

Abstract
Therapeutic hypothermia significantly improves survival without disability in near‐term and full‐term newborns with moderate to severe hypoxic–ischaemic encephalopathy. However, hypothermic neuroprotection is incomplete. The challenge now is to find ways to further improve outcomes. One major limitation to progress is that the specific mechanisms of hypothermia are only partly understood. Evidence supports the concept that therapeutic cooling suppresses multiple extracellular death signals, including intracellular pathways of apoptotic and necrotic cell death and inappropriate microglial activation. Thus, the optimal depth of induced hypothermia is that which effectively suppresses the cell death pathways after hypoxia–ischaemia, but without inhibiting recovery of the cellular environment. Thus mild hypothermia needs to be continued until the cell environment has recovered until it can actively support cell survival. This review highlights that key survival cues likely include the inter‐related restoration of neuronal activity and growth factor release. This working model suggests that interventions that target overlapping mechanisms, such as anticonvulsants, are unlikely to materially augment hypothermic neuroprotection. We suggest that further improvements are most likely to be achieved with late interventions that maximise restoration of the normal cell environment after therapeutic hypothermia, such as recombinant human erythropoietin or stem cell therapy.
Keywords: hypoxia‐ischaemia, encephalopathy, hypothermia, neuroprotection
Introduction
Therapeutic hypothermia is now standard care for infants with moderate to severe hypoxic–ischaemic encephalopathy (HIE) (Azzopardi et al. 2012), with compelling evidence from randomised controlled trials that it improves survival and neurological outcomes into middle childhood (Jacobs et al. 2013; Natarajan et al. 2016) and reduces brain damage on modern imaging (Shankaran et al. 2015). Hypothermic neuroprotection is significant but incomplete, reducing the combined risk of death and severe disabilities at 18 months of age by ∼12%, from 58 to 46% (Edwards et al. 2010). Thus, many infants still die or survive with major debilitating handicaps, despite therapeutic hypothermia.
The empirical parameters for optimal neuroprotection are now well established, as previously reviewed in detail (Wassink et al. 2014). Therapeutic hypothermia needs to be induced as soon as possible in the first 6 h after hypoxia–ischaemia (HI), optimally reducing brain temperature by no more than 3–5°C, and then continued for ∼72 h. Deeper cooling (by ∼8.5°C), or shorter or longer periods of cooling than 72 h reduced neuroprotection both in preclinical studies (Alonso‐Alconada et al. 2015; Davidson et al. 2015c , 2018) and in a randomised clinical trial (Shankaran et al. 2017). The precise mechanisms underlying these now well‐known empirical observations are still unclear. Further, given that current cooling protocols are near‐optimal, future progress depends on finding interventions that can complement hypothermia. In this review, we propose a mechanistic working model to help understand these parameters for hypothermic neuroprotection, and discuss which post‐insult phases and specific mechanisms should be targeted to further improve outcomes.
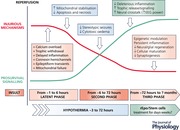
Hypoxic–ischaemic brain damage evolves over time
The seminal finding that underpinned the development and translation of therapeutic hypothermia is that perinatal brain damage after HI is a process that evolves over time rather than a ‘static’ event. Hope and colleagues first showed with magnetic resonance spectroscopy in term neonates with moderate to severe HIE that highly energetic substrates (i.e. phosphocreatine and ATP) often normalised shortly after birth but then deteriorated again (Hope et al. 1984; Azzopardi et al. 1989), despite sufficient cerebral oxygenation and perfusion. Studies in newborn piglets then demonstrated that cerebral energetic failure after HI corresponded with progressive neuronal death (Martin et al. 2000).
As illustrated by the Abstract Figure, during severe HI (the ‘primary’ phase), there is gradual depletion of high‐energy phosphate compounds and anoxic depolarisation. As energy‐dependent mechanisms that maintain cellular homeostasis (e.g. Na+/K+‐ATPase pumps) begin to fail, cytotoxic oedema (i.e. cellular swelling) and extracellular accumulation of excitatory amino acids occurs, with unregulated calcium influx into neurons. Neural energy metabolism and cell swelling typically recover to near‐normal values within 30–60 min after reperfusion and are then sustained during a ‘latent’ phase for the following ∼6 h (Hope et al. 1984; Azzopardi et al. 1989; Gunn et al. 1997; Bennet et al. 2007b ).
After moderate to severe HI, the latent phase is followed by delayed deterioration after ∼6–15 h (the ‘secondary phase’), with development of stereotypical seizures, accumulation of excitotoxins and oedema (Fig. 1), and gradual mitochondrial failure and spreading cell death (Gunn et al. 1997; Bennet et al. 2007b ). This triphasic pattern has been shown in multiple species, including rodents, piglets and humans (as reviewed by Wassink et al. 2014), and correlates with histological brain damage after HI (Williams et al. 1992; Blumberg et al. 1997; Vannucci et al. 2004). In newborn humans, the severity of loss of oxidative cerebral metabolism after HI is highly associated with death and adverse outcomes (Azzopardi et al. 1989; Roth et al. 1997). Finally, there is evidence of a ‘tertiary’ phase after HI, where chronic inflammation and epigenetics impair neural and glial regeneration, synaptogenesis and neurite outgrowth (Fleiss & Gressens, 2012).
Figure 1. The physiological effects of cerebral ischaemia for 30 min (from time zero), with or without cerebral cooling (indicated with the blue bar) induced from 3 until 72 h after reperfusion in term‐equivalent fetal sheep.
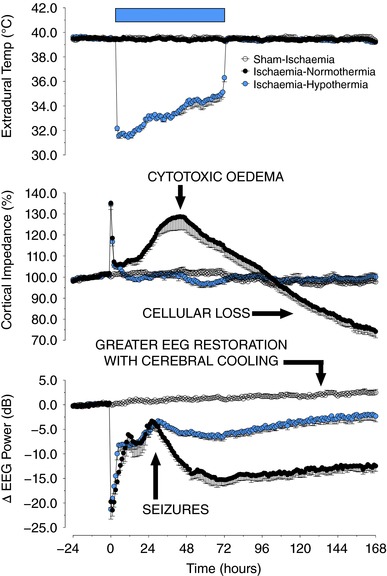
The panels show, in descending order, temporal changes in extradural temperature (°C), cortical impedance (i.e. cellular swelling, as a percentage from baseline), and electroencephalographic (EEG) power (decibels) in normothermia (black circles) and hypothermia groups (blue circles), compared to sham‐ischaemic animals (white circles). Treatment with hypothermia suppressed the delayed rise in cytotoxic oedema (as measured with cortical impedance), and improved recovery of EEG power after resolution of the secondary seizures.
How does hypoxic–ischaemic brain damage spread?
One of the striking features of HI‐mediated brain damage is that cell dysfunction and death spread over time from injured regions to areas that were originally intact (Thornton et al. 1998). The gap junctions that link adjacent cells to allow transport of small molecules, ions and second messengers (Davidson et al. 2015a ) are formed through docking of hexamer hemichannels (connexons). These hemichannels are active under physiological conditions, and signal via regulated ATP release.
There is increasing evidence that severe HI triggers transient, unregulated opening of these connexin hemichannels, resulting in disrupted resting membrane potential, release of damaging ATP and glutamate (Ye et al. 2003; Kang et al. 2008), and uptake of water leading to cell swelling and rupture (Quist et al. 2000; Rodriguez‐Sinovas et al. 2007). Supporting this concept, an intracerebroventricular infusion with a mimetic peptide that reversibly binds with the second extracellular binding loop on the connexin‐43 protein, at a dose that blocks hemichannels (O'Carroll et al. 2008), from 90 min until 25 h after profound asphyxia or cerebral ischaemia in preterm and near‐term fetal sheep, reduced status epilepticus, and improved restoration of electroencephalographic (EEG) power and neural and oligodendroglial survival (Davidson et al. 2012, 2014). These data show that connexin hemichannels have a critical role during the early latent phase in propagating damage after HI.
Mechanisms of delayed cellular death – programmed apoptosis
Multiple factors are involved in the delayed development of cell death following initial recovery of cerebral oxidative metabolism after HI. These include activation of cell death pathways, withdrawal of trophic factors and secondary inflammation. In particular, the cell death pathways are activated through unregulated influx of calcium during anoxic depolarization, exposure to reactive oxidative species during reperfusion and other factors (Thornton et al. 2017).
Apoptosis can be triggered through intracellular and extracellular pathways (Fig. 2; reviewed in detail by Thornton et al. 2017). The intracellular pathway involves excessive calcium influx and astrocytic growth factor withdrawal (Clawson et al. 1999), leading to increased translocation and interaction of pro‐apoptotic proteins at the neuronal mitochondria. These apoptotic proteins, such as the Bcl‐2‐associated X (Bax) and truncated BH3‐interacting‐domain death agonist (tBid) proteins (Raemy & Martinou, 2014), produce pores in the outer mitochondrial membrane. This releases several pro‐apoptogenic factors, including direct inhibitor of apoptosis‐binding protein with low pI (Diablo), also known as second mitochondria‐derived activator of caspases, apoptosis‐inducing factor (AIF) and cytochrome c from the mitochondrion (Wassink et al. 2014).
Figure 2. Flow chart to illustrate intracellular mechanisms associated with delayed programmed cell death after HI.
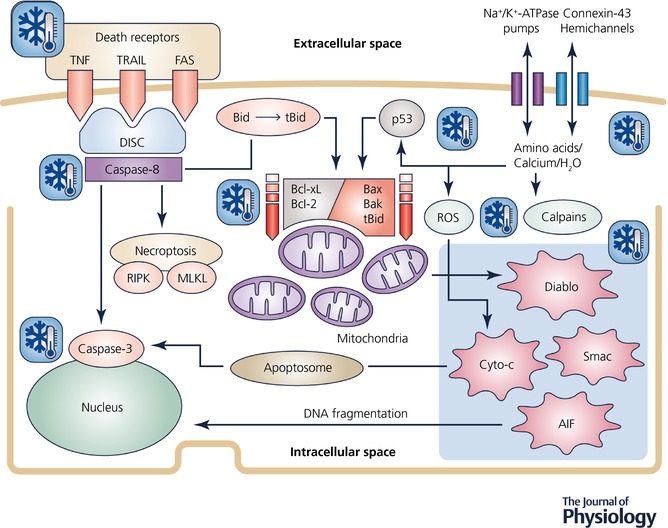
The snowflakes illustrate likely targets for therapeutic hypothermia. AIF, apoptosis inducing factor; APAF, apoptosis protease activating factor; Bid, BH3‐interacting domain death agonist; tBid, truncated BH3‐interacting domain death agonist; Bax, Bcl‐2‐associated X protein; Bak, Bcl‐2 antagonist/killer 1; Bcl‐2, B‐cell lymphoma 2 protein family; Bcl‐xL, B‐cell lymphoma‐extra‐large; Cyto‐c, cytochrome c; Diablo, direct inhibitor of apoptosis‐binding protein with low pI, also known as Smac, second mitochondria‐derived activator of caspases; DISC, death‐inducing signalling complex; Fas receptor, first apoptosis signal receptor; MLKL, mixed lineage kinase domain‐like pseudokinase; p53, tumour protein p53; ROS, reactive oxygen species; RIPK, receptor‐interacting serine/threonine‐protein kinase; TNF receptor, tumour necrosis factor receptor; TRAIL receptor, TNF‐related apoptosis‐inducing ligand receptor.
Intramitochondrial calcium overload also facilitates cytochrome c release through reactive oxygen species (Hagberg et al. 2014), and activates brain‐specific calpains that degrade intracellular structural and signalling proteins (Bevers & Neumar, 2008). In addition, HI activates extracellular death receptors that stimulate necroptosis or caspases‐8 and ‐3 (Giulian et al. 1993). These molecular mechanisms are detailed in Fig. 2. In neonatal rats, caspase, Bax and cytochrome c inhibitors all provide partial neuroprotection, supporting a pathological role for these intracellular mechanisms (Thornton et al. 2017).
Mechanisms of delayed cellular death – programmed necrosis
In the developing brain, necrosis after HI often demonstrates a variable morphology. This pattern typically involves cellular fragmentation, but there is increasing evidence that delayed necrotic cellular death is programmed (Northington et al. 2007). Necroptosis, for example, is mediated via interconnected mechanisms that involve caspase‐8, receptor‐interacting protein kinases (RIPK) 1 and 3 and the mixed lineage kinase domain‐like pseudokinase (MLKL) (Rodriguez et al. 2016). These proteins have multiple and often opposing roles that participate in both apoptosis and necrosis (Northington et al. 2007). For example, RIPKs activate the inflammasome, which might underlie the robust neuro‐inflammation triggered by HI (Man & Kanneganti, 2016), whereas MLKL has multiple functions that include facilitating pore formation that cause the cell membrane to rupture (Wang et al. 2014), culminating in cell death with a necrotic phenotype. Supporting these data, treatment with necrostatin‐1, a non‐selective necroptotic inhibitor, reduced necrotic cellular death and oxidative damage to proteins in post‐HI p10 mice (Northington et al. 2011a ).
Summary of the mechanisms of delayed cell death
Taken together, it is clear from these findings that brain metabolism can recover to normal or near‐normal levels after even severe HI, but multiple, inter‐related mechanisms are triggered that ultimately lead to delayed cellular death (Thornton et al. 2017).
The mechanisms of hypothermic neuroprotection
Induced hypothermia produces a graded reduction in cerebral metabolism of ∼5% °C−1 (Laptook et al. 1995). After resuscitation, tissue oxygenation and substrate delivery are restored (Gunn et al. 1997), and therefore it is improbable that reduced metabolism per se would be protective. However, it is important to reflect that the neuroprotective effects of cooling during HI are substantially greater than would be expected from a 15–20% reduction in metabolism. For example, in adult rats, cooling during cerebral ischaemia was associated with a dramatic reduction in major hippocampal neuronal loss compared with normothermia (6 ± 1% vs. 90 ± 17% dead neurons), for the same duration of neural depolarisation (Bart et al. 1998). This finding strongly indicates that hypothermia supports cell survival by suppressing active, intracellular cell death mechanisms rather than by reducing oxidative metabolism. There is considerable evidence that this interaction is critical for post‐resuscitation neuroprotection, as discussed next.
Hypothermia suppresses programmed cell death after hypoxia–ischaemia
There is increasing evidence that induced hypothermia suppresses apoptotic and necrotic processes triggered after HI (Wassink et al. 2014). For example, in vitro, intra‐hypoxic hypothermia reduced apoptotic and necrotic morphological death in developing neurons, and hypoxia‐driven protein formation (Bossenmeyer‐Pourie et al. 2000). Further, hypothermia also suppressed serum‐deprivation and H2O2‐induced neuronal apoptosis, with lower activation of caspases‐3, ‐8 and ‐9 and release of cytochrome c, consistent with depressed intracellular and receptor‐induced apoptosis (Xu et al. 2002; Li et al. 2012). Consistent with this, in adult rats, induced hypothermia after transient global ischaemia was associated with up‐regulated anti‐apoptotic B‐cell lymphoma 2 (Bcl‐2) protein, and down‐regulated pro‐apoptotic p53 protein (Zhang et al. 2010), with reduced neural necrosis and apoptosis. In adult rats with focal ischaemia, hypothermia also attenuated death receptor expression and caspase‐8 activation (Liu et al. 2008), supporting its interaction with extracellular apoptosis, and suppressed genes implicated in inflammation (Nagel et al. 2012).
In neonatal piglets, hypothermia started after severe HI reduced apoptotic but not necrotic cell death (Edwards et al. 1995), whereas hypothermic neuroprotection reduced caspase‐3 and microglial activation in term‐equivalent fetal sheep (Roelfsema et al. 2004). In neonatal rats, acute hypothermia after HI also reduced caspase‐3 and increased X‐linked inhibitor of apoptosis (XIAP) in the core ischaemic lesion, but not the penumbra, whereas AIF translocation was suppressed in both regions (Askalan et al. 2011), indicating that hypothermia interacts with both caspase‐dependent and ‐independent mechanisms. Finally, in neonatal rodents with HI, hypothermia attenuated macroscopic brain damage, with less necrotic and apoptotic neural death after 24 h, and suppressed cytochrome c release, caspase‐3 and calpain activation in the cortex, hippocampus, thalamus and striatum (Ohmura et al. 2005). Thus, taken together, these data suggest that hypothermic neuroprotection in the developing brain is likely achieved through both anti‐apoptotic and anti‐necrotic mechanisms (Northington et al. 2011b).
Hypothermia suppresses inflammation after hypoxia–ischaemia
Perinatal HI triggers an inflammation‐based cascade, which increases the release of cytokines and interleukins (Hagberg et al. 2015). These factors potentiate developing cellular damage, either through neurotoxically induced apoptosis or endothelial cell‐propagated inflammation, with leukocytes infiltrating the post‐ischaemic brain (Gunn et al. 2017). In experimental paradigms, post‐insult hypothermia inhibits microglial activation, chemotaxis, and interleukin and pro‐inflammatory cytokine release, which might provide mitochondrial protection (Wassink et al. 2014). For example, cytokine‐induced inducible nitric oxide synthase (iNOS) expression raises intracellular NO· levels, which competes with molecular oxygen for binding on cytochrome oxidase (Brown, 1997) and so depresses mitochondrial respiration. Tumour necrosis factor α‐ and interferon‐γ‐mediated iNOS production also caused apoptosis and DNA damage in cultured oligodendrocytes (Druzhyna et al. 2005). Critically, hypothermia has a differential effect on the glial reaction to ischaemia, demonstrating potent microglial suppression but little effect on astroglial proliferation (Si et al. 1997). This suggests that hypothermic neuroprotection results, in part, from reducing ‘bad’ inflammation while not suppressing astroglial recovery.
Hypothermia, excitotoxins and neuronal activity
In contrast to their role during the primary and reperfusion phases, the importance of excitotoxins after reperfusion is questionable given that extracellular levels rapidly return to baseline values (Tan et al. 1996; Thoresen et al. 1997). Early studies of anti‐excitotoxic agents found apparent protection but did not control for cerebral temperature (McDonald et al. 1987; Hattori et al. 1989). Critically, subsequent studies showed that glutamate blockade was associated with drug‐induced hypothermia and controlling for temperature abolished neuroprotection (Ikonomidou et al. 1989; Engidawork et al. 2001). In the adult rodent, Nurse and Corbett showed that the apparent neuroprotective effect of 2,3‐dihydroxy‐6‐nitro‐7‐sulfamoyl‐benzo[f] quinoxaline‐2,3‐dione (NBQX), a glutamate antagonist administered from 1 h after mild cerebral ischaemia, was directly associated with mild endogenous hypothermia for several days that developed an hour after drug administration (Nurse & Corbett, 1996), and that similar neuroprotection could be induced with application of the same hypothermia profile over 28 h. Conversely, NBQX ‘neuroprotection’ was effectively abolished by maintaining normothermia. Furthermore, anti‐excitotoxin therapy limited to the secondary phase did not reduce neuronal damage in the severely injured parasagittal cortex of fetal sheep, and had only limited neuroprotective effects in more mildly affected areas of the brain (Tan et al. 1992; Gressens et al. 2011).
Nevertheless, even with normal levels of extracellular glutamate, excitotoxicity may still play an indirect injurious role. Pathological hyperexcitability of glutamate receptors has been reported in P10 rats for many hours after HI, with improved neuronal outcome after receptor blockade (Jensen et al. 1998). Supporting this hypothesis, despite suppression of overall EEG activity for many hours after asphyxia, transient epileptiform activity was seen in the early recovery phase in preterm sheep fetuses that developed severe injury (George et al. 2004), which was correlated with the severity of neuronal loss in the striatum and hippocampus (Dean et al. 2006b ; Bennet et al. 2007c ). Suppression of these EEG transients with a glutamate receptor antagonist partially reduced cellular loss (Dean et al. 2006a ). Furthermore, neuroprotection with post‐asphyxial moderate cerebral hypothermia in the preterm fetal sheep was associated with a marked reduction in the numbers of epileptiform transients in the first 6 h after asphyxia, and reduced amplitude of delayed seizures (Bennet et al. 2007a ). The combination of glutamate receptor antagonist infusion and mild hypothermia after severe asphyxia in preterm fetal sheep, however, showed non‐additive neuroprotection, consistent with the suggestion that cooling is partly protective by attenuating this receptor hyperactivity (George et al. 2012). Further studies are needed to determine whether this is also the case after HI damage in the term‐equivalent brain.
Duration of cooling and recovery of EEG activity
Recent studies in near‐term fetal sheep have shown that when head cooling was started 3 h after ischaemia, cooling until 72 h was markedly more protective than cooling until 48 h (Fig. 3). Strikingly, rewarming at 48 h after cerebral ischaemia was associated with marked deterioration of EEG power over the next 24 h, and with greater numbers of microglia on histology at day 7 and substantially less improvement in overall neuronal survival compared to continued cooling until 72 h (Davidson et al. 2018). This suggests that deleterious inflammation is still continuing between 48 and 72 h after HI, and is reactivated or exacerbated by premature rewarming. It is of particular interest that in this animal study the spectral edge frequency of the EEG was still partially suppressed at 48 h, and did not reach control values until around 72 h. Conversely, we have shown that extending cooling from 72 to 120 h did not further improve EEG recovery, and indeed was associated with apparently impaired neuronal survival in some brain regions (Davidson et al. 2015c ). This suggests for the first time that normalization of EEG activity is an important biomarker for how long therapeutic hypothermia needs to be continued. Local neural interconnections, with shorter connections between neurons, lead to higher frequency activity. Thus increasing cortical EEG frequency strongly infers improved cortical function. More speculatively, it also seems to support the hypothesis that EEG activity, i.e. cross‐talk between neurons, represents an important aspect of gradual normalization of the cellular environment after HI.
Figure 3. The physiological effects of cerebral ischaemia for 30 min (from time zero), with or without cerebral cooling (indicated with blue symbols) induced from 3 h until either 48 or 72 h after reperfusion in term‐equivalent fetal sheep.
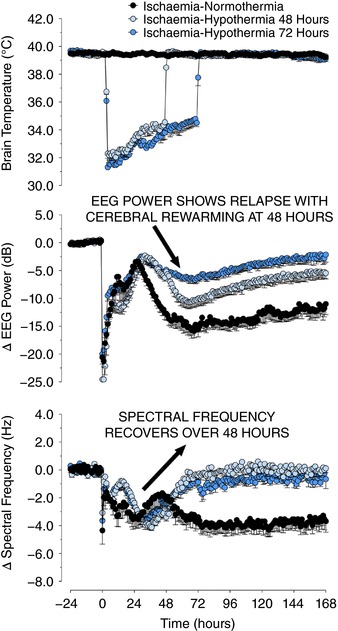
The panels show, in descending order, temporal changes in extradural temperature (°C), electroencephalographic (EEG) power (decibels) and spectral edge frequency (hertz) in ischaemia–normothermia (black circles), ischaemia–hypothermia 48 h (light blue circles) and ischaemia–hypothermia 72 h groups (dark blue circles). EEG activity was suppressed in all groups during and immediately after ischaemia followed by a transient increase during seizures from 8 to 48 h. EEG activity in the ischaemia–normothermia group remained low for the remainder of the experiment, whereas both hypothermia groups showed a significant recovery in power and spectral frequency from 24 to 72 h (P = 0.05). Rewarming at 48 h was associated with loss of EEG power in the ischaemia–48 h hypothermia group, which did not occur with rewarming at 72 h (P = 0.05). Data are means ± SEM.
Restoration of the neuronal environment: EEG activity and growth factors
The factors underlying recovery of brain activity after injury are incompletely understood. In part it is related to reversal of functional depression of injured cells, and restoration of signalling between interconnected structures (Glassman & Malamut, 1976). Neuronal activity itself is critical for cell viability and closely interacts with trophic growth factor release.
Electrical activity is a vital part of maintaining neuronal homeostasis in target neurons (Koike et al. 1989). Indeed there is some evidence that even abnormal activity can be beneficial in some settings. In rats, two electroconvulsive seizures within the first 24 h after contusion accelerated recovery of beam‐walking, with less cerebral necrosis (Feeney et al. 1987). Further, in cats, brief stimulation with d‐amphetamine after bilateral frontal cortex ablation was associated with persistent improvement in beam‐walking (Sutton et al. 1989). Conversely, the suppression of EEG activity with γ‐aminobutyric acid agonists such as diazepam and muscimol greatly impairs the recovery from cortical or striatal lesions (Schallert et al. 1990), which might relate to impaired synaptogenesis. Synaptogenesis is in part dependent on brain activity (Saneyoshi et al. 2008), whereas the inhibition of neuronal activity impairs synaptogenesis (van Huizen et al. 1985).
Endogenous growth factors play a complementary role with neural activity in supporting neural homeostasis. As well as the direct homeostatic effects of neuronal activity (Koike et al. 1989), neural stimulation also indirectly supports neuronal survival by promoting release of fibroblast growth factor (Mattson & Rychlik, 1990). Independently, during profound electrical suppression in vivo, endogenous growth factors help support neuronal survival (Anderson et al. 1988). After HI brain damage in neonatal rats, neurotrophic activity is initially suppressed (Clawson et al. 1999), but growth factor treatment markedly reduces post‐HI brain damage in rodents and fetal sheep (Guan et al. 2003). Endogenous growth factor activity increases from around 3–5 days, reaching maximum expression at 8–15 days (Nieto‐Sampedro et al. 1982; Guan et al. 2003). This induction of growth factors might help promote stabilization of the cellular environment and long‐term neurorepair.
Consistent with an important role for recovery of astrocytes in determining outcome of cerebral HI, there is some evidence in adult rodents that hypothermia after ischaemia and cardiac arrest is associated with increased expression of growth factors, including glial cell line‐derived neurotrophic factor (GDNF), and brain‐derived neurotropic factor (BDNF) and its tyrosine receptor kinase‐B, in a time‐ and region‐specific manner (Boris‐Moller et al. 1998; D'Cruz et al. 2002; Schmidt et al. 2003). Thus, at the least these data confirm that mild hypothermia does not suppress astroglial production of integral neurotrophins. Further research is needed to understand whether astroglial growth factor production is essential for long‐term neurodevelopmental recovery after therapeutic hypothermia.
A working model for hypothermic neuroprotection
Taken together, these experimental studies indicate that hypothermia actively prevents delayed cell death after profound HI by suppressing apoptotic and necrotic cellular death pathways and extracellular inflammation and thus stabilizing mitochondrial function. To achieve long‐term neuroprotection, this hypothermia‐induced suppression needs to be continued until the extracellular environment provides a sufficient level of pro‐survival cues.
Key survival cues are EEG activity and growth factors. Hypothermia in part achieves this by differentially depressing microglia more than astrocytes (Si et al. 1997) and so allows neurotrophin activity to recover after HI. Further, although induced hypothermia somewhat suppresses stereographic seizures, it does not significantly inhibit recovery of EEG activity (Davidson et al. 2018). Critically, as discussed above, there is now compelling evidence that optimally hypothermia should be continued until high frequency EEG activity has been restored (Davidson et al. 2018). It is intriguing to note that the timing of recovery of this EEG frequency to baseline values during cooling in this study at ∼72 h after ischaemia also corresponds broadly with the known time delay before endogenous growth factors begin to be induced after HI in adult and developing rodents (Guan et al. 2003).
This model is consistent with the empirical observation that optimally the brain should be cooled by 3–5°C, with loss of protection with deeper cooling (Alonso‐Alconada et al. 2015). This is likely, at least in part, related to the finding that mild cooling selectively suppresses microglial activation, whereas deeper cooling also suppresses astrocyte function and proliferation, and so might impair endogenous restoration of growth factors (Si et al. 1997). Potentially, it might also reflect greater suppression of neural function during deep hypothermia (Westover et al. 2015). This need to allow recovery of the cell environment before warming is consistent with the strong observation that cooling needs to be continued until normalization of EEG frequency (Davidson et al. 2015c , 2018).
The potential implications for combination therapies with hypothermia
This working model suggests that future combined therapies should focus on promoting cellular homeostasis after hypothermia through long‐term stimulation of survival cues like neurotrophins, differential suppression/stimulation of bad/good inflammation, plus functional integration of new neurons and oligodendroglial cells (i.e. with recombinant human erythropoietin (rEpo) or stem cell therapies). First, if EEG activity is indeed critical for restoration of the normal cell environment, then high dose anticonvulsant treatment, which suppresses background activity, is likely to overlap with the mechanisms of therapeutic hypothermia, and so not provide additional neuroprotection, but also has the potential to impair long‐term neural recovery.
Consistent with these concerns, there is good evidence that in adult rats diazepam therapy after cerebral ischaemia does not augment hypothermic neuroprotection (Davies et al. 2004) and, as discussed above, that prolonged suppression can impair functional recovery (Schallert et al. 1990). Supporting this, the anticonvulsant topiramate (Lee et al. 2000) also did not improve death or neurological disability in a small phase‐II trial in hypothermia‐treated neonates with HIE, compared with hypothermia‐treated babies alone (Filippi et al. 2018). Thus, there is an urgent need for highly targeted preclinical and clinical research that can resolve the real world impact.
Similarly, an increasing number of animal studies have shown non‐additive neuroprotection during immediate co‐treatment with hypothermia. For example, in fetal sheep after cerebral ischaemia, connexin hemichannel blockade reduced neuronal damage and restored EEG power (Davidson et al. 2012), but was non‐additive to mild hypothermia (Davidson et al. 2015b ). Intracerebral infusion with insulin‐like growth factor‐1 (IGF‐1) increased post‐ischaemic astroglial and oligodendrocyte survival in near‐term fetal sheep (Guan et al. 2001), but treatment with delayed IGF‐1 from 4.5 h after ischemia plus hypothermia from 5.5 to 72 h did not provide greater protection or caspase‐3 depression than cerebral cooling alone (George et al. 2011). The noble gas xenon, which has anti‐apoptotic effects through the N‐methyl‐d‐aspartate (NMDA) receptor (Zhuang et al. 2012), improved hypothermic protection in neonatal piglets after HI but not in a phase‐II clinical trial (Chakkarapani et al. 2010; Azzopardi et al. 2015). This study is not conclusive since xenon was not started until a median of 10 h after birth (range, 4.0–12.6). Nevertheless, these data are suggestive that non‐additive neuroprotection partially resulted from overlapping mechanisms of action.
By contrast, melatonin started 15 min after HI followed by hypothermia from 2 h improved histological outcomes and recovery of high energy phosphates on magnetic resonance spectroscopy compared with hypothermia alone (Robertson et al. 2013). This result likely reflects melatonin's potent anti‐free radical effects, which will have been maximal during reperfusion from HI (Miller et al. 2005), but it is unclear whether it would have been equally effective if it had been started at the same time as hypothermia. Nevertheless, a pilot trial in human babies with HIE reported that the combination of melatonin plus hypothermia was associated with improved survival at 6 months of age without neurological abnormalities compared to hypothermia alone (Aly et al. 2015). These preliminary findings are encouraging but need validation in larger trials.
Neuroprotection and neurorepair – rEpo and stem cell therapies
Residual or ‘persistent’ inflammation has been reported during or after hypothermia (Davidson et al. 2018). Thus, it is plausible that therapies with anti‐inflammatory and/or pro‐regenerative effects might augment hypothermic neuroprotection either during or after therapeutic hypothermia. In this respect, there is compelling preclinical evidence for benefit with rEpo and stem cells (Bennet et al. 2012; Juul & Pet, 2015). rEpo has anti‐apoptotic, anti‐oxidant, anti‐excitotoxic and anti‐inflammation effects in preclinical paradigms of neonatal brain damage (Rangarajan & Juul, 2014), promotes proliferation and maturation of oligodendrocytes and neurons (Sugawa et al. 2002; Iwai et al. 2007), and stimulates growth factors (BDNF and GDNF) and angiogenesis (Li et al. 2007; Juul & Pet, 2015), which is needed for neurorepair and normal neurodevelopment.
Multiple experimental studies have reported rEpo‐mediated neuroprotection with improved long‐term outcomes after HI (as reviewed by Wu & Gonzalez, 2015). For example, in preterm fetal sheep, rEpo infusion from 30 min until 72 h after asphyxia improved neuronal and oligodendroglial loss, and electrophysiological restoration (Wassink et al. 2017). In preterm infants, a recent meta‐analysis found that early, prophylactic rEpo improved neurodevelopmental outcomes at 18–24 months (Fischer et al. 2017). Moreover, small randomised clinical trials in term neonates with HIE have demonstrated improved outcomes on modern imaging and neurological measures after treating with rEpo (Zhu et al. 2009; Elmahdy et al. 2010; Malla et al. 2017). These and initial clinical phase II trials on co‐treatment with hypothermia are encouraging (Wu et al. 2016), but large definitive trials are awaited.
In addition, there is increasing evidence from in vitro and in vivo preclinical studies that stem/progenitor cells might have beneficial effects on outcomes after HI (as reviewed by Bennet et al. 2012). For example, in newborn rabbit kits that received intrauterine ischaemia at 0.7 gestation (Drobyshevsky et al. 2015), treatment with human umbilical cord blood cells at birth resulted in a dose‐dependent improvement in neurobehavioural outcomes. These stem cells improved functional outcomes without significant engraftment, suggesting that their effects were mediated by trophic or immunomodulation mechanisms. Similarly, in preterm fetal sheep, intranasal infusion with human amnion epithelial cells at 1, 3 and 10 days after HI reduced neuronal and white matter loss, and suppressed gliosis and caspase‐3, with improved maturation of the cortical EEG (van den Heuij et al. 2018). In postnatal day 7 rats, combined administration of mesenchymal stem cells with hypothermia, from 6 h after HI, was associated with greater improvement on imaging and behavioural tests than either intervention alone (Park et al. 2015).
Finally, one small double‐blind randomised placebo‐controlled trial in 96 children with cerebral palsy reported that treatment with umbilical cord blood plus rEpo attenuated neurocognitive and motor dysfunction at 6 months more than rehabilitation with or without rEpo (Min et al. 2013). Thus, stem cell therapies have potential as a treatment to improve recovery from HIE, whether in isolation or combined with hypothermia.
Conclusions and perspectives
The working model of the mechanisms of hypothermic neuroprotection presented here suggests that immediate co‐treatment of hypothermia with agents whose mechanisms overlap with those of hypothermia is unlikely to offer substantial benefit. Indeed, interventions such as high dose anticonvulsant therapy that suppress background neural activity may have the potential to impair long‐term neural recovery. We propose that research should focus on interventions that promote cellular homeostasis through long‐term stimulation of survival cues like neurotrophins, selective suppression/stimulation of bad/good inflammation, plus integration of new functional cells. Current evidence suggests that strategies that promote these outcomes, such as stem cells and erythropoietin, are the most likely to further improve the outcome of therapeutic hypothermia.
Additional information
Competing interests
The authors declare no potential conflict of interest in this article.
Author contributions
A.G., G.W. and J.D. conceptualised this topical review. G.W. undertook manuscript writing and preparation of figures. All authors reviewed and edited this manuscript. All authors have read and approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The authors’ studies discussed in this review received support from the Health Research Council of New Zealand (HRC ID no. 12/613 and 17/601) and Neurological Foundation of New Zealand (NF1715‐PG). J.D. holds a Sir Charles Hercus Fellowship from the Health Research Council of New Zealand.
Biographies
Guido Wassink is a research fellow working to understand why some fetuses survive oxygen deprivation and infection completely normal, whereas others suffer injury or die after apparently identical exposure. His studies have highlighted the factors determining the physiological resilience of the unborn child and the role of growth factors such as endogenous erythropoietin in helping to protect the brain after hypoxia‐ischaemia.
Alistair Jan Gunn is a Paediatrician‐scientist who has conducted groundbreaking basic research into ways of identifying compromised fetuses in labour, the mechanisms and treatment of asphyxial brain injury and the mechanisms of life threatening events in infancy. His research helped to establish mild cooling as the first ever technique to reduce brain injury due to low oxygen levels at birth.

Edited by: Ole Petersen & Janna Morrison
References
- Alonso‐Alconada D, Broad KD, Bainbridge A, Chandrasekaran M, Faulkner SD, Kerenyi A, Hassell J, Rocha‐Ferreira E, Hristova M, Fleiss B, Bennett K, Kelen D, Cady E, Gressens P, Golay X & Robertson NJ (2015). Brain cell death is reduced with cooling by 3.5°C to 5°C but increased with cooling by 8.5°C in a piglet asphyxia model. Stroke 46, 275–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aly H, Elmahdy H, El‐Dib M, Rowisha M, Awny M, El‐Gohary T, Elbatch M, Hamisa M & El‐Mashad AR (2015). Melatonin use for neuroprotection in perinatal asphyxia: a randomized controlled pilot study. J Perinatol 35, 186–191. [DOI] [PubMed] [Google Scholar]
- Anderson KJ, Dam D, Lee S & Cotman CW (1988). Basic fibroblast growth factor prevents death of lesioned cholinergic neurons in vivo. Nature 332, 360–361. [DOI] [PubMed] [Google Scholar]
- Askalan R, Wang C, Shi H, Armstrong E & Yager JY (2011). The effect of postischemic hypothermia on apoptotic cell death in the neonatal rat brain. Dev Neurosci 33, 320–329. [DOI] [PubMed] [Google Scholar]
- Azzopardi D, Robertson NJ, Bainbridge A, Cady E, Charles‐Edwards G, Deierl A, Fagiolo G, Franks NP, Griffiths J, Hajnal J, Juszczak E, Kapetanakis B, Linsell L, Maze M, Omar O, Strohm B, Tusor N & Edwards AD (2015). Moderate hypothermia within 6 h of birth plus inhaled xenon versus moderate hypothermia alone after birth asphyxia (TOBY‐Xe): a proof‐of‐concept, open‐label, randomised controlled trial. Lancet Neurol 15, 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzopardi D, Strohm B, Linsell L, Hobson A, Juszczak E, Kurinczuk JJ, Brocklehurst P & Edwards AD (2012). Implementation and conduct of therapeutic hypothermia for perinatal asphyxial encephalopathy in the UK – analysis of national data. PLoS One 7, e38504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzopardi D, Wyatt JS, Cady EB, Delpy DT, Baudin J, Stewart AL, Hope PL, Hamilton PA & Reynolds EO (1989). Prognosis of newborn infants with hypoxic‐ischemic brain injury assessed by phosphorus magnetic resonance spectroscopy. Pediatr Res 25, 445–451. [DOI] [PubMed] [Google Scholar]
- Bart RD, Takaoka S, Pearlstein RD, Dexter F & Warner DS (1998). Interactions between hypothermia and the latency to ischemic depolarization: implications for neuroprotection. Anesthesiology 88, 1266–1273. [DOI] [PubMed] [Google Scholar]
- Bennet L, Dean JM, Wassink G & Gunn AJ (2007a). Differential effects of hypothermia on early and late epileptiform events after severe hypoxia in preterm fetal sheep. J Neurophysiol 97, 572–578. [DOI] [PubMed] [Google Scholar]
- Bennet L, Roelfsema V, Dean J, Wassink G, Power GG, Jensen EC & Gunn AJ (2007b). Regulation of cytochrome oxidase redox state during umbilical cord occlusion in preterm fetal sheep. Am J Physiol Regul Integr Comp Physiol 292, R1569–R1576. [DOI] [PubMed] [Google Scholar]
- Bennet L, Roelfsema V, George S, Dean JM, Emerald BS & Gunn AJ (2007c). The effect of cerebral hypothermia on white and grey matter injury induced by severe hypoxia in preterm fetal sheep. J Physiol 578, 491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennet L, Tan S, Van den Heuij L, Derrick M, Groenendaal F, van Bel F, Juul S, Back SA, Northington F, Robertson NJ, Mallard C & Gunn AJ (2012). Cell therapy for neonatal hypoxia‐ischemia and cerebral palsy. Ann Neurol 71, 589–600. [DOI] [PubMed] [Google Scholar]
- Bevers MB & Neumar RW (2008). Mechanistic role of calpains in postischemic neurodegeneration. J Cereb Blood Flow Metab 28, 655–673. [DOI] [PubMed] [Google Scholar]
- Blumberg RM, Cady EB, Wigglesworth JS, McKenzie JE & Edwards AD (1997). Relation between delayed impairment of cerebral energy metabolism and infarction following transient focal hypoxia‐ischaemia in the developing brain. Exp Brain Res 113, 130–137. [DOI] [PubMed] [Google Scholar]
- Boris‐Moller F, Kamme F & Wieloch T (1998). The effect of hypothermia on the expression of neurotrophin mRNA in the hippocampus following transient cerebral ischemia in the rat. Mol Brain Res 63, 163–173. [DOI] [PubMed] [Google Scholar]
- Bossenmeyer‐Pourie C, Koziel V & Daval JL (2000). Effects of hypothermia on hypoxia‐induced apoptosis in cultured neurons from developing rat forebrain: comparison with preconditioning. Pediatr Res 47, 385–391. [DOI] [PubMed] [Google Scholar]
- Brown GC (1997). Nitric oxide inhibition of cytochrome oxidase and mitochondrial respiration: implications for inflammatory, neurodegenerative and ischaemic pathologies. Mol Cell Biochem 174, 189–192. [PubMed] [Google Scholar]
- Chakkarapani E, Dingley J, Liu X, Hoque N, Aquilina K, Porter H & Thoresen M (2010). Xenon enhances hypothermic neuroprotection in asphyxiated newborn pigs. Ann Neurol 68, 330–341. [DOI] [PubMed] [Google Scholar]
- Clawson TF, Vannucci SJ, Wang GM, Seaman LB, Yang XL & Lee WH (1999). Hypoxia‐ischemia‐induced apoptotic cell death correlates with IGF‐I mRNA decrease in neonatal rat brain. Biol Signals Recept 8, 281–293. [DOI] [PubMed] [Google Scholar]
- Davidson JO, Draghi V, Whitham S, Dhillon SK, Wassink G, Bennet L & Gunn AJ (2018). How long is sufficient for optimal neuroprotection with cerebral cooling after ischemia in fetal sheep? J Cereb Blood Flow Metab (in press; doi: 10.1177/0271678X17707671). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson JO, Drury PP, Green CR, Nicholson LF, Bennet L & Gunn AJ (2014). Connexin hemichannel blockade is neuroprotective after asphyxia in preterm fetal sheep. PLoS One 9, e96558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson JO, Green CR, Bennet L & Gunn AJ (2015a). Battle of the hemichannels – Connexins and Pannexins in ischemic brain injury. Int J Dev Neurosci 45, 66–74. [DOI] [PubMed] [Google Scholar]
- Davidson JO, Green CR, Nicholson LF, O'Carroll SJ, Fraser M, Bennet L & Gunn AJ (2012). Connexin hemichannel blockade improves outcomes in a model of fetal ischemia. Ann Neurol 71, 121–132. [DOI] [PubMed] [Google Scholar]
- Davidson JO, Rout AL, Wassink G, Yuill CA, Zhang FG, Green CR, Bennet L & Gunn AJ (2015b). Non‐additive effects of delayed connexin hemichannel blockade and hypothermia after cerebral ischemia in near‐term fetal sheep. J Cereb Blood Flow Metab 35, 2052–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson JO, Wassink G, Yuill CA, Zhang FG, Bennet L & Gunn AJ (2015c). How long is too long for cerebral cooling after ischemia in fetal sheep? J Cereb Blood Flow Metab 35, 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies LM, MacLellan CL, Corbett DR & Colbourne F (2004). Post‐ischemic diazepam does not reduce hippocampal CA1 injury and does not improve hypothermic neuroprotection after forebrain ischemia in gerbils. Brain Res 1013, 223–229. [DOI] [PubMed] [Google Scholar]
- D'Cruz BJ, Fertig KC, Filiano AJ, Hicks SD, DeFranco DB & Callaway CW (2002). Hypothermic reperfusion after cardiac arrest augments brain‐derived neurotrophic factor activation. J Cereb Blood Flow Metab 22, 843–851. [DOI] [PubMed] [Google Scholar]
- Dean JM, George SA, Wassink G, Gunn AJ & Bennet L (2006a). Suppression of post hypoxic‐ischemic EEG transients with dizocilpine is associated with partial striatal protection in the preterm fetal sheep. Neuropharmacology 50, 491–503. [DOI] [PubMed] [Google Scholar]
- Dean JM, Gunn AJ, Wassink G, George S & Bennet L (2006b). Endogenous α2‐adrenergic receptor‐mediated neuroprotection after severe hypoxia in preterm fetal sheep. Neuroscience 142, 615–628. [DOI] [PubMed] [Google Scholar]
- Drobyshevsky A, Cotten CM, Shi Z, Luo K, Jiang R, Derrick M, Tracy ET, Gentry T, Goldberg RN, Kurtzberg J & Tan S (2015). Human umbilical cord blood cells ameliorate motor deficits in rabbits in a cerebral palsy model. Dev Neurosci 37, 349–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druzhyna NM, Musiyenko SI, Wilson GL & LeDoux SP (2005). Cytokines induce nitric oxide‐mediated mtDNA damage and apoptosis in oligodendrocytes. Protective role of targeting 8‐oxoguanine glycosylase to mitochondria. J Biol Chem 280, 21673–21679. [DOI] [PubMed] [Google Scholar]
- Edwards AD, Brocklehurst P, Gunn AJ, Halliday H, Juszczak E, Levene M, Strohm B, Thoresen M, Whitelaw A & Azzopardi D (2010). Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: synthesis and meta‐analysis of trial data. BMJ 340, c363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AD, Yue X, Squier MV, Thoresen M, Cady EB, Penrice J, Cooper CE, Wyatt JS, Reynolds EO & Mehmet H (1995). Specific inhibition of apoptosis after cerebral hypoxia‐ischaemia by moderate post‐insult hypothermia. Biochem Biophys Res Commun 217, 1193–1199. [DOI] [PubMed] [Google Scholar]
- Elmahdy H, El‐Mashad AR, El‐Bahrawy H, El‐Gohary T, El‐Barbary A & Aly H (2010). Human recombinant erythropoietin in asphyxia neonatorum: pilot trial. Pediatrics 125, e1135–1142. [DOI] [PubMed] [Google Scholar]
- Engidawork E, Loidl F, Chen Y, Kohlhauser C, Stoeckler S, Dell'Anna E, Lubec B, Lubec G, Goiny M, Gross J, Andersson K & Herrera‐Marschitz M (2001). Comparison between hypothermia and glutamate antagonism treatments on the immediate outcome of perinatal asphyxia. Exp Brain Res 138, 375–383. [DOI] [PubMed] [Google Scholar]
- Feeney DM, Bailey BY, Boyeson MG, Hovda DA & Sutton RL (1987). The effect of seizures on recovery of function following cortical contusion in the rat. Brain Inj 1, 27–32. [DOI] [PubMed] [Google Scholar]
- Filippi L, Fiorini P, Catarzi S, Berti E, Padrini L, Landucci E, Donzelli G, Bartalena L, Fiorentini E, Boldrini A, Giampietri M, Scaramuzzo RT, la Marca G, Della Bona ML, Fiori S, Tinelli F, Bancale A, Guzzetta A, Cioni G, Pisano T, Falchi M & Guerrini R (2018). Safety and efficacy of topiramate in neonates with hypoxic ischemic encephalopathy treated with hypothermia (NeoNATI): a feasibility study. J Matern Fetal Neonatal Med 31, 973–980. [DOI] [PubMed] [Google Scholar]
- Fischer HS, Reibel NJ, Buhrer C & Dame C (2017). Prophylactic early erythropoietin for neuroprotection in preterm infants: a meta‐analysis. Pediatrics 139, e20164317. [DOI] [PubMed] [Google Scholar]
- Fleiss B & Gressens P (2012). Tertiary mechanisms of brain damage: a new hope for treatment of cerebral palsy? Lancet Neurol 11, 556–566. [DOI] [PubMed] [Google Scholar]
- George SA, Barrett RD, Bennet L, Mathai S, Jensen EC & Gunn AJ (2012). Nonadditive neuroprotection with early glutamate receptor blockade and delayed hypothermia after asphyxia in preterm fetal sheep. Stroke 43, 3114–3117. [DOI] [PubMed] [Google Scholar]
- George SA, Bennet L, Weaver‐Mikaere L, Fraser M, Bouwmans J, Mathai S, Skinner SJM & Gunn AJ (2011). White matter protection with insulin like‐growth factor 1 (IGF‐1) and hypothermia is not additive after severe reversible cerebral ischemia in term fetal sheep. Dev Neurosci 33, 280–287. [DOI] [PubMed] [Google Scholar]
- George S, Gunn AJ, Westgate JA, Brabyn C, Guan J & Bennet L (2004). Fetal heart rate variability and brainstem injury after asphyxia in preterm fetal sheep. Am J Physiol Regul Integr Comp Physiol 287, R925–R933. [DOI] [PubMed] [Google Scholar]
- Giulian D, Vaca K & Corpuz M (1993). Brain glia release factors with opposing actions upon neuronal survival. J Neurosci 13, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glassman RB & Malamut BL (1976). Recovery from electroencephalographic slowing and reduced evoked potentials after somatosensory cortical damage in cats. Behav Biol 17, 333–354. [DOI] [PubMed] [Google Scholar]
- Gressens P, Le Verche V, Fraser M, Rousset CI, Schwendimann L, Bennet L, George SA, Wang X, Mallard C, Tilley BC, Dournaud P, Gunn AJ, Hagberg H & Levison SW (2011). Pitfalls in the quest of neuroprotectants for the perinatal brain. Dev Neurosci 33, 189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J, Bennet L, George S, Wu D, Waldvogel HJ, Gluckman PD, Faull RL, Crosier PS & Gunn AJ (2001). Insulin‐like growth factor‐1 reduces postischemic white matter injury in fetal sheep. J Cereb Blood Flow Metab 21, 493–502. [DOI] [PubMed] [Google Scholar]
- Guan J, Bennet L, Gluckman PD & Gunn AJ (2003). Insulin‐like growth factor‐1 and post‐ischemic brain injury. Prog Neurobiol 70, 443–462. [DOI] [PubMed] [Google Scholar]
- Gunn AJ, Gunn TR, de Haan HH, Williams CE & Gluckman PD (1997). Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J Clin Invest 99, 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn AJ, Laptook AR, Robertson NJ, Barks JD, Thoresen M, Wassink G & Bennet L (2017). Therapeutic hypothermia translates from ancient history in to practice. Pediatr Res 81, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg H, Mallard C, Ferriero DM, Vannucci SJ, Levison SW, Vexler ZS & Gressens P (2015). The role of inflammation in perinatal brain injury. Nat Rev Neurol 11, 192–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg H, Mallard C, Rousset CI & Thornton C (2014). Mitochondria: hub of injury responses in the developing brain. Lancet Neurol 13, 217–232. [DOI] [PubMed] [Google Scholar]
- Hattori H, Morin AM, Schwartz PH, Fujikawa DG & Wasterlain CG (1989). Posthypoxic treatment with MK‐801 reduces hypoxic‐ischemic damage in the neonatal rat. Neurology 39, 713–718. [DOI] [PubMed] [Google Scholar]
- Hope PL, Costello AM, Cady EB, Delpy DT, Tofts PS, Chu A, Hamilton PA, Reynolds EO & Wilkie DR (1984). Cerebral energy metabolism studied with phosphorus NMR spectroscopy in normal and birth‐asphyxiated infants. Lancet 2, 366–370. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Mosinger JL & Olney JW (1989). Hypothermia enhances protective effect of MK‐801 against hypoxic/ischemic brain damage in infant rats. Brain Res 487, 184–187. [DOI] [PubMed] [Google Scholar]
- Iwai M, Cao G, Yin W, Stetler RA, Liu J & Chen J (2007). Erythropoietin promotes neuronal replacement through revascularization and neurogenesis after neonatal hypoxia/ischemia in rats. Stroke 38, 2795–2803. [DOI] [PubMed] [Google Scholar]
- Jacobs SE, Berg M, Hunt R, Tarnow‐Mordi WO, Inder TE & Davis PG (2013). Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst Rev 1, CD003311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen FE, Wang C, Stafstrom CE, Liu Z, Geary C & Stevens MC (1998). Acute and chronic increases in excitability in rat hippocampal slices after perinatal hypoxia in vivo. J Neurophysiol 79, 73–81. [DOI] [PubMed] [Google Scholar]
- Juul SE & Pet GC (2015). Erythropoietin and neonatal neuroprotection. Clin Perinatol 42, 469–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Kang N, Lovatt D, Torres A, Zhao Z, Lin J & Nedergaard M (2008). Connexin 43 hemichannels are permeable to ATP. J Neurosci 28, 4702–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike T, Martin DP & Johnson EM Jr (1989). Role of Ca2+ channels in the ability of membrane depolarization to prevent neuronal death induced by trophic‐factor deprivation: evidence that levels of internal Ca2+ determine nerve growth factor dependence of sympathetic ganglion cells. Proc Natl Acad Sci USA 86, 6421–6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laptook AR, Corbett RJ, Sterett R, Garcia D & Tollefsbol G (1995). Quantitative relationship between brain temperature and energy utilization rate measured in vivo using 31P and 1H magnetic resonance spectroscopy. Pediatr Res 38, 919–925. [DOI] [PubMed] [Google Scholar]
- Lee SR, Kim SP & Kim JE (2000). Protective effect of topiramate against hippocampal neuronal damage after global ischemia in the gerbils. Neurosci Lett 281, 183–186. [DOI] [PubMed] [Google Scholar]
- Li S, Zhang Z, Xue J, Liu A & Zhang H (2012). Cold‐inducible RNA binding protein inhibits H2O2‐induced apoptosis in rat cortical neurons. Brain Res 1441, 47–52. [DOI] [PubMed] [Google Scholar]
- Li Y, Lu Z, Keogh CL, Yu SP & Wei L (2007). Erythropoietin‐induced neurovascular protection, angiogenesis, and cerebral blood flow restoration after focal ischemia in mice. J Cereb Blood Flow Metab 27, 1043–1054. [DOI] [PubMed] [Google Scholar]
- Liu L, Kim JY, Koike MA, Yoon YJ, Tang XN, Ma H, Lee H, Steinberg GK, Lee JE & Yenari MA (2008). FasL shedding is reduced by hypothermia in experimental stroke. J Neurochem 106, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malla RR, Asimi R, Teli MA, Shaheen F & Bhat MA (2017). Erythropoietin monotherapy in perinatal asphyxia with moderate to severe encephalopathy: a randomized placebo‐controlled trial. J Perinatol 37, 596–601. [DOI] [PubMed] [Google Scholar]
- Man SM & Kanneganti TD (2016). Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat Rev Immunol 16, 7–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Brambrink AM, Price AC, Kaiser A, Agnew DM, Ichord RN & Traystman RJ (2000). Neuronal death in newborn striatum after hypoxia‐ischemia is necrosis and evolves with oxidative stress. Neurobiol Dis 7, 169–191. [DOI] [PubMed] [Google Scholar]
- Mattson MP & Rychlik B (1990). Glia protect hippocampal neurons against excitatory amino acid‐induced degeneration: involvement of fibroblast growth factor. Int J Dev Neurosci 8, 399–415. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Silverstein FS & Johnston MV (1987). MK‐801 protects the neonatal brain from hypoxic‐ischemic damage. Eur J Pharmacol 140, 359–361. [DOI] [PubMed] [Google Scholar]
- Miller SL, Yan EB, Castillo‐Melendez M, Jenkin G & Walker DW (2005). Melatonin provides neuroprotection in the late‐gestation fetal sheep brain in response to umbilical cord occlusion. Dev Neurosci 27, 200–210. [DOI] [PubMed] [Google Scholar]
- Min K, Song J, Kang JY, Ko J, Ryu JS, Kang MS, Jang SJ, Kim SH, Oh D, Kim MK, Kim SS & Kim M (2013). Umbilical cord blood therapy potentiated with erythropoietin for children with cerebral palsy: a double‐blind, randomized, placebo‐controlled trial. Stem Cells 31, 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel S, Papadakis M, Pfleger K, Grond‐Ginsbach C, Buchan AM & Wagner S (2012). Microarray analysis of the global gene expression profile following hypothermia and transient focal cerebral ischemia. Neuroscience 208, 109–122. [DOI] [PubMed] [Google Scholar]
- Natarajan G, Pappas A & Shankaran S (2016). Outcomes in childhood following therapeutic hypothermia for neonatal hypoxic‐ischemic encephalopathy (HIE). Semin Perinatol 40, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto‐Sampedro M, Lewis ER, Cotman CW, Manthorpe M, Skaper SD, Barbin G, Longo FM & Varon S (1982). Brain injury causes a time‐dependent increase in neuronotrophic activity at the lesion site. Science 217, 860–861. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Chavez‐Valdez R, Graham EM, Razdan S, Gauda EB & Martin LJ (2011a). Necrostatin decreases oxidative damage, inflammation, and injury after neonatal HI. J Cereb Blood Flow Metab 31, 178–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northington FJ, Chavez‐Valdez R & Martin LJ (2011b). Neuronal cell death in neonatal hypoxia‐ischemia. Ann Neurol 69, 743–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northington FJ, Zelaya ME, O'Riordan DP, Blomgren K, Flock DL, Hagberg H, Ferriero DM & Martin LJ (2007). Failure to complete apoptosis following neonatal hypoxia‐ischemia manifests as “continuum” phenotype of cell death and occurs with multiple manifestations of mitochondrial dysfunction in rodent forebrain. Neuroscience 149, 822–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurse S & Corbett D (1996). Neuroprotection after several days of mild, drug‐induced hypothermia. J Cereb Blood Flow Metab 16, 474–480. [DOI] [PubMed] [Google Scholar]
- O'Carroll SJ, Alkadhi M, Nicholson LF & Green CR (2008). Connexin 43 mimetic peptides reduce swelling, astrogliosis, and neuronal cell death after spinal cord injury. Cell Commun Adhes 15, 27–42. [DOI] [PubMed] [Google Scholar]
- Ohmura A, Nakajima W, Ishida A, Yasuoka N, Kawamura M, Miura S & Takada G (2005). Prolonged hypothermia protects neonatal rat brain against hypoxic‐ischemia by reducing both apoptosis and necrosis. Brain Dev 27, 517–526. [DOI] [PubMed] [Google Scholar]
- Park WS, Sung SI, Ahn SY, Yoo HS, Sung DK, Im GH, Choi SJ & Chang YS (2015). Hypothermia augments neuroprotective activity of mesenchymal stem cells for neonatal hypoxic‐ischemic encephalopathy. PLoS One 10, e0120893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quist AP, Rhee SK, Lin H & Lal R (2000). Physiological role of gap‐junctional hemichannels. Extracellular calcium‐dependent isosmotic volume regulation. J Cell Biol 148, 1063–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raemy E & Martinou JC (2014). Involvement of cardiolipin in tBID‐induced activation of BAX during apoptosis. Chem Phys Lipids 179, 70–74. [DOI] [PubMed] [Google Scholar]
- Rangarajan V & Juul SE (2014). Erythropoietin: emerging role of erythropoietin in neonatal neuroprotection. Pediatr Neurol 51, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson NJ, Faulkner S, Fleiss B, Bainbridge A, Andorka C, Price D, Powell E, Lecky‐Thompson L, Thei L, Chandrasekaran M, Hristova M, Cady EB, Gressens P, Golay X & Raivich G (2013). Melatonin augments hypothermic neuroprotection in a perinatal asphyxia model. Brain 136, 90–105. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Sinovas A, Cabestrero A, Lopez D, Torre I, Morente M, Abellan A, Miro E, Ruiz‐Meana M & Garcia‐Dorado D (2007). The modulatory effects of connexin 43 on cell death/survival beyond cell coupling. Prog Biophys Mol Biol 94, 219–232. [DOI] [PubMed] [Google Scholar]
- Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP, Oberst A, Quarato G, Low J, Cripps JG, Chen T & Green DR (2016). Characterization of RIPK3‐mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ 23, 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelfsema V, Bennet L, George S, Wu D, Guan J, Veerman M & Gunn AJ (2004). The window of opportunity for cerebral hypothermia and white matter injury after cerebral ischemia in near‐term fetal sheep. J Cereb Blood Flow Metab 24, 877–886. [DOI] [PubMed] [Google Scholar]
- Roth SC, Baudin J, Cady E, Johal K, Townsend JP, Wyatt JS, Reynolds EO & Stewart AL (1997). Relation of deranged neonatal cerebral oxidative metabolism with neurodevelopmental outcome and head circumference at 4 years. Dev Med Child Neurol 39, 718–725. [DOI] [PubMed] [Google Scholar]
- Saneyoshi T, Wayman G, Fortin D, Davare M, Hoshi N, Nozaki N, Natsume T & Soderling TR (2008). Activity‐dependent synaptogenesis: regulation by a CaM‐kinase kinase/CaM‐kinase I/βPIX signaling complex. Neuron 57, 94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schallert T, Jones TA & Lindner MD (1990). Multilevel transneuronal degeneration after brain damage. Behavioral events and effects of anticonvulsant gamma‐aminobutyric acid‐related drugs. Stroke 21, III143–146. [PubMed] [Google Scholar]
- Schmidt KM, D'Cruz BJ, DeFranco DB & Callaway CW (2003). Cardiac arrest and hypothermia increase GDNF in brain. Acad Emerg Med 10, 480. [Google Scholar]
- Shankaran S, Laptook AR, Pappas A, McDonald SA, Das A, Tyson JE, Poindexter BB, Schibler K, Bell EF, Heyne RJ, Pedroza C, Bara R, Van Meurs KP, Huitema CMP, Grisby C, Devaskar U, Ehrenkranz RA, Harmon HM, Chalak LF, DeMauro SB, Garg M, Hartley‐McAndrew ME, Khan AM, Walsh MC, Ambalavanan N, Brumbaugh JE, Watterberg KL, Shepherd EG, Hamrick SEG, Barks J, Cotten CM, Kilbride HW & Higgins RD (2017). Effect of depth and duration of cooling on death or disability at age 18 months among neonates with hypoxic‐ischemic encephalopathy: a randomized clinical trial. JAMA 318, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankaran S, McDonald SA, Laptook AR, Hintz SR, Barnes PD, Das A, Pappas A & Higgins RD (2015). Neonatal magnetic resonance imaging pattern of brain injury as a biomarker of childhood outcomes following a trial of hypothermia for neonatal hypoxic‐ischemic encephalopathy. J Pediatr 167, 987–993.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si QS, Nakamura Y & Kataoka K (1997). Hypothermic suppression of microglial activation in culture: inhibition of cell proliferation and production of nitric oxide and superoxide. Neuroscience 81, 223–229. [DOI] [PubMed] [Google Scholar]
- Sugawa M, Sakurai Y, Ishikawa‐Ieda Y, Suzuki H & Asou H (2002). Effects of erythropoietin on glial cell development; oligodendrocyte maturation and astrocyte proliferation. Neurosci Res 44, 391–403. [DOI] [PubMed] [Google Scholar]
- Sutton RL, Hovda DA & Feeney DM (1989). Amphetamine accelerates recovery of locomotor function following bilateral frontal cortex ablation in cats. Behav Neurosci 103, 837–841. [DOI] [PubMed] [Google Scholar]
- Tan WK, Williams CE, During MJ, Mallard CE, Gunning MI, Gunn AJ & Gluckman PD (1996). Accumulation of cytotoxins during the development of seizures and edema after hypoxic‐ischemic injury in late gestation fetal sheep. Pediatr Res 39, 791–797. [DOI] [PubMed] [Google Scholar]
- Tan WK, Williams CE, Gunn AJ, Mallard CE & Gluckman PD (1992). Suppression of postischemic epileptiform activity with MK‐801 improves neural outcome in fetal sheep. Ann Neurol 32, 677–682. [DOI] [PubMed] [Google Scholar]
- Thoresen M, Satas S, Puka‐Sundvall M, Whitelaw A, Hallstrom A, Loberg EM, Ungerstedt U, Steen PA & Hagberg H (1997). Post‐hypoxic hypothermia reduces cerebrocortical release of NO and excitotoxins. Neuroreport 8, 3359–3362. [DOI] [PubMed] [Google Scholar]
- Thornton C, Leaw B, Mallard C, Nair S, Jinnai M & Hagberg H (2017). Cell death in the developing brain after hypoxia‐ischemia. Front Cell Neurosci 11, 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton JS, Ordidge RJ, Penrice J, Cady EB, Amess PN, Punwani S, Clemence M & Wyatt JS (1998). Temporal and anatomical variations of brain water apparent diffusion coefficient in perinatal cerebral hypoxic‐ischemic injury: relationships to cerebral energy metabolism. Magn Reson Med 39, 920–927. [DOI] [PubMed] [Google Scholar]
- van den Heuij LG, Fraser M, Miller SL, Jenkin G, Wallace EM, Davidson JO, Lear CA, Lim R, Wassink G, Gunn AJ & Bennet L (2018). Delayed intranasal infusion of human amnion epithelial cells improves white matter maturation after asphyxia in preterm fetal sheep. J Cereb Blood Flow Metab (in press; doi: 10.1177/0271678X17729954). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Huizen F, Romijn HJ & Habets AM (1985). Synaptogenesis in rat cerebral cortex cultures is affected during chronic blockade of spontaneous bioelectric activity by tetrodotoxin. Brain Res 351, 67–80. [DOI] [PubMed] [Google Scholar]
- Vannucci RC, Towfighi J & Vannucci SJ (2004). Secondary energy failure after cerebral hypoxia‐ischemia in the immature rat. J Cereb Blood Flow Metab 24, 1090–1097. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS & Wang X (2014). Mixed lineage kinase domain‐like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 54, 133–146. [DOI] [PubMed] [Google Scholar]
- Wassink G, Davidson JO, Dhillon SK, Fraser M, Galinsky R, Bennet L & Gunn AJ (2017). Partial white and grey matter protection with prolonged infusion of recombinant human erythropoietin after asphyxia in preterm fetal sheep. J Cereb Blood Flow Metab 37, 1080–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassink G, Gunn ER, Drury PP, Bennet L & Gunn AJ (2014). The mechanisms and treatment of asphyxial encephalopathy. Front Neurosci 8, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westover MB, Ching S, Kumaraswamy VM, Akeju SO, Pierce E, Cash SS, Kilbride R, Brown EN & Purdon PL (2015). The human burst suppression electroencephalogram of deep hypothermia. Clin Neurophysiol 126, 1901–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CE, Gunn AJ, Mallard C & Gluckman PD (1992). Outcome after ischemia in the developing sheep brain: an electroencephalographic and histological study. Ann Neurol 31, 14–21. [DOI] [PubMed] [Google Scholar]
- Wu YW & Gonzalez FF (2015). Erythropoietin: a novel therapy for hypoxic‐ischaemic encephalopathy? Dev Med Child Neurol 57(Suppl 3), 34–39. [DOI] [PubMed] [Google Scholar]
- Wu YW, Mathur AM, Chang T, McKinstry RC, Mulkey SB, Mayock DE, Van Meurs KP, Rogers EE, Gonzalez FF, Comstock BA, Juul SE, Msall ME, Bonifacio SL, Glass HC, Massaro AN, Dong L, Tan KW, Heagerty PJ & Ballard RA (2016). High‐dose erythropoietin and hypothermia for hypoxic‐ischemic encephalopathy: a phase II trial. Pediatrics 137, e20160191. [DOI] [PubMed] [Google Scholar]
- Xu L, Yenari MA, Steinberg GK & Giffard RG (2002). Mild hypothermia reduces apoptosis of mouse neurons in vitro early in the cascade. J Cereb Blood Flow Metab 22, 21–28. [DOI] [PubMed] [Google Scholar]
- Ye ZC, Wyeth MS, Baltan‐Tekkok S & Ransom BR (2003). Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci 23, 3588–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Xu G, Zhang J, Murong S, Mei Y & Tong E (2010). Mild hypothermia reduces ischemic neuron death via altering the expression of p53 and bcl‐2. Neurol Res 32, 384–389. [DOI] [PubMed] [Google Scholar]
- Zhu C, Kang W, Xu F, Cheng X, Zhang Z, Jia L, Ji L, Guo X, Xiong H, Simbruner G, Blomgren K & Wang X (2009). Erythropoietin improved neurologic outcomes in newborns with hypoxic‐ischemic encephalopathy. Pediatrics 124, e218–226. [DOI] [PubMed] [Google Scholar]
- Zhuang L, Yang T, Zhao H, Fidalgo AR, Vizcaychipi MP, Sanders RD, Yu B, Takata M, Johnson MR & Ma D (2012). The protective profile of argon, helium, and xenon in a model of neonatal asphyxia in rats. Crit Care Med 40, 1724–1730. [DOI] [PubMed] [Google Scholar]