

Abstract
Key points
Fetal heart rate variability is a critical index of fetal wellbeing. Suppression of heart rate variability may provide prognostic information on the risk of hypoxic‐ischaemic brain injury after birth.
In the present study, we report the evolution of fetal heart rate variability after both mild and severe hypoxia‐ischaemia. Both mild and severe hypoxia‐ischaemia were associated with an initial, brief suppression of multiple measures of heart rate variability. This was followed by normal or increased levels of heart rate variability during the latent phase of injury.
Severe hypoxia‐ischaemia was subsequently associated with the prolonged suppression of measures of heart rate variability during the secondary phase of injury, which is the period of time when brain injury is no longer treatable.
These findings suggest that a biphasic pattern of heart rate variability may be an early marker of brain injury when treatment or intervention is probably most effective.
Abstract
Hypoxia‐ischaemia (HI) is a major contributor to preterm brain injury, although there are currently no reliable biomarkers for identifying infants who are at risk. We tested the hypothesis that fetal heart rate (FHR) and FHR variability (FHRV) would identify evolving brain injury after HI. Fetal sheep at 0.7 of gestation were subjected to either 15 (n = 10) or 25 min (n = 17) of complete umbilical cord occlusion or sham occlusion (n = 12). FHR and four measures of FHRV [short‐term variation, long‐term variation, standard deviation of normal to normal R‐R intervals (SDNN), root mean square of successive differences) were assessed until 72 h after HI. All measures of FHRV were suppressed for the first 3–4 h in the 15 min group and 1–2 h in the 25 min group. Measures of FHRV recovered to control levels by 4 h in the 15 min group, whereas the 25 min group showed tachycardia and an increase in short‐term variation and SDNN from 4 to 6 h after occlusion. The measures of FHRV then progressively declined in the 25 min group and became profoundly suppressed from 18 to 48 h. A partial recovery of FHRV measures towards control levels was observed in the 25 min group from 49 to 72 h. These findings illustrate the complex regulation of FHRV after both mild and severe HI and suggest that the longitudinal analysis of FHR and FHRV after HI may be able to help determine the timing and severity of preterm HI.
Keywords: fetus, hypoxia‐ischemia, fetal heart rate variability
Key points
Fetal heart rate variability is a critical index of fetal wellbeing. Suppression of heart rate variability may provide prognostic information on the risk of hypoxic‐ischaemic brain injury after birth.
In the present study, we report the evolution of fetal heart rate variability after both mild and severe hypoxia‐ischaemia. Both mild and severe hypoxia‐ischaemia were associated with an initial, brief suppression of multiple measures of heart rate variability. This was followed by normal or increased levels of heart rate variability during the latent phase of injury.
Severe hypoxia‐ischaemia was subsequently associated with the prolonged suppression of measures of heart rate variability during the secondary phase of injury, which is the period of time when brain injury is no longer treatable.
These findings suggest that a biphasic pattern of heart rate variability may be an early marker of brain injury when treatment or intervention is probably most effective.
Introduction
Antenatal and intrapartum hypoxia‐ischaemia (HI) is a major cause of brain injury in prematurely born infants, occurring in ∼5–9 per 1000 live births (Gopagondanahalli et al. 2016). The risks are greatest at lower gestational ages (Manuck et al. 2016). Infants born prematurely appear to have a very high burden of antenatal HI (de Vries et al. 1998; Becher et al. 2004; Low, 2004; Logitharajah et al. 2009). In particular, antenatal HI may be a significant contributor to cerebral palsy after preterm birth (McIntyre et al. 2013; Tan, 2014).
It is now well established that HI brain injury evolves over time. Initial restoration of cerebral metabolism is observed in the first ∼6–15 h (the latent phase of injury), followed by secondary failure of cerebral energy failure over the subsequent 48–72 h during which bulk neural cell loss occurs (the secondary phase of injury) (Wassink et al. 2014). A precise understanding of the timing of HI is important for the development and timely implementation of therapeutic interventions. For example, therapeutic hypothermia improves neurodevelopmental outcomes in term infants when started during the first 6 h after birth and continued until 72 h, although efficacy is lost if hypothermia is delayed (Gunn et al. 1997, 1998, 1999; Jacobs et al. 2013; Thoresen et al. 2013).
Fetal heart rate (FHR) and derivative indices such as FHR variability (FHRV) are routinely used to assess fetal wellbeing both before birth and in labour (Westgate et al. 2007; Lear et al. 2016). Clinical data from before and after birth suggest that HI is associated with suppression of heart rate variability (Phelan & Ahn, 1994; Goulding et al. 2017). However, experimental evidence suggests that this is a late event related to established brainstem injury (Ikeda et al. 1998; George et al. 2004). It is largely unknown whether FHRV could be a biomarker of earlier brain injury before the onset of secondary metabolic failure and thereby identify injury when it is still treatable.
In the present study, changes in FHR and FHRV were studied in fetal sheep at 0.7 of gestation after mild or severe HI. The neural maturation of the fetal sheep at this age is broadly equivalent to the human fetus at 28–32 weeks (McIntosh et al. 1979). Mild HI was induced by 15 min of complete umbilical cord occlusion (UCO), which is associated with only selective subcortical neuronal injury (Keogh et al. 2012). Severe HI was induced by 25 min of complete UCO, which is associated with a pattern of diffuse white matter injury and severe subcortical neuronal injury (Dean et al. 2006; Drury et al. 2013), consistent with the predominant pattern of injury seen in contemporary cohorts of preterm babies (Buser et al. 2012; de Vries et al. 2015). Changes in FHR and FHRV were investigated in relation to the well‐described phases of brain injury after HI: the latent phase (first 6 h after HI) is characterised by transient recovery of oxidative metabolism and represents the time when therapeutic intervention can mitigate the severity of secondary cell death (Bennet et al. 2006; Wassink et al. 2014). The secondary phase of injury (∼6–72 h after HI) represents the time when widespread secondary cell death occurs, along with the appearance of post‐HI seizures (Bennet et al. 2006). Current treatment strategies are unable to improve outcomes if initiated after the start of the secondary phase (Wassink et al. 2014).
Materials and methods
Ethical approval
All procedures were approved by the Animal Ethics Committee of the University of Auckland and were carried out in accordance with the New Zealand Animal Welfare Act 1999 and the University of Auckland's Code of Ethical Conduct for the use of animals for teaching and research, as approved by the Ministry of Primary Industries, Government of New Zealand. All procedures comply with the guidelines of the The Journal of Physiology (Grundy, 2015).
Surgical procedures
Thirty‐nine singleton Romney/Suffolk fetal sheep were surgically instrumented at 98–100 days of gestation (term = 147 days), as reported previously (Lear et al. 2014b). Food but not water was withdrawn 12–18 h before surgery. Ewes were given long acting oxytetracycline (20 mg kg−1; Phoenix Pharm Distributors, Auckland, New Zealand) i.m. 30 min before surgery for prophylaxis. Anaesthesia was induced by i.v. injection of propofol (5 mg kg−1; AstraZeneca, Auckland, New Zealand) and general anaesthesia was maintained using 2–3% isoflurane in oxygen. The depth of anaesthesia was monitored constantly by trained anaesthetic staff. Ewes received a slow infusion of isotonic saline (∼250 ml h−1) to maintain fluid balance.
A midline abdominal incision was made to expose the uterus and the fetus was partially exteriorised for instrumentation. Polyvinyl catheters (SteriHealth, Dandenong South, VIC, Australia) were placed in the left fetal femoral artery to measure blood pressure and right brachial artery for pre‐ductal blood sampling. An additional catheter was placed into the amniotic sac for the measurement of amniotic fluid pressure. A pair of electrodes (AS633‐3SSF; Cooner Wire, Chatsworth, CA, USA) was placed s.c. over the right shoulder and at the level of the fifth intercostal space to measure the fetal ECG from which FHR and FHRV were derived. Two pairs of electrodes (AS633‐7SSF; Cooner Wire) were placed over the parietal dura bilaterally, 5 mm and 10 mm anterior to bregma and 10 mm lateral to the sagittal suture, for the measurement of electroencephalographic (EEG) activity. A further two electrodes (AS633‐7SSF; Cooner Wire) were sewn into nuchal muscle for measurement of nuchal electromyographic (EMG) activity. A reference electrode was sewn over the occiput. An inflatable silicone occluder (OC16HD; In Vivo Metric, Healdsburg, CA, USA) was placed around the umbilical cord to allow UCO.
Gentamicin (80 mg; Pfizer, Auckland, New Zealand) was administered to the amniotic sac. The maternal midline skin incision was infiltrated with long‐acting analgesic (10 mL of 0.5% bupivacaine plus adrenaline; AstraZeneca). All fetal leads were exteriorised through the maternal flank and a maternal long saphenous vein was catheterised for postoperative care.
Postoperative care
After surgery, ewes were housed in separate metabolic cages with companion animals in a temperature‐controlled room (16 ± 1 °C, humidity 50 ± 10%) under a 12 12 h light/dark cycle (lights on 06.00 h) and ad libitum access to water and food. Ewes received i.v. antibiotics daily for 4 days (600 mg of benzylpenicillin sodium; Novartis, Auckland, New Zealand; 80 mg of gentamicin). Fetal vascular catheters were maintained patent by continuous infusion of heparinised saline (20 U mL−1 at 0.2 mL h−1). Three days after UCO, the ewes and their fetuses were killed by an i.v. overdose to the ewe of sodium pentobarbitone (9 g; Pentobarb 300; Chemstock International, Christchurch, New Zealand). Death was confirmed by the absence of cardiopulmonary function and deep maternal reflexes, including the corneal reflex. This method is fully consistent with the Animal Welfare Act of New Zealand.
Experimental recordings
Fetal mean arterial pressure (MAP), ECG, EEG amplitude and spectral edge frequency and nuchal EMG were recorded continuously from 24 h before until 72 h after UCO for offline analysis using customised LabVIEW‐based data acquisition software (National Instruments, Austin, TX, USA). Fetal MAP was recorded using Novatrans III Gold pressure transducers (MX860; Medex, Hilliard, OH, USA) and corrected for maternal movement by subtraction of amniotic fluid pressure. The pressure signals were amplified 500×, low‐pass filtered with a Butterworth filter set at 20 Hz and saved at 64 Hz. The raw ECG signal was analogue filtered with a first‐order high‐pass filter set at 1 Hz and an eight‐order low‐pass Bessel filter set at 100 Hz and saved at 512 and then used to derive FHR and FHRV (Koome et al. 2014; Lear et al. 2014a, 2016). EEG signals were amplified 10,000× and low‐pass filtered with a sixth‐order Butterworth filter set to 50 Hz and saved at 256 Hz. The intensity (power) was derived from the intensity spectrum signal between 1 and 20 Hz and normalised by log transformation (dB, 20 × log intensity). Spectral edge frequency was calculated as the frequency below which 90% of the intensity was present. The nuchal EMG signal was bandpass filtered between 100 Hz and 1 kHz, the signal was then integrated using a time constant of 0.1 s and digitalised at 512 Hz.
Experimental protocol
Experiments began between 09.00 and 09.30 h, 4–5 days after surgery when fetuses were at 104–105 days of gestation. Fetuses were randomly assigned to either the sham control (n = 12), mild HI (15 min UCO group, n = 10) or severe HI (25 min UCO group, n = 17). HI was induced by inflating the umbilical cord occluder with a volume of saline known to completely occlude the umbilical cord (Bennet et al. 1999) and released after 15 or 25 min. Successful UCO was confirmed by the rapid onset of bradycardia, a rise in MAP, and changes in pH and blood gas measurements (Bennet et al. 1999). The occluder was not inflated in the sham control group.
Fetal arterial blood samples
Baseline brachial artery blood samples (0.3 mL) were collected before the start of UCO. During UCO, samples were taken at 5 min after the start of UCO and either at 12 min (15 min UCO group) or 17 min (25 min UCO group). Post‐UCO samples were taken at 10 min and 1, 2, 4, 6, 24, 48 and 72 h after the end of UCO. Samples were analysed for fetal pH and blood gases (ABL 800; Radiometer, Copenhagen, Denmark) and glucose and lactate (model 2300; YSI, Yellow Springs, OH, USA).
Data analysis
Offline analysis of the physiological data was performed using customised LabVIEW‐based programs (National Instruments). Continuous RR intervals were extracted from the fetal ECG to calculate FHR and four time domain measures of FHRV [root mean square of successive differences (RMSSD), short‐term variation (STV), long‐term variation (LTV) and SD of normal to normal R‐R intervals (SDNN)]. All measures were assessed over 1 min continuous epochs. These epochs were averaged to 10 min means during the first 24 h after HI and to hourly means during the baseline period and from 24–72 h after HI. RMSSD was calculated as the root mean square of successive RR intervals during each 1 min epoch, providing a measure of beat‐to‐beat FHRV, which is sensitive to high frequency oscillations (Lear et al. 2016). The clinical measures STV and LTV were calculated by first dividing each 1 min epoch into 16 periods (3.75 s in duration) and the mean RR interval (ms) was calculated for each period. STV was calculated as the difference between successive periods averaged over each 1 min epoch (Street et al. 1991). LTV was calculated as the difference between the maximum and minimum period average for each 1 min epoch (Smith et al. 1987). SDNN was calculated as the SD of all RR intervals during each 1 min epoch, providing a measure of total FHRV irrespective of the frequency of oscillations (Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology, 1996). We additionally calculated the skewness (the third moment about the mean) of RR intervals as a measure of the asymmetry of the FHR trace (Griffin & Moorman, 2001). Skewness was calculated over 10 min continuous epochs. All other physiological parameters were calculated as hourly means.
Statistical analysis
Statistical analysis was performed using SPSS, version 23 (IBM Corp., Armonk, NY, USA). After HI, data were analysed in specific time bins consistent with the known phases of injury. Differences between groups were evaluated using two‐way ANOVA with time treated as a repeated measure and group as the independent factor. A Fisher's protected least‐significant difference post hoc test was subsequently performed when a significant overall effect was found. When assessing FHRV, the first 10 min period after UCO was excluded as a result of rapid decrease in RR interval during recovery from bradycardia. Arterial blood gas data were analysed by one‐way ANOVA with group as the independent factor. Baseline was used as a covariate where appropriate. Data are the mean ± SEM. P < 0.05 was considered statistically significant.
Results
Baseline parameters
Before UCO all fetuses were healthy, based on our laboratory standards including normal physiological and arterial blood gas parameters. There were no significant differences in any physiological parameters between the groups. All fetuses showed the discontinuous, mixed frequency EEG activity that is characteristic at this gestation (Davidson et al. 2011). Nuchal EMG activity in all three groups showed frequent, brief bursts of activity. The distribution of male and female fetuses in each group was: sham control (nine males, three females), 15 min UCO (six males, three females, one unknown) and 25 min UCO (three males, 14 females).
Umbilical cord occlusion
UCO was associated with sustained bradycardia and severe hypotension. The final extent of bradycardia and hypotension was more severe in the 25 min group compared to the 15 min group (Table 1). UCO was associated with profound hypoxaemia, hypoglycaemia and progressive respiratory and metabolic acidosis that was more severe in the 25 min group than in the 15 min group (Table 2).
Table 1.
Nadir of hypotension and bradycardia during complete umbilical cord occlusion
| Mean arterial pressure (mmHg) | Fetal heart rate (beats min–1) | |||
|---|---|---|---|---|
| Baseline | Occlusion | Baseline | Occlusion | |
| Control | 36.2 ± 0.7 | 36.3 ± 0.8 | 190.4 ± 3.7 | 187.9 ± 5.1 |
| 15 min | 34.8 ± 0.7 | 14.8 ± 1.3* | 192.0 ± 3.1 | 82.0 ± 2.7* |
| 25 min | 36.6 ± 0.5 | 11.2 ± 0.7#δ | 189.0 ± 2.3 | 57.6 ± 2.7#δ |
Baseline was taken as the average of the hour preceding the start of occlusion. The final extent of hypotension and bradycardia was calculated as the average over the final minute of umbilical cord occlusion. Data are the 1 min mean ± SEM. * P < 0.01, 15 min vs. control; # P < 0.01; 25 min vs. controls; δ P < 0.01, 15 min vs. 25 min.
Table 2.
Fetal pH, blood gases and metabolites before, during and after umbilical cord occlusion
| Group | Baseline | Occlusion | +1 h | +2 h | +4 h | +6 h | +24 h | +48 h | +72 h |
|---|---|---|---|---|---|---|---|---|---|
| pH | |||||||||
| Control | 7.37 ± 0.01 | 7.37 ± 0.01 | 7.37 ± 0.01 | 7.39 ± 0.01 | 7.37 ± 0.01 | 7.38 ± 0.01 | 7.37 ± 0.01 | 7.36 ± 0.01 | 7.36 ± 0.01 |
| 15 min | 7.36 ± 0.01 | 6.92 ± 0.01*δ | 7.33 ± 0.01*δ | 7.40 ± 0.01δ | 7.39 ± 0.01 | 7.37 ± 0.01 | 7.35 ± 0.01 | 7.36 ± 0.01 | 7.36 ± 0.00 |
| 25 min | 7.36 ± 0.01 | 6.84 ± 0.01# | 7.27 ± 0.02# | 7.32 ± 0.01# | 7.40 ± 0.01# | 7.40 ± 0.01 | 7.37 ± 0.01 | 7.37 ± 0.01 | 7.36 ± 0.01 |
| (mmHg) | |||||||||
| Control | 48.8 ± 1.2 | 47.0 ± 1.1 | 47.3 ± 1.1 | 47.4 ± 0.9 | 48.1 ± 1.1 | 47.5 ± 1.1 | 47.7 ± 1.1 | 47.6 ± 1.4 | 47.8 ± 1.5 |
| 15 min | 49.4 ± 0.9 | 114.5 ± 3.1*δ | 44.0 ± 0.6 | 45.6 ± 1.0 | 44.5 ± 1.1 | 46.7 ± 1.3 | 48.6 ± 1.0 | 48.4 ± 1.3 | 48.5 ± 1.3 |
| 25 min | 50.5 ± 1.3 | 141.6 ± 4.2# | 50.8 ± 3.6 | 48.2 ± 1.9 | 48.0 ± 1.3 | 47.4 ± 1.0 | 46.9 ± 1.1 | 47.1 ± 1.0 | 47.8 ± 1.1 |
| (mmHg) | |||||||||
| Control | 24.8 ± 0.7 | 24.5 ± 0.8 | 23.9 ± 0.9 | 24.9 ± 0.7 | 25.0 ± 0.6 | 24.7 ± 0.7 | 24.5 ± 1.0 | 24.6 ± 1.0 | 25.5 ± 1.2 |
| 15 min | 25.0 ± 0.6 | 7.6 ± 0.8* | 28.0 ± 0.6* | 26.5 ± 0.7 | 28.0 ± 0.7*δ | 26.1 ± 0.8 | 26.5 ± 0.6 | 25.6 ± 0.9 | 27.0 ± 0.9 |
| 25 min | 23.3 ± 0.6 | 9.4 ± 0.6# | 28.7 ± 2.2# | 25.9 ± 1.0 | 23.2 ± 0.9 | 24.1 ± 1.0 | 26.2 ± 1.1 | 27.9 ± 1.3 | 27.4 ± 1.2 |
| BE (mmol L−1) | |||||||||
| Control | 1.5 ± 0.5 | 1.6 ± 0.7 | 1.2 ± 0.7 | 2.7 ± 0.7 | 1.7 ± 1.0 | 2.4 ± 0.8 | 1.4 ± 0.8 | 1.1 ± 0.8 | 0.6 ± 0.6 |
| 15 min | 2.0 ± 0.4 | −9.6 ± 0.3*δ | −2.8 ± 0.4* | 2.6 ± 0.6δ | 1.7 ± 0.5 | 1.5 ± 0.6 | 0.8 ± 0.3 | 1.3 ± 0.7 | 1.3 ± 0.7 |
| 25 min | 2.3 ± 0.8 | −12.0 ± 0.6# | −4.0 ± 1.5# | −1.8 ± 1.0# | 3.2 ± 0.8 | 3.8 ± 0.7 | 0.8 ± 0.8 | 1.1 ± 0.8 | 0.6 ± 0.4 |
| Hct (%) | |||||||||
| Control | 26.7 ± 0.8 | 26.4 ± 1.0 | 26.8 ± 0.9 | 25.5 ± 2.1 | 26.3 ± 0.8 | 27.0 ± 1.0 | 27.2 ± 1.3 | 28.5 ± 1.8 | 29.6 ± 1.6 |
| 15 min | 25.3 ± 1.2 | 26.3 ± 1.6 | 23.9 ± 1.2δ | 25.7 ± 1.2 | 25.8 ± 1.1 | 25.4 ± 1.2 | 25.2 ± 1.3 | 26.3 ± 1.1 | 26.8 ± 1.4 |
| 25 min | 27.2 ± 0.9 | 27.7 ± 0.9 | 29.9 ± 1.6 | 28.5 ± 0.9 | 27.1 ± 0.8 | 27.6 ± 1.1 | 27.6 ± 1.1 | 26.6 ± 1.0 | 27.3 ± 1.1 |
| O2ct (mmol L−1) | |||||||||
| Control | 3.7 ± 0.1 | 6.0 ± 2.2 | 3.7 ± 0.2 | 3.9 ± 0.2 | 3.7 ± 0.2 | 3.8 ± 0.2 | 3.8 ± 0.2 | 5.9 ± 2.0 | 4.0 ± 0.2 |
| 15 min | 3.5 ± 0.2 | 0.3 ± 0.0*δ | 3.5 ± 0.2 | 3.8 ± 0.2 | 3.8 ± 0.2 | 3.7 ± 0.2 | 3.6 ± 0.2 | 3.7 ± 0.2 | 4.0 ± 0.3 |
| 25 min | 3.6 ± 0.1 | 0.5 ± 0.0# | 4.2 ± 0.3 | 4.0 ± 0.2 | 3.8 ± 0.2 | 3.9 ± 0.2 | 4.0 ± 0.2 | 4.1 ± 0.2 | 4.2 ± 0.2 |
| Lactate (mmol L−1) | |||||||||
| Control | 0.7 ± 0.1 | 0.8 ± 0.0 | 0.9 ± 0.1 | 0.8 ± 0.0 | 0.8 ± 0.1 | 0.9 ± 0.1 | 0.8 ± 0.0 | 0.8 ± 0.0 | 0.8 ± 0.1 |
| 15 min | 0.8 ± 0.1 | 5.2 ± 0.2*δ | 3.1 ± 0.1*δ | 1.5 ± 0.1δ | 1.0 ± 0.1δ | 1.0 ± 0.1δ | 0.8 ± 0.1δ | 0.9 ± 0.0 | 0.8 ± 0.0 |
| 25 min | 0.8 ± 0.1 | 7.0 ± 0.2# | 5.0 ± 0.5# | 4.3 ± 0.4# | 2.7 ± 0.4# | 2.3 ± 0.3# | 1.2 ± 0.1# | 0.9 ± 0.1 | 0.8 ± 0.1 |
| Glucose (mmol L−1) | |||||||||
| Control | 1.0 ± 0.1 | 1.0 ± 0.0 | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.0 ± 0.0 | 1.0 ± 0.1 |
| 15 min | 1.2 ± 0.0 | 0.7 ± 0.1* | 1.6 ± 0.1* | 1.7 ± 0.1*δ | 1.6 ± 0.01* | 1.6 ± 0.0* | 1.2 ± 0.1 | 1.2 ± 0.1* | 1.2 ± 0.1 |
| 25 min | 1.0 ± 0.1 | 0.8 ± 0.1# | 1.4 ± 0.1# | 1.4 ± 0.1# | 1.4 ± 0.1# | 1.5 ± 0.1# | 1.3 ± 0.1# | 1.2 ± 0.1# | 1.1 ± 0.1 |
Samples during occlusion were taken at either 12 min after the start of umbilical cord occlusion (15 min group) or 17 min after the start of umbilical cord occlusion (25 min group). , arterial partial pressure of carbon dioxide in arterial blood; , arterial partial pressure of oxygen in arterial blood; BE, base excess; Hct, haematocrit; O2ct, arterial blood oxygen content. Data are the mean ± SEM. * P < 0.05, 15 min vs. control; # P < 0.05, 25 min vs. controls; δ P < 0.05, 15 min vs. 25 min.
Post‐HI arterial blood pressure
There were significant differences in MAP between groups during the first 6 h after UCO (P < 0.001) (Fig. 1). Post hoc analysis suggested that MAP was mildly reduced in the 15 min group between 2 and 3 h after UCO compared to controls (P < 0.01), whereas MAP was significantly increased in the 25 min group for the first 6 h compared to controls (P < 0.005). Subsequently, there was no difference between groups.
Figure 1. Time sequence of changes in MAP, FHR and skewness.
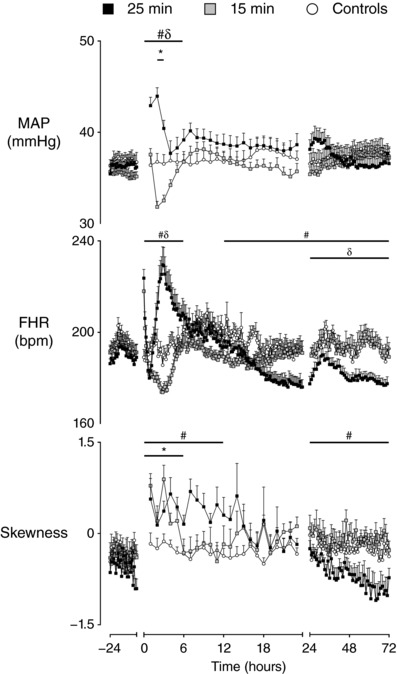
Time sequence of changes in MAP (mmHg), FHR (bpm) and skewness of R‐R intervals from 24 h before until 72 h after umbilical cord occlusion in sham controls (n = 12), 15 min group (n = 10) and 25 min group (n = 17). Data are the 1 h mean ± SEM. FHR is the 10 min mean ± SEM during the first 24 h after occlusion. * P < 0.05, 15 min vs. controls; # P < 0.05, 25 min vs. controls; δ P < 0.05, 15 min vs. 25 min.
Post‐HI EEG intensity, spectral edge frequency and nuchal EMG activity
There were significant differences in EEG power (P < 0.001) (Fig. 2) and spectral edge frequency (P < 0.001) between groups throughout the recovery period after UCO. Post hoc analysis suggested that EEG power in the 15 min group was significantly lower than the control group (P < 0.005) but significantly higher than the 25 min group (P < 0.01) in the first 6 h after UCO. Subsequently, EEG power in the 15 min group returned to control levels. Spectral edge frequency in the 15 min group was significantly lower than the control group for the first 24 h after UCO (P < 0.05) but subsequently returned to control levels. EEG power in the 25 min group was significantly lower than both the 15 min and control groups throughout the 72 h recovery period after UCO (P < 0.005). Spectral edge frequency in the 25 min group was significantly lower than controls throughout recovery (P < 0.001) and significantly lower than the 15 min group from 7 to 72 h after UCO (P < 0.001).
Figure 2. Time sequence of changes in EEG amplitude, spectral edge frequency and nuchal electromyogram.
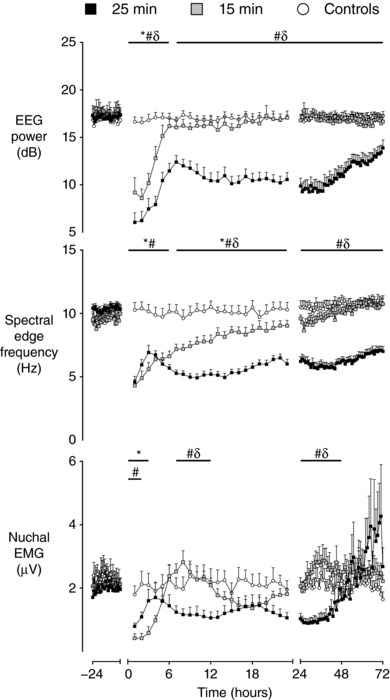
Time sequence of changes in EEG amplitude (dB), spectral edge frequency (Hz) and nuchal EMG (μV) from 24 h before until 72 h after umbilical cord occlusion in sham controls (n = 12), 15 min group (n = 10) and 25 min group (n = 17). Data are the 1 h mean ± SEM. * P < 0.05, 15 min vs. control; # P < 0.05, 25 min vs. controls; δ P < 0.05, 15 min vs. 25 min.
Nuchal EMG activity showed a complex pattern during recovery in both groups (Fig. 2). There was a significant interaction between group and time during the first 6 h after UCO (P < 0.001). In the 15 min group, post hoc analysis suggested that nuchal EMG activity was significantly reduced compared to sham controls for the first 3 h after the UCO (P < 0.01). Subsequently, EMG activity in the 15 min group returned to sham control levels and displayed frequent, brief bursts of activity. Nuchal EMG was also reduced in the 25 min group during the first 2 h after UCO compared to sham controls (P < 0.05), although average EMG activity returned to sham control levels between 3 and 6 h after UCO. However, this was mediated by sustained EMG activity rather than the brief bursts observed in the 15 min and sham control groups. There was a significant effect of group on nuchal EMG activity between 7–12 h (P < 0.05) and 25–48 h (P < 0.01) after UCO. Nuchal EMG was significantly reduced in the 25 min group compared to both the controls and 15 min group between 7–12 h (P < 0.05) and 25–48 h (P < 0.05) after UCO.
Post‐HI fetal heart rate
There was a significant effect of group on FHR from 0 to 6 h (P < 0.001) and from 13 to 72 h after UCO (P < 0.05) (Fig. 1). During the first 6 h after UCO, post hoc analysis suggested that FHR was not significantly different in the 15 min group compared to controls. By contrast, FHR was significantly increased in the 25 min group compared to both the controls (P < 0.01) and the 15 min group (P < 0.001), peaking at 3 h. FHR in the 25 min group then fell and was significantly reduced in the 25 min group compared to the controls from 13 to 72 h (P < 0.05) and compared to the 15 min group from 24 to 72 h (P < 0.05).
Fetal heart rate variability during the latent phase
An evolving pattern in FHRV was observed after HI across all measures. During the first 6 h after HI, a significant effect of group was observed for RMSSD (P < 0.05), STV (P < 0.01), SDNN (P < 0.01) and LTV (P < 0.05) (Figs 3 and 4). Post hoc analysis suggested that all measures of FHRV were suppressed in the 15 min group compared to controls from 0 to 3 h after UCO (P < 0.05, RMSSD; P < 0.005 STV, SDNN), although LTV was suppressed until 4 h after UCO (P < 0.001). The 25 min group showed brief suppression of FHRV compared to controls from 0 to 2 h after UCO (P < 0.001, SDNN, LTV), although STV was only suppressed from 0 to 1 h after UCO (P < 0.001). RMSSD showed no initial suppression in the 25 min group compared to controls.
Figure 3. Time sequence of changes in RMSSD and SDNN.
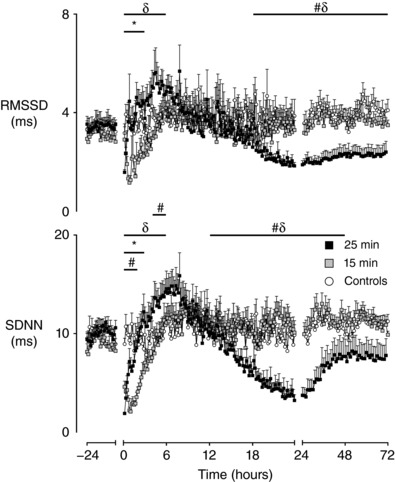
Time sequence of changes in RMSSD and SDNN from 24 h before until 72 h after umbilical cord occlusion in sham controls (n = 12), 15 min group (n = 10) and 25 min group (n = 17). Data the 10 min mean ± SEM during the first 24 h after occlusion and the 1 h mean ± SEM at all other time points. The first 10 min after occlusion is not shown * P < 0.05, 15 min vs. control; # P < 0.05, 25 min vs. controls; δ P < 0.05, 15 min vs. 25 min. The first 10 min bin is not shown.
Figure 4. Time sequence of changes in STV and LTV.
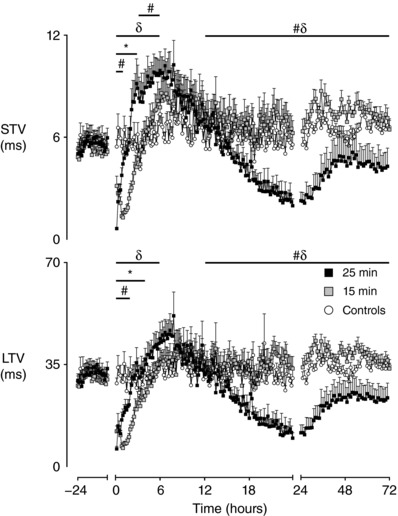
Time sequence of changes in STV and LTV from 24 h before until 72 h after umbilical cord occlusion in sham controls (n = 12), 15 min group (n = 10) and 25 min group (n = 17). Data are the 10 min mean ± SEM during the first 24 h after occlusion and the 1 h mean ± SEM at all other time points. The first 10 min after occlusion is not shown. * P < 0.05, 15 min vs. control; # P < 0.05, 25 min vs. controls; δ P < 0.05, 15 min vs. 25 min.
After this initial suppression, measures of FHRV in both the 15 min and 25 min groups progressively increased towards control levels, although the 25 min group continued to increase past control levels in two measures. STV in the 25 min group was significantly higher than controls from 3 to 6 h after UCO (P < 0.01). SDNN was significantly higher in the 25 min group compared to controls from 4 to 6 h after UCO (P < 0.05). Throughout the first 6 h after UCO, the 25 min group showed significantly higher levels in the measures of FHRV than the 15 min group (P < 0.005, RMSSD, STV, SDNN; P < 0.01, LTV).
Additionally, there was a significant effect of group on the skewness of RR intervals from 0 to 6 h after UCO (P < 0.001) (Fig. 1). Post hoc analysis suggested that both the 15 min (P < 0.005) and 25 min (P < 0.001) groups showed increased skewness of RR intervals from 0 to 6 h after UCO compared to controls.
Fetal heart rate variability during the secondary phase
The start of the secondary phase (7–12 h after HI) was associated with a significant effect of group on the skewness of RR intervals (P < 0.05) (Fig. 1); no significant differences were observed in any other measure of FHRV during this time. Post hoc analysis suggested that the 15 min group was not different from controls, whereas the 25 min group showed increased skewness of RR intervals from 7 to 12 h after UCO compared to both the 15 min group (P < 0.05) and controls (P < 0.05).
A significant effect of group was observed on SDNN from 13 to 48 h (P < 0.005), on STV and LTV from 13 to 72 h (P < 0.05), and on RMSSD from 19 to 72 h (P < 0.01). The measures of FHRV were not significant different in the 15 min group compared to controls. By contrast, the measures of FHRV became progressively suppressed in the 25 min group during the secondary phase and were significantly lower from 13 to 48 h after UCO compared to both the 15 min group (P < 0.05, STV; P < 0.005, SDNN, LTV) and controls (P < 0.005, SDNN, LTV; P < 0.05, STV). RMSSD in the 25 min group showed later suppression from 19 to 48 h after UCO compared to the 15 min (P < 0.05) and control groups (P < 0.005). A significant effect of group was also observed on the skewness of RR intervals from 25 to 72 h, resulting in the 25 min group having lower skewness of RR intervals than both the 15 min group (P < 0.01) (Fig. 1) and controls (P < 0.05).
There were no significant differences between the 15 min and control groups in any measure of FHRV at the end of the experiment. A partial recovery was observed in FHRV in the 25 min group from 49 to 72 h, with SDNN returning to control levels (Figs 3 and 4). However, the other measures remained significantly lower in the 25 min group from 49–72 h compared to the 15 min (P < 0.05, STV, LTV; P < 0.01, RMSSD) and control groups (P < 0.001, RMSSD; P = 0.056, LTV).
Discussion
In the present study, both mild (15 min) and severe (25 min) HI induced by complete UCO were associated with complex changes in FHR and FHRV that corresponded to the well‐described phases of injury (Bennet et al. 2006; Wassink et al. 2014). The four measures of FHRV assessed in the present study (STV, LTV, SDNN, RMSSD) were all initially suppressed in both the 15 min and 25 min groups during the latent phase. In the 15 min group, the measures of FHRV normalised by the end of the latent phase. By contrast, the initial suppression of FHRV measures in the 25 min group was followed by an earlier increase towards control levels. Two of the four measures (STV and SDNN) additionally suggested that there were increased levels of FHRV during the second half of the latent phase in the 25 min group.
The FHR response at this time was discordant between the groups. The 25 min group developed tachycardia for most of the latent phase compared to no significant change in the 15 min group. After the end of the latent phase, the 15 min group showed normal levels of FHRV across all measures until the end of the experiment. All measures of FHRV progressively declined in the 25 min group during the secondary phase of injury, reaching a nadir at 24 h, before showing a partial recovery towards the end of the experiment.
Fetal heart rate variability during the latent phase of injury
Heart rate variability reflects the complex integration of intrinsic pacemaker rhythms of the sinoatrial node with the sympathetic and parasympathetic activity of the autonomic nervous system (Dalton et al. 1983; Jensen et al. 2009; Lear et al. 2016). Autonomic outflow is in turn modulated by fetal behaviour including body and breathing movements (Dalton et al. 1977, 1983). The reduction in the indices of FHRV measured in the present study (STV, LTV, SDNN, RMSSD) during the latent phase could be related to the establishment of HI brain injury, particularly of the autonomic centres of the brainstem (George et al. 2004). Strikingly, however, the duration of suppression of these measures of FHRV was markedly longer in fetuses exposed to mild HI (15 min group) than those exposed to severe HI (25 min group). This suggests that suppression of FHRV after HI was not related to the severity of injury per se. Furthermore, the 25 min group showed rapid recovery and excitation of measures of FHRV within 6 h of HI, indicating that the brainstem was still able to increase autonomic outflow. It is therefore more probable that the reduction in measures of FHRV early after HI was related to an active suppression of autonomic activity in both the 15 min and 25 min groups.
More generally, the latent phase of injury is a period of relative quiescence and, in both groups, this phase was associated with suppression of EEG power, spectral edge frequency and body movements, as measured by nuchal EMG activity. This is an active, neuroprotective response that helps to minimise brain injury (Dean et al. 2006; Yawno et al. 2007, 2011). Suppression of the measures of FHRV is therefore probably a correlate of this generalised suppression of brain activity, secondary to inhibition of the upstream determinants of autonomic outflow. Consistent with this concept, normalization of the measures of FHRV in the 15 min group closely paralleled the recovery of EEG power and nuchal EMG activity at the end of the latent phase.
The period when measures of FHRV were suppressed was shorter in the 25 min group and all measures of FHRV returned to control levels by 2 h after HI despite continued profound suppression of EEG power and spectral edge frequency. A pattern of an abnormal, sustained increase in nuchal EMG activity was observed at this time, consistent with tonic flexion (George et al. 2004). This is very different from the normal episodic pattern of fetal body movements and probably did not contribute to the progressive increase in FHRV at this time. We did not measure breathing movements in the present study, although Ikeda et al. (1998) have previously reported that fetal gasping was associated with a ‘checkmark’ FHR pattern after severe HI in near‐term fetal sheep. The combination of abnormal nuchal EMG activity and increased measures of FHRV but continued suppression of EEG activity in the present study suggests a specific increase in brainstem activity and autonomic outflow.
We have previously shown that severe HI at 0.6 of gestation was associated with extensive injury to the medulla and nucleus tractus solitarii (George et al. 2004). It is therefore possible that the irregular autonomic outflow that led to the increase in measures of FHRV in the 25 min group is a reflection of evolving brainstem injury. We cannot rule out the possibility that reduced FHRV could, in part, reflect reduced cardiac responsiveness. However, this would not be not consistent with either the rapid recovery of blood pressure after release of occlusion or the longer suppression of FHRV after 15 min of occlusion compared to 25 min of occlusion.
The latent phase is critical because it represents the window of opportunity for the only neuroprotective strategy currently available for late preterm and term infants: therapeutic hypothermia (Gunn et al. 1997, 1998; Jacobs et al. 2013). The present study suggests that a pattern of high FHRV and tachycardia may identify the latent phase of injury after severe HI. By contrast, high FHRV is most typically associated with fetal wellbeing and even abnormal high amplitude FHR patterns such as pseudosinusoidal patterns can occur in healthy fetuses (Ayres‐de‐Campos et al. 2015). Moreover, the measures of FHRV assessed in the present study were no different from the control group for at least one‐third of the latent phase, making the two groups difficult to distinguish reliably.
The time‐domain based measures employed in the present study largely quantify the overall magnitude of FHRV and provide little direct information on the specific components or patterns present in the FHR trace. The duration of the epochs used to quantify FHRV can affect their magnitude (Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology, 1996). Reassuringly, however, studies in adult humans support a good correlation between the 1 min and 5 min parameters for time‐domain measures of FHRV (Nussinovitch et al. 2012). Additional measures of FHRV (e.g. frequency domain and non‐linear methods) may help to provide additional information on the physiological regulation of FHRV after HI (Bernardes et al. 2009; Frasch et al. 2012). For example, in the present study, we observed that both the 15 min and 25 min groups showed increased skewness after HI, which persisted into the early secondary phase in the 25 min group. Skewness provides a measure of the asymmetry of the FHR trace independent of the overall magnitude of oscillations. An increase in skewness suggests a relative predominance of downward FHR deflections (i.e. decelerations) in comparison to upward deflections (i.e. accelerations) (Lear et al. 2015, 2016). This finding suggests that HI was associated with alterations to the underlying FHR ‘architecture’ of FHR patterns, supporting the importance of characterizing additional measures of FHRV in the future.
Fetal heart rate patterns during the secondary phase of injury
The 15 min group showed near‐normalization of FHR and measures of FHRV to sham control levels after the end of the latent phase. Nuchal EMG and EEG power also recovered by the end of the latent phase and the spectral edge frequency recovered by 24 h. This combination of findings suggests that the upstream determinants of FHRV quickly recovered in the 15 min group.
By contrast, the 25 min group showed a progressive fall in both FHR and all measures of FHRV during the secondary phase, reaching similar nadirs at 24 h after HI. This period was associated with continued suppression of EEG power, spectral edge frequency and nuchal EMG activity (Fig. 2). Suppression of FHRV may therefore reflect withdrawal of the increased autonomic outflow seen in the latent phase, uncovering the overall reduction in fetal activity. All measures of FHRV showed similar suppression during this time and skewness was no different from control values, suggesting suppression of all FHRV components and frequencies. Our finding of suppression of FHRV during the secondary phase is consistent with findings in term neonates with HIE, who show greater suppression of heart rate variability in association with more severe encephalopathy (Goulding et al. 2017).
Stereotypic evolving seizures were observed during the secondary phase in the 25 min group but not in the 15 min group. At this gestation, seizures are discrete and occur at a rate of around three per hour (Quaedackers et al. 2004; Bennet et al. 2007; Lear et al. 2017). We have previously reported that individual seizures are associated with increased sympathetic activity, leading to intestinal vasoconstriction, hypertension and tachycardia, and also that 75% are associated with nuchal EMG activity (Quaedackers et al. 2004). Fetal seizures at term gestation have also previously been associated with an increase in FHRV (Westgate et al. 1999). We did not specifically investigate the effect of seizures on FHRV in the present study, although our findings most probably indicate profound suppression of FHRV between seizures.
Fetal heart rate variability 72 h after HI
The reduction in FHR and suppression of measures of FHRV in the 25 min group progressively resolved from 24 h onwards, in association with increasing EEG power and nuchal EMG activity. SDNN recovered to control levels by 72 h in the 25 min group. By contrast, RMSSD, STV and LTV remained significantly reduced at 72 h. A decrease in skewness was also observed at this time in the 25 min group, suggesting the FHR trace showed a relative predominance of accelerations, with loss of decelerations. These findings suggest that there may have been specific changes to the components of FHRV, along with an overall effect on the magnitude of FHRV. Future studies will need to assess whether these changes persist beyond 72 h.
Significance and perspectives
The aetiology of brain injury in premature infants is complex and multifactorial (Back, 2015; Gopagondanahalli et al. 2016). A significant proportion of preterm babies are exposed to antenatal infection (Combs et al. 2014). Biomarkers are urgently needed to identify such cases (Lear et al. 2014a, 2015). Although the timing of HI is poorly understood in preterm babies (Hayakawa et al. 1999), HI encephalopathy has recently been shown to be a significant burden in a large contemporary cohort of 8,334 singleton preterm babies (Manuck et al. 2016). Current understanding focuses on the utility of suppressed heart rate variability to identify HI. This study suggests this is too simplistic because complex patterns of FHRV were observed after HI. Although the secondary phase of injury was associated with prolonged suppression of all measures of FHRV, we found that these measures were either increased or near‐normal during the latent phase even after severe HI.
There are current trials of neuroprotective strategies (e.g. erythropoietin) for preterm babies at risk of HI brain injury (Juul & Pet, 2015). Unfortunately, without an accurate understanding of which biomarkers do and do not provide prognostic information for HI brain injury, it is difficult to reliably identify those who will probably benefit from such strategies. The present study suggests that an improved understanding of the dynamic changes in FHRV after severe HI may aid in the selection of preterm babies for neuroprotective strategies.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
These experiments were conducted in the Fetal Physiology and Neuroscience Group laboratory, at the University of Auckland. CAL, AJG, TI and LB conceived the hypotheses, experimental design and analysis protocols for the study. KY, CAL and LB were responsible for data collection. KY, CAL and MJB performed the physiological analysis. KY drafted the manuscript. KY and CAL contributed equally to this study and qualify as equal first authors. All authors were involved in data interpretation and critically reviewed the manuscript. All authors listed qualify for authorship and approved the final version of the manuscript submitted for publication.
Acknowledgements
The present study was funded by grants from the Health Research Council of New Zealand (grant numbers 12/613, 14/216 and 17/601), the Auckland Medical Research Foundation (grant number 1108004) and New Zealand Lottery Grants Board (grant numbers 209214 and 340855). Christopher Lear was supported by an Auckland Medical Research Foundation Doctoral Scholarship (grant number 1213003).
Biographies
Kyohei Yamaguchi is an obstetrician at Mie University Hospital, Japan. In parallel, he is undertaking PhD studies with Professors Laura Bennet, Alistair Gunn and Tomoaki Ikeda. His goals are to understand how to identify fetuses at risk of injury because of low oxygen levels before birth and whether it is possible to better use the heart rate records to select infants for new brain protective treatments.

Christopher Lear is a Research Fellow with the Fetal Physiology and Neuroscience Group, University of Auckland, New Zealand. His interests include the physiological adaptation to labour and how understanding this fundamental physiology can improve the identification of fetuses at risk of hypoxic‐ischaemic brain injury.
Edited by: Harold Schultz & Janna Morrison
Linked articles This article is highlighted in a Perspectives article by Frasch. To read this article, visit http://doi.org/10.1113/JP275776.
References
- Ayres‐de‐Campos D, Spong CY & Chandraharan E (2015). FIGO consensus guidelines on intrapartum fetal monitoring: cardiotocography. Int J Gynaecol Obstet 131, 13–24. [DOI] [PubMed] [Google Scholar]
- Back SA (2015). Brain injury in the preterm infant: new horizons for pathogenesis and prevention. Pediatr Neurol 53, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher JC, Bell JE, Keeling JW, McIntosh N & Wyatt B (2004). The Scottish perinatal neuropathology study: clinicopathological correlation in early neonatal deaths. Arch Dis Child Fetal Neonatal Ed 89, F399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennet L, Dean JM, Wassink G & Gunn AJ (2007). Differential effects of hypothermia on early and late epileptiform events after severe hypoxia in preterm fetal sheep. J Neurophysiol 97, 572–578. [DOI] [PubMed] [Google Scholar]
- Bennet L, Roelfsema V, Pathipati P, Quaedackers J & Gunn AJ (2006). Relationship between evolving epileptiform activity and delayed loss of mitochondrial activity after asphyxia measured by near‐infrared spectroscopy in preterm fetal sheep. J Physiol 572, 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennet L, Rossenrode S, Gunning MI, Gluckman PD & Gunn AJ (1999). The cardiovascular and cerebrovascular responses of the immature fetal sheep to acute umbilical cord occlusion. J Physiol 517, 247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardes J, Goncalves H, Ayres‐de‐Campos D & Rocha AP (2009). Sex differences in linear and complex fetal heart rate dynamics of normal and acidemic fetuses in the minutes preceding delivery. J Perinat Med 37, 168–176. [DOI] [PubMed] [Google Scholar]
- Buser JR, Maire J, Riddle A, Gong X, Nguyen T, Nelson K, Luo NL, Ren J, Struve J, Sherman LS, Miller SP, Chau V, Hendson G, Ballabh P, Grafe MR & Back SA (2012). Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann Neurol 71, 93–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs CA, Gravett M, Garite TJ, Hickok DE, Lapidus J, Porreco R, Rael J, Grove T, Morgan TK, Clewell W, Miller H, Luthy D, Pereira L, Nageotte M, Robilio PA, Fortunato S, Simhan H, Baxter JK, Amon E, Franco A, Trofatter K, Heyborne K & ProteoGenix/Obstetrix Collaborative Research Network (2014). Amniotic fluid infection, inflammation, and colonization in preterm labor with intact membranes. Am J Obstet Gynecol 210, 125. e121–e115. [DOI] [PubMed] [Google Scholar]
- Dalton KJ, Dawes GS & Patrick JE (1977). Diurnal, respiratory, and other rhythms of fetal heart rate in lambs. Am J Obstet Gynecol 127, 414–424. [DOI] [PubMed] [Google Scholar]
- Dalton KJ, Dawes GS & Patrick JE (1983). The autonomic nervous system and fetal heart rate variability. Am J Obstet Gynecol 146, 456–462. [DOI] [PubMed] [Google Scholar]
- Davidson JO, Quaedackers JS, George SA, Gunn AJ & Bennet L (2011). Maternal dexamethasone and EEG hyperactivity in preterm fetal sheep. J Physiol 589, 3823–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries LS, Benders MJ & Groenendaal F (2015). Progress in neonatal neurology with a focus on neuroimaging in the preterm infant. Neuropediatrics 46, 234–241. [DOI] [PubMed] [Google Scholar]
- de Vries LS, Eken P, Groenendaal F, Rademaker KJ, Hoogervorst B & Bruinse HW (1998). Antenatal onset of haemorrhagic and/or ischaemic lesions in preterm infants: prevalence and associated obstetric variables. Arch Dis Child Fetal Neonatal Ed 78, F51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean JM, Gunn AJ, Wassink G, George S & Bennet L (2006). Endogenous alpha(2)‐adrenergic receptor‐mediated neuroprotection after severe hypoxia in preterm fetal sheep. Neuroscience 142, 615–628. [DOI] [PubMed] [Google Scholar]
- Drury PP, Booth LC, Bennet L, Davidson JO, Wibbens B & Gunn AJ (2013). Dopamine infusion for post‐resuscitation blood pressure support after profound asphyxia in near‐term fetal sheep. Exp Physiol 98, 699–709. [DOI] [PubMed] [Google Scholar]
- Frasch MG, Frank B, Last M & Muller T (2012). Time scales of autonomic information flow in near‐term fetal sheep. Front Physiol 3, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George S, Gunn AJ, Westgate JA, Brabyn C, Guan J & Bennet L (2004). Fetal heart rate variability and brainstem injury after asphyxia in preterm fetal sheep. Am J Physiol Regul Integr Comp Physiol 287, R925–R933. [DOI] [PubMed] [Google Scholar]
- Gopagondanahalli KR, Li J, Fahey MC, Hunt RW, Jenkin G, Miller SL & Malhotra A (2016). Preterm hypoxic‐ischemic encephalopathy. Front Pediatr 4, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulding RM, Stevenson NJ, Murray DM, Livingstone V, Filan PM & Boylan GB (2017). Heart rate variability in hypoxic ischemic encephalopathy during therapeutic hypothermia. Pediatr Res 81, 609–615. [DOI] [PubMed] [Google Scholar]
- Griffin MP & Moorman JR (2001). Toward the early diagnosis of neonatal sepsis and sepsis‐like illness using novel heart rate analysis. Pediatrics 107, 97–104. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn AJ, Bennet L, Gunning MI, Gluckman PD & Gunn TR (1999). Cerebral hypothermia is not neuroprotective when started after postischemic seizures in fetal sheep. Pediatr Res 46, 274–280. [DOI] [PubMed] [Google Scholar]
- Gunn AJ, Gunn TR, de Haan HH, Williams CE & Gluckman PD (1997). Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J Clin Invest 99, 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn AJ, Gunn TR, Gunning MI, Williams CE & Gluckman PD (1998). Neuroprotection with prolonged head cooling started before postischemic seizures in fetal sheep. Pediatrics 102, 1098–1106. [DOI] [PubMed] [Google Scholar]
- Hayakawa F, Okumura A, Kato T, Kuno K & Watanabe K (1999). Determination of timing of brain injury in preterm infants with periventricular leukomalacia with serial neonatal electroencephalography. Pediatrics 104, 1077–1081. [DOI] [PubMed] [Google Scholar]
- Ikeda T, Murata Y, Quilligan EJ, Parer JT, Theunissen IM, Cifuentes P, Doi S & Park SD (1998). Fetal heart rate patterns in postasphyxiated fetal lambs with brain damage. Am J Obstet Gynecol 179, 1329–1337. [DOI] [PubMed] [Google Scholar]
- Jacobs SE, Berg M, Hunt R, Tarnow‐Mordi WO, Inder TE & Davis PG (2013). Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst Rev 1, CD003311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen EC, Bennet L, Guild SJ, Booth LC, Stewart J & Gunn AJ (2009). The role of the neural sympathetic and parasympathetic systems in diurnal and sleep state related cardiovascular rhythms in the late gestation ovine fetus. Am J Physiol Regul Integr Comp Physiol 297, R998–R1008. [DOI] [PubMed] [Google Scholar]
- Juul SE & Pet GC (2015). Erythropoietin and neonatal neuroprotection. Clin Perinatol 42, 469–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keogh MJ, Drury PP, Bennet L, Davidson JO, Mathai S, Gunn ER, Booth LC & Gunn AJ (2012). Limited predictive value of early changes in EEG spectral power for neural injury after asphyxia in preterm fetal sheep. Pediatr Res 71, 345–353. [DOI] [PubMed] [Google Scholar]
- Koome ME, Bennet L, Booth LC, Wassink G, Davidson JO, Gunning M & Gunn AJ (2014). Quantifying the power spectrum of fetal heart rate variability. Exp Physiol 99, 468. [DOI] [PubMed] [Google Scholar]
- Lear CA, Davidson JO, Booth LC, Wassink G, Galinsky R, Drury PP, Fraser M, Bennet L & Gunn AJ (2014a). Biphasic changes in fetal heart rate variability in preterm fetal sheep developing hypotension after acute on chronic lipopolysaccharide exposure. Am J Physiol Regul Integr Comp Physiol 307, R387–R395. [DOI] [PubMed] [Google Scholar]
- Lear CA, Davidson JO, Galinsky R, Yuill CA, Wassink G, Booth LC, Drury PP, Bennet L & Gunn AJ (2015). Subclinical decelerations during developing hypotension in preterm fetal sheep after acute on chronic lipopolysaccharide exposure. Sci Rep 5, 16201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lear CA, Davidson JO, Mackay GR, Drury PP, Galinsky R, Quaedackers JS, Gunn AJ & Bennet L (2017). Antenatal dexamethasone before asphyxia promotes cystic neural injury in preterm fetal sheep by inducing hyperglycemia. J Cereb Blood Flow Metab Jan 1:271678X17703124. 10.1177/0271678X17703124. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lear CA, Galinsky R, Wassink G, Mitchell CJ, Davidson JO, Westgate JA, Bennet L & Gunn AJ (2016). Sympathetic neural activation does not mediate heart rate variability during repeated brief umbilical cord occlusions in near‐term fetal sheep. J Physiol 594, 1265–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lear CA, Koome MM, Davidson JO, Drury PP, Quaedackers JS, Galinsky R, Gunn AJ & Bennet L (2014b). The effects of dexamethasone on post‐asphyxial cerebral oxygenation in the preterm fetal sheep. J Physiol 592, 5493–5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logitharajah P, Rutherford MA & Cowan FM (2009). Hypoxic‐ischemic encephalopathy in preterm infants: antecedent factors, brain imaging, and outcome. Pediatr Res 66, 222–229. [DOI] [PubMed] [Google Scholar]
- Low JA (2004). Reflections on the occurrence and significance of antepartum fetal asphyxia. Best Pract Res Clin Obstet Gynaecol 18, 375–382. [DOI] [PubMed] [Google Scholar]
- Manuck TA, Rice MM, Bailit JL, Grobman WA, Reddy UM, Wapner RJ, Thorp JM, Caritis SN, Prasad M, Tita AT, Saade GR, Sorokin Y, Rouse DJ, Blackwell SC & Tolosa JE (2016). Preterm neonatal morbidity and mortality by gestational age: a contemporary cohort. Am J Obstet Gynecol 215, 103.e101–e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh GH, Baghurst KI, Potter BJ & Hetzel BS (1979). Foetal brain development in the sheep. Neuropathol Appl Neurobiol 5, 103–114. [DOI] [PubMed] [Google Scholar]
- McIntyre S, Blair E, Badawi N, Keogh J & Nelson KB (2013). Antecedents of cerebral palsy and perinatal death in term and late preterm singletons. Obstet Gynecol 122, 869–877. [DOI] [PubMed] [Google Scholar]
- Nussinovitch U, Cohen O, Kaminer K, Ilani J & Nussinovitch N (2012). Evaluating reliability of ultra‐short ECG indices of heart rate variability in diabetes mellitus patients. J Diabetes Complications 26, 450–453. [DOI] [PubMed] [Google Scholar]
- Phelan JP & Ahn MO (1994). Perinatal observations in forty‐eight neurologically impaired term infants. Am J Obstet Gynecol 171, 424–431. [DOI] [PubMed] [Google Scholar]
- Quaedackers JS, Roelfsema V, Heineman E, Gunn AJ & Bennet L (2004). The role of the sympathetic nervous system in post‐asphyxial intestinal hypoperfusion in the preterm sheep fetus. J Physiol 557, 1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JH, Dawes GS & Redman CW (1987). Low human fetal heart rate variation in normal pregnancy. Br J Obstet Gynaecol 94, 656–664. [DOI] [PubMed] [Google Scholar]
- Street P, Dawes GS, Moulden M & Redman CW (1991). Short‐term variation in abnormal antenatal fetal heart rate records. Am J Obstet Gynecol 165, 515–523. [DOI] [PubMed] [Google Scholar]
- Tan S (2014). Fault and blame, insults to the perinatal brain may be remote from time of birth. Clin Perinatol 41, 105–117. [DOI] [PubMed] [Google Scholar]
- Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology (1996). Heart rate variability. Standards of measurement, physiological interpretation, and clinical use. Eur Heart J 17, 354–381. [PubMed] [Google Scholar]
- Thoresen M, Tooley J, Liu X, Jary S, Fleming P, Luyt K, Jain A, Cairns P, Harding D & Sabir H (2013). Time is brain: starting therapeutic hypothermia within three hours after birth improves motor outcome in asphyxiated newborns. Neonatology 104, 228–233. [DOI] [PubMed] [Google Scholar]
- Wassink G, Gunn ER, Drury PP, Bennet L & Gunn AJ (2014). The mechanisms and treatment of asphyxial encephalopathy. Front Neurosci 8, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westgate JA, Bennet L & Gunn AJ (1999). Fetal seizures causing increased heart rate variability during terminal fetal hypoxia. Am J Obstet Gynecol 181, 765–766. [DOI] [PubMed] [Google Scholar]
- Westgate JA, Wibbens B, Bennet L, Wassink G, Parer JT & Gunn AJ (2007). The intrapartum deceleration in center stage: a physiological approach to interpretation of fetal heart rate changes in labor. Am J Obstet Gynecol 197, e1–e11.236. [DOI] [PubMed] [Google Scholar]
- Yawno T, Yan EB, Hirst JJ & Walker DW (2011). Neuroactive steroids induce changes in fetal sheep behavior during normoxic and asphyxic states. Stress 14, 13–22. [DOI] [PubMed] [Google Scholar]
- Yawno T, Yan EB, Walker DW & Hirst JJ (2007). Inhibition of neurosteroid synthesis increases asphyxia‐induced brain injury in the late gestation fetal sheep. Neuroscience 146, 1726–1733. [DOI] [PubMed] [Google Scholar]