

Abstract
Focused on our interest to develop novel antiparasistic agents, the present study was aimed to evaluate the biological activity of an extract of Laurencia johnstonii collected in Baja California Sur, Mexico, against an Acantamoeba castellanii Neff strain. Bioassay-guided fractionation allowed us to identify the amoebicidal diastereoisomers α-bromocuparane (4) and α-isobromocuparane (5). Furthermore, bromination of the inactive laurinterol (1) and isolaurinterol (2) yielded four halogenated derivatives, (6)–(9), which improved the activity of the natural sesquiterpenes. Among them, the most active compound was 3α-bromojohnstane (7), a sesquiterpene derivative which possesses a novel carbon skeleton johnstane.
Keywords: brominated sesquiterpene, marine natural products, Laurencia johnstonii, johnstane, 3-bromojohnstane, anti-amoeboid activity, Acanthamoeba
1. Introduction
Free-living amoeba (FLA) are widely distributed protozoa in the environment [1,2,3]. These parasites present a life cycle with two different stages: the trophozoite and the resistant phase, the cyst. Among FLA, Acanthamoeba genus [4] has been isolated from air, soil, water, contact lenses, air conditioning units, clinical samples, among others [5]. These parasites are able to cause pathologies in humans such as Granulomatous Amoebic Encephalitis (GAE) and Amoebic Keratitis (AK) [1,2,3,5]. Regarding Acanthamoeba infections, an early diagnosis is crucial to achieve a successful treatment [3,6]. Antimicrobial chemotherapy is the most widely used method for treating Acanthamoeba-caused infections. Pentamidine, azoles, sulfonamides, and possibly flucytosine, are among the most frequently used drugs in successfully treated cases of GAE, whereas topical chlorhexidine or polyhexamethylene biguanide appear to be the most effective option in cases of AK [7]. However, the existing therapies are not fully effective against these organisms mainly due to the existence of the cyst phase, and also due to the presence of strains that are resistant to the currently used anti-amoebic drugs [2,3,8].
In the past, natural products have been used for the treatment of different parasitic diseases. Artemisinin, quinine, amphotericin B, and ivermectin are examples of important antiparasitic compounds isolated from plants and microorganisms [9,10]. From 1981 to 2014, 15 small molecules were approved as antiparasitic drugs; among them, two were obtained from natural sources and five are the result of semisynthetic derivatives of natural products [11], however, none of them came from marine sources. Many other natural products of diverse molecular structures have revealed antiparasitic potency in the laboratory and, thus, represent interesting lead structures for the development of new and urgently needed antiparasitic agents.
The genus Laurencia is one of the richest sources of novel secondary metabolites among red algae [12,13,14,15]. Although the chemistry of Laurencia species has been exhaustively investigated, the biological activity of the isolated secondary metabolites has not been studied in a systematic way. Furthermore, Laurencia metabolites have been revealed to possess antiparasitic properties against a number of parasites and their vectors. However, no activity has been previously reported against Acanthamoeba [10,16].
Focused on our interest to develop novel lead structures for the development of antiparasistic agents, the present study was aimed to evaluate the biological activity of an extract of Laurencia johnstonii against an Acanthamoeba castellanii Neff strain. To the best of our knowledge, johnstonol [17] and prepacifenol epoxide [18] are the only sesquiterpenes isolated from specimens of Laurencia johnstonii, both with a chamigrene carbon skeleton. In this paper, we report the isolation of the known brominated metabolites laurinterol (1), isolaurinterol (2), aplysin (3), and the antiparasitic diastereoisomers α-bromocuparane (4) and α-isobromocuparane (5), with cyclolaurane, laurane and cuparane backbones. (Figure 1). In addition, transformation of (1) and (2) has yielded four structural derivatives (6–9) which improved the antiparasitic activity with respect to the natural products.
Figure 1.

Structures of natural sesquiterpenes (1)–(5) isolated from Laurencia jonhstonii.
2. Results and Discussion
2.1. Isolation and Identification of Natural Brominated Sesquiterpenes
Laurencia johnstonii was collected off the coast of the Gulf of California, Mexico. Clean and dry specimens were extracted in ethanol to give a crude extract which showed moderate anti-Acanthamoeba activity with an IC50 value of 125.14 ± 4.5 µg/mL. The bioassay-guided fractionation of the ethanolic extract by Sephadex LH-20 led us to an active fraction (SF3). This fraction was further chromatographed over a silica gel column using a step-gradient from n-hexane to EtOAc to give seven subfractions. The most active was SF3.1 (n-hexane fraction) with an IC50 value of 101.29 ± 0.2 µg/mL. After a separation on a silica gel open column eluted with mixtures of n-hexane:EtOAc, fraction SF3.1 yielded aplysin (3) [19], and the active stereoisomers α-bromocuparane (4) and α-isobromocuparane (5) [20], which showed activity against Acanthamoeba castellanii Neff with IC50 values of 90.68 ± 1.5 and 64.25 ± 1.5 µg/mL, respectively. Additionally, laurinterol (1) [21], and isolaurinterol (2) [22] were isolated from the inactive fraction SF3.2. The NMR, mass spectrometry, and optical rotation data of (1)–(5) were compared with those previously reported in the literature to confirm their structures (Supplementary Materials).
2.2. Derivatization of Laurinterol (1) and Isolaurinterol (2)
Halogenation is a common biosynthetic strategy found in marine organisms as is the case of Laurencia species [23]. Halogen-containing natural products display a wide range of biological activities; therefore, they are interesting structures for medicinal chemistry studies. The presence of halogen substituents in many natural products also enhances their biological activities [24]. In order to evaluate structure−activity relationships of laurinterol and its congeners in antiparasitic assays, bromination of (1) and (2) were carried out to obtain their brominated derivatives. Hence, laurinterol (1) was dissolved in ethyl ether and treated with bromine. The reaction mixture was chromatographed on a silica gel column to obtain the brominated sesquiterpene derivatives (6) and (7) (Figure 2).
Figure 2.

Bromination reaction of natural laurinterol (1) and isolaurinterol (2).
Compound (6) was obtained as a colorless oil. Its molecular formula was established by HR-EI-MS (High-Resolution Electron Impact Mass Spectrometry) analysis as C15H18Br2O by the presence of three molecular ions [M]+ at m/z 371.9711/373.9702/375.9677 (calcd. 371.9724/373.9704/375.9683; ratio 50:100:48), indicative of two bromine atoms and six degrees of unsaturation. The structure of (6) was determined by comparison of its NMR data with those for aplysin (3) (Table 1). The main differences observed in their 1H NMR spectra were the absence of one of the two aromatic singlets of (3), H-8 (δH 6.59), and a deshielded aromatic methyl group H3-15 (δH 2.49) for (6) compared with that of 3 H3-15 (δH 2.30). These values agree with the presence of a bromine atom at C-8 in the aromatic ring as well as with the information provided by mass spectrometry.
Table 1.
1H and 13C NMR data for aplysin (3) and (6) (600 MHz, 150 MHz, CDCl3).
| Position | Aplysin (3) | 6 | ||
|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 1 | 54.3, C | 56.0, C | ||
| 2 | 99.8, C | 100.5, C | ||
| 3 | 46.0, CH | 1.75, ddd (12.8, 6.9, 6.5) | 46.4, CH | 1.79, ddd (13.3, 6.5, 6.5) |
| 4 | 31.1, CH2 | 1.62, m | 31.3, CH2 | 1.65, m |
| 1.15, m | 1.15, m | |||
| 5 | 42.5, CH2 | 1.85, dd (6.4, 12.1) | 42.7, CH2 | 1.86, dd (12.1, 6.2) |
| 1.58, d (6.2) | 1.64, ddd (12.1, 6.2, 6.2) | |||
| 6 | 136.2, C | 136.2, C | ||
| 7 | 158.1, C | 156.0 1, C | ||
| 8 | 110.8, CH | 6.59, s | 105.1, C | |
| 9 | 136.9, C | 136.0, C | ||
| 10 | 113.9, C | 114.1, C | ||
| 11 | 126.5, CH | 7.14, s | 125.4, CH | 7.11, s |
| 12 | 13.0, CH3 | 1.10, d (6.8) | 13.3, CH3 | 1.14, d (6.8) |
| 13 | 23.1, CH3 | 1.30, s | 20.4, CH3 | 1.34, s |
| 14 | 23.3, CH3 | 1.29, s | 23.2, CH3 | 1.32, s |
| 15 | 29.9, CH3 | 2.30, s | 23.5, CH3 | 2.49, s |
1 Chemical shift deduced from the HMBC experiment.
Aplysin (3) was easily obtained after treatment of laurinterol (1) with HBr (Figure 2). The specific rotations for (1) ([α] +17) and 3 ([α] −49) are consistent with those reported in the literature for both compounds (1): [α] +13 and 3: [α] −86, for which the absolute configuration was stablished by X-ray crystallography [21,25]. According to this, and supported by consistent chemical shifts and coupling constants of H3-12, H3-13, H3-14 and H-3 between 6 and aplysin (3) (Table 1), the absolute configurations of 6 are 1S, 2S, and 3S. Caccamese and Rinehart identified 6 as bromoaplysine in 1978 by GC-MS [26]. Later, in 2010, a patented methodology for the extraction of antiobesity sesquiterpenes from Laurencia species [27] refers 6 as 8-bromoaplysin, however no complete NMR data has been reported for this brominated sesquiterpene.
Compound 7 was obtained as a colorless oil. The HREIMS analysis provided four molecular ions [M]+ at m/z 449.8840/451.8800/453.8798/455.8758 (calcd. 449.8829/451.8809/453.8789/455.8768; ratio 26:78:67:26) suggesting a molecular formula C15H17OBr3 and six degrees of unsaturation. The structure of 7 was established based on the analysis of its spectroscopic data and comparison with those of 6. 1D and 2D NMR spectra evidenced three methyl, three methylenes, and two methine groups (one on sp2 carbon and one bearing heteroatom), in addition to five sp2 and two sp3 quaternary carbons. Similar to 6, the pentasubstituted phenyl ring was confirmed by the presence of a singlet at δH 7.14 (H-12) and the methyl group at δH 2.53 (H3-15) in the 1H NMR spectrum (Table 2), and the HMBC correlations from H3-13 (δH 2.54, s) and H3-14 (δH 1.04, s) to a quaternary carbon at δC 92.3 (C-2), and from H3-14 to C-7 (δC 136.3) confirming the presence of the same dihydrofuran ring found in 6. The main difference between both compounds was a COSY spin system from the diasterotopic protons H2-3 (δH 2.42/1.99), sequentially coupled with the bromomethine H-4 (δH 3.84), methylene H2-5 (δH 2.18/1.76), and methylene H2-6 (δH 2.18/1.54). HMBC correlations from H3-14 (δH 1.04) to C-6 (δC 32.8) and H3-13 (δH 2.54) to C-3 (δC 47.6) led to the connection of this substructure within the molecule and thus established the planar structure of the rearranged sesquiterpene 7 as shown in Figure 3.
Table 2.
NMR data for 3α-bromojohnstane (7) (600 MHz, 150 MHz, CDCl3).
| Position | 3α-Bromojohnstane (7) | |
|---|---|---|
| δC, Type | δH (J in Hz) | |
| 1 | 48.1, C | |
| 2 | 92.3, C | |
| 3 | 47.6, CH2 | β: 2.42 dd (3.6, 12.9) |
| α: 1.99 dd (12.6, 12.9) | ||
| 4 | 45.8, CH | 3.84 dddd (3.6, 3.6, 12.3, 12.6) |
| 5 | 34.2, CH2 | β: 2.18 dddd (3.2, 3.5, 3.6, 12.9) |
| α: 1.76 dddd (3.5, 12.3, 12.3, 12.6) | ||
| 6 | 32.8, CH2 | β: 1.54 ddd (3.5, 12.3, 12.9) |
| α: 2.18 ddd (3.2, 3.5, 12.9) | ||
| 7 | 136.3, C | |
| 8 | 155.3, C | |
| 9 | 107.6, C | |
| 10 | 136.6, C | |
| 11 | 115.5, C | |
| 12 | 124.6, CH | 7.14 s |
| 13 | 19.7, CH3 | 1.54 s |
| 14 | 26.7, CH3 | 1.04 s |
| 15 | 23.3, CH3 | 2.53 s |
Figure 3.

(a) Selected COSY and HMBC correlations, (b) key-NOE correlations and (c) 1D-NOE experiments of 3α-bromojohnstane (7).
NOE correlations observed from H3-13 to H3-14 and H-4, and from H-4 to the diastereotopic proton H-3β located all these protons on the same face of the molecule. Since the absolute configuration of laurinterol (1) has been confirmed and configuration at C-1 is conserved in 7, it is possible to establish the absolute configuration of its chiral centers as 1S, 2S, and 4S.
In 1972, Yamada et al. reported the synthesis of 2,3-dimethyl-3-(5-bromo-2-methoxy-4-methylphenyl)-cyclohexane from an anisole derivative as a key intermediate to obtain aplysin (3) and other related sesquiterpenes [28]. Also, synthetic efforts to obtain the sesquiterpene cuparene through radical cyclization exclusively yielded 1-(1,2-dimethylcyclohexyl)-4-methylbenzene [29].
It is noteworthy that, even though chemical investigations on the prolific genus Laurencia have yielded over 500 sesquiterpene metabolites belonging to more than 50 carbon frameworks [16], so far none has been reported to possess that of 7. Thus, for this carbon backbone we propose the name johnstane and the name 3α-bromojohnstane for 7 (Figure 4).
Figure 4.
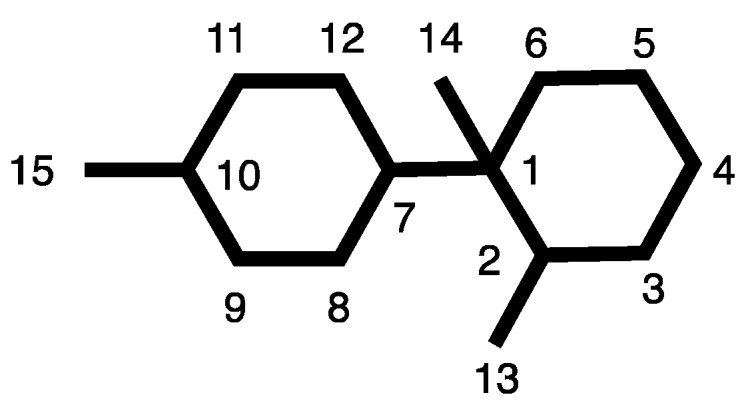
Johnstane carbon skeleton and numbering system.
In the same way, isolaurinterol (2) was dissolved in ethyl ether and treated with bromine to afford 6, and the known sesquiterpenes 8,10-dibromoisoaplysin (8) [30] as well as the 13-hydroxy derivative 9, also obtained after bromination of debromoaplysinol [31].
2.3. Antiparasitic Effect Against Acanthamoeba Castellanii Neff Strain
As well as in the case of natural sesquiterpenes 1–5, the antiamoeboid activity of the brominated sesquiterpene derivatives 6–9 was tested against Acanthamoeba castellanii Neff (Inhibitory Concentrations (IC50) shown in Table 3). Moreover, their toxicity (Cytotoxic Concentrations (CC50) at 24 h, Table 3) against murine macrophages J774.A1 (ATCC # TIB-67) at 24 h was evaluated as previously described [32,33].
Table 3.
Effect of Laurencia johnstonii ethanolic extract and 1–9 against Acanthamoeba castellanii Neff (IC50) and murine macrophage J774.A1 (CC50). * Reference compounds.
| Sample | IC50 (µg/mL) | IC50 (µM) | CC50 (µg/mL) |
|---|---|---|---|
| Crude extract | 125.14 ± 4.5 | n/d | |
| 1 | >100 | 23.65 ± 2.3 | |
| 2 | >100 | 7.25 ± 0.7 | |
| 3 | >100 | 323.69 ± 12.0 | |
| 4 | 90.674 ± 1.529 | 322.41 ± 5.44 | 33.04 ± 4.2 |
| 5 | 64.251 ± 3.492 | 228.46 ± 12.42 | 85.34 ± 10.9 |
| 6 | 24.559 ± 1.105 | 65.64 ± 2.95 | 32.880 ± 3.125 |
| 7 | 18.804 ± 0.198 | 41.51 ± 0.44 | 62.341 ± 2.589 |
| 8 | 22.818 ± 1.896 | 50.37 ± 4.19 | 70.365 ± 3.245 |
| 9 | 29.937 ± 2.918 | 76.74 ± 7.48 | 74.931 ± 2.769 |
| Chlorhexidine * | 1.526 ± 0.45 | 3.02 ± 0.89 | 6.64 ± 0.35 |
| Voriconazole * | 0.33 ± 0.1 | 0.94 ± 0.29 | 2.64 ± 0.27 |
As shown in Table 3, all brominated derivatives (6–9) improved the activity of the natural sesquiterpenes (1–5). Monobrominated compounds in the aromatic ring, 1–3, were inactive, thus suggesting that dibromination of the aromatic moiety is relevant to obtain antiamoeboid activity. On the other hand, natural diastereoisomers 4 and 5, which only differ in the configuration of the bromine atom attached to C-3 of cyclopentane ring, showed a differentiated activity. It is interesting to note that an α-oriented bromine substitution at C-3 in 5 increases the activity against A. castellanii while decreases the toxicity, a structural feature also found in compound 7. 3α-bromojohnstane (7) was the most active molecule against A. castellanii with an IC50 value of 41.51 µM, and one of those with lower toxicity values against murine macrophages among the assayed compounds.
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were measured in CH2Cl2 on a PerkinElmer 241 polarimeter (Waltham, MA, USA), by using a sodium lamp operating at 589 nm. NMR spectra were recorded on a Bruker AVANCE 500 MHz or 600 MHz, as required. NMR spectra were obtained dissolving samples in CDCl3 (99.9%) and chemical shifts are reported relative to solvent (δH 7.26 and δC 77.0 ppm) and TMS as internal pattern. Bruker AVANCE 600 MHz instrument is equipped with a 5 mm TCI inverse detection cryoprobe. Standard Bruker NMR pulse sequences were utilized. HR-ESI-MS (High-Resolution ElectroSpray Ionization Mass Spectrometry) and HR-EI-MS (High-Resolution Electron Impact Mass Spectrometry) data were obtained on a Waters LCT Premier XE Micromass (Manchester, UK) and VG-AutoSpec Micromass (Manchester, UK) spectrometers, respectively. IR spectra were recorded on a Bruker IFS66/S (Ettlingen, Germany) equipped with an ATR accessory using CH2Cl2 solutions. EnSpire® Multimode Reader (Perkin Elmer, Walt, MA, USA) using absorbance values of Alamar Blue® reagent. TLC (Thin layer chromatography) (Merck, Darmstadt, Germany) was visualized by UV light (254 nm) and spraying with cobalt chloride reagent (2% in sulfuric acid, 10%) and heating.
3.2. Biological Material
Laurencia johnstonii was collected by hand during June–July 2015 off the coast of Baja California Sur, Mexico (24°21′10.8′′ N, 110°16′58.8′′ W). A voucher specimen (code 13-003) was deposited at the Herbarium of the Laboratory of Marine Algae of the CICIMAR (Centro Interdisciplinario de Ciencias Marinas, Mexico). This seaweed was identified to genus and species level by Dr. Rafael Riosmena Rodríguez (Universidad Autónoma de Baja California Sur) using taxonomic keys for future reference.
3.3. Extraction and Isolation
Specimens of Laurencia johnstonii were cleaned of sand and epiphytes, and dried. Dried alga was crushed using a blender and extracted with EtOH for three days at 25 °C under gentle agitation. The EtOH was replaced (3 × 1200 mL), and the combined extracts were filtered through a Whatman no. 4 filter paper. Solvent was removed using a rotatory vacuum evaporator. 10.0 g of the resulting extract were chromatographed in Sephadex LH-20 (Sigma, St. Louis, MO, USA) (500 × 70 mm, CH3OH, 100%) to obtain five fractions. The active fraction SF3 was separated in flash Silicagel 0.2–0.5 mm (Sigma-Aldrich, St. Louis, MO, USA) (130 × 70 mm) using a stepwise gradient of n-hexane: ethyl acetate to obtain 7 fractions. Fractions SF3.1 (100% n-hexane) and SF3.2 (95% n-hexane) were separated on a normal phase open column (Silicagel, 0.2–0.5 mm, 300 × 50 mm, using a stepwise gradient of n-hexane: ethyl acetate) to yield pure compounds 5 (10.5 mg), 4 (7.2 mg) and 3 (45.9 mg) from SF3.1 and compounds 3 (10.2 mg), 2 (8.1 mg) and 1 (55.7 mg) from SF3.2. (Supplementary Materials, Scheme S1)
3.3.1. Laurinterol (1)
White crystal; [α] +17 (c 0.15, CH2Cl2); HRESIMS m/z 293.0531 [M − H]− (calc. C15H18O79Br, 293.0541), 295.0518 [M − H]− (calc. C15H18O81Br, 295.0521) 1H NMR (500 MHz, CDCl3) δ 0.55 (1H, dd, J = 7.9, 4.8 Hz, H-12), 0.58 (1H, t, J = 4.6 Hz, H-12), 1.15 (1H, dt, J = 8.1, 4.3 Hz, H-3), 1.28 (1H, m, H-5), 1.32 (3H, s, H-13), 1.41 (3H, s, H-14), 1.66 (1H, dd, J = 12.3, 8.0 Hz, H-4), 1.95 (1H, tdd, J = 12.3, 8.1, 4.4, H-4), 2.09 (1H, dd, J = 13.2, 8.1 Hz, H-5), 2.29 (3H, s, H-15), 5.26 (1H, br, s, 7-OH), 6,61 (1H, s, H-8), 7.61 (1H, s, H-11); 13C NMR (125 MHz, CDCl3) δ 16.2 (C-12), 18.6 (C-13), 22.2 (C-14), 23.5 (C-15), 24.4 (C-3), 25.3 (C-4), 29.6 (C-2), 35.9 (C-5), 114.9 (C-10), 118.8 (C-8), 132.3 (C-11), 134.0 (C-6), 135.9 (C-9), 153.3 (C-7).
3.3.2. Isolaurinterol (2)
Colorless amorphous solid; [α] −46 (c 0.14, CH2Cl2); HRESIMS m/z 293.0536 [M − H]− (calc. C15H18O79Br, 293.0541), 295.0528 [M − H]− (calc. C15H18O81Br, 295.0521) 1H NMR (500 MHz, CDCl3) δ 1.21 (3H, d, J = 7.0 Hz, H-12), 1.42 (1H, ddd, J = 12.8, 8.2, 6.6 Hz, H-5), 1.46 (3H, s, H-14), 1.59 (1H, dt, J = 12.9, 7.1, 7.1 Hz, H-4), 2.05 (1H, ddt J = 12.8, 8.5, 7.0, 7.0 Hz, H-3), 2.20 (1H, ddd, J = 13.0, 8.1 6.7 Hz, H-4), 2.31 (3H, s, H-15), 2.85 (1H, ddt, J = 9.1, 6.9, 6.9, 2.3, 2.3 Hz, H-5), 4.94 (1H, d, J = 2.2 Hz, H-13), 5.11 (1H, d, J = 2.2 Hz, H-13), 5.56 (1H, br, s, 7-OH), 6.73 (1H, s, H-8), 7.45 (1H, s, H-11); 13C NMR (125 MHz, CDCl3) δ 21.2 (C-14), 22.2 (C-15), 27.8 (C-12), 31.2 (C-4), 37.6 (C-3), 39.1 (C-5), 49.8 (C-1), 106.9 (C-13), 115.5 (C-10), 120.4 (C-8), 131.2 (C-11), 132.7 (C-6), 137.2 (C-9), 153.0 (C-7), 165.4 (C-2).
3.3.3. Aplysin (3)
Colorless needles; [α] −49 (c 0.19, CH2Cl2); HRESIMS m/z 293.0551 [M − H]− (calc. C15H18O79Br, 293.0541), 295.0529 [M − H]− (calc. C15H18O81Br, 295.0524). 1H and 13C NMR data (Table 1).
3.3.4. α-Bromocuparane (4)
White amorphous solid; [α] +21 (c 0.19, CH2Cl2); HREIMS [M − H]+ m/z 279.0744 (calc. for C15H2079Br, 279.0748), 282.0723 (calc. for C15H2081Br, 282.0728), [M − Br]+ m/z 201.1640 (calc. for C15H21, 201.1643); 1H NMR (500 MHz, CDCl3) δ 0.62 (3H, s, H-13), 1.09 (3H, s, H-12), 1.43 (3H, s, H-14), 1.96 (1H, m, H-5), 2.24 (1H, m, H-4), 2.27 (1H, m, H-5), 2.32 (3H, s, H-15), 2.52 (1H, ddt, J = 14.3, 9.3, 5.2, H-4), 4.07 (1H, t, J = 9.4 Hz, H-3), 7.09 (4H, m, H-7, H-8, H-10, H-11); 13C NMR (125 MHz, CDCl3) δ 20.7 (C-15), 20.9 (C-13), 22.4 (C-12), 25.0 (C-14), 33.1 (C-4), 36.4 (C-5), 47.4 (C-2), 48.4 (C-1), 62.0 (C-3), 127.3 (C-7, C-11), 128.1 (C-8, C-10), 135.4 (C-9), 143.9 (C-6).
3.3.5. α-Isobromocuparane (5)
Colorless oil; [α] +37 (c 0.14, CH2Cl2); HREIMS [M]+ m/z 280.0826 (calc. for C15H2179Br, 280.0827), 282.0801 (calc. for C15H2181Br, 282.0806), [M − Br]+ m/z 201.1649 (calc. for C15H21, 201.1643); 1H NMR (500 MHz, CDCl3) δ 0.65 (3H, s, H-13), 1.10 (3H, s, H-12), 1.28 (3H, s, H-14), 1.61 (1H, m, H-5), 2.20 (1H, m, H-4), 2.33 (3H, s, H-15), 2.51 (1H, dtd, J = 14.2, 9.5, 6.5 Hz, H-4), 2.71 (1H, td, J = 12.6, 6.4 Hz, H-5), 4.46 (1H, t, J = 9.2 Hz, H-3), 7.12 (1H, d, J = 8.1 Hz, H-10), 7.13 (1H, d, J = 8.1 Hz, H8), 7.27 (2H, d, J = 8.1, H-7, H-11); 13C NMR (125 MHz, CDCl3) δ 20.6 (C-15), 20.8 (C-13), 22.2 (C-12), 25.3 (C-14), 31.2 (C-4), 33.5 (C-5), 47.7 (C-2), 48.4 (C-1), 63.6 (C-3), 126.5 (C-7, C-11), 128.5 (C-8, C-10), 135.4 (C-9), 143.0 (C-6).
3.4. Transformaction of Natural Sesquiterpenes 1 and 2
Laurinterol (1, 8.0 mg, 0.027 mmol) was dissolved in ethyl ether (5 mL) and bromine (100 μL, 1.95 mmol) was added. The reaction was left under magnetic stirring for 15 min at room temperature, after which the solvent was evaporated in vacuo. The reaction mixture was purified on a silica gel 0.2–0.5 mm (Sigma-Aldrich, St. Louis, MO, USA) column (10 Ø × 50 mm) using a step gradient of n-Hex/EtOAc (100:0–95:5) to yield 8-bromoaplysin (6, 1.49 mg, 14.8%) and 7 (3.45 mg, 29.6%). Similarly, isolaurinterol (2, 5.0 mg, 0.017 mmol) was treated with bromine (100 μL, 1.95 mmol) in the same experimental conditions. After evaporation of the solvent, the reaction mixture was purified on a silica gel column (10 Ø × 50 mm; step gradient of n-Hex/EtOAc (100:0–95:5)) to give compound 6 (1.32 mg, 23.5%), 8 (0,38 mg, 6%) and 9 (1.81 mg, 27.3%).
Laurinterol (1, 10.0 mg, 0.034 mmol) was dissolved in ethyl ether (5 mL) and HBr (10 μL, 48%) was added. The reaction was left under magnetic stirring for 15 min at room temperature, after which the solvent was evaporated in vacuo. Quantitative conversion was observed to aplysin (3) which was purified by crystallization in n-hexane.
3.4.1. 8-Bromoaplysin (6)
Colorless oil; [α] −30 (c 0.12, CHCl3); IR υmax 3264, 1652, 1315, 1103, 1022, 904 cm−1; HREIMS [M]+ m/z 371.9711 (calc. for C15H18O79Br2, 371.9724), 373.9702 (calc. for C15H18O79Br81Br, 373.9704), 375.9677 (calc. for C15H18O81Br2, 375.9683); 1H and 13 C NMR data (Table 1).
3.4.2. 3α-Bromojohnstane (7)
Colorless oil; [α] −35 (c 0.19, CHCl3); IR υmax 3259, 2362, 2158, 2031, 1975, 906, 800 cm−1; HREIMS [M]+ m/z 449.8840 (calc. for C15H17O79Br3, 449.8829), 451.8800 (calc. for C15H17O79Br281Br, 451.8809), 453.8798 (calc. for C15H17O79Br81Br2, 453.8789), 455.8758 (calc. for C15H17O81Br3, 455.8768); 1H and 13 C NMR data (Table 2).
3.4.3. 8,10-Dibromoisoaplysin (8)
Colorless oil; [α] −17 (c 0.04, CHCl3); HREIMS [M]+ m/z 449.8836 (calc. for C15H17O79Br3, 449.8829), 451.8815 (calc. for C15H17O79Br281Br, 451.8809), 453.8783 (calc. for C15H17O79Br81Br2, 453.8789), 455.8764 (calc. for C15H17O81Br3, 455.8768).
3.4.4. 8,10-Dibromoaplysinol (9)
Colorless oil; [α] −6 (c 0.08, CHCl3); HREIMS [M]+ m/z 387.9680 (calc. for C15H18O279Br2, 387.9674), 389.9667 (calc. for C15H18O279Br81Br, 389.9653), 391.9639 (calc. for C15H18O281Br2, 391.9633).
3.5. Cell Culture
The amoeba strain used in this study was the type strain: Acanthamoeba castellanii Neff (ATCC 30010) which was axenically grown in PYG medium (0.75% (w/v) proteose peptone, 0.75% (w/v) yeast extract and 1.5% (w/v) glucose) containing 40 μg/mL of gentamicin (Biochrom AG, Cultek, Granollers, Barcelona, Spain).
The murine macrophages J774A.1 (ATCC# TIB-67) cell line was cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum at 37 °C and 5% CO2 atmosphere, was used for the cytotoxicity assays.
3.6. Anti-Acanthamoeba Activity
To evaluate the biological activity of the fractions and molecules, the anti-Acanthamoeba assays were determined using the alamarBlue® method as previously described [15,16]. Firstly, Acanthamoeba strain was seeded in triplicate on a 96-well microtiter plates with 50 μL from a stock solution of 104 cells/mL. Amoebae were left to adhere to the bottom of the well for 15 min, which was checked using a Leika DMIL inverted microscope (Leika, Wetzlar, Germany). After that, 50 μL of serial dilutions of the molecules were added to the wells (1% DMSO was used to dissolve the highest dose of the compounds with no effects on the parasites). As a control, we use chlorhexidine (chlorhexidine digluconate; Alfa Aesar), which is a standard antiseptic belonging to the biguanide family of antiseptics that is commonly used in contact lens maintenance solutions; and voriconazole (Sigma) is an inhibitor of ergosterol synthesis that has been proven previously to be highly effective against clinical strains of Acanthamoeba [26,27]. Finally, the alamarBlue Reagent® (Life Technologies, Madrid, Spain) was placed into each well at an amount equal to 10% of the final volume. Then, plates containing alamarBlue® were incubated for 96 h at 26 °C with a slight agitation.
3.7. Cytotoxicity Test
The cytotoxicity assay was carried out using the alamarBlue® method as previously described [15,16]. Firstly, the macrophages were seeded in triplicate on a 96-well microtiter plates with 50 μL from a stock solution of 2 × 105 cells/mL. Macrophages were left to adhere to the bottom of the well for 15 min, which was checked using a Leika DMIL inverted microscope (Leika, Wetzlar, Germany). After that, 50 μL of serial dilutions of the molecules were added to the wells. Finally, the alamarBlue Reagent® (Life Technologies, Madrid, Spain) was placed into each well at an amount equal to 10% of the final volume. Then, plates containing alamarBlue® were incubated for 24 h at 37 °C in presence of CO2 at 5%.
3.8. Statical Analysis
Briefly, the test plates were analyzed, during an interval of time between 24 and 96 h, on an EnSpire® Multimode Plate Reader (Perkin Elmer, Madrid, Spain) using fluorescence, a test wavelength of 570 nm and a reference wavelength of 630 nm. The percentage of the growth inhibition, 50% inhibitory concentration (IC50 or CC50) was calculated by linear regression analysis with 95% confidence limits using Sigma Plot 12.0 statistical analysis software (Systat Software). All experiments were performed three times, and the mean values were also calculated. A paired two-tailed t-test was used for analysis of the data. Values of p < 0.05 were considered significant.
4. Conclusions
To the best of our knowledge, red algae have never been evaluated as a potential source of amoebicidal agents. Furthermore, this is the first time that the anti-Acantamoeba activity of the brominated sesquiterpenes α-bromocuparane (4) and α-isobromocuparane (5), isolated from Laurencia johnstonii, is reported. On the other hand, after chemical modification of the inactive metabolites laurinterol (1) and isolaurinterol (2), a set of brominated derivatives, 6–9, that substantially improve the activity of the natural sesquiterpenes have been obtained. The addition of a second bromine atom in the aromatic ring of 6–9 seems to be relevant to increase the antiamoeboid activity. Whereas all obtained derivatives possess a laurane-type framework, the most active compound, 3α-bromojohnstane (7), possesses a different rearranged carbon skeleton, johnstane, never found among natural sesquiterpene metabolites isolated from Laurencia species so far. Our results suggest that Laurencia-based brominated sesquiterpenes could be a potential source of novel therapeutic agents against Acanthamoeba in the near future.
Acknowledgments
This research has been supported by Red de Investigation Colaborativa en Enfermedades Tropicales RICET (project no. RD16/0027/0001 of the Program of Redes Temáticas de Investigación Cooperativa). Authors thank funds from: CONACYT fellowship 504923/290666, (S.G.D.), Plan Propio ULL 2018, Agustín de Betancourt Program and Cabildo de Tenerife (A.R.D.M., I.S.), the use of General Research Support Services of University of La Laguna (SEGAI-ULL), and Rafael Riosmena Rodríguez (UABCS) for taxonomic classification of specimens of Laurencia johnstonii.
Supplementary Materials
Supplementary materials can be found at http://www.mdpi.com/1660-3397/16/11/443/s1. Scheme S1: Isolation process of sesquiterpenes 1–5 from Laurencia johnstonii; Figure S1:1H-NMR spectrum for laurinterol (1); Figure S2: 1H-NMR spectrum for isolaurinterol (2); Figure S3: 1H-NMR spectrum for aplysin (3); Figure S4: 1H-NMR spectrum for α-bromocuparane (4); Figure S5: 1H-NMR spectrum for α-isobromocuparane (5); Figure S6: 1H-NMR spectrum of 8-bromoaplysin (6); Figure S7: COSY spectrum of 8-bromoaplysin (6); Figure S8: HSQC-ed spectrum of 8-bromoaplysin (6); Figure S9: HMBC spectrum of 8-bromoaplysin (6); Figure S10: 13C NMR spectrum of 8-bromoaplysin (6); Figure S11: HREIMS spectrum of 8-bromoaplysin (6); Figure S12: 1H NMR spectrum of 3α-bromojohnstane (7); Figure S13: COSY spectrum of 3α-bromojohnstane (7); Figure S14: HSQC-ed spectrum of 3α-bromojohnstane (7); Figure S15: HMBC spectrum of 3α-bromojohnstane (7); Figure S16: 13C NMR spectrum of 3α-bromojohnstane (7); Figure S17: 1D-NOE experiments of 3α-bromojohnstane (7); Figure S18: HREIMS spectrum of 3α-bromojohnstane (7); Figure S19: 1H NMR spectrum of 8,10-dibromoisoaplysin 8; Figure S20: 1H NMR spectrum of 8,10-dibromoaplysinol 9.
Author Contributions
J.J.F., A.R.D.-M., and J.L.-M. conceived and designed the experiments; S.G.-D. and E.V.-V. collected the algae and prepared extracts; S.G.-D. performed isolation, purification and chemical experiments; J.L.-M., M.R.-B. and I.S. designed and performed the anti-Acanthamoeba activity assays and analyzed the activity data; J.E.P contributed to activity analysis tools and discussed activity data; J.J.F. and A.R.D.-M. analyzed the chemical data and wrote the paper. All authors contributed to the final version of the manuscript.
Funding
This research was funded by Ministerio de Economía y Competitividad: grant number CTQ2014-55888-C03-01/R.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Marciano-Cabral F., Cabral G. Acanthamoeba spp. as agents of disease in humans. Clin. Microbiol. Rev. 2003;16:273–307. doi: 10.1128/CMR.16.2.273-307.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siddiqui R., Khan N.A. Biology and pathogenesis of Acanthamoeba. Parasite. Vectors. 2012;5:6. doi: 10.1186/1756-3305-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lorenzo-Morales J., Martín-Navarro C.M., López-Arencibia A., Arnalich-Montiel F., Piñero J.E., Valladares B. Acanthamoeba keratitis: An emerging disease gathering importance worldwide? Trends Parasitol. 2013;29:181–187. doi: 10.1016/j.pt.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 4.Stothard D.R., Schroeder-Diedrich J.M., Awwad M.H., Gast R.J., Ledee D.R., Rodriguez-Zaragoza S., Dean C.L., Fuerst P.A., Byers T.J. he Evolutionary History of the Genus Acanthamoeba and the Identification of Eight New 18S rRNA Gene Sequence Types. J. Eukaryot. Microbiol. 1998;45:45–54. doi: 10.1111/j.1550-7408.1998.tb05068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lorenzo-Morales J., Khan N.A., Walochnik J. An update on Acanthamoeba keratitis: Diagnosis, pathogenesis and treatment. Parasite. 2015;22:10. doi: 10.1051/parasite/2015010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khan N.A. Acanthamoeba: Biology and increasing importance in human health. FEMS Microbiol. Rev. 2006;30:564–595. doi: 10.1111/j.1574-6976.2006.00023.x. [DOI] [PubMed] [Google Scholar]
- 7.Siddiqui R., Aqeel Y., Khan N.A. The development of drugs against Acanthamoeba infections. Antimicrob. Agents Chemother. 2016;60:6441–6450. doi: 10.1128/AAC.00686-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lorenzo-Morales J., Kliescikova J., Martinez-Carretero E., De Pablos L.M., Profotova B., Nohynkova E., Osuna A., Valladares B. Glycogen phosphorylase in Acanthamoeba spp.: Determining the role of the enzyme during the encystment process using RNA interference. Eukaryot. Cell. 2008;7:509–517. doi: 10.1128/EC.00316-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kayser O., Kiderlen A.F., Croft S.L. Natural products as antiparasitic drugs. Parasitol. Res. 2003;90(Suppl. 2):55–62. doi: 10.1007/s00436-002-0768-3. [DOI] [PubMed] [Google Scholar]
- 10.Young R.M., Pesce E.R. Marine Biomedicine from Beach to Bedside. CRC Press; Boca Raton, FL, USA: 2015. Screening for antiparasitic marine natural products. Chapter 9. [DOI] [Google Scholar]
- 11.Newman D.J., Cragg G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016;79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 12.Blunt J.W., Carroll A.R., Copp B.R., Davis R.A., Keyzers R.A., Prinsep M.R. Marine natural products. Nat. Prod. Rep. 2018;35:8–53. doi: 10.1039/C7NP00052A. [DOI] [PubMed] [Google Scholar]
- 13.Souto M.L., Manríquez C.P., Norte M., Leira F., Fernández J.J. The inhibitory effects of squalene-derived triterpenes on protein phosphatase PP2A. Bioorg. Med. Chem. Lett. 2003;13:1261–1264. doi: 10.1016/S0960-894X(03)00136-7. [DOI] [PubMed] [Google Scholar]
- 14.Gutiérrez-Cepeda A., Fernández J.J., Norte M., López-Rodríguez M., Brito I., Muller C.D., Souto M.L. Additional insights into the obtusallene family: components of Laurencia marilzae. J. Nat. Prod. 2016;79:1184–1188. doi: 10.1021/acs.jnatprod.5b01080. [DOI] [PubMed] [Google Scholar]
- 15.Morales-Amador A., de Vera C.R., Márquez-Fernández O., Daranas A.H., Padrón J.M., Fernández J.J., Souto M.L., Norte M. Pinnatifidenyne-derived ethynyl oxirane acetogenins from Laurencia viridis. Mar. Drugs. 2018;16 doi: 10.3390/md16010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harizani M., Ioannou E., Roussis V. The Laurencia paradox: An endless source of chemodiversity. Prog. Chem. Org. Nat. Prod. 2016;102:91–252. doi: 10.1007/978-3-319-33172-0_2. [DOI] [PubMed] [Google Scholar]
- 17.Sims J.J., Fenical W., Wing R.M., Radlick P. Marine natural products III. Johnstonol, an unusual halogenated epoxide from the red alga Laurencia johnstonii. Tetrahedron Lett. 1972;13:195–198. doi: 10.1016/S0040-4039(01)84278-7. [DOI] [Google Scholar]
- 18.Faulkner J.D., Stallard M.O., Ireland C. Prepacifenol epoxide, a halogenated sesquiterpene diepoxide. Tetrahedron Lett. 1974;15:3571–3574. doi: 10.1016/S0040-4039(01)91969-0. [DOI] [Google Scholar]
- 19.Yamamura S., Hirata Y. Structures of aplysin and aplysinol, naturally ocurring bromo-compounds. Tetrahedron. 1963;19:1485–1496. doi: 10.1016/S0040-4020(01)99222-1. [DOI] [Google Scholar]
- 20.Suzuki T., Suzuki M., Kuroswa E. α-bromocuparene and α-isobromocuparene, new bromo compounds from Laurencia species. Tetrahedron. 1975;35:3057–3058. doi: 10.1016/S0040-4039(00)75072-6. [DOI] [Google Scholar]
- 21.Irie T., Suzuki M., Kurosawa E., Masamune T. Laurinterol and debromolaurinterol, constituents from Laurencia intermedia. Tetrahedron Lett. 1966;17:1837–1840. doi: 10.1016/S0040-4039(00)90267-3. [DOI] [Google Scholar]
- 22.Irie T., Suzuki M., Kurosawa E., Masamune T. Laurinterol, debromolaurinterol and isolaurinterol, constituents of Laurencia intermedia Yamada. Tetrahedron. 1970;26:3271–3277. doi: 10.1016/S0040-4020(01)92906-0. [DOI] [Google Scholar]
- 23.Wang B.-G., Gloer J.B., Ji N.-Y., Zhao J.-C. Halogenated Organic Molecules of Rhodomelaceae Origin: Chemistry and Biology. Chem. Rev. 2013;113:3632–3685. doi: 10.1021/cr9002215. [DOI] [PubMed] [Google Scholar]
- 24.Gribble G.W. Biological Activity of Recently Discovered Halogenated Marine Natural Products. Mar. Drugs. 2015;13:4044–4136. doi: 10.3390/md13074044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cameron A.F., Ferguson G., Robertson J.M. The crystal structure and absolute stereochemistry of Laurinterol. The absolute stereochemistry of aplysin. Chem. Commun. 1967:271–272. doi: 10.1039/c19670000271. [DOI] [Google Scholar]
- 26.Erickson K.L. Constituents of Laurencia. In: Scheuer P.J., editor. Marine Natural Products, Chemical and Biological Perspectives. Volume V. Academic Press; New York, NY, USA: 1983. pp. 131–257. in reference to the original publication: Caccamese, S.; Rinehart, K. L. New compounds and activities from Laurencia species. In Drusgs and Food from the Sea. Myth or Reality? Kaul, P.N.; Sindermann, C.J., Eds.; University of Oklahoma: Norman, Oklahoma, 1978, pp. 187–197. [Google Scholar]
- 27.Guo Y., Wang H., Mao S., Zhang X. Extraction method of laurane-type sesquiterpenoids or their derivatives from seaweed of Laurencia and its application as antiobesity agent. CN101628855 A. Faming Zhuanli Shenqing. 2010 Jan 20;
- 28.Yamada K., Yazawa H., Hirata Y. The synthesis of 2,3-dimethyl-3-(5-bromo-2-methoxy-4-methylphenyl)-cyclohexene, a potentially useful intermediate for the syntheses of aplysin and related sesquiterpenes. Bull. Chem. Soc. Jpn. 1972;45:587–590. doi: 10.1246/bcsj.45.587. [DOI] [Google Scholar]
- 29.Srikrishna A., Sundarababu G. Radical cyclization strategies to terpenoids; syntheses of (±)-β-cuparenone, (±)-laurene and epilaurenes. Tetrahedron. 1991;47:481–496. doi: 10.1016/S0040-4020(01)90504-6. [DOI] [Google Scholar]
- 30.Yu X.Q., He W.F., Liu D.Q., Feng M.T., Fang Y., Wang B., Feng L.H., Guo Y.W., Mao S.C. A seco-laurane sesquiterpene and related laurane derivatives from the red alga Laurencia okamurai Yamada. Phytochemistry. 2014;103:162–170. doi: 10.1016/j.phytochem.2014.03.021. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki M., Kurata K., Kurosawa E. The structure of isoaplysin, a brominated rearranged cuparane-type sesquiterpenoid from the red alga Laurencia okamurai Yamada. Bull. Chem. Soc. Jpn. 1986;59:3981–3982. doi: 10.1246/bcsj.59.3981. [DOI] [Google Scholar]
- 32.Martín-Navarro C.M., Lorenzo-Morales J., Cabrera-Serra M.G., Rancel F., Coronado-Álvarez N.M., Piñero J.E., Valladares B. The potential pathogencity of chlorhexidine-sensitve Acanthamoeba strains isolates from contact lens from asyntomatic individuals in Tenerife, Canary Islands, Spain. J. Med. Microbiol. 2008;57:1399–1404. doi: 10.1099/jmm.0.2008/003459-0. [DOI] [PubMed] [Google Scholar]
- 33.McBride J., Ingram P.R., Henríquez F.L. Development of colorimetric microtiter plate assay for assesment of antiicrobials against Acanthamoeba. J. Clin. Microbiol. 2005;43:629–634. doi: 10.1128/JCM.43.2.629-634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.