF; (3) Bradbury et al. (1972), cisternal CSF; (4) Flexner (1938), cisternal CSF; (5) Rossdale et al. (1982), atlanto‐occipital subarachnoid CSF; (6) Stastny & Rychter (1976), ventricular CSF; (7) Anderson & Hazelwood (1969), cisternal CSF; (8) Heine et al. (1981), lumbar CSF; (9) Davson & Segal (1996), lumbar CSF. Note differences in units. The most appropriate are mmol L−1 or mEq kg−1 H2O as this allows for differences in protein concentration in CSF and plasma (see Davson, 1967, for discussion). These are markedly different early in development, as well as between CSF and plasma, see section on ‘Proteins in CSF’. *Not measured; †calculated from CSF/plasma ratios. CRL, crown rump length.
Figure 3. Ion gradients between CSF and plasma in developing and adult rat brain.
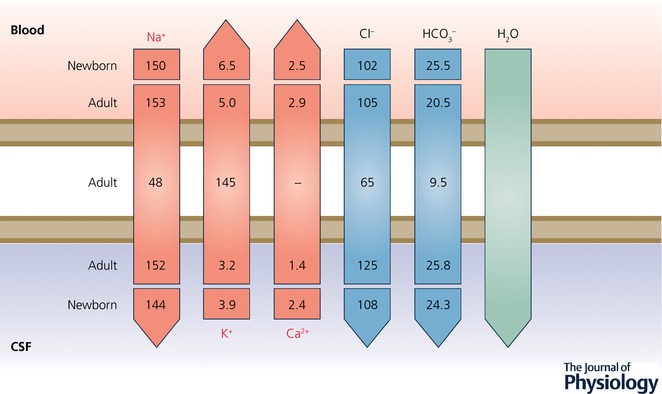
A characteristic of CSF is its stable ionic composition that differs from that of plasma to an extent that cannot be explained by ultrafiltration, as was once thought. Data for CSF and plasma (mEq L−1 H2O) are from Amtorp & Sørensen (1974) and for intracellular ions (mmol L−1 H2O) from Fig. 8 in Johanson & Murphy (1990). The gradients are the consequence of the complex interactions between enzymes (notably carbonic anhydrase) ion transporters and ion channels, as illustrated in Fig. 4. The CSF secretion rate in the embryo and newborn is much lower than in the adult (Bass & Lundborg, 1973; Johanson & Woodbury, 1974), which is perhaps explained by the much lower expression of carbonic anhydrase and ATPases in the developing choroid plexus, as indicated in Fig. 4. Redrawn from Saunders et al. (2016b).
A significant consideration in interpreting these results is that some ions (Ca2+ and Mg2+) bind to proteins in plasma and CSF, particularly to albumin, and the protein concentration in fetal CSF is much higher at a time when it is lower in plasma (see below). Protein concentrations in CSF and plasma are important because they affect the proportion of each fluid that the electrolytes are dispersed in. Thus accurate comparisons between electrolyte concentrations in the two fluids can only be made when the units take account of this (mEq kg−1 H2O). As can be seen in Table 1, most estimates have used units based on volume of CSF or plasma (which includes protein) and thus need to be interpreted with caution. Nevertheless it is clear from Table 1 that concentration gradients are established early in brain development. No values appear to be available for human embryos or fetuses; values for neonates and infant are rare. No reference values are given in textbooks of fetal and neonatal medicine and CSF electrolytes do not appear to be routinely measured in neonates (Royal Children's Hospital Melbourne; Royal Children's Hospital, Brisbane; Prof. R. Ariagno, Neonatologist, Stanford University). This misses the possibility of using ion gradients between CSF and plasma as a means of assessing choroid plexus and blood–brain barrier function in human neonates, as suggested many years ago by Bito & Myers (1970): ‘Existence of normal cation concentration gradients between CSF and blood may serve as a criterion for the normality of the (foetal) blood–brain barrier’.
More recently transcriptomic studies have shown that key ion channel and transporter genes are expressed very early in embryonic life in the choroid plexuses (Liddelow et al. 2012, 2013). These are summarized in Fig. 4. One puzzle is the astonishing number of some channel genes that are expressed in development when we have no idea whether they are functionally effective. Many of these channels are expressed at a higher level in the developing rat choroid plexus than in the adult. Numerous gene family members for K+ voltage gated channels were expressed at levels of between 2‐ and 210‐fold higher in E15 choroid plexus than adult. Seven Scn genes (voltage gated sodium channels), nine Trpv channels, two chloride channel genes (Clic1, Clic4) and two cyclic nucleotide gated channels (Cnga1 and Cnga3) were also expressed at a higher level in the developing choroid plexus. In addition there were six ion channel genes that were expressed only at E15 (Kcnmb2, Tmc5, Clcnka, Scnn1g, Kcnh8, Kcnmb1) and one in the adult (Kcnk9). Thirteen genes of the Cacn family of voltage gated Ca2+ subunits were expressed at higher levels in E15 choroid plexus than in the adult (Liddelow et al. 2013).
Figure 4. Localization of proteins for ion transporters, channels and associated enzymes and identification of their corresponding genes in adult and immature rat choroid plexus.
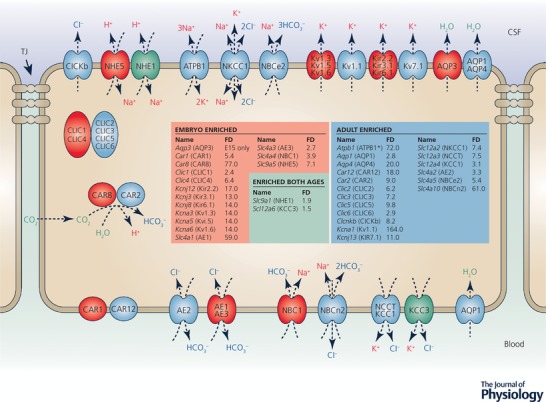
CSF secretion results from coordinated intracellular carbonic anhydrase activity and transport of ions and water from basolateral membrane to cytoplasm, then sequentially across apical membrane into the cerebral ventricles (Davson & Segal, 1996; Speake et al. 2001; Praetorius & Damkier, 2017). This process has only been studied in adult choroid plexus (Brown et al. 2004; Praetorius & Damkier, 2017). The membrane and intracellular locations of the ion channels, transporters and enzymes indicated are from Praetorius & Damkier (2017). Data from Liddelow et al. (2013) compares expression of these genes and other functionally related genes in E15 and adult rat lateral ventricular choroid plexus. Blue indicates the genes that are upregulated (enriched) in the adult. Light red indicates genes that are expressed at a higher level at E15. We have assumed the same cellular/membrane location for members of the same gene family. The genes all had substantial but variable transcript numbers in the RNA‐Seq analysis. In some cases where a gene was upregulated in the adult, the transcript number was also high in the embryo, suggesting this transporter or channel was likely to be functionally effective at both ages, e.g. the K+ channel Kcnj13 (Kir7.1), Slc12a2 (NKCC1) a Na+–K+–Cl− exchanger and Slc4a2 (NBCe2) a coupled Na+–HCO3 − pump. ATPB1 (Atpb1b1) is a Na+/K+‐ATPase. Green indicates genes that were expressed at similar levels at the two ages. There are many more channels that show age‐related differential expression in choroid plexus, the functions of which are unclear. Redrawn from Liddelow et al. (2016) with additional data from Liddelow et al. (2013).
Influx mechanisms
The main functional groups of influx transporters in brain barrier interfaces defined from physiological studies are those for glucose (GLUT‐1), amino acids (acidic, basic, neutral) monocarboxylic acids, peptides and ions, including metabolically important ions such as Fe2+, Cu2+ and Mg+. These mechanisms are summarized in Fig. 5. Most of the physiological studies were published many years ago, but this does not diminish their value in understanding the importance of such mechanisms in the developing brain. There is a fundamental difficulty in designing adequate experiments to study the mechanisms that control the influx of these molecules and ions into the brain and CSF because it is important, but technically difficult, to distinguish between transport and metabolic incorporation into different brain compartments and structures. This problem was solved by Oldendorf (1971) with the introduction of a short pass technique initially for studies in adult animals. Although technically more difficult, it has also been applied to a limited extent and with some modifications in developing animals (Braun et al. 1980; Cornford et al. 1982; Lefauconnier & Trouvé, 1983). The essence of the technique is that a radiolabelled compound is injected as close as possible to the cerebral circulation and the experiment is rapidly terminated after enough time for only one circuit through the cerebral circulation. Usually two labelled compounds are compared: one, which would be expected to enter almost instantaneously, as a reference (e.g. 3HOH), and a second that is the compound of interest, usually an amino acid or glucose. For those interested, we have reviewed these physiological experiments previously (Saunders et al. 2012, 2013).
Figure 5. Influx transporters at the blood–brain barrier.
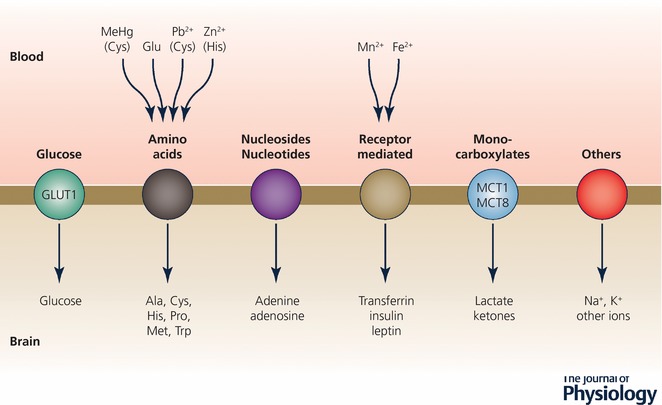
These are mainly SLC (solute carrier) transporters. See Hediger (2013) for comprehensive review. Only transporters for which there is physiological evidence for function are listed. As indicated in Table 2, transcripts for many more Slcs have been identified in molecular screens. Many of these genes are found in both endothelial cells of the blood–brain barrier and epithelial cells of the choroid plexuses. Others are unique to each interface as summarized in Table 2. Note, many metal ions that are potentially toxic can be carried in via some of these transporters.
Amino acids
It seems to have been assumed in most studies that entry into the brain was via the blood–brain barrier interface only and account was probably not taken of any entry via the choroid plexuses into the CSF. More importantly, in most studies it was not clear if the CSF and choroid plexuses had been removed prior to analysis of brain samples; any choroid plexus tissue or CSF included in the brain samples would have led to an overestimate of the contribution of blood–brain barrier transport of the amino acids into the brain, because at least some amino acids accumulate in the choroid plexuses (al‐Sarraf et al. 1997a) in addition to entering the CSF directly. There seems to have been only one series of studies of direct entry into CSF in the developing brain, which would mainly reflect entry across the blood–choroid plexus barrier (al‐Sarraf et al. 1995, 1997b). The results showed greater entry of some amino acids into neonatal rat CSF than that of the adult and in some cases the entry into CSF was greater than into the brain; thus entry via the choroid plexus appears to be more important than across cerebral blood vessels at this early stage of brain development. The higher entry is presumably a reflection of the metabolic requirements of a rapidly growing brain, although some have interpreted it as due to ‘immaturity’ of the blood–brain barrier (e.g. Watson et al. 2006). There is only a small amount of information about amino acids in CSF in children, but this indicates that the CSF/plasma concentration ratios are higher in 0‐ to 3‐year‐old children than in older children for some amino acids (serine, valine, histidine and arginine, but lower for glutamate, Akiyama et al. 2014). Scholl‐Burgi et al. (2008) and Jiménez et al. (2012) have published values for CSF and plasma amino acids in the first year of human life; they confirm that several amino acids are present in neonatal CSF at higher levels than later in life and with higher CSF/plasma ratios. This is consistent with the animal data showing higher transport early in brain development and is presumably a reflection of metabolic requirements at a time when the brain is still growing rapidly.
There is now substantial information about gene expression in the blood vessels and choroid plexus of the developing brain. These data are summarized for Slc (solute carrier) transporters in developing mouse brain in Table 2. There is a strikingly large number of Slc transcripts in almost all families that have been identified in cerebral endothelial cells, choroid plexus epithelial cells or both. There thus appears to be a large amount of redundancy and which precise Slcs are responsible for specific amino acid or other molecules is unclear. However, there are instances where a mutation in a single Slc gene has serious effects on brain development; for example in mice deletion of Slc7a5, a large neutral amino acid transporter, leads to severe neurological abnormalities. In a few patients deleterious homozygous mutations of this gene were associated with motor delay and autistic traits (Tarlungeanu et al. 2016).
Table 2.
Comparison of Slc gene expression in developing mouse and rat brain endothelial cells and choroid plexus epithelial cells
| Family | Transporter function | Choroid plexus epithelial cells | Present in both cell types | Cerebral endothelial cells |
|---|---|---|---|---|
| Slc1 | High affinity glutamate and neutral AA transporter | — | a3, a4, a5 | a1, a4rg‐ps, a2, a6 |
| Slc2 | GLUT‐1 transporter | — | a1, a3, a4, a5, a6, a8, a10, a12, a13 | a9 |
| Slc3 | Heavy subunits of heterodimeric AA transporters | ra1 | a2 | |
| Slc4 | Bicarbonate transporter | a1 | a2, a3, a4, a5, a8, a10, a11 | a7, a1ap |
| Slc5 | Na+/glucose co‐transporter | a1, ra8, ra10 | a3, a5, a7, a6, a10 | a3 |
| Slc6 | Na+‐ and Cl−‐dependent Na+/neurotransmitter symporters | a11, a13, a14, a15, a20b | a1, a4, a6, a8, a9, a13, a15, a17, a20a | a2, a1, a7, a11, a20a |
| Slc7 | Cationic AA transporter/glycoprotein‐associated | a2, a7,a10, a11, ra3, ra12 | a1, a5, a6, a7, a10, a11, | a3, a4, a8 a2, a14, a6os |
| Slc8 | Na+/Ca2+ exchanger | — | a1, a3 | a2 |
| Slc9 | Na+/H+ exchanger | ra3 | a1, a2, a3r1, a3r2, a5, a6, a7, a8, a9 | — |
| Slc10 | Sodium bile salt co‐transport | — | a3, a4, a6 | a7 |
| Slc11 | Proton coupled metal ion transport | — | a2, a1 | — |
| Slc12 | Electroneutral cation/Cl− co‐transporter | ra1, ra3 | a2, a4, a6, a7, a8, a9 | a5 |
| Slc13 | Na2SO4/carboxylate co‐transporter | — | a4, a5 | a3 |
| Slc14 | Urea transporter | a2_v1 | a2 | — |
| Slc15 | Proton oligopeptide co‐transporter | — | a2, a3 | a4 |
| Slc16 | Monocarboxylate transporter | ma3 | a1, a2, a3, a4, a6, a7, a8, a9, a10, a12, a13, a14 | a11 |
| Slc17 | Vesicular glutamate transporter | a6, ra9 | a5‐8 | — |
| Slc18 | Synaptic vesicular amine transporter | — | a2 | a3 |
| Slc19 | Folate/thiamine transporter | — | a1‐3 | |
| Slc20 | Na+/PO4 3− co‐transporter | — | a1, a2 | — |
| Slco | Organic anion transporter | m1a5, m5a1 | 1a4, 1c1, 2a1 | 2b1, 3a1, 4a1, 5a1 |
| Slc22 | Organic cation/anion/zwitterion transporter | a6, a23, ma17, ma21, ra2, ra7, ra9, ra18, ra25 | a5, a17, a18, a8, a12, a15 | a2, a3, a4, a21, a23, |
| Slc23 | Na+‐dependent ascorbic acid transporter | ra3 | a2, ra1 | |
| Slc24 | Na+/(Ca2+/K+) exchanger | a3 | a4, a5, a6 | a2 |
| Slc25 | Mitochondrial carriers | a18, a21, ra31‐32, ra40, ra44, ra46 | a1, a3, a4, a5, a10, a12, a14, a15, a16, a17, a20, a22, a24, a26, a27, a28, a29, a30, a32, a33, a35 a37, a38, a39, a45 | a2, a11, a13, a18, a19, a23, a25, a34, a36, a40, a42, a44, a46, a47, a51, a53 |
| Slc26 | Multifunctional anion exchanger | a7, ra3‐4 | a2 | a6, a1, a7, a10, a11 |
| Slc27 | Fatty acid transport | ma2, ma3 | a1, a3, a4, a6 | a4, a2 |
| Slc28 | Na+‐coupled nucleoside transport | a3, ra2 | — | — |
| Slc29 | Facilitative nucleoside transporter | a2 | a4, a2, ra3 | a1 |
| Slc30 | Zn2+ efflux | ma10, ra2 | a3, a4, a5, a6, a9, ra10 | a1, a7 |
| Slc31 | Cu2+ transporter | — | a1, a2 | — |
| Slc32 | Vesicular inhibitory amino acid transporter | — | ra1 | — |
| Slc33 | Acetyl‐CoA transporter | ma1 | ra1 | — |
| Slc34 | Sodium‐dependent phosphate transport protein 2B | ra2 | — | — |
| Slc35 | Nucleoside‐sugar transporter | e2, f1, f3 | a1, a2, a3, a4, d2, e4, f2, f5 | a5, b1, b2, e1, e3 |
| Slc36 | Proton‐coupled AA transporter | — | a1, a4 | |
| Slc37 | Sugar‐phosphate/phosphate exchanger | a1, a2 | a3, a4 | |
| Slc38 | Sodium‐coupled neutral AA transporter | ma4, a11, ra1, ra7, ra8 | a1, a3, a5, a4 | a2, a6, a7, a9 |
| Slc39 | Metal ion transporter | ma4, ma12 | a8, a10, a11, a14, a1, a3, a7,a12, a13 | a6, a5, a9, a10, a14 |
| Slc40 | Basolateral Fe2+ transporter | a1 | — | |
| Slc41 | Mg2+ transporter | — | a1, a2 | a3 |
| Slc43 | Na+‐independent, AA transporter | ma1, ma2 | ra1, ra2, ra3 | — |
| Slc44 | Choline‐like transporter | a3, ra4 | a1 | a2, a5 |
| Slc45 | Sugar transporter | — | a4, a1, a3 | a2 |
| Slc46 | Folate transporter | a1 | a3 | — |
| Slc47 | Multidrug and toxin extrusion protein 1 | a1 | — | — |
| Slc48 | Heme transporter | a1 | — | a1 |
| Slc50 | Sugar efflux | ma1 | — | a1 |
| Slc52 | Novel riboflavin transporter family | — | — | a2 |
Data from Affymetrix mouse genechip arrays for choroid plexus (E15, Liddelow et al. 2011a) and cerebral endothelial cells (Daneman et al. 2010a). Data from RNA‐Seq Saunders et al. (2015a) rat E15 choroid plexus; Whish et al. (2015) E17 mouse ventricular zone (would have contained transcripts from endothelial and non endothelial cells). Transcripts that were present at both the blood–CSF and blood–brain barriers are shown in middle column. Transcripts that are present only in cerebral endothelial cells and not peripheral endothelial cells are underlined. AA, amino acid; m, mouse only; r, rat only.
When many Slcs are involved in transport of the same amino acids it is difficult to assign specific Slcs to each amino acid class. In Table 3 we indicate Slc genes that may correspond to the transporters for molecules where there is evidence of their entry from blood into the developing brain, but many others may also be involved.
Table 3.
Comparison of Slc transporter gene expression and function in embryonic mouse (E15) choroid plexus and embryonic mouse (E17) neuroepithelium
| Transporter | E15–adult mouse CP fold change | E17–adult mouse VZ fold change | Transport function | Reference |
|---|---|---|---|---|
| Slc16a10 | 66.8 | 1.2 | Iodothyronines T3, T4 | Porterfield & Hendrich (1992) |
| Slc16a2 | n.d. | −2.4 | Monocarboxylates | |
| Slc6a15 | 11.4 | −2.7 | Neutral amino acids | Lefauconnier & Trouvé (1983) |
| Slc40a1 * | 9.6 | 1.1 | Fe2+ | Morgan & Moos (2002) |
| Slc7a11 | 7.1 | 5.1 | Cysteine, glutamate | Lefauconnier & Trouvé (1983) |
| Slc4a1 | 5.5 | n.d. | Anion transporter (Cl−/HCO3 − exchange) | |
| Slc6a13 | 4.6 | 2.4 | GABA transporter | Al‐Sarraf (2002) |
| Slc1a4 | 4.4 | −1.6 | Glutamate, neutral amino acids | Al‐Sarraf et al. (1997a) |
| Slc38a4 | 4.2 | 0.94 | Acidic and neutral amino acids | |
| Slc6a6 | 4.1 | 0.90 | Taurine | Lefauconnier & Trouvé (1983) |
| Slc4a4 | 4.1 | 0.22 | Na+–HCO3 − cotransporter | Damkier et al. (2010) |
| Slc7a1 | 4.1 | 1.99 | Acidic amino acids | Lefauconnier & Trouvé (1983) |
| Slc39a8 | 3.3 | 2.63 | Zn2+ | Chowanadisai et al. (2005) |
| Slc39a10 | 2.8 | n.d. | Zn2+ | Chowanadisai et al. (2005) |
| Slc25a37 | 2.4 | 5.5 | Fe2+ | Morgan & Moos (2002) |
| Slc14a2 | 2.4 | n.d. | Urea | Johanson & Woodbury (1978) |
| Slc7a7 | 2.3 | n.d. | Dibasic and neutral amino acids | Lefauconnier & Trouvé (1983) |
| Slc39a11 | 2.3 | n.d. | Zn2+ | Chowanadisai et al. (2005) |
| Slc43a2 | 2.2 | 3.9 | Large neutral amino acids | Oldendorf (1973) |
| Slc1a3 | 2.2 | 3.1 | Glutamate | Al‐Sarraf et al. (1997a) |
Transcript fold change ratios compared to adult. References are for evidence of transport (blood to brain or CSF) in physiological experiments. Because of the large number of genes often involved in transport of similar classes of molecules, this is only an indication that genes located at these sites are functional. Data from Liddelow et al. (2012) and Whish et al. (2015). There are many more Slc genes that are expressed at a higher level in adult than in embryo, not listed here. *Gene product ferroportin‐1 identified in choroid plexus. n.d., not detected.
Monocarboxylates
A family of monocarboxylate transporters (MCTs) is involved in transport of monocarboxylates (e.g. pyruvate, lactate and ketone bodies) across plasma membranes, some of which are proton linked. These are now designated as members of the SLC16 family, of which there are 14 (Halestrup, 2013a ). Thus far only four have been shown to be involved in monocarboxylate transport in humans (Halestrap, 2013b ): SLC16A1 (MCT1), SLC16A3 (MCT4), SLC16A7 (MCT2) and SLC16A8 (MCT3). SLC16A1 (MCT1) is involved in transport of monocarboxylates across the endothelial cells of the blood–brain barrier (Halestrup, 2013a , b ). Slc16a1 (MCT1), a2 (MCT8), a6 (MCT7), a8 (MCT3), a9 (MCT9), a12 (MCT12) and a13 (MCT13) genes have been identified in adult mouse choroid plexus (Koehler‐Stec et al. 1998; Marques et al. 2011; Saunders et al. 2015a). Slc16a2 is a thyroid hormone transporter, which is expressed at similar levels in embryonic and adult choroid plexus; the others are all monocarboxylate transporters and expressed at a lower level in the rat embryonic plexus compared to the adult (Saunders et al. 2015a). Only Slc16a10 (MCT10) is expressed at a higher level in mouse and rat embryonic choroid plexus compared to adult (Liddelow et al. 2012; Saunders et al. 2015a). Slc16a10 transports tyrosine, the amino acid precursor of the thyroid hormones tri‐ and tetraiodothyronine. The protein product of Slc16a10 has been shown to have much stronger immunohistochemical staining in embryonic compared to adult choroid plexus (Saunders et al. 2015a). This suggests that the very high expression of this transporter reflects an important role in thyroid hormone transport in early brain development.
Triiodothyronine (T3) and thyroxine (T4) are essential for normal brain development. Inadequate delivery of T4 to the developing brain is usually due to iodine deficiency; it may result in cretinism (Rivas & Naranjo, 2007; Skeaff, 2011). The choroid plexuses in the embryonic brain are prominent compared to vascularization of the rest of the brain and it has been suggested to be the main portal of entry into the developing brain (Johansson et al. 2008); this is consistent with high expression of Slc16a10 (MCT10) in the choroid plexuses early in development. Transthyretin (TTR) a thyroid hormone carrier highly expressed throughout development, is the major mechanism previously thought to deliver thyroxine to the brain in early stages of its development, whereas Slco1c1, which is the main thyroid hormone transporter expressed at the blood–brain interfaces in the adult, is expressed at only a low level in the developing brain (Kratzer et al. 2013). Notwithstanding that Slc16a2 (MCT8) is expressed at similar levels in the developing and adult brain (in rodents), in humans mutations of this gene cause an X‐linked syndrome of psychomotor retardation and altered thyroid hormone levels (López‐Espíndola et al. 2014). Thus this gene is critical for normal human brain development. It seems not to be known what the expression level of Slc16a10 (MCT10) is in the human fetus nor have mutations been reported. Slc16a2 (MCT8) appears not to be critical for rodent brain development, as there is no neurological phenotype in knock‐out animals (Visser, 2016). Thus there appear to be species differences in the relative importance of thyroid hormone transporters for brain development.
Glucose
GLUT‐1 (SLC2a1, solute‐linked carrier, SLC transporters) was the first glucose transporter to be described; its gene, Slc2a1, is expressed in both cerebral endothelial cells (Daneman et al. 2010a) and choroid plexus epithelial cells (Liddelow et al. 2012, 2013). GLUT‐1 is a facilitative transporter. It is the main member of this family in cerebral endothelial cells (Enerson & Drewes, 2006). Slc2a3, a8, a12 and a13 expression has also been identified in mouse cerebral endothelial cells (Table 2). Slc2a1 is expressed in rodent choroid plexus (Table 2); in the embryo it is expressed at a slightly higher level (Liddelow et al. 2013). Of the other glucose‐transporting genes in this family that have been identified in rat choroid plexus (Slc2a3, a4, a8, a12, a13 and a15, see Table 2) only Slc2a12 is expressed at a level that is likely to be functionally significant (at five times higher in the adult, Liddelow et al. 2013).
As summarized in Table 2, the SLC5 transporters are sodium–glucose co‐transporters. Slc5a1, a5 and a6 have been identified in mouse cerebral endothelial cells and Slc5a6 and Slc5a10 are present in mouse choroid plexus.
There is good evidence from physiological experiments for glucose transport into the brain in both adult and neonatal rats and rabbits (Oldendorf, 1971; Cornford et al. 1982; Cornford & Cornford, 1986; Vannucci et al. 1994). Much of this is probably mediated by GLUT‐1 (SLC2a1). As indicated above with respect to amino acid transporters there is a general problem, yet to be resolved, where many transporters for the same substrate have evolved; there are currently no methods available for unequivocally linking a specific transporter to a specific contribution to transport. However, it is clear that at least in the case of GLUT‐1 this apparent redundancy does not compensate for the loss of GLUT‐1 function (Ito et al. 2015). So perhaps the redundancy is there to entertain molecular biologists rather than having particular biological relevance.
A comprehensive study of gene expression profiles in embryonic mouse brain in bulk tissue samples and separated endothelial cells has recently been published (Hupe et al. 2017). Unfortunately its value is limited because the authors do not appear to have excluded choroid plexuses from their tissue samples and the method used to separate endothelial cells would have also included those from the choroid plexuses; these are considerably more vascularized in embryonic brain than is the brain itself (Johansson et al. 2008).
Efflux mechanisms protecting the developing brain
The developing brain is protected by a combination of morphological features and cellular efflux mechanisms, as is the adult brain. An important practical and biological question is to what extent these are functional in the embryo, fetus and newborn. In particular how vulnerable is the developing brain to drugs and toxins reaching it from the mother across the placenta or via the milk? This will be the subject of a review to be published later in 2018 (Saunders et al., Annual Review of Pharmacology and Toxicology); this aspect of brain barrier mechanisms will only be dealt with in outline here.
The main mechanisms that remove or exclude metabolic and potentially toxic compounds such as drugs across brain barriers in the adult brain are ATP‐binding cassette (ABC) efflux transporters (Hartz & Bauer 2011) and some of the Slc families of transporters: Slc21 now re‐designated as Slco1 (solute carrier organic anion transporter), Slc15 and Slc22 (Strazielle & Ghersi‐Egea, 2015). The main efflux transporters at the adult blood–brain barrier are illustrated in Fig. 6. These are: ABCB1 (P‐glycoprotein or MDR1) and ABCG2 (breast cancer resistance protein; BCRP). ABCC2 (multidrug resistance‐associated protein 2; MRP2), ABCC4 (MRP4) have also been demonstrated at the blood–brain barrier interface (Strazielle & Ghersi‐Egea, 2015). In cerebral capillary endothelial cells they export compounds into the blood. At the blood–CSF interface ABCC1 (multidrug resistance‐associated protein 1; MRP1) is the main efflux transporter, but ABCC4 (MRP4) and ABCG2 (BCRP) are also present (Ek et al. 2010; Strazielle & Ghersi‐Egea, 2015). They export compounds into the stroma of the plexus (Maliepaard et al. 2001; Ek et al. 2010). BCRP and MRP transport compounds that have been conjugated to specific transport motifs (glutathione, glucuronic acid or sulphated), which confer a wide range of substrate specificity and considerable overlap between transporters (see Fig. 6 and Löscher & Potschka, 2005). There are probably species differences in the level of expression and functional capacity of these various efflux transporters. From limited studies of the developing brain it seems that expression changes with age during brain development at both interfaces (Schumacher & Møllgård, 1997; Virgintino et al. 2008; Ek et al. 2010; Daneman et al. 2010b; Kratzer et al. 2013); however, there are few functional studies dealing with the problem of whether these transporters are functionally effective. Even less is known about the presence of ABC transporters in brain barriers of the human embryo and fetus. We have published an immunohistochemical study of three of these transporters (P‐glycoprotein, BCRP and MRP1) from 5 weeks’ post‐conception to mid‐gestation (Møllgård et al. 2017) that reveals some striking differences between these transporters. Thus P‐glycoprotein was not detectable at any barrier interface at 5 weeks’ post‐conception (but was detectable later in gestation), whereas the other two were detectable from the earliest ages but with differences in their distribution patterns (Møllgård et al. 2017).
Figure 6. Efflux transporters at the blood–brain barrier.
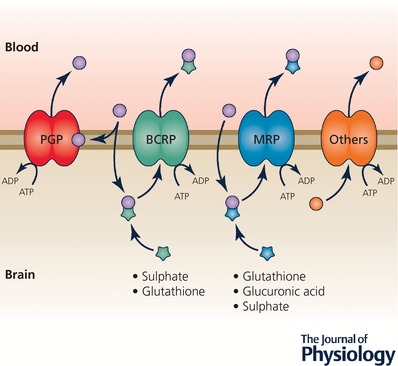
These are mainly ATP‐binding cassette (ABC) transporters. Some, e.g. P‐glycoprotein (PGP; ABCB1), reduce entry into cells. Others, e.g. multidrug resistance protein (MRPs; ABCCs), ligand (drug or toxin) combines with glutathione, glucuronic acid or sulphate in cells before efflux. BCRP, breast cancer resistance protein (ABCG2). ‘Others’ include SLC efflux (SLCO) transporters.
The Slc22 gene family of efflux transporters includes organic anion and cation transporters (OAT, OCT and OCTN proteins). Slco (SLC21) is a subfamily of organic anion transporting polypeptides (OATPs). The homology between rodent and human Slco genes is inexact. Judging from their cellular localization some may function as influx rather than efflux transporters (Strazielle & Ghersi‐Egea, 2015). In rat choroid plexus most Slco and efflux Slc genes are expressed at similar or higher levels than in the adult, except for Slc22a8, which is expressed at a lower level in the embryo and neonate. Slc15a2 is well expressed in both brain and choroid plexus at similar levels in developing and adult choroid plexus. Comparison of the developmental profiles of ABC and SLC efflux transporters in rats shows the adult pattern is achieved earlier in the choroid plexus than in the blood–brain barrier (Strazielle & Ghersi‐Egea, 2015). Nothing seems to be known about the expression or presence of SLC efflux transporters in the human fetal brain.
The extent to which gene or protein expression of an ABC transporter in the developing human brain corresponds to function in vivo cannot be determined experimentally. Thus animal studies are required, but very few studies of drug entry related to efflux transporter function in the developing brain have been performed; notable exceptions are the papers of Staud and colleagues (Staud et al. 2006; Cygalova et al. 2008). Knowledge of the presence and effectiveness of efflux transporters in the developing brain is essential for assessing the risk to fetuses when drugs are administered to a mother. Systematic understanding of efflux transporter expression and function would allow a rational approach to safer prescription of drugs in pregnant women.
Proteins in fetal and newborn CSF
The protein concentration in fetal CSF is much higher than in the adult (Dziegielewska & Saunders, 1988; Saunders et al. 1999). The first quantitative estimates of protein concentration in CSF of embryos appear to be those of Klosovski (1963) in embryonic and postnatal cats. He used an uncalibrated spectrophotometric method and reported that the protein concentration in 10 cm (E48, term is E63–70, Evans & Sack, 1973) embryos was 21.6 times higher than in the adult. Wahle et al. (2014) recorded a median value of 10 mg (100 mL)−1 for adult cat, which if assumed for Klosovski's data gives 216 mg (100 mL)−1 at a post‐conception age of about 60 days (cf. Fig. 7). This is somewhat below the values for sheep and pig fetuses at this age but much higher than species with smaller brains (Fig. 7). It has been suggested that a possible function of the high concentration of protein in CSF of the developing brain is to provide an oncotic pressure gradient, which expands the ventricular system (Saunders, 1992). Deflation of the ventricular system in the chick embryo resulted in very abnormal brain development (Desmond & Jacobsen, 1977). Thus a higher concentration of protein in the ventricular system of larger brains may be a component of the mechanisms required for achieving the larger brain, which essentially grows around the expanding ventricles. The presence of strap junctions between cells of the neuroepithelium that line the ventricular system are also likely to be an important structural component of this mechanism. They have been shown to be impermeable to proteins and other large molecules early in brain development (Fossan et al. 1985; Whish et al. 2015). Without such a diffusion impediment, the oncotic pressure generated by proteins in CSF would be ineffective. The peak in CSF protein in the various species studied (Fig. 7) coincides with the time when the developing brain is generating the neurons that form the cortical plate, the forerunner of the neocortical layers that form the neocortex (see Table 1 in Dziegielewska et al. 2000). The diffusion restriction provided by the strap junctions of the neuroepithelium may provide an important mechanism for regulation of the local environment to which the cells of the neuroepithelium are exposed.
Figure 7. Total protein concentration in CSF of various species at different post‐conceptional ages.
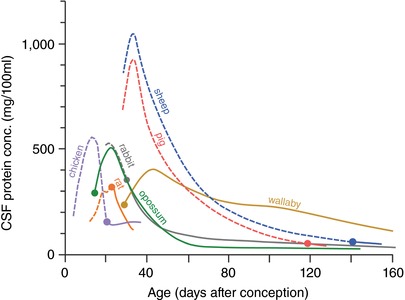
Ordinate: total protein concentration mg (100 mL CSF)−1. Abscissa: post‐conceptional age in days. Filled circles indicate time of birth. Data for sheep from Dziegielewska et al. (1980a); pig (Cavanagh et al. 1982); rat (Dziegielewska et al. 1981; Checiu et al. 1984); tammar wallaby, Macropus eugenii (Dziegielewska et al. 1986); opossum, Monodelphis domestica (Dziegielewska et al. 1989); chick, (Birge et al. 1974); rabbit (Ramey & Birge (1979). Adult values not shown. For mammalian species, including human (see Davson, 1967) and marsupials mean values are between 23 and 31 mg (100 mL)−1. Chicken is 141 mg (100 mL)−1.
The protein composition of CSF in the developing brains has been reported for a number of species (reviewed in Dziegielewska & Saunders, 1988). These studies used immunological methods to characterize and quantify major proteins in CSF from developing brains. More recently proteomic studies of the composition of CSF at very early stages of brain development, including before the differentiation of the choroid plexuses, have been published (chick and rat embryos, Parada et al. 2005; rat and human embryos, Zappaterra et al. 2007). These comprehensive methods identified many more proteins in CSF from developing brain in these species. In a comparison of amniotic fluid and early CSF from mouse embryos, Chau et al. (2015) showed that amniotic fluid at E8.5, the time of neural tube closure, contained many more proteins (764) than eCSF at E10.5 (504) or E14.5 (410). In the initial stages of development after neural tube closure there is a decline in the total protein concentration in eCSF, followed by an increase around the time of differentiation of the choroid plexuses as previously shown (Dziegielewska et al. 1981). These studies of eCSF coupled with ones that begin to relate specific properties of CSF to particular features of early brain development (e.g. Gato & Desmond, 2009; Lehtinen & Walsh, 2011; Lehtinen et al. 2011, 2013; Gato et al. 2014) are opening a new era of understanding of blood–brain and blood–CSF barrier properties and how they relate to brain development.
There is much less information about proteins in human fetal CSF. Adinolfi and colleagues have published some valuable data on this and have shown that as in animal species the total protein concentration is very high compared to the neonate and adult. Thus at 14 weeks’ gestation the CSF total protein concentration was about 120 mg (100 mL)−1 increasing to over 560 mg (100 mL)−1 at 20–24 weeks’ gestation (Adinolfi & Haddad, 1977). The dominant protein was albumin with a substantial contribution from α‐fetoprotein. However, as the samples were from aborted fetuses it is uncertain whether the state of the fetuses would have affected the protein content of the CSF sampled. In the newborn period CSF is sampled in investigation of a number of pathological conditions, but some normal values have been published; the level in preterm infants is higher than at term and than in adults (see Table 1 in Saunders, 1977; Bonadio, 1992; Srinivasan et al. 2012). It has been suggested that the higher CSF protein concentration in neonates reflects increased permeability of the blood–brain barrier (Bonadio, 1992; Srinivasan et al. 2012). CSF protein concentration does not reach adult values until about 6 months after birth (Adinolfi, 1985), which has been taken by some as the age when the blood–brain barrier is ‘mature’ (Rodier, 1995; Watson et al. 2006). In reality the concentration of protein in neonatal CSF is but one of many indicators of blood–brain or more accurately blood–CSF barrier function. Rather than indication of a more permeable blood–brain barrier a more critical determinant is likely to be the turnover of CSF, which is less in the developing brain.
Adinolfi & Haddad (1977) and Adinolfi (1985) interpreted the high concentration of protein in human fetal CSF as evidence that ‘permeability of the blood–CSF barrier is ‘incomplete’ or ‘immature’ in humans; from permeability experiments with 125I‐labelled proteins in newborn rats they drew the same conclusion (Adinoffi & Haddad, 1977). From measurements of total protein concentration in fetal and newborn rabbits Ramey & Birge (1979) concluded that ‘the blood–cerebrospinal fluid barrier to proteins begins to function by 18 to 20 days of gestation’. From their data on total protein concentration in CSF of chick embryos, Birge et al. (1974) concluded ‘a specialized restrictive barrier to protein does not operate between plasma and CSF in chick embryos of 5–10 days.’ However, Ramey & Birge 1979 make the important point that because the turnover of CSF in the developing brain is much less than in the adult (Bass & Lundborg, 1973; Johanson & Woodbury, 1974), this could account for the higher level of protein in CSF.
Transfer of plasma proteins across choroid plexus epithelial cells
The levels of proteins in fetal and newborn CSF may be set by the turnover of CSF, but there is good experimental evidence that their entry into CSF from plasma is a result of specific transcellular transfer of plasma proteins across choroid plexus epithelial cells (Dziegielewska et al. 1980b, 1991; Habgood et al. 1992; Knott et al. 1997; Liddelow et al. 2009, 2011b). Johansson et al. (2008) have suggested that it may be the total amount of protein in CSF that is important as a reflection of transport capacity rather than the concentration of protein. Counts have been made of the number of cells in choroid plexuses immunostained for individual proteins, e.g. albumin, α‐fetoprotein and transferrin, at different stages of brain development in different species (Jacobsen et al. 1982a, b , 1983; Liddelow et al. 2009). The proteins derive from plasma and cross the plexus epithelial cells by what appears to be a specific transfer mechanism. This has best been studied extensively for albumin (Dziegielewska et al. 1980b, 1991; Habgood et al. 1992; Knott et al. 1997; Liddelow et al. 2009, 2011b). Depending upon the animal species, different choroid plexuses can distinguish different species of albumin (Dziegielewska et al.1980b, 1991; Habgood et al. 1992; Knott et al. 1997). Thus fetal sheep choroid plexus distinguishes between on the one hand its own albumin and bovine albumin compared to human albumin, which is transferred into CSF to only about one third of the other albumins (Dziegielewska et al. 1980b, 1991) as illustrated in Fig. 8. The proportion of cells that are immunostained for individual proteins is much higher in species with large brains and ventricular systems. For example in human fetuses about 35–40% of the plexus cells in the lateral and third ventricle are positive for albumin and this declines to about 10% at birth (Jacobsen et al. 1982a). In fetal sheep at E30–40, 40–50% of cells in the lateral ventricle are immunopositive for albumin, this falls to about 1% in late gestation (Jacobsen et al. 1983) when the brain is more mature than in human fetuses. In Monodelphis domestica at P1 when the lateral ventricular choroid plexus first appears, < 1% of cells are positive for albumin. This increases to about 3% in young adults. We have not systematically counted albumin positive cells in rodents, but it is clear in our material that the proportion of these cells is small in rats and mice. For total plasma proteins (to which albumin contributes only a fraction) in rat embryos at E13–16, some 14–17% of cells were protein positive and this declined to 10% by E18. The ventricular system in rodents is larger than in Monodelphis.
Figure 8. Penetration of human plasma proteins from blood into CSF of 60‐day sheep fetuses.
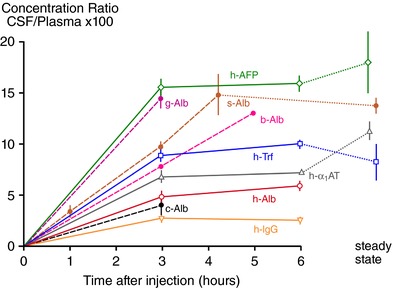
Human plasma was injected intravenously and blood was sampled to give an estimate of mean plasma concentration. At the times indicated CSF was sampled from cisterna magna. Concentrations of individual proteins in CSF and plasma were estimated by radial immunodiffusion assay. Abscissa: time in hours following i.v. injection; ordinate: CSF concentration/plasma concentration × 100. Steady state indicates CSF/plasma ratio for naturally occurring sheep proteins. Mean ± SEM for three to six experiments. All injected proteins were human (h‐) except for s‐Alb (35S‐sheep albumin) g‐Alb (goat albumin) and b‐Alb (bovine albumin) measured using sheep anti‐goat or anti‐bovine albumin antiserum). Experimental details and data are from Dziegielewska et al. (1980a,1980b, 1991). α1AT, α1‐antitrypsin; AFP, α‐fetoprotein; Alb, albumin; IgG, immunoglobulins; Trf, transferrin. The sheep fetus does not possess any IgG of its own, and hence no steady state ratio is shown. Note (i) there is an apparent relation between molecular size and permeability (the largest molecule, IgG, has the lowest ratio and the smallest molecule, AFP, has the largest ratio; however, all of these ratios except for IgG are higher than would be expected from passive diffusion (Saunders, 1992); and (ii) albumin from different species may have different ratios, which suggests that there is a selective mechanism that transports proteins from plasma to CSF. The route of protein transfer appears to be via the epithelial cells of the choroid plexus (Jacobsen et al. 1983; Dziegielewska et al. 1991).
A screen of transcriptomic data from mouse choroid plexus, confirmed by single cell PCR, identified several albumin binding molecules which were then shown to be localized in choroid plexus epithelial cells: glycophorin A (GYPA) and SPARC (secreted protein acidic and rich in cysteine, also known as osteonectin/BM‐40/culture‐shock protein). These albumin binding molecules might be involved in transcellular transport of albumin in the choroid plexus (Liddelow et al. 2012). By using an in situ proximity ligation assay (Duolink©) we have been able to show intracellular co‐localization of mouse albumin and Sparc in choroid plexus cells of postnatal mice (Fig. 9); there was no such co‐localization for injected human albumin confirming the species specificity of the co‐localization (Liddelow et al. 2014). An additional step in the process may be involvement of vesicle‐associated membrane proteins, which are members of a family of SNARE proteins (soluble NSF attachment protein receptors), mostly involved in vesicle fusion. The genes for three of these vesicle‐associated membrane proteins (vamp1, vamp5 and vamp8) were identified in the transcriptomic study of mouse choroid plexus and the localization in these cells demonstrated by immunohistochemistry (Liddelow et al. 2012). The ultrastructure of a potential transcellular route for albumin in fetal choroid plexus cells was previously demonstrated using a combination of transmission electron microscopy (EM), high voltage EM and gold labelling EM immunocytochemistry; it has been designated as a system of tubulocisternal endoplasmic reticulum (Møllgård & Saunders, 1975, 1977; Balslev et al. 1997a). Figure 10 and Fig. 11 illustrate how these various components of a transcellular albumin transfer system might operate.
Figure 9. Cellular distribution of mouse albumin–SPARC (A–C) and human albumin–SPARC (D–F).
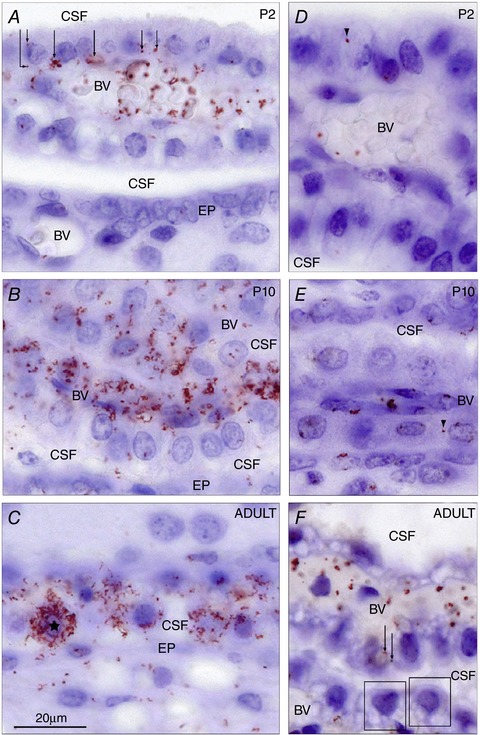
Demonstrated by in situ Proximity Ligation Assay (in situ PLA) signals in the lateral ventricular choroid plexus at P2 (A and D), P10 (B and E) and adult (C and F). Note at P2 that most of the signal was distributed within blood vessels (BV), often associated with red blood cells. Under this magnification it is possible to distinguish positive signals distributed in the basolateral cytoplasm of choroid plexus epithelial cells (arrows). In contrast to the mouse albumin–SPARC signal, the human albumin–SPARC signal (D) was very rarely found and nearly always only associated with blood vessels (BV). Only one positive signal was found and it appears to be located in the extended extracellular space (arrowhead). At P10 (B and E) a very strong signal was visible for mouse albumin–SPARC (B) in many plexus cells distributed throughout the whole cytoplasm, blood vessels (BV) and also in the CSF. The human albumin–SPARC signal (E) was generally only present in blood vessels (BV) but a very occasional signal was detected in the apparent extended extracellular space (arrowhead). The CSF space was negative. In the adult (C and F) a mouse albumin–SPARC signal was distributed clearly throughout the cytoplasm of some choroid plexus epithelial cells (one cell marked with an asterisk). The positive signal was also detected in the ependymal (EP) and subependymal layers of the brain. The human albumin–SPARC signal (F) was visible in blood vessels (BV) but not in the CSF and only very sporadically in the plexus epithelium (two positive red dots are indicated by arrows). Otherwise plexus epithelial cells (boxes) showed no in situ PLA signal. BV, blood vessels; CSF, cerebrospinal fluid; EP, ependymal. Same magnification, scale bar is 20 μm. From Liddelow et al. (2014).
Figure 10. Proposed transepithelial pathway for albumin through choroid plexus epithelial cells.
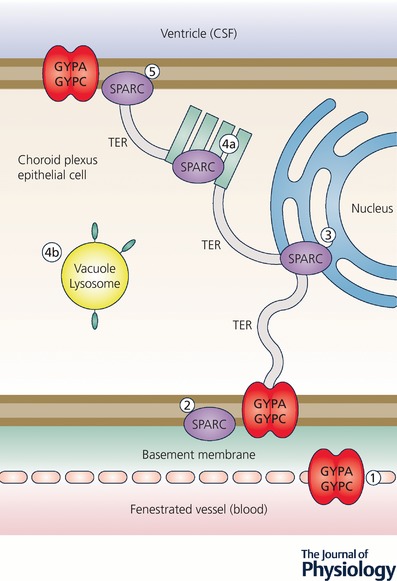
Cartoon of suggested routes of albumin transfer from plasma into CSF across the choroid plexus epithelium. GYPA/C in the endothelial cells may deliver albumin to the basement membrane (1) from where it can be taken up into the plexus epithelium by GYPA/C or SPARC (2). From here albumin may travel along a SPARC‐specific pathway through the tubulocisternal endoplasmic reticulum (3) (see Fig. 11) and Golgi (4a), or via a VAMP‐mediated pathway in vacuoles, lysosomes or multivesicular bodies (4b). On the apical surface of the plexus epithelium, GYPA/C may be involved in efflux of protein from the cell into the CSF of the ventricles (5), as validated by extensive GYPA immunoreactivity in embryonic plexus (Liddelow et al. 2012). In the adult, the lack of immunoreactivity in the endoplasmic reticulum and Golgi (Liddelow et al. 2012) along with increased expression of gene products for VAMP molecules suggests that the majority of transport possibly occurs via VAMP‐mediated vesicular/lysosomal transport such as shown in (4b). CSF, cerebrospinal fluid; GYPA, glycophorin A; GYPC, glycophorin C; SPARC, secreted protein acidic and rich in cysteine; VAMP, vesicle‐associated membrane proteins. Redrawn from Liddelow et al. (2012).
Figure 11. Tubulocisternal endoplasmic reticulum (TER) in fetal sheep choroid plexus epithelial cells.
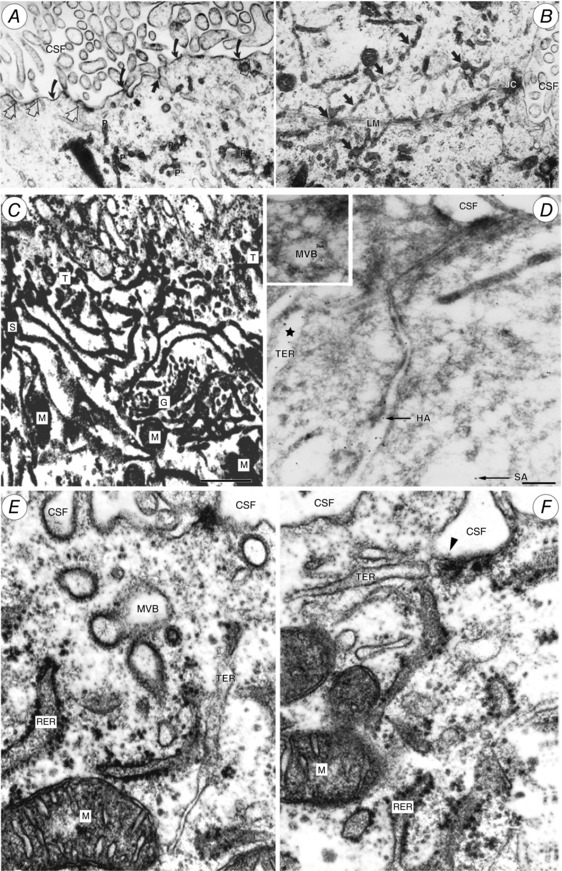
A and B, electron micrographs of E60 fetal sheep choroid plexus. Alcian blue in Krebs solution was injected i.v. 10 min prior to fixation. Alcian blue is electron dense and binds to plasma albumin. Particulate precipitate (P in A) is visible within tubular endoplasmic reticulum (TER; dark arrows in B), which extends close to the lateral cell membrane (LM). JC, junctional complex separating the lateral intercellular space from lateral ventricular CSF. Curved arrows in A indicate precipitated Alcian blue on apical cell membrane; open arrows indicate close contact of TER with apical cell membrane, exposed to CSF. From Møllgård & Saunders (1975). C, high voltage double impregnation thick section EM of E60 fetal sheep choroid plexus. Note the extensive network of TER with contacts to CSF surface (uppermost in micrograph) and close association of TER with Golgi complex (G) and mitochondria (M). From Møllgård & Saunders (1977). D, electron micrograph of ultracryosection from E60 fetal sheep choroid plexus immunolabelled for human (HA) 6 nm gold particles and sheep albumin (SA) 12 nm gold particles (arrows). Gold particles labelling each of the albumins are colocalized within the same TER‐cistern (star) and a multivesicular body (MVB; inset). Scale bar, 0.2 μm. From Balslev et al. (1997a). E and F, fetal sheep choroid plexus (E60) double impregnation technique. Profiles of rough endoplasmic reticulum (RER) and TER system. Note TER termination on apical plasma membrane via a caveola (arrowhead in F). MVB, multivesicular body; M, mitochondrion. From Møllgård & Saunders (1977).
Misconceptions and controversies
Continued use of the term ‘immature’ to indicate a functional deficiency in brain barrier mechanisms
Examples of continued use of the term ‘immature’ to indicate a functional deficiency in brain barrier mechanisms can be found in Allen (2015), Oberdick et al. (2016), Panfoli et al. (2016) and Amaraneni et al. (2017). This usually turns out to be a consequence of poor experimentation or an assumption that because a mechanism is different from that in the adult it is in some way deficient. In our view such differences are more likely to reflect an important match between appropriate barrier function and particular features of the stage of brain development. This concept has the merit of suggesting a way forward in future research whereas dismissing something as ‘immature’ is a dead end for both understanding and future enquiry. It seems reasonable to write in terms of maturation of blood–brain barrier mechanisms when comparing the embryo and adult (Hupe et al. 2017), but this should not be taken to imply deficient function. That needs to be studied in the context of the relevant aspects of brain development in different brain regions. At present there is not much information on this approach as it has rarely been examined (but see above section on ‘Proteins in fetal and newborn CSF’ and also below).
Do the first blood vessels that grow into the brain anlage have functionally effective tight junctions?
Or is there ‘tightening’ of the blood–brain barrier as brain development proceeds (e.g. Cullen et al. 2011)? Early developing cerebral blood vessels may not be as fully developed in their molecular and morphological characteristics compared to the adult as they are operating at much lower blood pressures. This might influence the molecular composition of junctional complexes but does not mean that their permeability properties are different. Ek et al. (2006) have shown that biotin ethylenediamine (molecular mass 286 Da) and 3000 Da biotin dextran are excluded from entering the neocortex of Monodelphis domestica at P5 (a stage when blood vessels are first entering the neocortex) by functionally effective tight junctions. Umans et al. (2017) have shown that in the CNS of zebrafish angiogenesis and blood–brain barrier characteristics (barriergenesis) occur simultaneously as the first blood vessels grow into the neural tissue. They carried out some ingenious experiments in vivo in which they compared the first appearance of the glucose transporter Glut‐1 and an angiogenesis marker, plvap:EGFP (plasmalemma vesicle‐associated protein: enhanced green fluorescent protein) in a double transgenic fish. Glut‐1 is an important early characteristic of cerebral endothelial cells, but its presence in itself does not tell anything about the permeability of the vessels. This could be investigated by immunostaining for a protein in plasma, as a test of large molecule permeability or injection of a labelled small molecule such as biotin ethylenediamine. Fleming et al. (2013) have published permeability experiments and EM observations at very early stages of zebrafish development. They claim that Evans blue entered the brain at 3 days post‐fertilization (dpf) but not at 5 dpf or subsequently; exclusion from the brain of sodium fluorescein was not observed until 10 dpf. The images provided are unconvincing. Evans blue is an unsuitable marker for blood–brain barrier studies (Saunders et al. 2015b); the sodium fluorescein solution used (10%) is strongly hyperosmolar and therefore likely to be damaging to fragile blood vessels. The EM evidence of tight junctions was either ‘not illustrated’ or unconvincing in poor quality micrographs. Perhaps no evidence is better than poor quality evidence.
Van Leewen et al. (2018) have used a similar approach to Umans et al. (2017) and report that the tight junction protein claudin 5 is present in both cerebral endothelial cells and choroid plexus epithelial cells. In developing rodent choroid plexuses the only expression and immunostaining for claudin 5 is in blood vessels; in rodents claudin 5 is regarded as specific for the blood–brain barrier (Kratzer et al. 2012). Van Leewen et al. (2018) provide additional evidence for the proposal that in early development vascularization of the choroid plexuses compared to that of the brain indicates that the plexuses are likely to be a more important route of entry into the brain early on (Johansson et al. 2008).
Ben‐Zvi et al. (2014) conducted experiments in mice at E13.5, E14.5 and E15.5 in which 10 kDa dextran marker was injected into the liver of the embryos. In the youngest embryos dextran was identified in cells in the CNS parenchyma and it was suggested that this had crossed the cerebral vessel walls, thus indicating that the vessels were permeable. It was not clear whether the route was intercellular or transcellular. An alternative explanation is that the dextran may have been transferred across the choroid plexus cells into CSF and taken up from there into parenchymal cells. At E14.5 it was reported that there was a small amount of dextran in the interstitial space around some vessels, although it was not apparent in the micrographs provided. At E15.5 the dextran was found confined to the cerebral blood vessels and on this basis it was suggested that the blood–brain barrier in the mouse becomes ‘functional’ at E15.5. It would require ultrastructural studies with a suitable small marker to confirm this (cf. Ek et al. 2003). At 5 weeks post‐conception in human embryos, Møllgård et al. (2017) found that α‐fetoprotein was confined to the lumen of the first blood vessels entering the CNS tissue and the specific marker for cerebral endothelial cell tight junctions, claudin 5, was already present. These observations suggest the presence of a functionally effective blood–brain barrier to protein in the human embryo in the earliest vessels growing into the brain parenchyma.
Is increased permeability (leakiness) to dyes and small molecular mass markers due to ‘breakdown’ of tight junctions?
Examples can be found in Krizbai et al. (2005), Doherty et al. (2016) and Reinhold & Rittner (2017). To demonstrate this requires electron microscopy, which is rarely done. In the absence of EM evidence, the ‘leakiness’ could as well be transcellular via caveaoli/vesicular transport rather than being due to disrupted tight junctions. This has been demonstrated when electron microscopy was employed (Povlishock et al. 1978; Ben‐Zvi et al. 2014; Krueger et al. 2015; Nahirney et al. 2016; Andreaone et al. 2017). However, it seems that whether or not changes in the ultrastructure of tight junctions occur in pathological conditions may depend on the nature of the disorder (Castejón, 2012). An additional potential source of confusion is that it often seems to be assumed that a change in tight junction protein or gene expression is synonymous with tight junction dysfunction (Sun et al. 2013; Leclerq et al. 2017). Of relevance to blood–brain barrier development is that it seems that transcellular transport may be important in developing cerebral vessels but appears to be suppressed by the gene major facilitator super family domain containing 2a (Mfsd2a) (Ben‐Zvi et al. 2014). See also ‘Permeability of the paracellular pathway’, below.
Does greater accumulation of a molecule in developing brain and CSF reflect greater permeability of barriers in the developing brain?
Such apparent permeability may be due to the use of brain/plasma or CSF/plasma ratios without allowing for the progressive decline in blood level of a marker following injection: compare Ferguson & Woodbury (1969) in which this was not done and Dziegielewska et al. (1979) in which it was. These studies gave very different values for radiolabelled inulin. It might have been hoped that articles such as Ferguson & Woodbury (1969) would have disappeared into the obscurity of unsatisfactory papers, but it was cited recently by Amaraneni et al. (2017) implying that it was evidence of a progressive decrease in blood–brain barrier permeability with age. The accumulation of inulin in brain and CSF in the fetal sheep experiments of Dziegielewska et al. (1979) did indeed decline with age (although the actual levels were much less that in Ferguson & Woodbury (1969)), but this decline in what we might call ‘apparent permeability’ was more likely to have been due to the marked increase in CSF secretion (sink effect) that occurs as brain development progresses (see Johansson et al. 2008).
What is the role of astrocytes in formation (initiation) and maintenance of blood–brain barrier properties?
The controversy surrounding the possible role in the formation of tight junctions stems from the paper of Janzer & Raff (1987) and numerous in vitro studies of cerebral endothelial cells, which have shown that the presence of astroctyes in the culture system was essential for development of some features of monolayers of cerebral endothelial cells that it was hoped would mimic essential features of the blood–brain barrier. The relevance of in vitro studies of these endothelial cells for understanding blood–brain barrier mechanisms in vivo seems doubtful, given that cells change their properties and gene expression when isolated (Szmydynger‐Chodobska et al. 2007; Lyck et al. 2009; Zhang et al. 2016); this will not be discussed further. Janzer & Raff (1987) claimed that following injection of astrocytes into the anterior eye chamber of the rat or on the chorioallantoic membrane of the chicken embryo, blood vessels formed that were impermeable to injected dye because of the formation of tight junctions between the cells of what were assumed to be blood vessels. However, demonstration of tight junctions requires electron microscopy, which was not performed. When this was done in a replication of the study (Holash et al. 1993), it was shown that the dye‐containing structures were not blood vessels and no tight junctions were observed. Unfortunately the Janzer & Raff (1987) paper continues to be cited as evidence of the involvement of astrocytes in the formation of tight junctions in vessels of the developing brain (Abbott, 2002; Engelhardt, 2003; Siegenthaler et al. 2013; Rosas‐Hernandez et al. 2018). This is perhaps an example of when author eminence and the supposed standing of a journal trumps high quality evidence (Vazire, 2017). It seems to be frequently overlooked that no astrocytes have differentiated at the time when the brain is first vascularized and the capillaries are impermeable to even small lipid‐insoluble molecules because of the presence of functionally effective tight junctions (Ek et al. 2006). Furthermore, capillaries in the subarachnoid space have tight junctions (Balslev et al. 1997b), but there are no astrocytes in this location. Also controversial is the role that astrocytes may play in postnatal development and more particularly maintenance of a range of blood brain barrier properties (Abbott, 2002; Obermeier et al. 2013; Engelhardt & Liebner, 2014; Saunders et al. 2016a).
Permeability of the paracellular pathway
The concept of the paracellular pathway arose from experiments of Frömter & Diamond (1972) in which they measured transepithelial resistance in a variety of epithelia in vitro by passing a microelectrode over the external surface of the epithelium. There was a marked drop in resistance across the epithelium when the electrode tip was over the region between adjacent cells. It was proposed that this indicated a low resistance pathway across the epithelium for water and ion flow. Diamond (1974) later suggested that this was also the route by which small lipid insoluble molecules, such as sucrose, crossed epithelia. The concept was later extended to cerebral endothelial cells. The physical basis for the paracellular pathway is the tight junction component of the junctional complex between adjacent cells of an epithelial interface (Farquar & Palade, 1963). Brightman & Reese, (1969) using horseradish peroxidase and transmission EM showed that horseradish peroxides (actually its reaction product) did not permeate through the tight junctions of either choroid plexus epithelial or cerebral endothelial cells. More recent studies using small lipid insoluble molecules that are visualizable at the EM level (286 Da biotin ethylenediamine and 10 kDa biotin‐labelled dextran) showed that these molecules did not permeate tight junctions of cerebral endothelial or choroid plexus epithelial cells in Monodelphis domestica, even in the newborn period when the brain is first vascularized and the choroid plexuses are first apparent; rather these molecules traversed plexus epithelial cells by a transcellular route (Ek et al. 2003, 2006; Liddelow et al. 2009). Thus in respect of small lipid‐insoluble molecules, Diamond's (1974) proposal appears to be incorrect although it is still widely believed as a fundamental property of epithelia and cerebral endothelium (Abbott et al. 2010). In the case of water and ions there is no direct evidence that they cross epithelial interfaces through a paracellular pathway as proposed by Frömter & Diamond (1972) as water and ions cannot yet be visualized at a sufficiently high resolution to demonstrate this. An important limitation of the experiments of Frömter & Diamond (1972) is that the dimension of the tip of the microelectrodes used was about 5 μm. This compares to the intercellular space at the level of tight junctions, which is measured in nanometres, if not zero where adjacent cell membranes are fused. Thus an alternative explanation for the low resistance pathway is that there is an intracellular pathway close to the border of the cell membrane between adjacent cells. Diamond (1974) seems to have acknowledged this possibility as he suggested that in some epithelia the low resistance pathway might be due to ‘leaky’ cells, although he thought this would be exceptional. The studies of Ek et al. (2003, 2006) require independent replication, preferably in several different species, but they indicate that the earliest vessels growing into the brain are structurally well enough developed for their tight junctions to be impermeable to even very small molecules. Similarly, in the early developing choroid plexus the paracellular route appears to be closed to small lipid‐insoluble molecules. Thus a low resistance transcellular pathway across epithelial cells for ions and water, such as in the choroid plexus, is a plausible alternative explanation to the paracellular route envisaged by Frömter & Diamond (1972); these alternatives will only be resolved when suitable high resolution microscopical methods become available. The current dependence on in vitro systems (the permeability properties of which are generally not representative of those in intact vessels, Curry, 2005), modelling (Curry, 2005), cartoon biology (Gunzel, 2017) and deductions from pathological conditions (Mankertz & Schulzke, 2007) to support the functional importance of the paracellular pathway may be misplaced.
Different animal species used for brain barrier studies
For a large number of problems, there will be some animal of choice on which it can be most conveniently studied (Krogh, 1929)
Effective research not only depends upon the convenience of the species but also on its relevance to the problem being studied. Thus the animal species to be chosen depends to a large degree on the questions asked. In developmental studies it is often a trade‐off between the limits imposed by the size of the species (for example where adequate physiological monitoring is essential) and the requirement to study as early stages of development as possible.
Table 4 provides information about developmental milestones and ages at which experiments have been performed in the key mammalian species that have been used for developmental brain barrier studies. We have divided the species into four groups based on stages of brain development at birth. Group I are marsupial species, which are born with a two‐layered undifferentiated neocortex; their choroid plexuses are only just beginning to appear and all four are present within a week postnatal. These animals are very small (10 mm crown–rump length) at birth but have the considerable advantage compared to eutherian mammals that they are born at such an early stage of development that they can be studied without having to work in utero. They also survive much better than rodents at equivalent stages of brain development. Tammars are seasonal breeders and usually have only one young; they require substantial infrastructure for maintaining a colony. Monodelphis will breed all the year round and have multiple young that are exposed on the mother's abdomen; they are pouchless marsupials. They can easily be bred within a standard animal house, although they require a higher room temperature and humidity than rodents (Fadem et al. 1982). Some have questioned the value of marsupial species for developmental studies on the grounds of lack of research tools compared to rodents. However the genomes of both species have now been sequenced (Mikkelsen et al. 2007; Renfree et al. 2011) and in our experience antibodies to human antigens cross‐react well with proteins in these species.
Table 4.
Developmental milestones of mammalian species commonly used for blood‐bran and blood–CSF barrier studies
| Group I | Group II | Group III | Group IV | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Short gestation, at birth cortex undifferentiated | Short gestation, at birth neurogenesis near complete | Intermediate gestation, brain more mature than Group II | Long gestation, larger brain at birth neurogenesis & gliogenesis mostly complete | ||||||
| Opossum | Wallaby | Mouse | Rat | Rabbit | Guinea | Sheep | Pig | Human | |
| Days post‐conception | 14 | 28 | 20–21 | 21–22 | 30–32 | 67–70 | 147 | 115 | 266 |
| Neural tube closure | E10.5* | ∼E18† | E9–E9.5‡ | E11‡ | E9‡ | 14‡ | E21‡ | E17–E18‡ | E26 |
| Published permeability§ studies |
|
P0–3 |
|
E13 | E28–30 a | E60–65 b |
|
E20¶ | E35¶ |
*McCrady (1938). †Ullmann et al. (1997). ‡Evans & Sack (1973). §Permeability studies include blood–brain and blood–CSF barriers. ¶Blood–brain barrier impermeability to plasma proteins demonstrated by immunohistochemistry only. Data sources for permeability studies: opossum (Monodelphis domestica), Knott et al. (1997); tammar wallaby (Macropus eugenii), Dziegielewska et al. (1986); mouse, Bauer et al. (1995); rat, Johansson et al. (2006); sheep, Evans et al. (1974); Dziegielewska et al. (1979, 1980b); Reynolds et al. 1983); pig, Cavanagh & Møllgård (1985); human, Møllgård et al. (2017); rabbit, aSandberg et al. (1996); guinea pig, bBissonette et al. (1991).
Group II are neonatal rats and mice, which are as suitable as marsupials for barrier studies at later stages of development. At birth the neurogenesis is mostly complete while gliogenesis is beginning. In utero studies are more difficult technically because of small size, fragility and difficulty of maintaining fetuses in a physiologically normal state. Group III animals are appreciably larger at birth and more advanced in brain development. In utero experiments are also difficult although a few studies have been published (e.g. Bissonnette et al. 1991; Sandberg et al. 1996). Group IV is the sheep fetus, which since it was introduced by Barcroft and Barron (Barcroft et al. 1936, 1940) has been the traditional species for a whole range of developmental physiological studies. Sheep placenta is made up of around 100 cotyledons, each with its own circulation (Dawes, 1968). Thus access to the fetal circulation can be obtained with minimal disruption of the placental circulation, which is a significant problem in rodents, where there is a single placenta for each embryo. Permeability studies in fetal sheep under monitored and reasonably well‐controlled physiological conditions have been possible as early as E50 (Evans et al. 1974). Most blood–brain barrier studies have been conducted at older ages (E60: Evans et al. 1974; Dziegielewska et al. 1979, 1980b b, 1991) or later (e.g. E87 to term: Stonestreet et al. 1996). The monitored studies in fetal sheep are relatively expensive and require more space and instrumentation, but they provide important background validation to studies in species where monitoring has so far not been technically possible.
Not included in Table 4 are non‐mammalian species such as chick and zebrafish embryos. These have the considerable advantages of accessibility at very early stages of brain development. Zebrafish have the so‐far‐unique advantage of being transparent so various processes can be visualized real‐time with time‐lapse microscopy (Chang & Sive, 2012; Umans et al. 2017). More detailed information on brain development milestones in some of these species is to be found in Clancy et al. (2001, 2007), Clowry et al. (2010) and Semple et al. (2013).
In conclusion, because each species comes with inherent limitations, it is preferable to study barrier problems in several animal species. This extends the range of developmental ages that can be studied and provides complimentary evidence on a particular question, rather than mere replication. This is an approach that has been described as ‘triangulation’ (Munafò & Smith, 2018).
Future studies
Further work will be required to resolve the controversies outlined above. In addition, there are several topics that have been barely touched on in studies so far. These include when the proposed cerebral lymphatic system (Johnston et al. 2004; Iliff & Nedergaard, 2013) develops (Koh et al. 2006) and whether the ‘glymphatic’ system (Plog & Nedergaard, 2017) is functional in the developing brain. Major areas for future research are the following.
Drugs that enter the developing brain: do they affect brain development and behaviour?
The efflux mechanisms summarized above limit the entry of drugs into the adult brain. But the effectiveness of these protective mechanisms in the developing brain when drugs are administered to pregnant women or to neonates is largely unknown. Systematic studies in the human fetus that define which efflux transporters are present in the developing brain and when they appear (e.g. Møllgård et al. 2017) are required as a prerequisite for functional studies of these transporters in an appropriate animal model. Fortunately it seems likely that only a limited number of the ABC transporters are involved. Thousands of drugs and toxins are kept out or eliminated from the brain; this is because many drugs are substrates for individual transporters. Thus if key drugs that are known to be substrates for each efflux transporter are selected, then the effectiveness of each transporter can be assessed at different stages of brain development. However, this assessment is complicated by a number of factors. Any one drug may be a substrate for more than one efflux transporter. Many of the transporters are also functional in the placenta. Thus placental function needs to be taken into account when assessing the possible entry of drugs into the brain. On the other hand for prematurely born infants that have lost the protection of the placenta early, it is possible that the brain will be more susceptible to the entry of drugs administered in the neonatal period. There is some evidence, although as yet controversial, that some transporters up‐regulate in the face of continued exposure to their substrates. Thus for pregnant women on regular medication, for example anti‐epileptics, this may affect the entry of drugs into the fetal brain. Once it is established which classes of drugs can enter the developing brain and when, a large field will open up for systematic studies of the short‐ and long‐term consequences of drug exposure in pregnancy and the neonatal period. At present only limited studies have been carried out and these are often poorly designed, resulting in unnecessary alarm in patients. To give a few recent examples: poor quality epidemiological studies of paracetamol in pregnancy (Stergiakouli et al. 2016, see comments in Beale, 2017; Saunders & Habgood, 2017); clinically improbable doses of paracetamol in pregnant mice (Hay‐Schmidt et al. 2017) and administration of penicillin to mice during a large proportion of pregnancy and the postnatal period (Leclerq et al. 2017) in which there was a lack of randomization, lack of blinding of behavioural studies, dubious statistical analysis and dubious interpretation of results of tight junction protein expression.
Composition, origin and functions of CSF in mammalian embryos
It is worth emphasizing the likely importance of CSF in the development of the CNS. Consideration of this can conveniently be divided into two stages. (a) after the closure of the neural tube, which traps amniotic fluid within the tube and before choroid plexuses appear. Its composition is subsequently modified although different emphasis has been placed on the importance of materials emanating from the neuropepithelium (Zappaterra et al. 2007; Chau et al. 2015) and those entering from outside the CNS (Bueno et al. 2014; Gato et al. 2014). (b) the period from differentiation of the choroid plexuses onwards. There is evidence that proteins are transferred from the circulating blood to the CSF across the plexus epithelial cells (see ‘Transfer of plasma proteins across choroid plexus epithelial cells’, above) but other constituents of the CSF are likely to originate from the choroid plexuses themselves or from the neuroepithelial tissues lining the ventricular system. It is worth emphasizing that in the first stage (after the closure of the neural tube) the neural tissue is not vascularized and in the early period of the second stage (from differentiation of the choroid plexuses onwards) there is little vascularization of the CNS; it is only after birth, for example in rodents, that the neocortex develops most of its vasculature (Caley & Maxwell, 1970). Thus non‐vascular sources of the constituents of CSF dominate in the early stages of brain development. Features of brain development that have so far been studied in the context of specific aspects of brain development are neurogenesis (Alonso et al. 2011; Lehtinen & Walsh, 2011; Johansson, 2014) and cell proliferation (Miyan et al. 2006; Lehtinen et al. 2011). Zappaterra & Lehtinen (2012) have reviewed a range of developmental processes in which CSF constituents may be involved.
Additional information
Competing interests
None declared.
Author contributions
All authors have read and approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Acknowledgements
We should like to thank the numerous undergraduate and graduate students as well as the many collaborators who have contributed to this work over nearly five decades. Without their input very little of this could have been achieved. We also acknowledge financial support from many grant bodies, which included MRC (UK), AFRC (UK), Action Research for the Crippled Child (UK), The Wellcome Trust, The Leverhulme Trust, National Institutes of Health (USA), Australian Research Council, National Health & Medical Research Council, Australia and European Union‐FP7.
Biographies
Norman Saunders is a Professorial Fellow in Neuroscience at the University of Melbourne. He studied medicine (MBBS) and physiology (PhD) at University College London.

Katarzyna Dziegielewska is an Honorary Associate Professor at the University of Melbourne and studied at the University of Warsaw (MSc) and University College London (PhD).
Kjeld Møllgård is professor at the University of Copenhagen, where he studied medicine (MD). He was then postdoc at University of California, Berkeley, and at University of Lund, Sweden (PhD).
Mark Habgood, Senior Research Fellow, studied at the Universities of Canterbury, New Zealand, BSc), Auckland (New Zealand, MSc) and Southampton (PhD). We have worked together since the early 1970s–80s on barrier mechanisms in the developing brain.
Edited by: Ole Petersen and Laura Bennet
References
- Abbott NJ (2002). Astrocyte‐endothelial interactions and blood‐brain barrier permeability. J Anat 200, 629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott NJ, Patabendige AA, Dolman DE, Yusof SR & Begley DJ (2010). Structure and function of the blood‐brain barrier. Neurobiol Dis 37, 13–25. [DOI] [PubMed] [Google Scholar]
- Adinolfi M (1985). The development of the human blood‐CSF‐brain barrier. Dev Med Child Neurol 27, 532–537. [DOI] [PubMed] [Google Scholar]
- Adinolfi M & Haddad SA (1977). Levels of plasma proteins in human and rat fetal CSF and the development of the blood‐CSF barrier. Neuropadiatrie 8, 345–353. [DOI] [PubMed] [Google Scholar]
- Akiyama T, Kobayashi K, Higashikage A, Sato J & Yoshinaga H (2014). CSF/plasma ratios of amino acids: reference data and transports in children. Brain Dev 36, 3–9. [DOI] [PubMed] [Google Scholar]
- Allen KA (2015). Is prenatal lead exposure a concern in infancy? What is the evidence? Adv Neonatal Care 15, 416–420. [DOI] [PubMed] [Google Scholar]
- Alonso MI, Martín C, Carnicero E, Bueno D & Gato A (2011). Cerebrospinal fluid control of neurogenesis induced by retinoic acid during early brain development. Dev Dyn 240, 1650–1659. [DOI] [PubMed] [Google Scholar]
- al‐Sarraf H, Preston JE & Segal MB (1995). The entry of acidic amino acids into brain and CSF during development, using in situ perfusion in the rat. Brain Res Dev Brain Res 90, 151–158. [DOI] [PubMed] [Google Scholar]
- al‐Sarraf H, Preston JE & Segal MB (1997a). Acidic amino acid accumulation by rat choroid plexus during development. Brain Res Dev Brain Res 102, 47–52. [DOI] [PubMed] [Google Scholar]
- al‐Sarraf H, Preston JE & Segal MB (1997b). Changes in the kinetics of the acidic amino acid brain and CSF uptake during development in the rat. Dev Brain Res 102, 127–134. [DOI] [PubMed] [Google Scholar]
- al‐Sarraf H (2002). Transport of 14C-gamma-aminobutyric acid into brain, cerebrospinal fluid and choroid plexus in neonatal and adult rats. Brain Res Dev Brain Res 139, 121–129. [DOI] [PubMed] [Google Scholar]
- Amaraneni M, Pang J, Mortuza TB, Muralidhara S, Cummings BS, White CA, Vorhees CV, Zastre J & Bruckner JV (2017). Brain uptake of deltamethrin in rats as a function of plasma protein binding and blood‐brain barrier maturation. Neurotoxicology 62, 24–29. [DOI] [PubMed] [Google Scholar]
- Amtorp O & Sørensen SC (1974). The ontogenetic development of concentration differences for protein and ions between plasma and cerebrospinal fluid in rabbits and rats. J Physiol 243, 387–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DK & Hazelwood RL (1969). Chicken cerebrospinal fluid: normal composition and response to insulin administration. J Physiol 202, 83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreone BJ, Chow BW, Tata A, Lacoste B, Ben‐Zvi A, Bullock K, Deik AA, Ginty DD, Clish CB & Gu C (2017). Blood‐brain barrier permeability is regulated by lipid transport‐dependent suppression of caveolae‐mediated transcytosis. Neuron 94, 581–594.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balslev Y, Dziegielewska KM, Møllgård K & Saunders NR (1997a). Intercellular barriers to and transcellular transfer of albumin in the fetal sheep brain. Anat Embryol (Berl) 195, 229–236. [DOI] [PubMed] [Google Scholar]
- Balslev Y, Saunders NR & Møllgård K (1997b). Ontogenetic development of diffusional restriction to protein at the pial surface of the rat brain: an electron microscopical study. J Neurocytol 26, 133–148. [DOI] [PubMed] [Google Scholar]
- Barcroft J, Barron DH & Windle WF (1936). Some observations on genesis of somatic movement in sheep embryos. J Physiol 87, 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcroft J, Barron DH, Cowie AT & Forsham PH (1940). The oxygen supply of the foetal brain of the sheep and the effect of asphyxia on foetal respiratory movement. J Physiol 97, 338–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass NH & Lundborg P (1973). Postnatal development of bulk flow in the cerebrospinal fluid system of the albino rat: clearance of carboxyl‐(14C)inulin after intrathecal infusion. Brain Res 52, 323–332. [DOI] [PubMed] [Google Scholar]
- Bauer H, Sonnleitner U, Lametschwandtner A, Steiner M, Adam H & Bauer HC (1995). Ontogenic expression of the erythroid‐type glucose transporter (Glut 1) in the telencephalon of the mouse: correlation to the tightening of the blood‐brain barrier. Brain Res Dev Brain Res 86, 317–325. [DOI] [PubMed] [Google Scholar]
- Beale DJ (2017). Acetaminophen in pregnancy and adverse childhood neurodevelopment. JAMA Pediatr 171, 394–395. [DOI] [PubMed] [Google Scholar]
- Ben‐Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H & Gu C (2014). Mfsd2a is critical for the formation and function of the blood‐brain barrier. Nature 509, 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birge WJ, Rose AD, Haywood JR & Doolin PF (1974). Development of the blood‐cerebrospinal fluid barrier to proteins and differentiation of cerebrospinal fluid in the chick embryo. Dev Biol 41, 245–254. [DOI] [PubMed] [Google Scholar]
- Bissonnette JM, Hohimer AR & Chao CR (1991). Unidirectional transport of glucose and lactate into brain of fetal sheep and guinea‐pig. Exp Physiol 76, 515–523. [DOI] [PubMed] [Google Scholar]
- Bito LZ & Myers RE (1970). The ontogenesis of haematoencephalic cation transport processes in the rhesus monkey. J Physiol 208, 153–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonadio WA (1992). The cerebrospinal fluid: physiologic aspects and alterations associated with bacterial meningitis. Pediatr Infect Dis J 11, 423–431. [PubMed] [Google Scholar]
- Bradbury MW, Crowder J, Desai S, Reynolds JM, Reynolds M & Saunders NR (1972). Electrolytes and water in the brain and cerebrospinal fluid of the foetal sheep and guinea‐pig. J Physiol 227, 591–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun LD, Cornford EM & Oldendorf WH (1980). Newborn rabbit blood‐brain barrier is selectively permeable and differs substantially from the adult. J Neurochem 34, 147–152. [DOI] [PubMed] [Google Scholar]
- Brightman MW & Reese TS (1969). Junctions between intimately apposed cell membranes in the vertebrate brain. J Cell Biol 40, 648–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brøchner CB, Holst CB & Møllgård K (2015). Outer brain barriers in rat and human development. Front Neurosci 9, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PD, Davies SL, Speake T & Millar ID (2004). Molecular mechanisms of cerebrospinal fluid production. Neuroscience 129, 957–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno D, Parvas M, Hermelo I & Garcia‐Fernàndez J (2014). Embryonic blood‐cerebrospinal fluid barrier formation and function. Front Neurosci 8, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caley DW & Maxwell DS (1970). Development of the blood vessels and extracellular spaces during postnatal maturation of rat cerebral cortex. J Comp Neurol 138, 31–47. [DOI] [PubMed] [Google Scholar]
- Castejón OJ (2012). Ultrastructural pathology of endothelial tight junctions in human brain oedema. Folia Neuropathol 50, 118–129. [PubMed] [Google Scholar]
- Cavanagh ME, Cornelis ME, Dziegielewska KM, Luft AJ, Lai PC, Lorscheider FL & Saunders NR (1982). Proteins in cerebrospinal fluid and plasma of fetal pigs during development. Dev Neurosci 5, 492–502. [DOI] [PubMed] [Google Scholar]
- Cavanagh ME & Møllgård K (1985). An immunocytochemical study of the distribution of some plasma proteins within the developing forebrain of the pig with special reference to the neocortex. Brain Res 349, 183–194. [DOI] [PubMed] [Google Scholar]
- Chang JT & Sive H (2012). Manual drainage of the zebrafish embryonic brain ventricles. J Vis Exp 16, e4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau KF, Springel MW, Broadbelt KG, Park HY, Topal S, Lun MP, Mullan H, Maynard T, Steen H, LaMantia AS & Lehtinen MK (2015). Progressive differentiation and instructive capacities of amniotic fluid and cerebrospinal fluid proteomes following neural tube closure. Dev Cell 35, 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checiu I, Prelipceanu O & Popescu O (1984). The role of the cerebrospinal fluid during embryonic development. A biochemical study. Morphol Embryol (Bucur) 30, 243–250. [PubMed] [Google Scholar]
- Cho C, Smallwood PM, Nathans J (2017). Reck and Gpr124 are essential receptor cofactors for Wnt7a/Wnt7b‐specific signaling in mammalian CNS angiogenesis and blood‐brain barrier regulation. Neuron 95, 1056–1073.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowanadisai W, Kelleher SL & Lönnerdal B (2005). Zinc deficiency is associated with increased brain zinc import and LIV‐1 expression and decreased ZnT‐1 expression in neonatal rats. J Nutr 135, 1002–1007. [DOI] [PubMed] [Google Scholar]
- Clancy B, Darlington RB & Finlay BL (2001). Translating developmental time across mammalian species. Neuroscience 105, 7–17. [DOI] [PubMed] [Google Scholar]
- Clancy B, Finlay BL, Darlington RB & Anand KJ (2007). Extrapolating brain development from experimental species to humans. Neurotoxicology 28, 931–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clowry G, Molnár Z & Rakic P (2010). Renewed focus on the developing human neocortex. J Anat 217, 276–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornford EM, Braun LD & Oldendorf WH (1982). Developmental modulations of blood‐brain barrier permeability as an indicator of changing nutritional requirements in the brain. Pediatr Res 16, 324–328. [DOI] [PubMed] [Google Scholar]
- Cornford EM & Cornford ME (1986). Nutrient transport and the blood brain barrier in developing animals. Federation Proc 45, 2065–2072. [PubMed] [Google Scholar]
- Cullen M, Elzarrad MK, Seaman S, Zudaire E, Stevens J, Yang MY, Li X, Chaudhary A, Xu L, Hilton MB, Logsdon D, Hsiao E, Stein EV, Cuttitta F, Haines DC, Nagashima K, Tessarollo L & St Croix B (2011). GPR124, an orphan G protein‐coupled receptor, is required for CNS‐specific vascularization and establishment of the blood‐brain barrier. Proc Natl Acad Sci U S A 108, 5759–5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curry FR (2005). Microvascular solute and water transport. Microcirculation 12, 17–31. [DOI] [PubMed] [Google Scholar]
- Cygalova L, Ceckova M, Pavek P & Staud F (2008). Role of breast cancer resistance protein (Bcrp/Abcg2) in fetal protection during gestation in rat. Toxicol Lett 178, 176–180. [DOI] [PubMed] [Google Scholar]
- Damkier HH, Brown PD & Praetorius J (2010). Epithelial pathways in choroid plexus electrolyte transport. Physiology (Bethesda) 25, 239–249. [DOI] [PubMed] [Google Scholar]
- Daneman R & Prat A (2015). The blood-brain barrier. Cold Spring Harb Perspect Biol 7, a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Agalliu D, Cahoy JD, Kausal A & Barres BA (2010a). The mouse blood‐brain barrier transcriptome: A new resource for understanding the development and function of brain endothelial cells. PLoS One 5, e13741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA & Barres BA (2010b). Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature 468, 562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davson H (1967). Physiology of the Cerebrospinal Fluid, p35 Churchill, London. [Google Scholar]
- Davson H & Segal MB (1996). Physiology of the CSF and Blood‐Brain Barriers. CRC Press, Boca Raton. [Google Scholar]
- Dawes GS (1968). Foetal and Neonatal Physiology, Ch 2. Year Book Medical Publishers Inc., Chicago. [Google Scholar]
- Desmond ME & Jacobsen AG (1977). Embryonic brain enlargement requires cerebrospinal fluid pressure. Dev Biol 57, 188–198. [DOI] [PubMed] [Google Scholar]
- Diamond JM (1974). Tight and leaky junctions of epithelia: a perspective on kisses in the dark. Fed Proc 33, 2220–2224. [PubMed] [Google Scholar]
- Doherty CP, O'Keefe E, Wallace E, Loftus T, Keaney J, Kealy J, Humphries MM, Molloy MG, Meaney JF, Farrell M & Campbell M (2016). Blood‐brain barrier dysfunction as a hallmark pathology in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 75, 656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziegielewska KM & Saunders NR (1988). The development of the blood‐brain barrier: proteins in fetal and neonatal CSF, their nature and origins In Handbook of Human Growth and Developmental Biology, vol 1, Neural, Sensory, Motor and Integrative Development, ed. Meisami E. & Timiras PS, pp. 169–191. CRC Press, Boca Raton. [Google Scholar]
- Dziegielewska KM, Evans CA, Fossan G, Lorscheider FL, Malinowska DH, Møllgård K, Reynolds ML, Saunders NR & Wilkinson S (1980a). Proteins in cerebrospinal fluid and plasma of fetal sheep during development. J Physiol 300, 441–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziegielewska KM, Evans CAN, Lai PCW, Lorscheider FL, Malinowska DH, Møllgård K & Saunders NR (1981). Proteins in cerebrospinal fluid and plasma of fetal rats during development. Dev Biol 83, 192–200. [DOI] [PubMed] [Google Scholar]
- Dziegielewska KM, Evans CA, Malinowska DH, Møllgård K, Reynolds JM, Reynolds ML & Saunders NR (1979). Studies of the development of brain barrier systems to lipid insoluble molecules in fetal sheep. J Physiol 292, 207–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziegielewska KM, Evans CA, Malinowska DH, Møllgård K, Reynolds ML & Saunders NR (1980b). Blood‐cerebrospinal fluid transfer of plasma proteins during fetal development in the sheep. J Physiol 300, 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziegielewska KM, Habgood M, Jones SE, Reader M & Saunders NR (1989). Proteins in cerebrospinal fluid and plasma of postnatal Monodelphis domestica (grey short‐tailed opossum). Comp Biochem Physiol B 92, 569–576. [DOI] [PubMed] [Google Scholar]
- Dziegielewska KM, Habgood MD, Møllgård K, Stagaard M & Saunders NR (1991). Species‐specific transfer of plasma albumin from blood into different cerebrospinal fluid compartments in the fetal sheep. J Physiol 439, 215–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziegielewska KM, Hinds LA, Sarantis ME, Saunders NR & Tyndale‐Biscoe CH (1986). Proteins in cerebrospinal fluid and plasma of the pouch young tammar wallaby (Macropus eugenii) during development. Comp Biochem Physiol B 83, 561–567. [DOI] [PubMed] [Google Scholar]
- Dziegielewska KM, Knott GW & Saunders NR (2000). The nature and composition of the internal environment of the developing brain. Cell Mol Neurobiol 20, 41–56. [DOI] [PubMed] [Google Scholar]
- Ek CJ, Dziegielewska KM, Stolp H & Saunders NR (2006). Functional effectiveness of the blood‐brain barrier to small water‐soluble molecules in developing and adult opossum (Monodelphis domestica). J Comp Neurol 496, 13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ek CJ, Habgood MD, Dziegielewska KM & Saunders NR (2003). Structural characteristics and barrier properties of the choroid plexuses in developing brain of the opossum (Monodelphis Domestica). J Comp Neurol 460, 451–464. [DOI] [PubMed] [Google Scholar]
- Ek CJ, Wong A, Liddelow SA, Johansson PA, Dziegielewska KM & Saunders NR (2010). Efflux mechanisms at the developing brain barriers: ABC‐transporters in the fetal and postnatal rat. Toxicol Lett 197, 51–59. [DOI] [PubMed] [Google Scholar]
- Enerson BE & Drewes LR (2006). The rat blood‐brain barrier transcriptome. J Cereb Blood Flow Metab 26, 959–973. [DOI] [PubMed] [Google Scholar]
- Engelhardt B (2003). Development of the blood‐brain barrier. Cell Tissue Res 314, 119–129. [DOI] [PubMed] [Google Scholar]
- Engelhardt B & Liebner S (2014). Novel insights into the development and maintenance of the blood‐brain barrier. Cell Tissue Res 355, 687–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt B, & Sorokin L (2009). The blood‐brain and the blood‐cerebrospinal fluid barriers: function and dysfunction. Semin Immunopathol 31, 497–511. [DOI] [PubMed] [Google Scholar]
- Errede M, Girolamo F, Rizzi M, Bertossi M, Roncali L & Virgintino D (2014). The contribution of CXCL12‐expressing radial glia cells to neuro‐vascular patterning during human cerebral cortex development. Front Neurosci 8, 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CAN, Reynolds JM, Reynolds ML, Saunders NR & Segal MB (1974). The development of a blood‐brain barrier mechanism in foetal sheep. J Physiol 238, 371–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans HE & Sack WO (1973). Prenatal development of domestic and laboratory mammals: Growth curves, external features and selected references. Anat Histol Embryol 2, 11–45. [DOI] [PubMed] [Google Scholar]
- Fadem BH, Trupin GL, Maliniak E, VandeBerg JL & Hayssen V (1982). Care and breeding of the gray, short‐tailed opossum (Monodelphis domestica). Lab Anim Sci 32, 405–409. [PubMed] [Google Scholar]
- Farquhar MG & Palade GE (1963). Junctional complexes in various epithelia. J Cell Biol 17, 375–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson RK & Woodbury DM (1969). Penetration of 14C‐inulin and 14C‐sucrose into brain, cerebrospinal fluid, and skeletal muscle of developing rats. Exp Brain Res 7, 181–194. [DOI] [PubMed] [Google Scholar]
- Fleming A, Diekmann H & Goldsmith P (2013). Functional characterisation of the maturation of the blood‐brain barrier in larval zebrafish. PLoS One 8, e77548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flexner LB (1938). Changes in the chemistry and nature of the cerebrospinal fluid during fetal life in the pig. Am J Physiol 124, 131–135. [Google Scholar]
- Fossan G, Cavanagh ME, Evans CA, Malinowska DH, Møllgård K, Reynolds ML & Saunders NR (1985). CSF‐brain permeability in the immature sheep fetus: a CSF‐brain barrier. Brain Res 350, 113–124. [DOI] [PubMed] [Google Scholar]
- Frömter E & Diamond J (1972). Route of passive ion permeation in epithelia. Nat New Biol 235, 9–13. [DOI] [PubMed] [Google Scholar]
- Gato A, Alonso MI, Martín C, Carnicero E, Moro JA, De la Mano A, Fernández JM, Lamus F & Desmond ME (2014). Embryonic cerebrospinal fluid in brain development: neural progenitor control. Croat Med J 55, 299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gato A & Desmond ME (2009). Why the embryo still matters: CSF and the neuroepithelium as interdependent regulators of embryonic brain growth, morphogenesis and histiogenesis. Dev Biol 327, 263–272. [DOI] [PubMed] [Google Scholar]
- Günzel D (2017). Claudins: vital partners in transcellular and paracellular transport coupling. Pflugers Arch 469, 35–44. [DOI] [PubMed] [Google Scholar]
- Habgood MD, Sedgwick JE, Dziegielewska KM & Saunders NR (1992). A developmentally regulated blood‐cerebrospinal fluid transfer mechanism for albumin in immature rats. J Physiol 456, 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP (2013a). The SLC16 gene family – structure, role and regulation in health and disease. Mol Aspects Med 34, 337–349. [DOI] [PubMed] [Google Scholar]
- Halestrap AP (2013b). Monocarboxylic acid transport. Compr Physiol 3, 1611–1643. [DOI] [PubMed] [Google Scholar]
- Hartz AM & Bauer B (2011). ABC transporters in the CNS – an inventory. Curr Pharm Biotechnol 12, 656–673. [DOI] [PubMed] [Google Scholar]
- Hay‐Schmidt A, Finkielman OTE, Jensen BAH, Høgsbro CF, Bak Holm J, Johansen KH, Jensen TK, Andrade AM, Swan SH, Bornehag CG, Brunak S, Jegou B, Kristiansen K & Kristensen DM (2017). Prenatal exposure to paracetamol/acetaminophen and precursor aniline impairs masculinisation of male brain and behaviour. Reproduction 154, 145–152. [DOI] [PubMed] [Google Scholar]
- Hediger MA (2013). The ABCs of membrane transporters in health and disease (SLC series). Mol Aspects Med 34, 95–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine W, Hobusch D & Drescher U (1981). [Cerebrospinal fluid protein levels and blood‐cerebrospinal fluid ratio of glucose and electrolytes in infants and children]. Helv Paediatr Acta 36, 217–227 [German]. [PubMed] [Google Scholar]
- Holash JA, Noden DM & Stewart PA (1993). Re‐evaluating the role of astrocytes in blood‐brain barrier induction. Dev Dyn 197, 14–25. [DOI] [PubMed] [Google Scholar]
- Hupe M, Li MX, Kneitz S, Davydova D, Yokota C, Kele‐Olovsson J, Hot B, Stenman JM & Gessler M (2017). Gene expression profiles of brain endothelial cells during embryonic development at bulk and single‐cell levels. Sci Signal 10, eaag2476. [DOI] [PubMed] [Google Scholar]
- Iliff JJ & Nedergaard M (2013). Is there a cerebral lymphatic system? Stroke 44(6 Suppl 1), S93–S95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Takahashi S, Kagitani‐Shimono K, Natsume J, Yanagihara K, Fujii T & Oguni H (2015). Nationwide survey of glucose transporter‐1 deficiency syndrome (GLUT‐1DS) in Japan. Brain Dev 37, 780–789. [DOI] [PubMed] [Google Scholar]
- Jacobsen M, Clausen PP, Jacobsen GK, Saunders NR & Møllgård K (1982a). Intracellular plasma proteins in human fetal choroid plexus during development. I. Developmental stages in relation to the number of epithelial cells which contain albumin in telencephalic, diencephalic and myelencephalic choroid plexus. Brain Res 255, 239–250. [DOI] [PubMed] [Google Scholar]
- Jacobsen M, Jacobsen GK, Clausen PP, Saunders NR & Møllgård K (1982b). Intracellular plasma proteins in human fetal choroid plexus during development. II. The distribution of prealbumin, albumin, α‐fetoprotein, transferrin, IgG, IgA, IgM, and α1‐antitrypsin. Brain Res 255, 251–262. [DOI] [PubMed] [Google Scholar]
- Jacobsen M, Møllgård K, Reynolds ML & Saunders NR (1983). The choroid plexus in fetal sheep during development with special reference to intracellular plasma proteins. Dev Brain Res 8, 77–88. [Google Scholar]
- Janzer RC & Raff MC (1987). Astrocytes induce blood‐brain barrier properties in endothelial cells. Nature 325, 253–257. [DOI] [PubMed] [Google Scholar]
- Jimenez E, Ormazabal A, Serrano M, Ortez‐Gonzalez CI, Artuch R, Garcia‐Cazorla A & Campistol J (2012). [Amino acids in cerebrospinal fluid and plasma: its usefulness in the study of neuropaediatric diseases]. Rev Neurol 54, 394–398 [Spanish]. [PubMed] [Google Scholar]
- Johanson CE & Murphy VA (1990). Acetazolamide and insulin alter choroid plexus epithelial cell [Na+], pH, and volume. Am J Physiol 258, F1538–F1546. [DOI] [PubMed] [Google Scholar]
- Johanson CE & Woodbury DM (1974). Changes in CSF flow and extracellular space in the developing rat In Drugs and the Developing Brain, ed. Vernadakis A. & Weiner N, pp 281–287. Plenum Press, New York. [Google Scholar]
- Johansson CE & Woodbury DM (1978). Uptake of [14C]urea by the in vivo choroid plexus–cerebrospinal fluid–brain system: identification of sites of molecular sieving. J Physiol 275, 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson PA (2014). The choroid plexuses and their impact on developmental neurogenesis. Front Neurosci 8, 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson PA, Dziegielewska KM, Ek CJ, Habgood MD, Liddelow SA, Potter AM, Stolp HB & Saunders NR (2006). Blood‐CSF barrier function in the rat embryo. Eur J Neurosci 24, 65–76. [DOI] [PubMed] [Google Scholar]
- Johansson PA, Dziegielewska KM, Liddelow SA & Saunders NR (2008). The blood‐CSF barrier explained: when development is not immaturity. Bioessays 30, 237–248. [DOI] [PubMed] [Google Scholar]
- Johnston M, Zakharov A, Papaiconomou C, Salmasi G & Armstrong D (2004). Evidence of connections between cerebrospinal fluid and nasal lymphatic vessels in humans, non‐human primates and other mammalian species. Cerebrospinal Fluid Res 1, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klosovski BN (1963). The Development of the Brain and its Disturbance by Harmful Factors, p. 94 Pergamon Press, Oxford. [Google Scholar]
- Knott GW, Dziegielewska KM, Habgood MD, Li ZS & Saunders NR (1997). Albumin transfer across the choroid plexus of South American opossum (Monodelphis domestica). J Physiol 499, 179–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler‐Stec EM, Simpson IA, Vannucci SJ, Landschulz KT & Landschulz WH (1998). Monocarboxylate transporter expression in mouse brain. Am J Physiol 275, E516–E524. [DOI] [PubMed] [Google Scholar]
- Koh L, Zakharov A, Nagra G, Armstrong D, Friendship R & Johnston M (2006). Development of cerebrospinal fluid absorption sites in the pig and rat: connections between the subarachnoid space and lymphatic vessels in the olfactory turbinates. Anat Embryol (Berl) 211, 335–344. [DOI] [PubMed] [Google Scholar]
- Korogod N, Petersen CC & Knott GW (2015). Ultrastructural analysis of adult mouse neocortex comparing aldehyde perfusion with cryo fixation. Elife 4, e05793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratzer I, Liddelow SA, Saunders NR, Dziegielewska KM, Strazielle N & Ghersi‐Egea JF (2013). Developmental changes in the transcriptome of the rat choroid plexus in relation to neuroprotection. Fluids Barriers CNS 10, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratzer I, Vasiljevic A, Rey C, Fevre‐Montange M, Saunders N, Strazielle N, Ghersi‐Egea JF (2012). Complexity and developmental changes in the expression pattern of claudins at the blood‐CSF barrier. Histochem Cell Biol 138, 861–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizbai IA, Lenzser G, Szatmari E, Farkas AE, Wilhelm I, Fekete Z, Erdos B, Bauer H, Bauer HC, Sandor P & Komjati K (2005). Blood‐brain barrier changes during compensated and decompensated hemorrhagic shock. Shock 24, 428–433. [DOI] [PubMed] [Google Scholar]
- Krogh A (1929). The progress of physiology. Am J Physiol 90, 243–251. [Google Scholar]
- Lange ECM (2013). Utility of CSF in translational neuroscience. J Pharmacokinet Pharmacodyn 40, 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger M, Bechmann I, Immig K, Reichenbach A, Härtig W & Michalski D (2015). Blood‐brain barrier breakdown involves four distinct stages of vascular damage in various models of experimental focal cerebral ischemia. J Cereb Blood Flow Metab 35, 292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclercq S, Mian FM, Stanisz AM, Bindels LB, Cambier E, Ben‐Amram H, Koren O, Forsythe P & Bienenstock J (2017). Low‐dose penicillin in early life induces long‐term changes in murine gut microbiota, brain cytokines and behavior. Nat Commun 8, 15062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefauconnier J‐M & Trouvé R (1983). Developmental changes in the pattern of amino acid transport at the blood‐brain barrier in rats. Dev Brain Res 6, 175–182. [DOI] [PubMed] [Google Scholar]
- Lehtinen MK, Bjornsson CS, Dymecki SM, Gilbertson RJ, Holtzman DM & Monuki ES (2013). The choroid plexus and cerebrospinal fluid: emerging roles in development, disease, and therapy. J Neurosci 33, 17553–17559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK & Walsh CA (2011). Neurogenesis at the brain‐cerebrospinal fluid interface. Annu Rev Cell Dev Biol 27, 653–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Zappaterra MW, Chen X, Yang YJ, Hill AD, Lun M, Maynard T, Gonzalez D, Kim S, Ye P, D'Ercole AJ, Wong ET, LaMantia AS & Walsh CA (2011). The cerebrospinal fluid provides a proliferative niche for neural progenitor cells. Neuron 69, 893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Dziegielewska KM, Ek CJ, Habgood MD, Bauer H, Bauer HC, Lindsay H, Wakefield MJ, Strazielle N, Kratzer I, Møllgård K, Ghersi‐Egea JF & Saunders NR (2013). Mechanisms that determine the internal environment of the developing brain: a transcriptomic, functional and ultrastructural approach. PLoS One 8, e65629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Dziegielewska KM, Ek CJ, Habgood MD, Bauer H, Bauer H‐C, Lindsay H, Wakefield MJ, Strazielle N, Kratzer I, Møllgård K, Ghersi‐Egea JF & Saunders NR (2016). Correction: Mechanisms that determine the internal environment of the developing brain: A transcriptomic, functional and ultrastructural approach. PLoS One 11, e0147680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Dziegielewska KM, Ek CJ, Johansson PA, Potter AM & Saunders NR (2009). Cellular transfer of macromolecules across the developing choroid plexus of Monodelphis domestica . Eur J Neurosci 29, 253–266. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Dziegielewska KM, Møllgård K, Phoenix TN, Temple S, Vandeberg JL & Saunders NR (2011a). SPARC/osteonectin, an endogenous mechanism for targeting albumin to the blood‐cerebrospinal fluid interface during brain development. Eur J Neurosci 34, 1062–1073. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Dzięgielewska KM, Møllgård K, Whish SC, Noor NM, Wheaton BJ, Gehwolf R, Wagner A, Traweger A, Bauer H, Bauer HC & Saunders NR (2014). Cellular specificity of the blood‐CSF barrier for albumin transfer across the choroid plexus epithelium. PLoS One 9, e106592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Dziegielewska KM, VandeBerg JL, Noor NM, Potter AM & Saunders NR (2011b). Modification of protein transfer across blood/cerebrospinal fluid barrier in response to altered plasma protein composition during development. Eur J Neurosci 33, 391–400. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Temple S, Møllgård K, Gehwolf R, Wagner A, Bauer H, Bauer HC, Phoenix TN, Dziegielewska KM & Saunders NR (2012). Molecular characterisation of transport mechanisms at the developing mouse blood‐CSF interface: a transcriptome approach. PLoS One 7, e33554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López‐Espíndola D, Morales‐Bastos C, Grijota‐Martínez C, Liao XH, Lev D, Sugo E, Verge CF, Refetoff S, Bernal J & Guadaño‐Ferraz A (2014). Mutations of the thyroid hormone transporter MCT8 cause prenatal brain damage and persistent hypomyelination. J Clin Endocrinol Metab 99, E2799–E2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W & Potschka H (2005). Blood‐brain barrier active efflux transporters: ATP‐binding cassette gene family. NeuroRx 2, 86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery LA & Sive H (2009). Totally tubular: the mystery behind function and origin of the brain ventricular system. Bioessays 31, 446–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun MP, Monuki ES & Lehtinen MK (2015). Development and functions of the choroid plexus‐cerebrospinal fluid system. Nat Rev Neurosci 16, 445–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyck R, Ruderisch N, Moll AG, Steiner O, Cohen CD, Engelhardt B, Makrides V & Verrey F (2009). Culture‐induced changes in blood‐brain barrier transcriptome: implications for amino‐acid transporters in vivo . J Cereb Blood Flow Metab 29, 1491–1502. [DOI] [PubMed] [Google Scholar]
- Madsen JK & Møllgård K (1979). The tight epithelium of the Mongolian gerbil subcommissural organ as revealed by freeze‐fracturing. J Neurocytol 8, 481–491. [DOI] [PubMed] [Google Scholar]
- Maliepaard M, Scheffer GL, Faneyte IF, van Gastelen MA, Pijnenborg AC, Schinkel AH, van De Vijver MJ, Scheper RJ & Schellens JH (2001). Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res 61, 3458–3464. [PubMed] [Google Scholar]
- Mankertz J & Schulzke JD (2007). Altered permeability in inflammatory bowel disease: pathophysiology and clinical implications. Curr Opin Gastroenterol 23, 379–383. [DOI] [PubMed] [Google Scholar]
- Marques F, Sousa JC, Coppola G, Gao F, Puga R, Brentani H, Geschwind DH, Sousa N, Correia‐Neves M & Palha JA (2011). Transcriptome signature of the adult mouse choroid plexus. Fluids Barriers CNS 8, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrady E Jr (1938). The embryology of the opossum American Anatomical Memoirs No. 16, 5–225. The Wistar Institute of Anatomy and Biology, Philadelphia, PA, USA. [Google Scholar]
- Mikkelsen TS, Wakefield MJ, Aken B, Amemiya CT, Chang JL, Duke S, Garber M, Gentles AJ, Goodstadt L, Heger A, Jurka J, Kamal M, Mauceli E, Searle SM, Sharpe T, Baker ML, Batzer MA, Benos PV, Belov K, Clamp M, Cook A, Cuff J, Das R, Davidow L, Deakin JE, Fazzari MJ, Glass JL, Grabherr M, Greally JM, Gu W, Hore TA, Huttley GA, Kleber M, Jirtle RL, Koina E, Lee JT, Mahony S, Marra MA, Miller RD, Nicholls RD, Oda M, Papenfuss AT, Parra ZE, Pollock DD, Ray DA, Schein JE, Speed TP, Thompson K, VandeBerg JL, Wade CM, Walker JA, Waters PD, Webber C, Weidman JR, Xie X, Zody MC, Broad Institute Genome Sequencing Platform , Broad Institute Whole Genome Assembly Team , Graves JA, Ponting CP, Breen M, Samollow PB, Lander ES & Lindblad‐Toh K (2007). Genome of the marsupial Monodelphis domestica reveals innovation in non‐coding sequences. Nature 447, 167–177. [DOI] [PubMed] [Google Scholar]
- Miyan JA, Zendah M, Mashayekhi F & Owen‐Lynch PJ (2006). Cerebrospinal fluid supports viability and proliferation of cortical cells in vitro, mirroring in vivo development. Cerebrospinal Fluid Res 3, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møllgård K, Balslev Y, Lauritzen B & Saunders NR (1987). Cell junctions and membrane specializations in the ventricular zone (germinal matrix) of the developing sheep brain: a CSF‐brain barrier. J Neurocytol 16, 433–444. [DOI] [PubMed] [Google Scholar]
- Møllgård K, Dziegielewska KM, Holst CB, Habgood MD & Saunders NR (2017). Brain barriers and functional interfaces with sequential appearance of ABC efflux transporters during human development. Sci Rep 7, 11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møllgård K & Saunders NR (1975). Complex tight junctions of epithelial and of endothelial cells in early foetal brain. J Neurocytol 4, 453–468. [DOI] [PubMed] [Google Scholar]
- Møllgård K & Saunders NR (1977). A possible transepithelial pathway via endoplasmic reticulum in foetal sheep choroid plexus. Proc R Soc Lond B Biol Sci 199, 321–326. [DOI] [PubMed] [Google Scholar]
- Monafò MR & Smith GD (2018). Repeating experiments is not enough. Nature 553, 399–401.29368721 [Google Scholar]
- Morgan EH & Moos T (2002). Mechanism and developmental changes in iron transport across the blood‐brain barrier. Dev Neurosci 24, 106–113. [DOI] [PubMed] [Google Scholar]
- Nahirney PC, Reeson P & Brown CE (2016). Ultrastructural analysis of blood‐brain barrier breakdown in the peri‐infarct zone in young adult and aged mice. J Cereb Blood Flow Metab 36, 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwelt EA (2004). Mechanisms of disease: the blood‐brain barrier. Neurosurgery 54, 131–140; discussion 141–142. [DOI] [PubMed] [Google Scholar]
- Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S (2003). Size‐selective loosening of the blood‐brain barrier in claudin‐5‐deficient mice. J Cell Biol 161, 653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberdick J, Ling Y, Phelps MA, Yudovich MS, Schilling K & Sadee W (2016). Preferential delivery of an opioid antagonist to the fetal brain in pregnant mice. J Pharmacol Exp Ther 358, 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermeier B, Daneman R & Ransohoff RM (2013). Development, maintenance and disruption of the blood‐brain barrier. Nat Med 19, 1584–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldendorf WH (1971). Brain uptake of radiolabeled amino acids, amines, and hexoses after arterial injection. Am J Physiol 221, 1629–1639. [DOI] [PubMed] [Google Scholar]
- Oldendorf WH (1973). Stereospecificity of blood-brain barrier permeability to amino acids. Am J Physiol 224, 967–969. [DOI] [PubMed] [Google Scholar]
- Panfoli I, Cassanello M, Bruschettini M, Colella M, Cerone R, Ravera S, Calzia D, Candiano G & Ramenghi L (2016). Why do premature newborn infants display elevated blood adenosine levels? Med Hypotheses 90, 53–56. [DOI] [PubMed] [Google Scholar]
- Parada C, Gato A & Bueno D (2005). Mammalian embryonic cerebrospinal fluid proteome has greater apolipoprotein and enzyme pattern complexity than the avian proteome. J Proteome Res 4, 2420–2428. [DOI] [PubMed] [Google Scholar]
- Parvas M & Bueno D (2010). The embryonic blood‐CSF barrier has molecular elements to control E‐CSF osmolarity during early CNS development. J Neurosci Res 88, 1205–1212. [DOI] [PubMed] [Google Scholar]
- Plog BA & Nedergaard M (2017). The glymphatic system in central nervous system health and disease: Past, present, and future. Annu Rev Pathol 13, 379–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porterfield SP & Hendrich CE (1992). Tissue iodothyronine levels in fetuses of control and hypothyroid rats at 13 and 16 days gestation. Endocrinology 131, 195–200. [DOI] [PubMed] [Google Scholar]
- Povlishock JT, Becker DP, Sullivan HG & Miller JD (1978). Vascular permeability alterations to horseradish peroxidase in experimental brain injury. Brain Res 153, 223–239. [DOI] [PubMed] [Google Scholar]
- Praetorius J & Damkier HH (2017). Transport across the choroid plexus epithelium. Am J Physiol Cell Physiol 312, C673–C686. [DOI] [PubMed] [Google Scholar]
- Ramey BA & Birge WJ (1979). Development of cerebrospinal fluid and the blood‐cerebrospinal fluid barrier in rabbits. Dev Biol 68, 292–298. [DOI] [PubMed] [Google Scholar]
- Reinhold AK & Rittner HL (2017). Barrier function in the peripheral and central nervous system‐a review. Pflugers Arch 469, 123–134. [DOI] [PubMed] [Google Scholar]
- Renfree MB, Papenfuss AT, Deakin JE, Lindsay J, Heider T, Belov K, Rens W, Waters PD, Pharo EA, Shaw G, Wong ES, Lefèvre CM, Nicholas KR, Kuroki Y, Wakefield MJ, Zenger KR, Wang C, Ferguson‐Smith M, Nicholas FW, Hickford D, Yu H, Short KR, Siddle HV, Frankenberg SR, Chew KY, Menzies BR, Stringer JM, Suzuki S, Hore TA, Delbridge ML, Patel HR, Mohammadi A, Schneider NY, Hu Y, O'Hara W, Al Nadaf S, Wu C, Feng ZP, Cocks BG, Wang J, Flicek P, Searle SM, Fairley S, Beal K, Herrero J, Carone DM, Suzuki Y, Sugano S, Toyoda A, Sakaki Y, Kondo S, Nishida Y, Tatsumoto S, Mandiou I, Hsu A, McColl KA, Lansdell B, Weinstock G, Kuczek E, McGrath A, Wilson P, Men A, Hazar‐Rethinam M, Hall A, Davis J, Wood D, Williams S, Sundaravadanam Y, Muzny DM, Jhangiani SN, Lewis LR, Morgan MB, Okwuonu GO, Ruiz SJ, Santibanez J, Nazareth L, Cree A, Fowler G, Kovar CL, Dinh HH, Joshi V, Jing C, Lara F, Thornton R, Chen L, Deng J, Liu Y, Shen JY, Song XZ, Edson J, Troon C, Thomas D, Stephens A, Yapa L, Levchenko T, Gibbs RA, Cooper DW, Speed TP, Fujiyama A, Graves JA, O'Neill RJ, Pask AJ, Forrest SM & Worley KC (2011). Genome sequence of an Australian kangaroo, Macropus eugenii, provides insight into the evolution of mammalian reproduction and development. Genome Biol 12, R81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds ML, Møllgård K & Saunders NR (1983). The distribution of plasma proteins during early embryonic development in the sheep. Anat Embryol (Berl) 168, 227–240. [DOI] [PubMed] [Google Scholar]
- Rivas M & Naranjo JR (2007). Thyroid hormones, learning and memory. Genes Brain Behav 6(Suppl 1), 40–44. [DOI] [PubMed] [Google Scholar]
- Rodier PM (1995). Developing brain as a target of toxicity. Environ Health Perspect 103 (Suppl 6), 73–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas‐Hernandez H, Cuevas E, Lantz S, Imam SZ, Paule MG, Ali SF (2018). Blood–brain barrier: Physiological and functional considerations In Handbook of Developmental Neurotoxicology, 2nd edn, ed. Slikker W, Jr, Paule MG. & Wang C, Ch 20, pp. 229–236. Academic Press. [Google Scholar]
- Rossdale PD, Cash RS, Leadon DP & Jeffcott LB (1982). Biochemical constituents of cerebrospinal fluid in premature and full term foals. Equine Vet J 14, 134–138. [DOI] [PubMed] [Google Scholar]
- Sandberg JA, Duhart HM, Lipe G, Binienda Z, Slikker W Jr & Kim CS (1996). Distribution of 2,4‐dichlorophenoxyacetic acid (2,4‐D) in maternal and fetal rabbits. J Toxicol Environ Health 49, 497–509. [DOI] [PubMed] [Google Scholar]
- Saunders NR (1977). Ontogeny of the blood‐brain barrier. Exp Eye Res 25 (Suppl), 523–550. [DOI] [PubMed] [Google Scholar]
- Saunders NR, Daneman R, Dziegielewska KM & Liddelow SA (2013). Transporters of the blood-brain and blood-CSF interfaces in development and in the adult. Mol Aspects Med 34, 742–752. [DOI] [PubMed] [Google Scholar]
- Saunders NR (1992). Ontogenetic development of brain barrier mechanisms In Handbook of Experimental Pharmacology, vol. 103, Physiology and Pharmacology of the Blood Brain‐Barrier, ed. Bradbury MWB, pp. 327–369. Springer‐Verlag, Berlin. [Google Scholar]
- Saunders NR, Dreifuss JJ, Dziegielewska KM, Johansson PA, Habgood MD, Møllgård K & Bauer HC (2014). The rights and wrongs of blood‐brain barrier permeability studies: a walk through 100 years of history. Front Neurosci 8, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders NR, Dziegielewska KM, Møllgård K & Habgood MD (2015b). Markers for blood‐brain barrier integrity: how appropriate is Evans blue in the twenty‐first century and what are the alternatives? Front Neurosci 9, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders NR, Dziegielewska KM, Møllgård K, Habgood MD, Wakefield MJ, Lindsay H, Stratzielle N, Ghersi‐Egea JF, Liddelow SA (2015a). Influx mechanisms in the embryonic and adult rat choroid plexus: a transcriptome study. Front Neurosci 9, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders NR, Dziegielewska KM, Unsicker K & Ek CJ (2016a). Delayed astrocytic contact with cerebral blood vessels in FGF‐2 deficient mice does not compromise permeability properties at the developing blood‐brain barrier. Dev Neurobiol 76, 1201–1212. [DOI] [PubMed] [Google Scholar]
- Saunders NR & Habgood MD (2017). Acetaminophen in pregnancy and adverse childhood neurodevelopment. JAMA Pediatr 171, 395. [DOI] [PubMed] [Google Scholar]
- Saunders NR, Habgood MD & Dziegielewska KM (1999). Barrier mechanisms in the brain, II. Immature brain. Clin Exp Pharmacol Physiol 26, 85–91. [DOI] [PubMed] [Google Scholar]
- Saunders NR, Habgood MD, Møllgård K & Dziegielewska KM (2016b). The biological significance of brain barrier mechanisms: help or hindrance in drug delivery to the central nervous system? F1000Res 5, F1000 Faculty Rev‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders NR, Liddelow SA & Dziegielewska KM (2012). Barrier mechanisms in the developing brain. Front Pharmacol 3, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl‐Bürgi S, Haberlandt E, Heinz‐Erian P, Deisenhammer F, Albrecht U, Sigl SB, Rauchenzauner M, Ulmer H & Karall D (2008). Amino acid cerebrospinal fluid/plasma ratios in children: influence of age, gender, and antiepileptic medication. Pediatrics 121, e920–e926. [DOI] [PubMed] [Google Scholar]
- Schumacher U & Møllgård K (1997). The multidrug‐resistance P‐glycoprotein (Pgp, MDR1) is an early marker of blood‐brain barrier development in the microvessels of the developing human brain. Histochem Cell Biol 108, 179–182. [DOI] [PubMed] [Google Scholar]
- Semple BD, Blomgren K, Gimlin K, Ferriero DM & Noble‐Haeusslein LJ (2013). Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol 106–107, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegenthaler JA, Sohet F & Daneman R (2013). ‘Sealing off the CNS’: cellular and molecular regulation of blood‐brain barriergenesis. Curr Opin Neurobiol 23, 1057–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeaff SA (2011). Iodine deficiency in pregnancy: the effect on neurodevelopment in the child. Nutrients 3, 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speake T, Whitwell C, Kajita H, Majid A & Brown PD (2001). Mechanisms of CSF secretion by the choroid plexus. Microsc Res Tech 52, 49–59. [DOI] [PubMed] [Google Scholar]
- Srinivasan L, Shah SS, Padula MA, Abbasi S, McGowan KL & Harris MC (2012). Cerebrospinal fluid reference ranges in term and preterm infants in the neonatal intensive care unit. J Pediatr 161, 729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stastný F & Rychter Z (1976). Effect of hydrocortisone on the growth of choroid plexus and composition of cerebrospinal fluid in the developing chick embryo. Acta Neurol 53, 260–274. [DOI] [PubMed] [Google Scholar]
- Staud F, Vackova Z, Pospechova K, Pavek P, Ceckova M, Libra A, Cygalova L, Nachtigal P & Fendrich Z (2006). Expression and transport activity of breast cancer resistance protein (Bcrp/Abcg2) in dually perfused rat placenta and HRP‐1 cell line. J Pharmacol Exp Ther 319, 53–62. [DOI] [PubMed] [Google Scholar]
- Stergiakouli E, Thapar A & Davey Smith G (2016). Association of acetaminophen use during pregnancy with behavioral problems in childhood: Evidence against confounding. JAMA Pediatr 170, 964–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strazielle N & Ghersi‐Egea JF (2015). Efflux transporters in blood‐brain interfaces of the developing brain. Front Neurosci 9, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stonestreet BS, Patlak CS, Pettigrew KD, Reilly CB & Cserr HF (1996). Ontogeny of blood‐brain barrier function in ovine fetuses, lambs, and adults. Am J Physiol 271, R1594–R1601. [DOI] [PubMed] [Google Scholar]
- Sun H, Tang Y, Guan X, Li L & Wang D (2013). Effects of selective hypothermia on blood‐brain barrier integrity and tight junction protein expression levels after intracerebral hemorrhage in rats. Biol Chem 394, 1317–1324. [DOI] [PubMed] [Google Scholar]
- Szmydynger‐Chodobska J, Pascale CL, Pfeffer AN, Coulter C & Chodobski A (2007). Expression of junctional proteins in choroid plexus epithelial cell lines: a comparative study. Cerebrospinal Fluid Res 4, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tărlungeanu DC, Deliu E, Dotter CP, Kara M, Janiesch PC, Scalise M, Galluccio M, Tesulov M, Morelli E, Sonmez FM, Bilguvar K, Ohgaki R, Kanai Y, Johansen A, Esharif S, Ben‐Omran T, Topcu M, Schlessinger A, Indiveri C, Duncan KE, Caglayan AO, Gunel M, Gleeson JG & Novarino G (2016). Impaired amino acid transport at the blood brain barrier is a cause of autism spectrum disorder. Cell 167, 1481–1494.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullmann SL, Shaw G, Alcorn GT & Renfree MB (1997). Migration of primordial germ cells to the developing gonadal ridges in the tammar wallaby Macropus eugenii . J Reprod Fertil 110, 135–143. [DOI] [PubMed] [Google Scholar]
- Umans RA, Henson HE, Mu F, Parupalli C, Ju B, Peters JL, Lanham KA, Plavicki JS & Taylor MR (2017). CNS angiogenesis and barriergenesis occur simultaneously. Dev Biol 425, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen LM, Evans RJ, Jim KK, Verboom T, Fang X, Bojarczuk A, Malicki J, Johnston SA & van der Sar AM (2018). A transgenic zebrafish model for the in vivo study of the blood and choroid plexus brain barriers using claudin 5. Biol Open 7, bio030494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannucci SJ, Seaman LB, Brucklacher RM & Vannucci RC (1994). Glucose transport in developing rat brain: glucose transporter proteins, rate constants and cerebral glucose utilization. Mol Cell Biochem 140, 177–184. [DOI] [PubMed] [Google Scholar]
- Vazire S (2017). Our obsession with eminence warps research. Nature 547, 7. [DOI] [PubMed] [Google Scholar]
- Virgintino D, Errede M, Girolamo F, Capobianco C, Robertson D, Vimercati A, Serio G, Di Benedetto A, Yonekawa Y, Frei K & Roncali L (2008). Fetal blood‐brain barrier P‐glycoprotein contributes to brain protection during human development. J Neuropathol Exp Neurol 67, 50–61. [DOI] [PubMed] [Google Scholar]
- Visser TJ (2016). Cellular uptake of thyroid hormones In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000–2016 Jun 15. [Google Scholar]
- Wahle AM, Brühschwein A, Matiasek K, Putschbach K, Wagner E, Mueller RS & Fischer A (2014). Clinical characterization of epilepsy of unknown cause in cats. J Vet Intern Med 28, 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson RE, Desesso JM, Hurtt ME & Cappon GD (2006). Postnatal growth and morphological development of the brain: a species comparison. Birth Defects Res B Dev Reprod Toxicol 77, 471–484. [DOI] [PubMed] [Google Scholar]
- Whish S, Dziegielewska KM, Møllgård K, Noor NM, Liddelow SA, Habgood MD, Richardson SJ & Saunders NR (2015). The inner CSF‐brain barrier: developmentally controlled access to the brain via intercellular junctions. Front Neurosci 9, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappaterra MW & Lehtinen MK (2012). The cerebrospinal fluid: regulator of neurogenesis, behavior, and beyond. Cell Mol Life Sci 69, 2863–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappaterra MD, Lisgo SN, Lindsay S, Gygi SP, Walsh CA & Ballif BA (2007). A comparative proteomic analysis of human and rat embryonic cerebrospinal fluid. J Proteome Res 6, 3537–3548. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, Duncan JA 3rd, Cheshier SH, Shuer LM, Chang EF, Grant GA, Gephart MG & Barres BA (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Nelson AR, Betsholtz C & Zlokovic BV (2015). Establishment and dysfunction of the blood‐brain barrier. Cell 163, 1064–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]