

Abstract
Key points
Maternal obesity (MO) and exposure to a high‐fat, high‐simple‐carbohydrate diet during pregnancy predisposes offspring to obesity, metabolic and cardiovascular disorders in later life.
Underlying molecular pathways and potential epigenetic factors that are dysregulated in MO were identified using unbiased transcriptomic methods.
There was increased lipid accumulation and severe steatosis in the MO baboon fetal liver suggesting that these offspring are on an early trajectory of non‐alcoholic fatty liver disease and non‐alcoholic steatohepatitis.
Abstract
Maternal obesity (MO) increases offspring cardiometabolic disease risk. Altered fetal liver development in response to the challenge of MO has metabolic consequences underlying adverse offspring life‐course health outcomes. Little is known about the molecular pathways and potential epigenetic changes regulating primate fetal liver responses to MO. We hypothesized that MO would induce fetal baboon liver epigenetic changes resulting in dysregulation of key metabolic pathways that impact lipid metabolism. MO was induced prior to pregnancy by a high‐fat, high‐fructose diet. Unbiased gene and microRNA (small RNA Seq) abundance analyses were performed on fetal baboon livers at 0.9 gestation and subjected to pathway analyses to identify fetal liver molecular responses to MO. Fetal baboon liver lipid and glycogen content were quantified by the Computer Assisted Stereology Toolbox. In response to MO, fetal livers revealed dysregulation of TCA cycle, proteasome, oxidative phosphorylation, glycolysis and Wnt/β‐catenin signalling pathways together with marked lipid accumulation supporting our hypothesis that multiple pathway dysregulation detrimentally impacts lipid management. This is the first study of MO programming of the non‐human primate fetal liver using unbiased transcriptome analysis to detect changes in hepatic gene expression levels and identify potential microRNA epigenetic regulators of metabolic disruption.
Keywords: maternal obesity, fetal liver, high fat/sugar diet, gene expression, microRNA integration, biological pathways, lipid metabolism
Key points
Maternal obesity (MO) and exposure to a high‐fat, high‐simple‐carbohydrate diet during pregnancy predisposes offspring to obesity, metabolic and cardiovascular disorders in later life.
Underlying molecular pathways and potential epigenetic factors that are dysregulated in MO were identified using unbiased transcriptomic methods.
There was increased lipid accumulation and severe steatosis in the MO baboon fetal liver suggesting that these offspring are on an early trajectory of non‐alcoholic fatty liver disease and non‐alcoholic steatohepatitis.
Introduction
The incidence of obesity and overweight has reached epidemic proportions in the developed world, with approximately 64% of women of childbearing age in the USA being overweight (BMI = 25–30 kg m−2) or obese (BMI ≥30 kg m−2) (Wilson & Messaoudi, 2015). Both an obesogenic nutritional environment and a sedentary lifestyle contribute to the risk of developing obesity. A growing body of evidence links early life nutritional adversity to development of long‐term metabolic disorders (Li et al. 2011; Regnault et al. 2013). Therefore, early life exposure of offspring to environmental stimuli, including altered nutrition during critical periods of development, can program alterations in organogenesis, tissue development and metabolism, predisposing offspring to obesity, metabolic and cardiovascular disorders in later life (Segovia, 2014; Carreras‐Badosa et al. 2017). In particular, the increasing prevalence of maternal obesity (MO) and excess maternal weight gain has been associated with additional risk of obesity in offspring (Nathanielsz et al. 2007). A number of studies have focused on maternal nutrition restriction and health outcomes (Armitage et al. 2004; Cox et al. 2006; Pereira et al. 2015); however, the majority of our population is experiencing an environment of caloric excess, and the increase in MO is a major cause of concern for the health of offspring from these pregnancies (McCurdy et al. 2009).
Human epidemiological evidence (Boerschmann et al. 2010; Krishnaveni et al. 2010; Mills et al. 2010; Gaillard 2015) and data from carefully controlled animal studies (Armitage et al. 2004; Alfradhi & Ozanne, 2011; Zambrano & Nathanielsz, 2013) have demonstrated that MO has long‐term consequences for offspring, predisposing or ‘programming’ them to develop cardiometabolic disease in adulthood. MO‐induced developmental programming has been validated in mouse, rat, sheep and non‐human primates (NHPs) (Shankar et al. 2008; McCurdy et al. 2009; Long et al. 2012; Cox et al. 2013; Maloyan et al. 2013).
In the light of these studies, we hypothesized that MO would induce epigenetic changes in fetal baboon (Papio hamadryas) liver resulting in dysregulation of key metabolic pathways that impact lipid metabolism. The baboon is a well‐characterized animal model sharing many physiological, metabolic and genetic characteristics with humans (Penfold & Ozanne, 2015), allowing direct translation of findings to human pregnancy.
Fetal liver development during MO programming is a central metabolic regulator and thus important for understanding the role of adverse maternal nutrition on offspring life course health. One study on fetal Japanese macaques (130 days’ gestation) revealed that maternal exposure to a chronic high‐fat diet influences lipid accumulation in the liver, oxidative stress and apoptosis (McCurdy et al. 2009). Heerwagen et al. (2013) demonstrated that fetuses of obese dams had higher fatty liver triacylglycerol deposition than lean dams at embryonic day 18.5 in mice.
Emerging evidence indicates that microRNAs (miRNAs) are major regulators of gene expression and key players in control of various metabolic processes and diseases (Bartel, 2004; Maloyan et al. 2013). miRNAs are a class of small (18–25 nucleotides) non‐coding RNA molecules identified as epigenetic regulators in the transduction of MO into metabolic outcomes in the offspring (Benatti et al. 2014; Nicholas et al. 2016). Several authors have reported altered expression of placental miRNAs associated with adverse pregnancy conditions such as preeclampsia (Hromadnikova et al. 2013; Li et al. 2013) and intrauterine growth restriction (Hromadnikova et al. 2013), MO with pre‐ and post‐natal growth (Segovia 2014), gestational hypertension (Hromadnikova et al. 2013) and congenital heart defects (Zhu et al. 2013). Maloyan et al. (2013) reported significant alterations in cardiac miRNA expression in fetuses of obese baboons. Recently, Tessitore et al. (2016) and Tian et al. (2016) identified epigenetic dysregulation of Wnt//β‐catenin in the development of non‐alcoholic fatty liver disease (NAFLD)‐associated hepatocellular carcinoma.
To date, only targeted gene expression analysis (qRT‐PCR and immunohistochemistry) has been performed in fetal livers of NHPs (McCurdy et al. 2009), but underlying molecular pathways and potential epigenetic changes due to miRNA were not identified. To our knowledge, no studies have been performed to explore the association of dysregulated miRNAs and the genes they regulate in fetal liver with obese mothers. This is the first study where unbiased transcriptome analysis, including gene expression and miRNA expression, has been used to identify potential miRNA epigenetic regulators of metabolic disruption in NHP fetal MO livers.
Methods
Ethical approval
All animal procedures were approved by the Texas Biomedical Research Institute (TBRI) Institutional Animal Care and Use Committee (IACUC) and conducted in Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) international‐approved facilities. The present work complies with the animal ethical principles of The Journal of Physiology and Grundy's (2015) checklist.
Animal model
Baboons were maintained in groups of up to 16 in custom‐built outdoor facilities allowing full socialization and free movement. Healthy, non‐pregnant female nulliparous baboons of similar weight, body dimensions and age were randomly assigned to either an ad libitum regular/normal diet (RD) (n = 6) or an ad libitum combination of the RD and high‐fat/high‐fructose diet (HFD) with free access to a sugar‐containing drink (Kool Aid) (n = 5) at least 9 months before pregnancy (Nathanielsz et al. 2015). Recording of individual measurements and other related information is described elsewhere (Maloyan et al. 2013; Schlabritz‐Loutsevitch et al. 2004).
The basic composition of RD was 12% energy from fat, 0.29% from glucose and 0.32% from fructose with an energy content of 3.07 kcal g−1 (Purina Monkey Diet, Purina LabDiets, St Louis, MO, USA). The basic composition of HFD was 45% energy from fat, 4.62% from glucose, 5.64% from fructose and 2.32% from sucrose, with an energy content of 4.03 kcal g−1. Protein, all essential minerals and vitamins were equalized for the RD and HFD regimens. The dietary regimens were maintained throughout pregnancy.
Caesarean sections and tissue collection
Pregnant baboons underwent Caesarean section at 165 days of gestation (0.9 gestation (G); term ∼184 days) to obtain the fetal liver samples. Caesarean sections/necropsies were performed using a standard sterile surgical technique (Schlabritz‐Loutsevitch et al. 2004). Briefly, baboons were pre‐medicated with ketamine hydrochloride (10 mg kg−1 i.m.), intubated and maintained at a surgical plane of anaesthesia with isoflurane (2%) throughout surgery (Schlabritz‐Loutsevitch et al. 2004). The only change from previous published procedures was that after hysterotomy, the umbilical cord was identified and then used for fetal exsanguination with maternal and fetal baboon under general anaesthesia as approved by the American Veterinary Medical Association Panel on Euthanasia. This change has been made since we have demonstrated fewer tissue artefacts following exsanguination than following administration of euthanasia solutions, which produce marked histological changes, especially in the liver (Grieves et al. 2008). Morphometric measurements were collected and tissue samples obtained immediately after the placenta and fetus were removed from the uterus. The right lobe of the fetal liver was immediately snap frozen in liquid nitrogen and stored at −80°C. The fetal liver samples obtained from mothers fed RD were called experimental control fetal liver samples. The fetal liver samples obtained from mothers fed HFD were called MO fetal liver samples. Buprenorphine hydrochloride (buprenorphine HCl injection; Hospira, Inc., Lake Forest, IL, USA), 0.015 mg kg day−1 split as two doses for 3 days, was administered for postoperative maternal analgesia (Schlabritz‐Loutsevitch et al. 2004). After recovery from anaesthesia, baboons were individually caged for the initial post‐operative period and then group‐housed for 90 days with a vasectomized male to prevent pregnancy before the surgical site was totally healed.
RNA isolation from tissue
Total RNA was isolated from fetal livers (control (CON) = 6, 3 males and 3 females; MO = 5, 2 males and 3 females) using TRI Reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's instructions. Genomic DNA in the sample was sheared by passing the homogenate several times through a 22‐gauge needle attached to a 1 ml syringe. The homogenized samples were incubated for 5 min at 25°C. To each sample, 200 μl of chloroform was added, and the samples were shaken vigorously by hand for 15 s and incubated at 25°C for 3 min. Samples were then centrifuged at 4°C and 14,000 g for 10 min. The aqueous phase containing RNA was transferred to a fresh tube and one volume 70% ethanol was mixed thoroughly with the sample. This mix was cleaned and concentrated with the Qiagen RNeasy MinElute spin column kit (Qiagen, Germantown, MD, USA). The RNA was resuspended in 30 μl RNase‐free water and stored at −80°C. RNA integrity and concentration were determined by an Agilent 2100 Bioanalyzer and UV‐Vis Spectrophotometer (Nanodrop 2000), respectively (Agilent Technologies, Santa Clara, CA, USA).
Total RNA, including small RNAs, was isolated from the same tissue samples described above using Qiazol Lysis Reagent according to the manufacturer's instructions (Qiagen). Briefly, approximately 5 mg of frozen liver was homogenized in 1 ml Qiazol Lysis Reagent using a BioSpec (Bartlesville, OK, USA) BeadBeater with 1 × 6.35 mm stainless steel beads for three 30 s cycles with a 15 s pause between cycles. The homogenized samples were incubated for 5 min at 25°C and centrifuged through a Qiashredder tube (Qiagen) at 17,000 g for 2 min. To each sample, 200 μl of chloroform was added, and the samples were shaken vigorously by hand for 15 s and incubated at 25°C for 3 min. Samples were then centrifuged at 4°C and 14,000 g for 15 min. The aqueous phase containing RNA was transferred to a fresh tube and 1.5 volumes 100% ethanol was mixed thoroughly with the sample. This mix was cleaned and concentrated with the Qiagen RNeasy Mini spin column kit. The RNA was resuspended in 60 μl RNase free water and stored at −80°C. RNA integrity and concentration were determined as detailed above.
Quantification of gene expression
Complementary RNA (cRNA) was synthesized from total RNA using the Illumina total prep 96 RNA Amplification Kit (Illumina, Inc., San Diego, CA, USA). Individual cRNA samples were used to interrogate each BeadChip (human HT‐12 v3) (CON = 6, 3 males and 3 females; MO = 5, 2 males and 3 females) as described (Shi et al. 2012). Gene expression data were processed and analysed using GenomeStudio (Illumina) and GeneSifter (Perkin Elmer, Waltham, MA, USA) as detailed in Shi et al. (2012) and Kerr et al. (2000) (GEO accession no: GSE99718) (Supporting information, Supplementary Table S1).
Quantification of miRNA expression
Small RNA sequencing libraries were prepared using the Illumina Truseq Small RNA Sample Prep Kit. Each library included a bar code adapter and libraries were pooled after cDNA synthesis. cDNA libraries were used for cluster generation on Illumina's Cluster Station and sequenced using the Illumina GAIIx sequencer. Raw sequence reads were obtained using Illumina's Pipeline v1.5 software following sequencing image analysis by the Pipeline Firecrest Module and base calling by the Pipeline Bustard Module. The extracted sequence reads were normalized, annotated and abundance determined using mirDeep2 (Friedländer et al. 2012) (GEO accession no: GSE99718) (Supporting information, Supplementary Table S2).
Pathway analysis
Differently expressed genes were overlaid onto gene ontological (GO) (Ashburner et al. 2000) pathways and Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al. 2004) pathways using GeneSifter.
We used an unbiased end‐of‐pathway gene expression approach to identify pathways dysregulated in MO baboon fetal liver that were likely relevant to the MO phenotype. We hypothesized that a pathway may only be relevant to the liver phenotype if gene expression profiles at the end of the pathway were consistent with the overall pathway change. Furthermore, if there were no differentially expressed genes at the end of a pathway, that pathway was not considered relevant to the phenotype (Nijland et al. 2007). After we found gene expression profiles at the end of the pathway that were consistent with the overall pathway change, we investigated pathways downstream of each of these pathways (Nijland et al. 2007).
MicroRNA Target Filter Analysis in IPA (Ingenuity® Pathway Analysis, Qiagen) was used to integrate miRNA expression with gene expression to search for miRNAs that regulate differentially expressed genes in KEGG pathways that passed the end‐of‐pathway analysis. The analysis used TargetScan human and Ingenuity Expert Findings from moderate and high confidence predicted sources as filters. We further prioritized identifying the miRNAs that exhibited differential expression (P ≤ 0.05) and showed inverse expression with the targeted mRNAs.
Histological analyses
Frozen fetal liver tissues (CON, n = 16, 8 males and 8 females; MO, n = 16, 8 males and 8 females) were cut into 5 μm sections, simultaneously thawed and fixed in 10% neutral buffered formalin, stained with Oil Red O (ORO) for lipid content and counterstained with haematoxylin. Periodic acid–Schiff (PAS) stain was used to detect glycogen on fixed liver tissue.
Lipid and glycogen contents in fetal livers were quantified using the Computer Assisted Stereology Toolbox (CAST) 2.0 system (Visiopharm, Ballerup, Denmark) coupled with an Olympus BX61 microscope (Olympus Corporation of the Americas, Center Valley, PA, USA). Utilizing this stereology approach (Mandarim‐de‐Lacerda 2003, we calculated the percentage area of ORO‐stained liver tissues through the use of lines and points. One liver section from each animal was used to quantify lipid content. On average, we analysed 48 fields per slide with each field selected randomly from the whole section using CAST‐directed sampling. In each field, point counting of total fetal liver tissue was performed at the magnification of ×40 to calculate the percentage area of lipid component; 180 points were inserted in each field and an average of 8028 points were used per slide. Cell percentage areas within each liver section were calculated using the following formula: (CP/TP) × 100, where CP is points that hit specified liver cells and TP is total liver points (Quinn et al. 2012, Guardado et al. 2009).
In addition, an experienced hepatopathologist evaluated the ORO‐, haematoxylin and eosin (H&E)‐, Mason trichrome‐, and PAS‐stained liver slides. The hepatopathologist was blinded to the identity of the condition of the groups (CON and MO). The Kleiner scoring system was used to assess the degree of steatosis (Kleiner et al. 2005). The ORO‐ and H&E‐stained slides were used for assessing degree of steatosis in fetal livers. Individual scoring and categorization of steatosis in fetal liver was evaluated on the following features based on ORO%: steatosis (grade 0 (<5%), grade 1 (5–33%), grade 2 (34–66%), or grade 4 (67–100%)) of the examined liver tissue at ×40 magnification. Other histological features of liver injury seen with NAFLD were also assessed including lobular and portal inflammation, hepatocyte ballooning and fibrosis.
Statistical analysis
Array data were all‐median normalized and log2 transformed using GeneSifter software. Statistical analyses of array data were performed by Student's unpaired t test using GeneSifter software. We did not find significant differences between the sexes and so we pooled the data from male and female fetuses within the dietary treatment groups (CON and MO) for the analysis.
Histological lipid and glycogen content data are expressed as means ± SEM. Lipid and glycogen differences between CON and MO groups were performed by unpaired t test using Prism 7 (GraphPad Software, La Jolla, CA, USA). Fetal and maternal morphometric and clinical data are expressed as means ± SEM and the differences between CON and MO were performed by unpaired t test using Prism 7. P < 0.05 was considered statistically significant.
Results
Weight and clinical measures during pre‐pregnancy period
Prior to pregnancy, weight gain was significantly higher (P < 0.001) in the MO mothers fed HFD than CON fed RD (Nathanielsz et al. 2015). During pre‐pregnancy, the dams fed HFD showed greater waist‐hip circumferences, higher plasma low density lipoprotein (LDL)‐cholesterol and triglyceride concentrations, and a greater percentage of body fat (P < 0.05) than age matched CON (Nathanielsz et al. 2015).
Weight changes during pregnancy
Baseline body weight was higher (P < 0.05) in MO than CON mothers (Table 1). During pregnancy, weight gain in CON mothers fed RD was greater than MO (P < 0.05) mothers fed HFD (Table 1).
Table 1.
Morphometric measurements
| Weights | CON | MO | P |
|---|---|---|---|
| Maternal basal weight (kg) | 17.3 ± 0.4 | 19.9 ± 0.6 | <0.05 |
| Maternal C‐section weight (kg) | 19.2 ± 0.6 | 20.3 ± 0.6 | 0.18 |
| Maternal weight gain (kg) | 1.9 ± 0.4 | 0.4 ± 0.5 | <0.05 |
| Placenta weight (g) | 220.7 ± 10.0 | 195.5 ± 15.0 | 0.20 |
| Placenta size (% fetal weight) | 26.8 ± 0.9 | 27.3 ± 1.7 | 0.79 |
| Fetal weight (g) | 813.1 ± 31.0 | 768.0 ± 23.0 | 0.26 |
| Fetal liver weight (g) | 24.1 ± 1.1 | 23.8 ± 0.9 | 0.85 |
Data are means ± SEM; CON, n = 16; MO, n = 16.
Maternal and fetal blood measures at 0.9G
Table 2 presents maternal and fetal clinical measurements from CON and MO dams on Caesarean section day 0.9G. MO dams exhibited significantly greater blood triglycerides (P = 0.03), glucose (P = 0.02) and insulin (P = 0.01) concentrations compared with CON dams. Fetuses of MO dams exhibited significantly increased blood insulin concentrations compared to CON (P = 0.03), with no differences in other fetal clinical measures (Table 2). We did not find sex differences for the fetal morphometrics and clinical measures in plasma samples.
Table 2.
Maternal and fetal clinical measurements at 0.9G
| Parameter | CON | MO | P value |
|---|---|---|---|
| Maternal | |||
| Cholesterol (mg dl−1) | 70.3 ± 5.2 | 79.0 ± 5.5 | 0.25 |
| Triglycerides (mg dl−1) | 49.6 ± 5.9 | 71.2 ± 8.1 | 0.03 |
| HDL (mg dl−1) | 34.6 ± 3.5 | 35.9 ± 2.5 | 0.76 |
| LDL (mg dl−1) | 26.6 ± 2.0 | 28.9 ± 5.3 | 0.69 |
| Glucose (mg dl−1) | 56.4 ± 6.3 | 92.9 ± 14.6 | 0.02 |
| Insulin (μIU ml−1) | 29.5 ± 9.2 | 73.7 ± 14.5 | 0.01 |
| ALT (U l−1) | 32.3 ± 4.3 | 33.9 ± 4.8 | 0.80 |
| AST (U l−1) | 20.9 ± 2.4 | 18.6 ± 2.7 | 0.53 |
| Fetal | |||
| Cholesterol (mg dl−1) | 61.5 ± 3.2 | 59.9 ± 3.4 | 0.74 |
| Triglycerides (mg dl−1) | 21.3 ± 4.5 | 21.7 ± 1.8 | 0.94 |
| HDL (mg dl−1) | 23.4 ± 1.3 | 20.5 ± 1.3 | 0.15 |
| LDL (mg dl−1) | 33.9 ± 3.3 | 35.1 ± 3.0 | 0.78 |
| Glucose (mg dl−1) | 68.7 ± 14.9 | 39.3 ± 6.5 | 0.08 |
| Insulin (μIU ml−1) | 10.2 ± 1.4 | 28.2 ± 8.1 | 0.03 |
| ALT (U l−1) | 20.1 ± 14.3 | 5.31 ± 1.7 | 0.31 |
| AST (U l−1) | 20.9 ± 2.4 | 18.6 ± 2.7 | 0.54 |
Data are means ± SEM; CON, n = 16; MO, n = 16. ALT, alanine transaminase; AST, aspartate transaminase; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein.
Gene expression profiling
Pairwise comparisons showed 933 differentially expressed genes between MO and CON fetal liver samples at 0.9G: 350 genes were up‐regulated and 583 were down‐regulated in MO compared with CON livers. Graphical representation of this analysis provides an overall view of the variation within groups and between groups for this dataset. No significant differences were observed between the differentially expressed genes between the sexes.
Differentially expressed genes were overlaid onto KEGG pathways to identify up‐regulated and down‐regulated biological pathways between MO and CON fetal livers. Table 3 lists the 11 pathways that were up‐regulated and 29 pathways that were down‐regulated. Included in Table 3 are the number of differentially expressed genes, the number of up‐regulated genes, the number of down‐regulated genes, the total number of genes in that pathway that were detected with a quality signal using baboon cRNA on the human gene array, and the z‐score (greater than 2.0 or less than −2.0) for the listed pathway.
Table 3.
KEGG pathways enriched for differentially expressed genes for 0.9G MO versus CON fetal livers
| Diff genes | Up | Down | Array | z‐score | |
|---|---|---|---|---|---|
| Up‐regulated pathways | |||||
| Butirosin and neomycin biosynthesis | 1 | 1 | 0 | 5 | 4.07 |
| Lysine degradation | 8 | 3 | 5 | 44 | 3.67 |
| Galactose metabolism | 4 | 2 | 2 | 26 | 3.24 |
| Ribosome | 6 | 4 | 2 | 87 | 3.17 |
| GnRH signalling pathway | 4 | 4 | 0 | 98 | 2.87 |
| Wnt signalling pathway | 8 | 5 | 3 | 149 | 2.69 |
| Primary immunodeficiency | 2 | 2 | 0 | 35 | 2.64 |
| Bile secretion | 3 | 3 | 0 | 71 | 2.55 |
| Non‐homologous end‐joining | 1 | 1 | 0 | 13 | 2.29 |
| ABC transporters | 4 | 2 | 2 | 44 | 2.21 |
| Amino sugar and nucleotide sugar metabolism | 3 | 2 | 1 | 46 | 2.13 |
| Down regulated pathways | |||||
| Tricarboxylic acid cycle | 9 | 0 | 9 | 30 | 7.16 |
| Pyruvate metabolism | 10 | 1 | 9 | 40 | 5.88 |
| Metabolic pathways | 82 | 5 | 77 | 1069 | 5.66 |
| Proteasome | 8 | 0 | 8 | 44 | 4.72 |
| Pentose and glucuronate interconversions | 5 | 0 | 5 | 21 | 4.56 |
| Ascorbate and aldarate metabolism | 4 | 0 | 4 | 16 | 4.22 |
| Oxidative phosphorylation | 12 | 0 | 12 | 114 | 3.49 |
| Arginine and proline metabolism | 7 | 0 | 7 | 53 | 3.35 |
| Valine, leucine and isoleucine degradation | 6 | 0 | 6 | 42 | 3.34 |
| Glycosphingolipid biosynthesis – ganglio series | 3 | 0 | 3 | 15 | 3.11 |
| SNARE interactions in vesicular transport | 5 | 0 | 5 | 35 | 3.04 |
| Renin–angiotensin system | 3 | 0 | 3 | 17 | 2.82 |
| Glycolysis/gluconeogenesis | 8 | 1 | 7 | 65 | 2.72 |
| Parkinson's disease | 10 | 0 | 10 | 111 | 2.63 |
| Starch and sucrose metabolism | 6 | 1 | 5 | 41 | 2.62 |
| Lysine biosynthesis | 1 | 0 | 1 | 3 | 2.55 |
| Porphyrin and chlorophyll metabolism | 5 | 1 | 4 | 31 | 2.47 |
| Glycine, serine and threonine metabolism | 4 | 0 | 4 | 32 | 2.4 |
| Propanoate metabolism | 4 | 0 | 4 | 32 | 2.4 |
| Folate biosynthesis | 2 | 0 | 2 | 11 | 2.35 |
| Cysteine and methionine metabolism | 4 | 0 | 4 | 35 | 2.19 |
| Peroxisome | 9 | 2 | 7 | 78 | 2.18 |
| Phenylalanine, tyrosine and tryptophan biosynthesis | 1 | 0 | 1 | 4 | 2.11 |
| Sulfur metabolism | 2 | 0 | 2 | 13 | 2.05 |
| Phagosome | 11 | 0 | 11 | 149 | 2.04 |
| Focal adhesion | 5 | 3 | 2 | 198 | −2.24 |
| Vascular smooth muscle contraction | 2 | 2 | 0 | 122 | −2.31 |
| Neuroactive ligand‐receptor interaction | 8 | 6 | 2 | 309 | −3.15 |
| Olfactory transduction | 3 | 3 | 0 | 379 | −4.17 |
‘Up pathways’ denote KEGG pathways that are significantly up‐regulated in MO compared with CON samples. ‘Down pathways’ denote KEGG pathways that are significantly down‐regulated in MO compared with CON samples. ‘Diff genes’ denotes the total number of differentially expressed genes between CON and MO fetal liver RNA on the array. ‘Up’ denotes the number of genes up‐regulated in MO compared with CON. ‘Down’ denotes the number of genes down‐regulated in MO compared with CON. ‘Array’ denotes the total number of genes in this pathway that are included on the array and give a signal with baboon RNA. ‘z‐score’ denotes the z‐score for the list pathway. GnRH, gonadotropin‐releasing hormone.
The end‐of‐pathway analysis revealed five pathways in which gene expression at the end of the pathway was consistent with the overall pathway changes in MO compared with CON fetal livers. Of the five pathways, the tricarboxylic acid (TCA) cycle, proteasome, oxidative phosphorylation and glycolysis/gluconeogenesis were down‐regulated, and Wnt/β‐catenin signalling was up‐regulated in MO compared with CON fetal livers.
In the TCA cycle, dihydrolipoamide S‐succinyltransferase (DLST), dihydrolipoamide dehydrogenase (DLD), succinate‐coA ligase ADP‐forming β‐subunit (SUCLA2) and oxoglutarate dehydrogenase (OGDH) were down‐regulated (Table 4). This end‐of‐pathway analysis expression of DLST, DLD, SUCLA2 and OGDH genes is consistent with the overall pathway activity of the tricarboxylic acid cycle.
Table 4.
Expression profiles of end‐of‐pathway genes comparing MO and CON fetal livers at 0.9G
| Pathway, direction, z‐score) | End of pathway genes | Direction of change | Downstream pathway |
|---|---|---|---|
| Wnt signalling, up, 2.7 | Peroxisome proliferator activated receptor delta, PPARδ | Up | Cell cycle |
| Tricarboxylic acid cycle, down, 7.2 | Dihydrolipoamide S‐succinyltransferase, DLD | Down | NADH |
| Dihydrolipoamide dehydrogenase, DLST | Down | ||
|
Down | ||
| Proteasome, down, 4.7 | Proteasome 26S subunit ATPase 5, PSMC5 | Down | Protein degradation |
| Proteasome 26S subunit ATPase 1, PSMC1 | Down | ||
|
Down | ||
| Oxidative phosphorylation, down, 3.5 | NADH: ubiquinone oxidoreductase subunit B3, NDUFB3 | Down | Mitochondrial complex I |
| NADH: ubiquinone oxidoreductase core subunit B2, NDUFB2 | Down | ||
| Succinate dehydrogenase complex flavoprotein subunit A, SDHA | Down | Mitochondrial complex II | |
| Cytochrome c oxidase subunit 6B2, COX6B2 | Down | Mitochondrial complex IV | |
| ATP synthase, ATP5L | Down | Mitochondrial complex V | |
| Glycolysis, down, 2.7 | Pyruvate dehydrogenase (lipoamide) alpha 1, PDHA1 | Down | Amino acid biosynthesis |
| Pyruvate kinase, PKLR | Down | Pyruvate |
The end‐of‐pathway analysis of the proteasome pathway showed expression of four genes (Table 4), proteasome 26S subunit ATPase 5 (PSMC5), proteasome 26S subunit ATPase 1 (PSMC1), proteasome 26S subunit ATPase 1 (PSMD6) and thimet oligopeptidase 1 (THOP1), to be down‐regulated. The changed expression of these genes is consistent with the down‐regulation of activity of the proteasome pathway.
In the oxidative phosphorylation, gene expression of NADH:ubiquinone oxidoreductase subunit B3 (NDUFB3), NADH:ubiquinone oxidoreductase core subunit B2 (NDUFB2), succinate dehydrogenase complex flavoprotein subunit A (SDHA), cytochrome c oxidase subunit 6B2 (COX6B2) and ATP synthase (ATP5L) was down‐regulated (Table 4), consistent with down‐regulation of the oxidative phosphorylation pathway.
The end‐of‐pathway analysis of the glycolysis/gluconeogenesis pathway showed pyruvate dehydrogenase (lipoamide) α 1 (PDHA1) and pyruvate kinase (PKLR) gene expression to be down‐regulated (Table 4), and these end‐of‐pathway expression changes are consistent with the overall down‐regulation pathway activity.
In the Wnt/β‐catenin signalling pathway, peroxisome proliferator‐activated δ (PPARδ) was up‐regulated (Table 4). This changed end‐of‐pathway expression of PPARδ is consistent with increased overall pathway activity. Along with the above‐mentioned pathways, it is interesting to note here that the cysteine and methionine metabolism pathway is down‐regulated in our study, which is critical for important metabolic processes involved in developmental programming during pregnancy (Nathanielsz et al. 2015).
miRNA inversely expressed with target genes
We identified 67 miRNAs that were differentially expressed (P < 0.05) between CON and MO fetal livers. Of these 49 miRNAs were predicted to target 543 genes. We used miRNA target filter in IPA to search for targets that regulate differentially expressed genes in TCA cycle, proteasome, oxidative phosphorylation, glycolysis and Wnt/β‐catenin signalling pathways. A total of 31 miRNAs targeting 34 mRNAs were filtered based on Targetscan human and ingenuity expert findings and predicted confidence sources. To further prioritize the genes, we identified those targeted by miRNAs that exhibited differential expression and showed inverse expression between the targeted genes and miRNAs. As a result, we identified 11 miRNAs targeting 13 genes in these pathways. Differential expression of genes and the corresponding targeting miRNAs are shown in Table 5.
Table 5.
Differentially expressed microRNAs inversely expressed with gene targets in key KEGG pathways
| Pathway | Gene ID | Gene CON | Gene MO | mRNA fold change | miRNA ID | miRNA CON | miRNA MO | P | Fold change |
|---|---|---|---|---|---|---|---|---|---|
| Wnt/β‐catenin signalling | PPARδ | 0.43 ± 0.03 | 0.61 ± 0.04 | 1.13 | miR‐185‐5p | 31.56 ± 4.16 | 18.69 ± 4.31 | 9.91 × 10−3 | −2.20 |
| 1.13 | miR‐194‐3p | 28.37 ± 1.96 | 18.66 ± 2.08 | 1.89 × 10−3 | −2.06 | ||||
| SMAD4 | 2.46 ± 0.12 | 2.01 ± 0.13 | −1.36 | miR‐145‐3p | 39.15 ± 3.44 | 84.08 ± 15.70 | 2.95 × 10−4 | 2.67 | |
| −1.36 | miR‐183‐5p | 15.38 ± 3.44 | 32.24 ± 5.47 | 7.55 × 10−4 | 3.26 | ||||
| Tricarboxylic acid cycle | DLD | 2.53 ± 0.15 | 2.09 ± 0.10 | −1.35 | miR‐130a‐3p | 113.56 ± 14.17 | 162.76 ± 16.97 | 6.14 × 10−3 | 1.66 |
| SUCLA2 | 3.3 ± 0.10 | 3.0 ± 0.07 | −1.38 | miR‐186‐5p | 3148.88 ± 97.73 | 3343.14 ± 149.18 | 1.32 × 10−2 | 1.13 | |
| OGDH | 1.4 ± 0.07 | 1.1 ± 0.06 | −1.13 | miR‐96‐5p | 1.09 ± 0.63 | 2.44 ± 1.14 | 3.23 × 10−3 | 5.66 | |
| Proteasome | UBC | 6.6 ± 0.14 | 6.01 ± 0.16 | −1.51 | miR‐130a‐3p | 113.56 ± 14.17 | 162.76 ± 16.97 | 6.14 × 10−3 | 1.66 |
| PSMD5 | 1.19 ± 0.04 | 1.01 ± 0.06 | −1.14 | miR‐143‐3p | 67954.63 ± 1353.79 | 85384.32 ± 7097.08 | 2.67 × 10−2 | 1.30 | |
| PSMC1 | 4.77 ± 0.09 | 4.32 ± 0.05 | −1.36 | miR‐1285‐3p | 2.01 ± 0.58 | 2.91 ± 0.70 | 2.31 × 10−2 | 2.19 | |
| THOP1 | 1.91 ± 0.07 | 1.59 ± 1.11 | −1.25 | miR‐1285‐3p | 2.01 ± 0.58 | 2.91 ± 0.70 | 2.31 × 10−2 | 2.19 | |
| THOP1 | 1.91 ± 0.07 | 1.59 ± 1.11 | −1.25 | miR‐130a‐3p | 113.56 ± 14.17 | 162.76 ± 16.97 | 6.14 × 10−3 | 1.66 | |
| PSMD6 | 2.37 ± 0.19 | 1.81 ± 0.07 | −1.47 | miR‐1285‐3p | 2.01 ± 0.58 | 2.91 ± 0.70 | 2.31 × 10−2 | 2.19 | |
| Oxidative phosphorylation | NDUFS2 | 1.21 ± 0.09 | 0.85 ± 0.10 | −1.28 | miR‐199a‐5p | 344.72 ± 38.65 | 455.01 ± 41.88 | 3.98 × 10−2 | 1.42 |
| ATP5L | 5.68 ± 0.07 | 5.38 ± 0.07 | −1.24 | miR‐182‐5p | 121.51 ± 14.53 | 197.85 ± 19.45 | 4.66 × 10−3 | 2.31 | |
| Glycolysis | TPI1 | 1.02 ± 0.14 | 0.63 ± 0.05 | −1.39 | miR‐1285‐3p | 2.01 ± 0.58 | 2.91 ± 0.70 | 2.31 × 10−2 | 2.19 |
We found three TCA cycle genes differentially expressed in MO are targeted by three differentially expressed miRNAs, and these miRNAs were inversely expressed with their target genes. For example, miR‐130a‐3p is predicted to target DLD, miR‐186‐5p is predicted to target SUCLA2, and miR‐96‐5p is predicted to target OGDH (Table 5).
A total of five differentially expressed proteasome pathway genes in MO were targeted by four differentially expressed miRNAs, and these miRNAs were inversely expressed with their target genes. For example, miR‐130a‐3p is predicted to target UBC and THOP1, miR‐143‐3p is predicted to target PSMD5, and miR‐1285‐3p is predicted to target PSMC1, PSMD6 and THOP1 (Table 5).
We found two oxidative phosphorylation pathway genes differentially expressed in MO were targeted by two differentially expressed miRNAs and these miRNAs were inversely expressed with their target genes. For example, miR‐199a‐5p is predicted to target NDUFS2 and miR‐182‐5p is predicted to target ATP5L. Lastly, we found the TPI1 gene from the glycolysis pathway to be targeted by miR‐1285‐3p, which is inversely expressed in response to MO (Table 5).
Two miRNAs targeted the PPARδ and SMAD4 genes in the Wnt/β‐catenin signalling pathway. For example, the PPARδ gene is up‐regulated in response to MO and is targeted by miR‐185‐5p and miR‐194‐3p, which are down‐regulated in response to MO. SMAD4, which is down‐regulated in response to MO, is targeted by miR‐145‐3p and miR‐183‐5p miRNAs, which are up‐regulated in response to MO (Table 5).
Fetal liver lipid and glycogen content
The MO fetal livers showed three‐fold more hepatic lipid accumulation than the CON fetal livers (P = 0.02) (Fig. 1).
Figure 1. Lipid content in 0.9G baboon fetal livers.
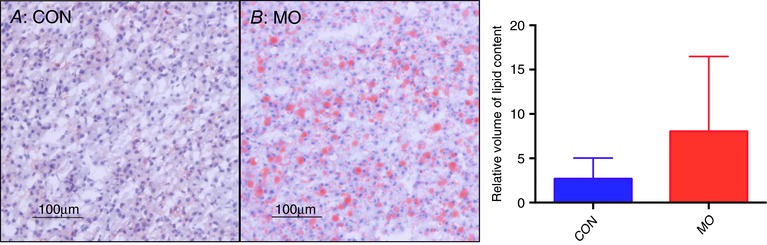
Sections were stained with Oil Red O (ORO) and quantified with stereomicroscopy; red indicates lipid. Lipid content quantified by CAST. CON = 16 and MO = 16, error bars indicate SD, P = 0.02.
Hepatic glycogen content showed a marginal decrease in MO fetal livers compared with CON (P = 0.09) (Fig. 2).
Figure 2. Glycogen content in 0.9G baboon fetal livers.
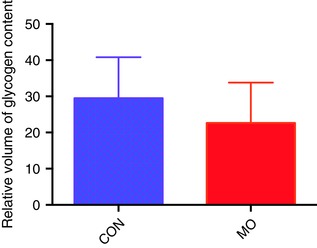
Glycogen content quantified by CAST. CON = 16 and MO = 16, error bars indicate SD, P = 0.09. [Color figure can be viewed at http://wileyonlinelibrary.com]
Histological assessment
In the present study, the control fetal livers showed mild steatosis (8 out of 16; 50%) but did not progress to severe steatosis (Table 6). However, in MO fetal livers, 11 out of 16 MO slides (68.7%) progressed to severe steatosis (Table 6). Overall, 56.2% (9/16) of MO fetal livers showed severe steatosis (Table 6). We found a significant (P < 0.0001) correlation (r = 69%) between ORO lipid content and steatosis score but no significant correlation was found between liver weight and steatosis score. Other features of liver injury in NAFLD including lobular and portal inflammation, hepatocyte ballooning and fibrosis were absent in both groups.
Table 6.
Histopathological features of steatosis grades in stained fetal livers at 0.9G
| Fetal liver stained slides | Grade 0 (<5%) | Grade 1 (5–33%) | Grade 2 (34–66%) | Grade 3 severe >66% |
|---|---|---|---|---|
| CON (n = 16) | 8 | 8 | 0 | 0 |
| MO (n = 16) | 5 | 2 | 2 | 7 |
Discussion
The aim of this study was to examine the molecular mechanisms by which MO impacts the developing fetal primate liver. There is increasing evidence that MO and/or consumption of an unhealthy diet by the mother during pregnancy adversely affects the long‐term offspring health in rodents (Zambrano et al. 2010; Alfaradhi et al. 2014), sheep (Long et al. 2012; Shasa et al. 2015), NHPs (Grant et al. 2011) and humans (Gillman et al. 2008, Gaillard, 2015). The present study used the baboon as a model of MO with dams fed a high‐fat, high‐fructose diet before and during pregnancy. Compared to CON dams, MO dams exhibited greater waist circumference and hip circumference, dyslipidaemia (elevated LDL‐cholesterol and triglycerides (TGs)), and greater percentage body fat. In addition fetal methionine, glycine, serine and taurine were lower in the fetuses of MO mothers, showing dysregulation of methionine function (Nathanielsz et al. 2015).
To our knowledge, this is the first NHP study of MO fetal livers using unbiased transcriptome analysis to quantify gene expression, identify dysregulated signalling pathways and identify potential miRNA epigenetic regulators of metabolic disruption.
We found evidence that MO impacts the fetal liver through pathways central to metabolism with dysregulation of TCA cycle, proteasome, oxidative phosphorylation, glycolysis and Wnt/β‐catenin signalling pathways. These well‐characterized, interconnected pathways have been shown to be important for liver metabolic function (Liu et al. 2011). The TCA cycle and oxidative phosphorylation result in oxidation of nutrients to produce energy in the form of ATP. The proteasome is a multi‐catalytic enzyme complex that plays an important role in degradation of both normal and damaged proteins (Tomaru et al. 2012). Proteasomes regulate many essential cellular processes, including proliferation, cell cycle, gene transcription, apoptosis, antioxidant responses and immune function (Coux et al. 1996). The Wnt/β‐catenin signalling pathway plays an important role in development, tissue homeostasis, oxidative stress, hepatic fibrosis and hepatocellular carcinoma (Thompson & Monga, 2007; Garcia‐Ruiz et al. 2014). In liver, the stability of β‐catenin is regulated by the ubiquitin–proteasome pathway (Zhou et al. 2014). Precise control of protein degradation by the ubiquitin–proteasome system is critical for signal transduction, transcriptional regulation, response to stress and receptor function (Aberle et al. 1997). Degradation of β‐catenin by the ubiquitin–proteasome pathway regulates turnover of β‐catenin and affects multiple aspects of hepatic lipid metabolism, including fatty acid oxidation, ketogenesis and lipogenesis (Geng et al. 2012).
To provide support for dysregulation of key metabolic pathways, and identify potential epigenetic mechanisms responsible for dysregulation of these pathways, we used unbiased methods to quantify miRNAs and identify those inversely expressed with predicted target genes in the pathways. We found the following miRNAs: miR‐130a‐3p, miR‐186‐5p and miR‐96, which play roles in the regulation of insulin sensitivity, liver steatosis (Jin et al. 2009; Xiao et al. 2014) and cholesterol metabolism (Wang et al. 2016) and are predicted to target the TCA cycle genes DLD, SUCLA2 and OGDH; miR‐130a‐3p, miR‐143‐3p and miR‐1285‐3p, which are predicted to target genes UBC, PSMD5, PSMC1, THOP1 and PSMD1 in the proteasome pathway; miR‐199a‐5p and miR‐182‐5p, which are associated with progression of liver fibrosis (Liu et al. 2015, Murakami et al. 2011; Ceccarelli et al. 2013) and target oxidative phosphorylation genes NDUFS2 and ATP5L; miR‐1285‐3p, which targets TPI1 and is associated with hepatic insulin sensitivity and liver steatosis in db/db mice (Behari et al. 2014); miR‐185‐5p and miR‐194‐3p, which target PPARδ in the Wnt/β‐catenin signalling pathway, which plays an important role in lipid metabolism in mice (Leti et al. 2015) and hepatic fibrosis in rats (Wang et al. 2014); and miR‐145‐3p and miR‐183‐5p target SMAD4 in the Wnt/β‐catenin signalling pathway, and are known to regulate hepatic cell proliferation and differentiation (Venugopal et al. 2010; Wojcicka et al. 2014; Tian et al. 2016). These fetal hepatic miRNA–gene interactions suggest that MO influences these interconnected pathways regulating cell proliferation, liver steatosis, hepatic fibrosis and lipid metabolism through epigenetic mechanisms.
Based on our findings that pathways central to lipid and glucose metabolism were altered by MO, we quantified lipid and glycogen content in an expanded cohort of CON and MO fetal livers. We found a three‐fold increase in lipid content in MO fetal livers, demonstrating that MO results in lipid accumulation in the NHP fetal liver consistent with studies in Japanese macaques (McCurdy et al. 2009) and mice (Goeppert et al. 2010). Using targeted qRT‐PCR and immunohistochemistry, McCurdy et al. (2009) demonstrated that macaque dams exposed to a chronic high‐fat diet during pregnancy showed increased gene expression of four key enzymes associated with gluconeogenesis in fetal liver. In addition, they showed increased abundance of hepatic oxidative stress markers and TGs. It has been suggested that MO results in excess exposure of the fetal liver to TGs, lipids and adipokines causing alterations in gene expression that up‐regulate lipogenesis and down‐regulate lipolysis, contributing to hepatic lipid accumulation and inflammation (Segovia 2014).
In addition to lipid accumulation, our study shows that in utero exposure to MO leads to severe steatosis in the fetal liver that has not progressed to NASH or fibrosis. McCurdy et al. (2009) demonstrated that third trimester fetal Japanese macaque from MO pregnancies showed signs of NAFLD, including hepatic inflammation, oxidative stress and/or damage, TG accumulation and premature gluconeogenic gene activation. Bruce et al. (2009) provided evidence that maternal fat intake contributes to the development of NAFLD in adult mouse offspring. Brumbaugh and Friedman (2014) and McCurdy et al. (2009) suggested that early‐life hepatic fat accumulation is not simply an early manifestation of NAFLD but also an independent pathophysiological event that leads to progressive fatty liver disease in later life (McCurdy et al. 2009; Brumbaugh & Friedman 2014). Our results showing increased lipid accumulation in MO baboon fetal liver suggest that these offspring are on an early trajectory to develop NAFLD and NASH.
Our finding of increased Wnt/β‐catenin signalling differs from studies of adult livers in animal models. One study demonstrated a link between Wnt/β‐catenin signalling and lipid accumulation using liver‐specific β‐catenin knockout mice (Behari et al. 2014). Another study using obese Zucker male rats found decreased Wnt/β‐catenin signalling and 3.3‐fold increased lipid levels in the livers of obese compared to control rats (Zhou et al. 2014). However, these rodent studies are based on immunohistochemistry of postnatal livers, so it is not clear whether our observation of increased Wnt/β‐catenin signalling is due to species differences between rodents and primates, or differences in fetal versus postnatal livers.
In summary, our study shows that fetal exposure to the MO intrauterine environment results in dysregulation of fetal hepatic genes central to metabolism including TCA cycle, proteasome function, oxidative stress, glycolysis and Wnt/β‐catenin signalling. These findings were further supported by identification of miRNAs that were inversely expressed with key genes in these pathways that have been shown to be regulated by these miRNAs. These miRNA–gene interactions suggest important early molecular mechanisms by which MO programs fetal hepatic lipid metabolism. In addition, our study supports prior studies indicating that fetal exposure to MO in primates leads to hepatic lipid accumulation prior to birth. Future studies are required in MO post‐natal offspring to determine the extent to which the fetal phenotype persists, and the degree to which this increases the offspring's risk of cardiometabolic disorders in later life.
Additional information
Competing interests
The authors have no potential conflict of interest, financial or otherwise, to disclose.
Author contributions
The author's responsibilities were as follows: S.P. analysed the molecular and histological data and wrote the first draft of manuscript; C.L. was involved with study design and sample and data collection; J.P.G. performed gene array analyses and small RNA‐Seq including data cleaning and quantification of transcript abundance; R.S. and S.G. assisted with histological assessment of fetal livers; A.Q., P.J. and E.J.D. assisted with histological analysis of samples; L.A.C. and P.W.N. designed the study and contributed to interpretation of results and writing of the manuscript. All authors have read and approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was supported by NIH R24 5R24OD010916. This investigation used resources that were supported by the Southwest National Primate Research Centre grant P51 RR013986 from the National Centre for Research Resources, NIH, currently supported by the Office of Research Infrastructure Programs (ORIP), NIH, through grant P51 OD011133. This investigation was conducted in facilities constructed with support from ORIP through grant numbers C06 RR14578, C06 RR15456, C06 RR013556 and C06 RR017515.
Translational perspective.
Maternal obesity (MO) has adverse effects on offspring development, predisposing to chronic disease including metabolic and cardiovascular disease (CVD) (Bouret et al. 2015). Offspring of women with a high BMI die earlier of CVD than offspring of women with normal BMI (Godfrey et al. 2017). Programming of offspring liver disease by MO (Mouralidarane et al. 2015) is a major health concern given the epidemic of obesity among women of reproductive age. Most studies are in rodents, whose maternal nutritional burden and offspring early life development differ greatly from humans (Rabadán‐Diehl & Nathanielsz, 2013). In our well‐characterized obese pregnant baboon model, we hypothesized that fetal exposure to the MO intrauterine environment programs offspring metabolism. In MO fetuses we observed dysregulation of fetal hepatic genes central to metabolism and lipid accumulation with a trend towards hepatic steatosis. These findings provide information on epigenetic mechanisms underlying fetal metabolic programming by MO. Fetal liver development during MO programming is a central metabolic regulator, important for understanding effects of poor maternal nutrition on offspring life course health (McCurdy et al. 2009; Heerwagen et al. 2013). The baboon is a primate, sharing many human physiological and molecular characteristics, making it a strong model for translation to humans. Our results indicate important molecular mechanisms linking early life exposures and health outcomes to assist identification of genetic and epigenetic mechanisms that regulate hepatic metabolism and inflammation. Further postnatal studies will determine the extent to which the altered fetal phenotype persists and progresses, contributing to offspring risk of chronic later life disease.
Supporting information
Disclaimer: Supporting information has been peer‐reviewed but not copyedited.
Supplementary Table S1. Differentially expressed genes between MO and CON fetal livers at 0.9G
Supplementary Table S2. Differentially expressed known and novel microRNAs between MO and CON fetal livers at 0.9G
Acknowledgements
We acknowledge contributions by Ms Renee Escalona for ORO staining of liver sections.
Biography
Sobha Puppala's area of research is focused on identifying the genetic basis of complex diseases such as obesity, diabetes and metabolic syndrome. She is also interested in the mechanisms underlying the impact of maternal obesity, intake of high‐fat‐high‐sugar diets during pregnancy and predisposition of children to obesity and other chronic metabolic and cardiovascular diseases. She plans to investigate metabolic and cardiovascular health of baboon offspring of obese mothers, including liver function, to determine whether the consequences of maternal obesity persist. Her major contributions include the identification of susceptibility genes for gallbladder disease, diabetic nephropathy‐related traits, metabolic syndrome and cardiovascular disease, and hypertension in human populations.

Edited by: Laura Bennet & Dino Giussani
References
- Aberle H, Bauer A, Stappert J, Kispert A & Kemler R (1997). β‐catenin is a target for the ubiquitin‐proteasome pathway. EMBO J 16, 3797–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfaradhi MZ & Ozanne SE (2011). Developmental programming in response to maternal overnutrition. Front Genet 2, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfaradhi MZ, Fernandez‐Twinn DS, Martin‐Gronert MS, Musial B, Fowden A & Ozanne SE (2014). Oxidative stress and altered lipid homeostasis in the programming of offspring fatty liver by maternal obesity. Am J Physiol Regul Integr Comp Physiol 307, R26–R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage JA, Khan IY, Taylor PD, Nathanielsz PW & Poston L (2004). Developmental programming of the metabolic syndrome by maternal nutritional imbalance: how strong is the evidence from experimental models in mammals? J Physiol 561, 355–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel‐Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM & Sherlock G (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–97. [DOI] [PubMed] [Google Scholar]
- Behari J, Li H, Liu S, Stefanovic‐Racic M, Alonso L, O'Donnell CP, Shiva S, Singamsetty S, Watanabe Y, Singh VP & Liu Q (2014). β‐catenin links hepatic metabolic zonation with lipid metabolism and diet‐induced obesity in mice. Am J Pathol 184, 3284–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benatti RO, Melo AM, Borges FO, Ignacio‐Souza LM, Simino LA, Milanski M, Velloso LA, Torsoni MA & Torsoni AS (2014). Maternal high‐fat diet consumption modulates hepatic lipid metabolism and microRNA‐122 (miR‐122) and microRNA‐370 (miR‐370) expression in offspring. Br J Nutr 111, 2112–2122. [DOI] [PubMed] [Google Scholar]
- Boerschmann H, Pflüger M, Henneberger L, Ziegler AG & Hummel S (2010). Prevalence and predictors of overweight and insulin resistance in offspring of mothers with gestational diabetes mellitus. Diabetes Care 33, 1845–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouret S, Levin BE & Ozanne SE (2015). Gene‐environment interactions controlling energy and glucose homeostasis and the developmental origins of obesity. Physiol Rev 95, 47–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce KD, Cagampang FR, Argenton M, Zhang J, Ethirajan PL, Burdge GC, Bateman AC, Clough GF, Poston L, Hanson MA, McConnell JM & Byrne CD (2009). Maternal high‐fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology 50, 1796–1808. [DOI] [PubMed] [Google Scholar]
- Brumbaugh DE & Friedman JE (2014). Developmental origins of nonalcoholic fatty liver disease. Pediatr Res 75, 140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreras‐Badosa G, Bonmatí A, Ortega FJ, Mercader JM, Guindo‐Martínez M, Torrents D, Prats‐Puig A, Martinez‐Calcerrada JM, DE Zegher F, Ibáñez L, Fernandez‐Real JM, Lopez‐Bermejo A & Bassols J (2017). Dysregulation of placental miRNA in maternal obesity is associated with pre and post‐natal growth. J Clin Endocrinol Metab 102, 2584–2594. [DOI] [PubMed] [Google Scholar]
- Ceccarelli S, Panera N, Gnani D & Nobili V (2013). Dual role of microRNAs in NAFLD. Int J Mol Sci 14, 8437–8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coux O, Tanaka K & Goldberg AL (1996). Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem 65, 801–847. [DOI] [PubMed] [Google Scholar]
- Cox LA, Comuzzie AG, Havill LM, Karere GM, Spradling KD, Mahaney MC, Nathanielsz PW, Nicolella DP, Shade RE, Voruganti S & VandeBerg JL (2013). Baboons as a model to study genetics and epigenetics of human disease. ILAR J 54, 106–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox LA, Schlabritz‐Loutsevitch N, Hubbard GB, Nijland MJ, McDonald TJ & Nathanielsz PW (2006). Gene expression profile differences in left and right liver lobes from mid‐gestation fetal baboons: a cautionary tale. J Physiol 572, 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedländer MR, Mackowiak SD, Li N, Chen W & Rajewsky N (2012). miRDeep2 accurately identifies known and hundreds of novel microRNA genes in sevenanimal clades. Nucleic Acids Res 40, 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard R (2015). Maternal obesity during pregnancy and cardiovascular development and disease in the offspring. Eur J Epidemiol 30, 1141–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Ruiz I, Solís‐Muñoz P, Fernández‐Moreira D, Grau M, Colina F, Muñoz‐Yagüe T & Solís‐Herruzo JA (2014). High‐fat diet decreases activity of the oxidative phosphorylation complexes and causes nonalcoholic steatohepatitis in mice. Dis Model Mech 7, 1287–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng F, Wenzel S & Tansey WP (2012). Ubiquitin and proteasomes in transcription. Annu Rev Biochem 81, 177–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillman MW, Rifas‐Shiman SL, Kleinman K, Oken E, Rich‐Edwards JW & Taveras EM (2008). Developmental origins of childhood overweight: potential public health impact. Obesity (Silver Spring) 16, 1651–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey KM, Reynolds RM, Prescott SL, Nyirenda M, Jaddoe VW, Eriksson JG & Broekman BF (2017). Influence of maternal obesity on the long‐term health of offspring. Lancet Diabetes Endocrinol 5, 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeppert B, Schmezer P, Dutruel C, Oakes C, Renner M, Breinig M, Warth A, Vogel MN, Mittelbronn M, Mehrabi A, Gdynia G, Penzel R, Longerich T, Breuhahn K, Popanda O, Plass C, Schirmacher P & Kern MA (2010). Down‐regulation of tumor suppressor A kinase anchor protein 12 in human hepatocarcinogenesis byepigenetic mechanisms. Hepatology 52, 2023–2033. [DOI] [PubMed] [Google Scholar]
- Grant WF, Gillingham MB, Batra AK, Fewkes NM, Comstock SM, Takahashi D, Braun TP, Grove KL, Friedman JE & Marks DL (2011). Maternal high fat diet is associated with decreased plasma n‐3 fatty acids and fetal hepaticapoptosis in nonhuman primates. PLoS One 6, e17261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieves JL, Dick EJ Jr, Schlabritz‐Loutsevich NE, Butler SD, Leland MM, Price SE, Schmidt CR, Nathanielsz PW & Hubbard GB (2008). Barbiturate euthanasia solution‐induced tissue artifact in nonhuman primates. J Med Primatol 37, 154–161. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardado M, Davalli AM, Chavez AO, Hubbard GB & Dick EJ (2009). Pancreatic islet amyloidosis, β‐cell apoptosis, and α‐cell proliferation are determinants of islet remodeling in type‐2 diabetic baboons. Proc Natl Acad Sci USA 106, 13992–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerwagen MJ, Stewart MS, de la Houssaye BA, Janssen RC & Friedman JE (2013). Transgenic increase in n‐3/n‐6 fatty acid ratio reduces maternal obesity‐associated inflammation and limits adverse developmental programming in mice. PLoS One 8, e67791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hromadnikova I, Kotlabova K, Ondrackova M, Kestlerova A, Novotna V, Hympanova L, Doucha J & Krofta L (2013). Circulating C19MC microRNAs in preeclampsia, gestational hypertension, and fetal growth restriction. Mediators Inflamm 2013, 186041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Ye YF, Chen SH, Yu CH, Liu J & Li YM (2009). MicroRNA expression pattern indifferent stages of nonalcoholic fatty liver disease. Dig Liver Dis 41, 289–97. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Kawashima S, Okuno Y & Hattori M (2004). The KEGG resource for deciphering the genome. Nucleic Acids Res 32, D277–D280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr MK, Martin M & Churchill GA (2000). Analysis of variance for gene expression microarray data. J Comput Biol 7, 819–837. [DOI] [PubMed] [Google Scholar]
- Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp‐Arida A, Yeh M, McCullough AJ & Sanyal AJ (2005). Nonalcoholic steatohepatitis clinical research network. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321. [DOI] [PubMed] [Google Scholar]
- Krishnaveni GV, Veena SR, Hill JC, Kehoe S, Karat SC & Fall CH (2010). Intrauterine exposure to maternal diabetes is associated with higher adiposity and insulin resistance and clustering of cardiovascular risk markers in Indian children. Diabetes Care 33, 402–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leti F, Malenica I, Doshi M, Courtright A, Van Keuren‐Jensen K, Legendre C, Still CD, Gerhard GS & DiStefano JK (2015). High‐throughput sequencing reveals altered expression of hepatic microRNAs in nonalcoholic fatty liver disease‐related fibrosis. Transl Res 166, 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ge Q, Guo L & Lu Z (2013). Maternal plasma miRNAs expression in pre‐eclamptic pregnancies. Biomed Res Int 2013, 970265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Sloboda DM & Vickers MH (2011). Maternal obesity and developmental programming of metabolic disorders in offspring: evidence from animal models. Exp Diabetes Res 2011, 592408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Fergusson MM, Wu JJ, Rovira II, Liu J, Gavrilova O, Lu T, Bao J, Han D, Sack MN & Finkel T (2011). Wnt signaling regulates hepatic metabolism. Sci Signal 1, ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yan J, Zhou C, Ma Q, Jin Q & Yang Z (2015). miR‐1285‐3p acts as a potential tumor suppressor miRNA via downregulating JUN expression in hepatocellularcarcinoma. Tumour Biol 36, 219–225. [DOI] [PubMed] [Google Scholar]
- Long NM, Rule DC, Zhu MJ, Nathanielsz PW & Ford SP (2012). Maternal obesity upregulates fatty acid and glucose transporters and increases expression of enzymes mediating fatty acid biosynthesis in fetal adipose tissue depots. J Anim Sci 90, 2201–2210. [DOI] [PubMed] [Google Scholar]
- Maloyan A, Muralimanoharan S, Huffman S, Cox LA, Nathanielsz PW, Myatt L & Nijland J (2013). Identification and comparative analyses of myocardial miRNAs involved in the fetal response to maternal obesity. Physiol Genomics 45, 889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandarim‐de‐Lacerda CA (2003). Stereological tools in biomedical research. An Acad Bras Cienc 75, 469–486. [DOI] [PubMed] [Google Scholar]
- McCurdy CE, Bishop JM, Williams SM, Grayson BE, Smith MS, Friedman JE & Grove KL (2009). Maternal high‐fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest 119, 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JL, Troendle J, Conley MR, Carter T & Druschel CM (2010). Maternal obesity and congenital heart defects: a population‐based study. Am J Clin Nutr 91, 1543–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouralidarane A, Soeda J, Sugden D, Bocianowska A, Carter R, Ray S, Saraswati R, Cordero P, Novelli M, Fusai G, Vinciguerra M, Poston L, Taylor PD & Oben JA (2015). Maternal obesity programs offspring non‐alcoholic fatty liver disease through disruption of 24‐h rhythms in mice. Int J Obes (Lond) 39, 1339–1348. [DOI] [PubMed] [Google Scholar]
- Murakami Y, Toyoda H, Tanaka M, Kuroda M, Harada Y, Matsuda F, Tajima A, Kosaka N, Ochiya T & Shimotohno K (2011). The progression of liver fibrosis is related with overexpression of the miR‐199 and 200 families. PLoS One 6, e16081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanielsz PW, Poston L & Taylor PD (2007). In utero exposure to maternal obesity and diabetes: animal models that identify and characterize implications for future health. Clin Perinatol 34, 515–526. [DOI] [PubMed] [Google Scholar]
- Nathanielsz PW, Yan J, Green R, Nijland M, Miller JW, Wu G, McDonald TJ & Caudill MA (2015). Maternal obesity disrupts the methionine cycle in baboon pregnancy. Physiol Rep 3, e12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas LM, Morrison JL, Rattanatray L, Zhang S, Ozanne SE, McMillen IC & Nicholas LM (2016). The early origins of obesity and insulin resistance: timing, programming and mechanisms. Int J Obes (Lond) 40, 229–238. [DOI] [PubMed] [Google Scholar]
- Nijland MJ, Schlabritz‐Loutsevitch NE, Hubbard GB, Nathanielsz PW & Cox LA (2007). Non‐human primate fetal kidney transcriptome analysis indicates mammalian target of rapamycin (mTOR) is a central nutrient‐responsive pathway. J Physiol 579, 643–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penfold NC & Ozanne SE (2015). Developmental programming by maternal obesity in 2015: Outcomes, mechanisms, and potential interventions. Horm Behav 76, 43–52. [DOI] [PubMed] [Google Scholar]
- Pereira SP, Oliveira PJ, Tavares LC, Moreno AJ, Cox LA, Nathanielsz PW & Nijland MJ (2015). Effects of moderate global maternal nutrient reduction on fetal baboon renal mitochondrial gene expression at 0.9 gestation. Am J Physiol Renal Physiol 308, F1217–F1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn AR, Blanco CL, Perego C, Finzi G, La Rosa S, Capella C, Guardado‐Mendoza R, Casiraghi F, Gastaldelli A, Johnson M, Dick EJ Jr & Folli F (2012). The ontogeny of the endocrine pancreas in the fetal/newborn baboon. J Endocrinol 214, 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabadán‐Diehl C & Nathanielsz P (2013). From Mice to Men: research models of developmental programming. J Dev Orig Health Dis 4, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnault TR, Gentili S, Sarr O, Toop CR & Sloboda DM (2013). Fructose, pregnancy and later life impacts. Clin Exp Pharmacol Physiol 40, 824–837. [DOI] [PubMed] [Google Scholar]
- Schlabritz‐Loutsevitch NE, Howell K, Rice K, Glover EJ, Nevill CH, Jenkins SL, Bill Cummins L, Frost PA, McDonald TJ & Nathanielsz PW (2004). Development of a system for individual feeding of baboons maintained in an outdoor group social environment. J Med Primatol 33, 117–126. [DOI] [PubMed] [Google Scholar]
- Segovia SA (2014). Maternal obesity, inflammation, and developmental programming. Biomed Res Int 2014, 418975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar K, Harrell A, Liu X, Gilchrist JM, Ronis MJ & Badger TM (2008). Maternal obesity at conception programs obesity in the offspring. Am J Physiol Regul Integr Comp Physiol 294, R528–R538. [DOI] [PubMed] [Google Scholar]
- Shasa DR, Odhiambo JF, Long NM, Tuersunjiang N, Nathanielsz PW & Ford SP (2015). Multigenerational impact of maternal overnutrition/obesity in the sheep on the neonatal leptin surge in granddaughters. Int J Obes (Lond) 39, 695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Cox LA, Hodara V, Wang XL & VandeBerg JL (2012). Repertoire of endothelial progenitor cells mobilized by femoral artery ligation: a nonhuman primate study. J Cell Mol Med 16, 2060–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessitore A, Cicciarelli G, Del Vecchio F, Gaggiano A, Verzella D, Fischietti M, Mastroiaco V, Vetuschi A, Sferra R, Barnabei R, Capece D, Zazzeroni F & Alesse E (2016). MicroRNA expression analysis in high fat diet‐induced NAFLD‐NASH‐HCC progression study on C57BL/6J mice. BMC Cancer 16, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MD & Monga SP (2007). WNT/β‐catenin signaling in liver health and disease. Hepatology 45, 1298–1305. [DOI] [PubMed] [Google Scholar]
- Tian Y, Mok MT, Yang P & Cheng AS (2016). Epigenetic activation of Wnt/β‐catenin signaling in NAFLD‐associated hepatocarcinogenesis. Cancers (Basel) 8, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaru U, Takahashi S, Ishizu A, Miyatake Y, Gohda A, Suzuki S, Ono A, Ohara J, Baba T, Murata S, Tanaka K & Kasahara M (2012). Decreased proteasomal activity causes age‐related phenotypes and promotes the development of metabolic abnormalities. Am J Pathol 180, 963–972. [DOI] [PubMed] [Google Scholar]
- Venugopal SK, Jiang J, Kim TH, Li Y, Wang SS, Torok NJ, Wu J & Zern MA (2010). Liver fibrosis causes downregulation of miRNA‐150 and miRNA‐194 in hepaticstellate cells, and their overexpression causes decreased stellate cell activation. Am J Physiol Gastrointest Liver Physiol 298, G101–G106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XC, Zhan XR, Li XY, Yu JJ & Liu XM (2014). MicroRNA‐185 regulates expression of lipid metabolism genes and improves insulin sensitivity in mice with non‐alcoholic fatty liver disease. World J Gastroenterol 20, 17914–17923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KJ, Zhao X, Liu YZ, Zeng QT, Mao XB, Li SN, Zhang M, Jiang C, Zhou Y, Qian C, Feng KG, Guan HQ, Tang TT, Cheng X & Chen ZJ (2016). Circulating MiR‐19b‐3p, MiR‐134‐5p and MiR‐186‐5p are promising novel biomarkers for early diagnosis of acute myocardial infarction. Cell Physiol Biochem 38, 1015–1029. [DOI] [PubMed] [Google Scholar]
- Wilson RM & Messaoudi I (2015). The impact of maternal obesity during pregnancy on offspring immunity. Mol Cell Endocrinol 418, 134–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcicka A, Swierniak M, Kornasiewicz O, Gierlikowski W, Maciag M, Kolanowska M, Kotlarek M, Gornicka B, Koperski L, Niewinski G, Krawczyk M & Jazdzewski K (2014). Next generation sequencing reveals microRNA isoforms in liver cirrhosis and hepatocellular carcinoma. Int J Biochem Cell Biol 53, 208–217. [DOI] [PubMed] [Google Scholar]
- Xiao F, Yu J, Liu B, Guo Y, Li K, Deng J, Zhang J, Wang C, Chen S, Du Y, Lu Y, Xiao Y, Zhang Z & Guo F (2014). A novel function of microRNA 130a‐3p in hepatic insulin sensitivity and liver steatosis. Diabetes 63, 2631–2642. [DOI] [PubMed] [Google Scholar]
- Zambrano E, Martínez‐Samayoa PM, Rodríguez‐González GL & Nathanielsz PW (2010). Dietary intervention prior to pregnancy reverses metabolic programming in male offspring of obese rats. J Physiol 588, 1791–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrano E & Nathanielsz PW (2013). Mechanisms by which maternal obesity programs offspring for obesity: evidence from animal studies. Nutr Rev 71(Suppl 1), S42–S54. [DOI] [PubMed] [Google Scholar]
- Zhou D, Lezmi S, Wang H, Davis J, Banz W & Chen H (2014). Fat accumulation in thel iver of obese rats is alleviated by soy protein isolate through β‐catenin signaling. Obesity (Silver Spring) 22, 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Cao L, Zhu J, Kong L, Jin J, Qian L, Zhu C, Hu X, Li M, Guo X, Han S & Yu Z (2013). Identification of maternal serum microRNAs as novel non‐invasive biomarkers for prenatal detection of fetal congenital heart defects. Clin Chim Acta 424, 66–72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supporting information has been peer‐reviewed but not copyedited.
Supplementary Table S1. Differentially expressed genes between MO and CON fetal livers at 0.9G
Supplementary Table S2. Differentially expressed known and novel microRNAs between MO and CON fetal livers at 0.9G