

Abstract
Aim
Prior to the discontinuation of bococizumab's clinical development, it was considered advantageous to develop an infrequent dosing regimen (eg, monthly). Therefore, we conducted a phase 1 study to evaluate the pharmacokinetics, pharmacodynamics, and safety of bococizumab when administered in co‐mixture with recombinant human hyaluronidase (rHuPH20).
Method
Healthy subjects (N = 60) were randomized equally among 4 groups that received a single subcutaneous dose of either bococizumab 150, 300, or 450 mg co‐mixed with rHuPH20 or bococizumab 300 mg alone. Bioavailability and lipid‐lowering effect of bococizumab were evaluated by using ANCOVA models.
Results
In the groups administered bococizumab co‐mixed with rHuPH20, dose‐normalized C max and AUCinf were 26.6 to 39.1% and 18.3 to 36.6% greater, respectively, compared with bococizumab 300 mg alone. Despite these increases, mean percent reductions from baseline in low‐density lipoprotein cholesterol were smaller in the bococizumab 300 mg + rHuPH20 group than in the bococizumab 300‐mg group at Day 21 (52.2% and 59.5%, respectively) and were similar at Day 29 (51.7% and 49.6%, respectively). Compared with the group administered bococizumab 300 mg alone, the bococizumab 300 mg + rHuPH20 group did not show a significantly altered AUEC85 (ratio of adjusted means: 102.5%, 90% confidence interval: 96.1‐109.3%) but did show a higher MaxELDL‐C (ratio of adjusted means: 125.4%, 90% confidence interval: 103.3‐152.2%), indicating diminution of efficacy. The most frequent adverse events were injection‐site erythema, injection‐site bruising, and nasopharyngitis; all injection‐site adverse events were mild.
Conclusion
Co‐mixture with rHuPH20 increased the bioavailability of bococizumab without proportional increase in pharmacodynamic effect.
Trial Registration
ClinicalTrials.gov, NCT02667223.
Keywords: bococizumab, hyaluronidase, hypercholesterolemia, PCSK9 inhibitor, pharmacodynamics, pharmacokinetics
Short abstract
Aim Prior to the discontinuation of bococizumab's clinical development, it was considered advantageous to develop an infrequent dosing regimen (eg, monthly). Therefore, we conducted a phase 1 study to evaluate the pharmacokinetics, pharmacodynamics, and safety of bococizumab when administered in co‐mixture with recombinant human hyaluronidase (rHuPH20).
Methods Healthy subjects (N = 60) were randomized equally among 4 groups that received a single subcutaneous dose of either bococizumab 150, 300, or 450 mg co‐mixed with rHuPH20 or bococizumab 300 mg alone. Bioavailability and lipid‐lowering effect of bococizumab were evaluated by using ANCOVA models.
Results In the groups administered bococizumab co‐mixed with rHuPH20, dose‐normalized C max and AUCinf were 26.6 to 39.1% and 18.3 to 36.6% greater, respectively, compared with bococizumab 300 mg alone. Despite these increases, mean percent reductions from baseline in low‐density lipoprotein cholesterol were smaller in the bococizumab 300 mg + rHuPH20 group than in the bococizumab 300‐mg group at Day 21 (52.2% and 59.5%, respectively) and were similar at Day 29 (51.7% and 49.6%, respectively). Compared with the group administered bococizumab 300 mg alone, the bococizumab 300 mg + rHuPH20 group did not show a significantly altered AUEC85 (ratio of adjusted means: 102.5%, 90% confidence interval: 96.1‐109.3%) but did show a higher MaxELDL‐C (ratio of adjusted means: 125.4%, 90% confidence interval: 103.3‐152.2%), indicating diminution of efficacy. The most frequent adverse events were injection‐site erythema, injection‐site bruising, and nasopharyngitis; all injection‐site adverse events were mild.
Conclusion Co‐mixture with rHuPH20 increased the bioavailability of bococizumab without proportional increase in pharmacodynamic effect.
1. INTRODUCTION
Atherosclerotic cardiovascular disease (ASCVD) is the leading cause of death in industrialized countries.1 Epidemiologic studies have shown that hypercholesterolemia, and especially hypercholesterolemia caused by elevated levels of low‐density lipoprotein cholesterol (LDL‐C), is a direct and strong risk factor for ASCVD.2, 3, 4 Despite the widespread availability of effective lipid‐lowering agents, such as statins, a significant proportion of patients at high risk of ASCVD continue to be managed suboptimally due to insufficient therapeutic efficacy, safety concerns regarding higher doses of statins, and poor compliance.5, 6 Therefore, there is a medical need for additional lipid‐lowering options capable of minimizing the residual cardiovascular risk of patients who display a suboptimal response and/or intolerance to current first‐line agents.7
Bococizumab is a humanized monoclonal antibody against proprotein convertase subtilisin/kexin type 9 (PCSK9),8, 9, 10, 11, 12, 13, 14, 15, 16, 17 which is a protein that regulates the availability of the LDL receptor (LDLR) expressed predominantly on hepatocytes. Binding of PCSK9 to the LDLR leads to the receptor's internalization for degradation, which ultimately results in elevated circulating levels of LDL‐C due to a reduction in LDLR‐mediated uptake.18, 19, 20 By binding to PCSK9 and preventing its interaction with the LDLR, bococizumab and other anti‐PCSK9 antibodies are able to prevent LDLR internalization and consequently reduce the level of circulating LDL‐C via enhanced receptor‐mediated clearance. Phase 2 studies showed bococizumab to be well tolerated and capable of providing substantial reductions in fasting LDL‐C levels when administered as single or multiple doses, either alone or against a background of ongoing lipid‐lowering therapy.12, 13, 14 The drug was subsequently investigated in the multinational phase 3 SPIRE (Studies of PCSK9 Inhibition and the Reduction of vascular Events) clinical development program as an add‐on therapy for primary hyperlipidemia and mixed dyslipidemia in patients at high and very high risk of cardiovascular events.15, 16, 17 The dosing regimen investigated in the SPIRE program was a 1.5‐mL subcutaneous injection of bococizumab 150 mg every 2 weeks. From a patient compliance and convenience perspective, however, it was also desirable to develop a less frequent dosing regimen. To achieve this goal, we proposed concurrent delivery of bococizumab with recombinant human hyaluronidase (rHuPH20), which is the 61‐kDa active ingredient in the commercial product Hylenex® recombinant (hyaluronidase human injection) and an enzyme that transiently and locally increases the dispersion and absorption of subcutaneously delivered therapeutic agents via degradation of hyaluronan, which is a major glycosaminoglycan constituent of the extracellular matrix.21, 22 We hypothesized that administering bococizumab in co‐mixture with rHuPH20 would enable delivery of a larger volume in a single subcutaneous injection, thus allowing an increase in dose and potentially improving the bioavailability of bococizumab. We further hypothesized that an increase in the bioavailability of bococizumab or the delivery of a higher dose would, in turn, lead to a greater reduction in LDL‐C of sufficient magnitude to support a dosing interval less frequent than every 2 weeks.
The aims of the current study were to characterize the pharmacokinetic (PK), pharmacodynamic (PD), and safety profiles of a single subcutaneous dose of bococizumab 150, 300, or 450 mg in co‐mixture with rHuPH20, as well as a single subcutaneous dose of bococizumab 300 mg alone, in healthy adult subjects, and to compare the bioavailability and PD effect of bococizumab when administered alone or in co‐mixture with rHuPH20.
2. MATERIALS AND METHODS
2.1. Study design
This was a phase 1, open‐label, single‐dose, randomized, dose‐escalation study in healthy adult volunteers (http://clinicaltrials.gov identifier: NCT02667223). A total of 60 subjects were randomized equally among 4 dosing groups (15 subjects/group) that received bococizumab 150 mg in co‐mixture with 2 kU of rHuPH20 in a total injection volume of 1.2 mL, bococizumab 300 mg in co‐mixture with 4 kU of rHuPH20 in a total injection volume of 2.3 mL, bococizumab 450 mg in co‐mixture with 6 kU of rHuPH20 in a total injection volume of 3.4 mL, or bococizumab 300 mg in a total injection volume of 2 mL. The clinical study protocol also planned to enroll a dosing group of 15 subjects with hypercholesterolemia and on statin therapy, but this group was not recruited at the sponsor's discretion because prohibitive operational challenges were encountered and because the scientific objectives of the study were adequately addressed without the inclusion of this group.
Complete lists of the inclusion and exclusion criteria for this study are included in the supplementary material. The main inclusion criteria were healthy men and women aged 18 to 65 years, total body weight from 60 to 120 kg and body mass index (BMI) ≤35 kg/m2, fasting LDL‐C from 70 to 190 mg/dL at both screening visits and with the 2 screening values within 20% of one another, and fasting triglycerides ≤400 mg/dL at the first screening visit.
2.1.1. Sample size calculations
A total of 15 evaluable subjects in each group were estimated as sufficient to provide 90% confidence intervals (CIs) for intergroup differences of ±0.3795 in the estimated, natural log‐transformed, total plasma exposure to bococizumab (AUCinf) and ±0.3686 in the natural log‐transformed, maximal plasma concentration of bococizumab (C max) with 90% coverage probability. These calculations were based on standard deviation (SD) estimates of 0.525 and 0.510 for AUCinf and C max, respectively, which were obtained from a previous phase 1 study of bococizumab.11 A total of 15 evaluable subjects in each dosing group were also estimated as sufficient to provide 90% CIs for intergroup differences of ±0.0983 in the natural log‐transformed area under the LDL‐C concentration‐time profile from time zero to Day 85 (AUEC85) and of ±0.2024 in the natural log‐transformed maximal reduction in LDL‐C (MaxELDL‐C) with 90% coverage probability. These calculations were based on SD estimates of 0.136 and 0.280 for AUEC85 and MaxELDL‐C, respectively, which were obtained from the previous phase 1 study of bococizumab mentioned above.
2.2. Study conduct
The study protocol and informed consent documentation were reviewed and approved by the independent ethics committee at the participating investigational center, which was the Pfizer clinical research unit (CRU) in Brussels, Belgium. The CRU was responsible for medical and clinical monitoring and, in addition to following all local regulatory requirements, the study was conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and in compliance with all International Conference on Harmonization Good Clinical Practice guidelines. The principal investigator explained the nature, purpose, and risks of the study to each participating subject. A signed and dated informed consent was required before any screening procedures were undertaken.
The initial screening of subjects took place 14 to 28 days prior to dosing, with additional screening procedures performed 7 days before dosing. Enrolled subjects were given a unique electronic data capture and management system identification and, prior to dosing, were also allocated a randomization number that was retained throughout the study. Each subject's randomization number corresponded to a treatment schedule determined by a sponsor‐generated randomization code. Subjects reported to the CRU on Day 0, received a single subcutaneous dose of bococizumab on Day 1, and remained confined through the completion of study procedures on Day 2. The subjects then returned to the CRU as outpatients on Days 3, 4, 5, 6, 8, 15, 22, 29, 36, 43, 50, 57, 64, 71, and 85 for follow‐up safety assessments and collection of blood or urine samples for safety laboratory tests and PK, PD, and immunogenicity evaluations. The first visit of the first subject occurred on February 2, 2016, and the final visit of the final subject took place on July 13, 2016. Clinical laboratory evaluations were performed by DCL Medical Laboratories, Inc. (Indianapolis, IN, USA).
2.3. Study treatment, sampling, and measurement of analytes
2.3.1. Treatment and sampling
Following an overnight fast of at least 12 hours, the subjects were administered the study medication by qualified and trained CRU personnel as a subcutaneous injection within a radius of approximately 10 cm around the center of the abdominal periumbilical region at approximately 08:00 am (±2 h). Blood samples for all assessments were taken prior to dosing on Day 1 and in the morning following a fast of at least 12 hours on subsequent visits. Every effort was made to obtain samples at the exact nominal time relative to dosing, but samples could be taken within a window of ±1 day for the Day 15, 22, 29, 36, 43, 50, 57, 64, and 71 samples and within a window of ±2 days for the Day 85 sample.
2.3.2. Bococizumab analysis
Plasma samples were stored at −70°C until analysis. Bococizumab plasma concentrations were measured by ICON Laboratory Services, Inc. (Whitesboro, NY, USA) by using a validated, sensitive, and specific enzyme‐linked immunosorbent assay, which has been described previously.16 The lower limit of quantification for the assay was 400 ng/mL. Samples with bococizumab concentrations above the upper limit of quantification (12 800 ng/mL) were adequately diluted into the range of quantification. The overall interassay accuracy, expressed as percent relative error, in the measurement of control samples ranged from 0.625 to 8.36%, and the overall interassay precision, expressed as percent coefficient of variation (%CV) of the mean estimated concentrations of control samples, was ≤6.54%.
2.3.3. LDL‐C analysis
Low‐density lipoprotein cholesterol concentrations used to characterize lipid‐lowering effects were obtained by a direct‐measurement assay (not calculated). Blood samples were analyzed for LDL‐C concentrations by using the ADVIA® Chemistry LDL Cholesterol Direct (DLDL) assay (catalogue number: 74028; Siemens Healthineers, Huizingen, Belgium) on the ADVIA Chemistry XPT System (Siemens Healthineers, Huizingen, Belgium) at the Institut de Biologie Clinique (Watermael‐Boitsfort, Belgium). Baseline values for lipid parameters were defined as the average of the values obtained on Day 7 and before bococizumab administration on Day 1.
2.3.4. Analysis of anti‐bococizumab and anti‐rHuPH20 antibodies and nAbs
Titers of bococizumab antidrug antibodies (ADAs) within blood samples were determined by using a semiquantitative electrochemiluminescence immunoassay with a %CV of ≤8.21% for control samples, while anti‐bococizumab neutralizing antibodies (nAbs) were measured by using an immunoassay with a %CV of ≤12.5% for control samples. Analyses of bococizumab ADAs and bococizumab nAbs were provided by ICON Laboratory Services, Inc. (Whitesboro, NY, USA). The methodologies for these assays have been described previously.16 Samples with a bococizumab concentration > 1 μg/mL were not analyzed by using the nAb assay due to potential interference and were instead classified as “inconclusive.” Titers of anti‐rHuPH20 antibodies within blood samples were determined by using a semiquantitative electrochemiluminescence immunoassay with a %CV of ≤16.9% for control samples, while anti‐rHuPH20 nAbs were measured by using a semiquantitative, in vitro hyaluronidase enzymatic immunoassay. Analyses of anti‐rHuPH20 antibodies were provided by Eurofins Pharma Bioanalytics Services US, Inc. (St. Charles, MO, USA), and analyses of anti‐rHuPH20 nAbs were provided by MicroConstants, Inc. (San Diego, CA, USA). The methodologies for these assays have been described previously.23
2.4. Data analysis
2.4.1. PK parameters
Parameters serving as PK endpoints were the AUCinf, C max, median time to Cmax (T max), terminal half‐life (t ½), apparent clearance (CL/F), and apparent volume of distribution (V z/F) of bococizumab. All randomized and dosed subjects who had at least 1 recorded value for these PK parameters were included in the PK analysis population. Plasma samples with bococizumab concentrations below the lower limit of quantification were assigned a value of 0 for the statistical analysis. Dose‐normalized AUCinf, AUClast, and C max were analyzed by using an ANCOVA model with treatment as a fixed effect and BMI as a covariate. Estimates of the adjusted means and corresponding 90% CIs were obtained from the model and exponentiated to provide estimates of the ratios between test and reference groups. Calculation of PK parameters was performed by using an internally validated software system (electronic noncompartmental analysis version 2.2.4), and statistical analysis was performed by using SAS® 9.4.
2.4.2. PD parameters
Parameters serving as PD endpoints were the absolute and percent changes from baseline in LDL‐C at Day 29 following administration of bococizumab, as well as the AUEC85 for LDL‐C. Additional measures included MaxELDL‐C and the time taken to reach MaxELDL‐C (T max,LDL‐C). PD parameters were analyzed by using an ANCOVA model with treatment as a fixed effect, and baseline LDL‐C and BMI as covariates. Estimates of the adjusted means and corresponding 90% CIs were obtained from the model and exponentiated to provide estimates of the ratios between test and reference groups. Calculation of PD parameters and statistical analysis was performed by using SAS 9.4.
2.5. Safety evaluations
Safety evaluations included the incidence and severity of treatment‐emergent adverse events (AEs) and serious AEs (SAEs), the relationship of AEs and SAEs to study medication, and the incidence of abnormal and clinically relevant results from clinical laboratory tests, vital signs measurements, electrocardiograms, and physical examinations. Immunogenicity was assessed by monitoring the incidence and titer of bococizumab ADAs and rHuPH20 ADAs. A tiered ADA‐testing strategy was used: All samples that were positive in the screening assay were confirmed for antibody specificity, with confirmed‐positive samples further characterized for titer and tested in the nAb assay if appropriate.
2.6. Ethics statement
The final study protocol and informed consent documentation were reviewed and approved by the independent ethics committee at the participating investigational center. In addition to following all local regulatory requirements, the study was conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and in compliance with all International Conference on Harmonization Good Clinical Practice guidelines.
3. RESULTS
3.1. Study population
Sixty healthy adult subjects were recruited and randomized equally among the 4 dosing groups (Table 1). All randomized subjects completed the study, and data from all 60 subjects were included in the PK, PD, and safety analyses. The study included more men than women (66.7% vs 33.3%), and most were of white race (91.7%). The mean (SD) age and mean (SD) BMI of all subjects were 41.5 (12.4) years and 25.1 (2.7) kg/m2, respectively. Mean baseline LDL‐C levels ranged from 116.0 to 125.2 mg/dL across the 4 dosing groups.
Table 1.
Baseline demographics
| Variable | Bococizumab 150 mg + rHuPH20 (N = 15) | Bococizumab 300 mg (N = 15) | Bococizumab 300 mg + rHuPH20 (N = 15) | Bococizumab 450 mg + rHuPH20 (N = 15) | Total (N = 60) |
|---|---|---|---|---|---|
| Sex, n (%) | |||||
| Male | 15 (100) | 9 (60.0) | 9 (60.0) | 7 (46.7) | 40 (66.7) |
| Female | 0 (0.0) | 6 (40.0) | 6 (40.0) | 8 (53.3) | 20 (33.3) |
| Age, years | |||||
| Mean (SD) | 46.5 (12.8) | 40.5 (11.7) | 40.2 (13.4) | 38.8 (11.2) | 41.5 (12.4) |
| Range | 26‐64 | 25‐61 | 20‐59 | 20‐56 | 20‐64 |
| Raceb, n (%) | |||||
| White | 13 (86.7) | 14 (93.3) | 15 (100) | 13 (86.7) | 55 (91.7) |
| Black | 1 (6.7) | 1 (6.7) | 0 (0.0) | 1 (6.7) | 3 (5.0) |
| Asian | 1 (6.7) | 0 (0.0) | 0 (0.0) | 1 (6.7) | 2 (3.3) |
| Weight, kg | |||||
| Mean (SD) | 79.3 (8.4) | 78.3 (11.9) | 73.0 (12.5) | 74.6 (13.3) | 76.3 (11.7) |
| Range | 65.5‐96.3 | 62.1‐97.0 | 60.0‐104.6 | 60.6‐104.4 | 60.0‐104.6 |
| Body mass indexa, kg/m2 | |||||
| Mean (SD) | 24.8 (1.9) | 26.0 (2.3) | 24.4 (3.2) | 25.0 (3.1) | 25.1 (2.7) |
| Range | 22.7‐29.1 | 21.1‐30.0 | 20.2‐32.3 | 20.9‐30.2 | 20.2‐32.3 |
| Baseline LDL‐C, mg/dL | |||||
| Mean (SD) | 121.2 (21.49) | 125.2 (22.92) | 121.0 (23.87) | 116.0 (20.98) | Not calculated |
| Range | 86‐158 | 92‐176 | 74‐167 | 73‐140 | 73‐176 |
LDL‐C, low‐density lipoprotein cholesterol; rHuPH20, recombinant human hyaluronidase; SD, standard deviation.
Defined as weight/(height × 0.01)2.
Self‐reported.
3.2. PK profile of bococizumab with and without rHuPH20 co‐mixture
The rate of absorption of bococizumab into the systemic circulation was similar across the 4 dosing groups, with median T max from 4.00 to 5.05 days (Figure 1 and Table 2). The t ½ of bococizumab ranged from 6.59 to 7.77 days across the 4 dosing groups. The CL/F ranged from 0.67 to 0.80 L/day across the 3 dosing groups that received bococizumab co‐mixed with rHuPH20, which was lower than the value observed in the group administered bococizumab 300 mg alone (1.02 L/day). The V z/F ranged from 7.04 to 7.59 L across the dosing groups that received bococizumab co‐mixed with rHuPH20, which was also lower than the value observed in the group administered bococizumab 300 mg alone (10.15 L).
Figure 1.
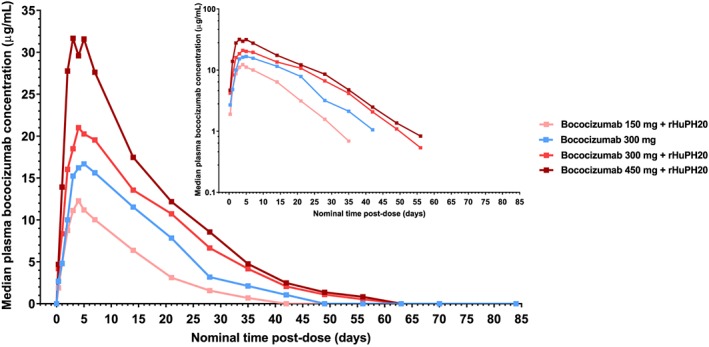
Linear and semilogarithmic (inset) concentration‐time profiles of bococizumab following a single subcutaneous dose of bococizumab alone or in co‐mixture with rHuPH20. rHuPH20, recombinant human hyaluronidase
Table 2.
Descriptive statistical summary of pharmacokinetic parameters following a single subcutaneous dose of bococizumab alone or in co‐mixture with rHuPH20
| Pharmacokinetic Parametera | Bococizumab 150 mg + rHuPH20 (N = 15) | Bococizumab 300 mg (N = 15) | Bococizumab 300 mg + rHuPH20 (N = 15) | Bococizumab 450 mg + rHuPH20 (N = 15) |
|---|---|---|---|---|
| AUCinf, μg. day/mL | 187.5 (29) | 294.6 (37) | 449.0 (40) | 648.2 (40) |
| AUCinf (DN), μg. day/mL/mg | 1.251 (29) | 0.9810 (37) | 1.498 (40) | 1.440 (40) |
| C max, μg/mL | 11.95 (27) | 16.54 (29) | 22.66 (32) | 32.58 (34) |
| C max (DN), μg/mL/mg | 0.07958 (27) | 0.05514 (29) | 0.07553 (32) | 0.07238 (34) |
| T max, days | 4.05 (1.99‐6.98) | 4.06 (2.94‐7.04) | 5.05 (2.97‐7.02) | 4.00 (2.96‐6.99) |
| t ½, days | 6.589 ± 1.099 | 6.952 ± 0.7996 | 7.342 ± 0.8102 | 7.765 ± 1.848 |
| CL/F, L/day | 0.7997 (29) | 1.018 (37) | 0.6681 (40) | 0.6938 (40) |
| V z/F, L | 7.504 (28) | 10.15 (31) | 7.035 (39) | 7.593 (43) |
AUCinf, area under the plasma concentration‐time profile from time zero extrapolated to infinite time; CL/F, apparent clearance; C max, maximum plasma concentration; DN; dose‐normalized to a 300‐mg dose; rHuPH20, recombinant human hyaluronidase; T max, time to C max; V z/F, apparent volume of distribution; t ½, terminal half‐life.
Data are presented as geometric mean (% coefficient of variation), except T max data which are median (range) and t ½ data which are arithmetic mean ± standard deviation.
There was a dose‐dependent increase in total plasma exposure to bococizumab across the dosing groups: Dose‐normalized AUCinf showed a slightly supraproportional increase across the dosing groups that received bococizumab in co‐mixture with rHuPH20, whereas dose‐normalized C max showed a slightly subproportional increase across these groups (Table 2). Based on the dose‐normalized AUCinf and C max values, plasma exposure to bococizumab was higher in the dosing groups that received bococizumab in co‐mixture with rHuPH20 compared with the group administered bococizumab 300 mg alone.
Model‐based estimates of the ratios between test and reference dosing groups in dose‐normalized AUCinf, AUClast, and C max are shown in Table 3. Relative to the dosing group administered bococizumab 300 mg alone (the reference group), the ratios (90% CI) for dose‐normalized AUCinf in the groups that received bococizumab 150, 300, or 450 mg in co‐mixture with rHuPH20 (the test groups) were 118.3% (97.9‐143.0%), 136.6% (112.8‐165.5%), and 136.4% (112.9‐164.8%), respectively, while the ratios for dose‐normalized C max were 139.1% (116.7‐166.0%), 129.8% (108.6‐155.1%), and 126.6% (106.2‐151.0%), respectively. Ratios for AUClast were similar to those for AUCinf.
Table 3.
Comparison of the dose‐normalized pharmacokinetic parameters of bococizumab when administered alone or in co‐mixture with rHuPH20
| Comparison and Pharmacokinetic Parameters | Testa | Referencea | Test/Reference Ratio, % (90% Confidence Interval) |
|---|---|---|---|
|
Test: Bococizumab 150 mg + rHuPH20 Reference: Bococizumab 300 mg | |||
| AUCinf (DN), μg. day/mL/mg | 1.238 | 1.047 | 118.32 (97.93‐142.95) |
| AUClast (DN), μg. day/mL/mg | 1.197 | 1.023 | 117.03 (96.59‐141.81) |
| C max (DN), μg/mL/mg | 0.07919 | 0.05691 | 139.14 (116.67‐165.95) |
|
Test: bococizumab 300 mg + rHuPH20 Reference: bococizumab 300 mg | |||
| AUCinf (DN), μg. day/mL/mg | 1.430 | 1.047 | 136.64 (112.81‐165.50) |
| AUClast (DN), μg. day/mL/mg | 1.405 | 1.023 | 137.37 (113.09‐166.86) |
| C max (DN), μg/mL/mg | 0.07385 | 0.05691 | 129.76 (108.55‐155.11) |
|
Test: bococizumab 450 mg + rHuPH20 Reference: bococizumab 300 mg | |||
| AUCinf (DN), μg. day/mL/mg | 1.428 | 1.047 | 136.42 (112.92‐164.81) |
| AUClast (DN), μg. day/mL/mg | 1.407 | 1.023 | 137.56 (113.54‐166.67) |
| C max (DN), μg/mL/mg | 0.07208 | 0.05691 | 126.64 (106.19‐151.03) |
AUCinf, area under the plasma concentration‐time profile from time zero extrapolated to infinite time; AUClast, area under the plasma concentration‐time profile from time zero to the time of the last quantifiable concentration; C max, maximum plasma concentration; DN; dose‐normalized to a 300‐mg dose; rHuPH20, recombinant human hyaluronidase.
Data are presented as natural log‐transformed, adjusted geometric means obtained from an ANCOVA model with body mass index as a covariate.
3.3. PD effect of bococizumab with and without rHuPH20 co‐mixture
The time at which the reduction in LDL‐C was maximal (T max,LDL‐C) was approximately 21 days in the 3 dosing groups that received bococizumab 150 or 300 mg, while it was 28 days in the group administered bococizumab 450 mg + rHuPH20 (Figure 2). At Day 21, the group administered bococizumab 300 mg alone showed a greater mean percent reduction in LDL‐C from baseline compared with the bococizumab 300 mg + rHuPH20 group (59.5% vs 52.2%, respectively; Table 4), but the mean percent reductions observed in these 2 groups at Day 29 were similar (49.6% vs 51.7%, respectively).
Figure 2.
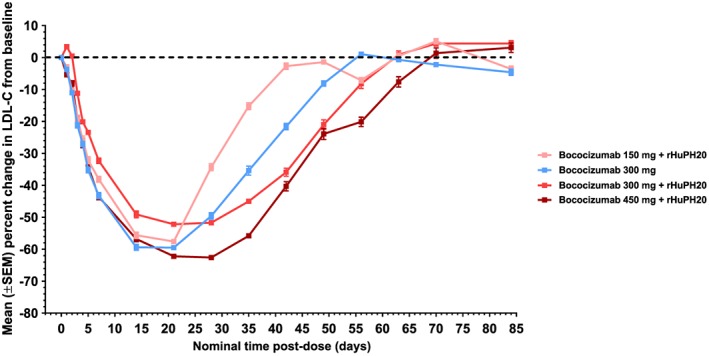
Time course of percent change in LDL‐C from baseline following a single subcutaneous dose of bococizumab alone or in co‐mixture with rHuPH20. LDL‐C, low‐density lipoprotein cholesterol; rHuPH20, recombinant human hyaluronidase; SEM, standard error of the mean
Table 4.
Descriptive statistical summary of pharmacodynamic parameters following a single subcutaneous dose of bococizumab alone or in co‐mixture with rHuPH20
| Pharmacodynamic Parametera | Bococizumab 150 mg + rHuPH20 (N = 15) | Bococizumab 300 mg (N = 15) | Bococizumab 300 mg + rHuPH20 (N = 15) | Bococizumab 450 mg + rHuPH20 (N = 15) |
|---|---|---|---|---|
| Change in LDL‐C From Baseline at Day 21 | ||||
| Absolute, mg/dL | −69.47 (17.02) | −74.77 (21.75) | −63.50 (21.78) | −72.23 (17.06) |
| Percent | −57.6 (11.47) | −59.5 (10.05) | −52.2 (11.38) | −62.2 (10.04) |
| Change in LDL‐C from baseline at Day 29 | ||||
| Absolute, mg/dL | −41.53 (19.50) | −62.63 (24.09) | −62.83 (20.19) | −72.63 (16.67) |
| Percent | −34.3 (16.19) | −49.6 (15.28) | −51.7 (11.29) | −62.6 (8.81) |
| AUEC85 (absolute), mg. Day/dL | 8,447.48 (22) | 7,984.11 (18) | 7,837.87 (26) | 6,698.05 (20) |
| MaxELDL‐C (absolute), mg/dL | 47.21 (35) | 42.80 (35) | 52.73 (30) | 40.27 (32) |
| T max,LDL‐C (absolute), day | 20.94 (13.9–21.0) | 20.95 (13.9–35.0) | 21.00 (14.0–42.0) | 27.95 (20.9–41.9) |
AUEClast, area under the LDL‐C concentration‐time profile from time zero to Day 85; LDL‐C, low‐density lipoprotein cholesterol; MaxELDL‐C, maximum reduction in LDL‐C; rHuPH20, recombinant human hyaluronidase; T max,LDL‐C, time to MaxELDL‐C.
Data for changes in LDL‐C from baseline are presented as arithmetic mean (standard deviation), data for MaxELDL‐C and AUEC85 are arithmetic mean (percent coefficient of variation), and data for T max,LDL‐C are median (range).
Ratios of the exponentiated means for AUEC85 and MaxELDL‐C between test and reference groups are shown in Table 5. The dosing group administered bococizumab 300 mg + rHuPH20 did not show a significantly altered AUEC85 (ratio of adjusted means: 102.5%, 90% CI: 96.1‐109.3%) but did show a higher MaxELDL‐C, indicating a diminution of efficacy in reducing LDL‐C compared with the group administered bococizumab 300 mg alone (ratio of adjusted means: 125.4%, 90% CI: 103.3‐152.2%).
Table 5.
Comparison of the pharmacodynamic parameters of bococizumab 300 mg when administered alone or in co‐mixture with rHuPH20
| Pharmacodynamic Parameter | Bococizumab 300 mg + rHuPH20a | Bococizumab 300 mga | Ratio of Adjusted Means, % (90% Confidence Interval) |
|---|---|---|---|
| AUEC85, mg. day/dL | 7822.75 | 7634.94 | 102.46 (96.08‐109.26) |
| MaxELDL‐C, mg/dL | 50.80 | 40.52 | 125.37 (103.27‐152.20) |
AUEC85, area under the LDL‐C concentration‐time profile from time zero to Day 85; LDL‐C, low‐density lipoprotein cholesterol; MaxELDL‐C, maximum reduction in LDL‐C; rHuPH20, recombinant human hyaluronidase.
Data are natural log‐transformed, adjusted geometric means obtained from an ANCOVA model with baseline LDL‐C and body mass index as covariates.
3.4. Safety
There were no deaths, severe AEs, or discontinuations due to an AE in this study (Table 6). Two SAEs of “arthralgia” and “condition aggravated,” both of which were considered to be unrelated to study medication by the investigator, were reported in one subject in the bococizumab 300‐mg group. Overall, AEs were reported most frequently in subjects receiving bococizumab 450 mg + rHuPH20 (14/15 subjects; 93.3%) and least frequently in subjects receiving bococizumab 150 mg + rHuPH20 (8/15 subjects; 53.3%). Most (121/147; 82.3%) of the reported AEs were mild in severity, with the remaining 26 categorized as moderate. Most (99/147; 67.3%) of the treatment‐emergent AEs were considered treatment‐related by the investigator. Overall, the most frequently reported AEs were injection‐site erythema (21/60 subjects; 35.0%), injection‐site bruising (14/60 subjects; 23.3%), and nasopharyngitis (14/60 subjects; 23.3%). The most frequently reported AE within a single dosing group was injection‐site erythema, which occurred in 10/15 subjects (66.7%) in both the bococizumab 300 mg + rHuPH20 and bococizumab 450 mg + rHuPH20 dosing groups. All injection‐site AEs were considered mild in severity, and their overall incidence was highest in the group receiving bococizumab 450 mg + rHuPH20 (13/15 subjects; 86.7%). There were no results from laboratory tests, vital signs measurements, electrocardiograms, or physical examinations that were considered clinically significant or reported as AEs by the investigator.
Table 6.
Summary of safety data (treatment‐emergent, all‐cause adverse events)
| Variablea | Bococizumab 150 mg + rHuPH20 (N = 15) | Bococizumab 300 mg (N = 15) | Bococizumab 300 mg + rHuPH20 (N = 15) | Bococizumab 450 mg + rHuPH20 (N = 15) |
|---|---|---|---|---|
| Number of deaths | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Subjects experiencing a serious adverse event | 0 (0.0) | 0 (0.0)b | 0 (0.0) | 0 (0.0) |
| Subjects experiencing a severe adverse event | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Total number of adverse events | 21 | 41 | 43 | 42 |
| Number of subjects experiencing an adverse event | 8 (53.3) | 12 (80.0) | 12 (80.0) | 14 (93.3) |
| Subjects discontinuing treatment due to adverse events | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Most frequent adverse events (≥2 subjects in any dosing group) | ||||
| Injection site erythema | 0 (0.0) | 1 (6.7) | 10 (66.7) | 10 (66.7) |
| Injection site bruising | 1 (6.7) | 3 (20.0) | 2 (13.3) | 8 (53.3) |
| Nasopharyngitis | 3 (20.0) | 5 (33.3) | 3 (20.0) | 3 (20.0) |
| Headache | 0 (0.0) | 7 (46.7) | 2 (13.3) | 0 (0.0) |
| Injection site pain | 2 (13.3) | 3 (20.0) | 2 (13.3) | 0 (0.0) |
| Injection site pruritus | 0 (0.0) | 3 (20.0) | 1 (6.7) | 3 (20.0) |
| Injection site swelling | 0 (0.0) | 0 (0.0) | 3 (20.0) | 3 (20.0) |
| Diarrhea | 2 (13.3) | 0 (0.0) | 2 (13.3) | 0 (0.0) |
| Vomiting | 1 (6.7) | 2 (13.3) | 0 (0.0) | 0 (0.0) |
| Abdominal pain | 2 (13.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Back pain | 0 (0.0) | 0 (0.0) | 2 (13.3) | 0 (0.0) |
| Dry skin | 0 (0.0) | 0 (0.0) | 2 (13.3) | 0 (0.0) |
Data are n (%).
A 36‐year‐old man in the bococizumab 300‐mg group reported 2 nontreatment‐emergent serious adverse events (SAEs) of “arthralgia” and “condition aggravated” (investigator reported terms: worsening knee pain due to previous meniscus tear). The subject had experienced intermittent knee pain since a knee injury more than 3 months prior to the administration of study medication and went on to have an arthroscopic meniscus repair. Both SAEs were considered unrelated to study medication by the investigator.
3.4.1. Immunogenicity
A total of 13 subjects (21.7% of those exposed) developed bococizumab ADAs (4 [26.7%] in the bococizumab 300 mg group, 6 [40.0%] in the bococizumab 300 mg + rHuPH20 group, and 3 [20.0%] in the bococizumab 450 mg + rHuPH20 group). Of the subjects with detectable ADAs, 7 had nAbs (3 [20.0%] in the bococizumab 300‐mg group and 4 [26.7%] in the bococizumab 300 mg + rHuPH20 group; 11.7% of the total number of subjects exposed to bococizumab). There were 4 subjects (8.9% of those exposed) who developed anti‐rHuPH20 antibodies (2 [13.3%] each in the bococizumab 300 mg + rHuPH20 and bococizumab 450 mg + rHuPH20 groups), with none of these found to be neutralizing. No subjects in the bococizumab 150 mg + rHuPH20 group developed bococizumab ADAs or anti‐rHuPH20 antibodies. The small numbers of ADA‐positive and nAb‐positive subjects precluded meaningful comparisons with the antibody‐negative subjects, but the AEs observed did not suggest any marked imbalance between these groups for either anti‐bococizumab or anti‐rHuPH20 antibodies.
4. DISCUSSION
Our data demonstrate that the subcutaneous administration of bococizumab in co‐mixture with rHuPH20 increased the bioavailability (both Cmax and AUCinf) of bococizumab compared with the administration of bococizumab alone. Despite the observed increase in bioavailability, however, co‐mixture with rHuPH20 did not enhance the lipid‐lowering PD effect of a 150‐ or 300‐mg dose of bococizumab by an extent considered sufficient to support a dosing regimen less frequent than every 2 weeks. The reason why the increase in bioavailability of bococizumab did not translate into an enhanced LDL‐C response is not known at this time. Comparing the time course profiles of LDL‐C reductions in the two 300‐mg dosing groups showed that co‐mixture with rHuPH20 appeared to reduce the nadir of LDL‐C reduction but prolong the duration of effect. Our data also show that a larger‐volume, 450‐mg dose of bococizumab co‐mixed with rHuPH20 was tolerable and reduced LDL‐C by 62.6% at Day 29 following a single injection. A similar magnitude and duration of LDL‐C response was observed following the administration of a 420‐mg dose of evolocumab when given as 3 separate 1‐mL subcutaneous injections,24 which is a dosing method that would seem suboptimal with regard to patient compliance.
Prior simulations using a population PK/PD model predicted that, following a 300‐mg dose, an increase in the bioavailability of bococizumab of approximately 30% or greater would result in an LDL‐C reduction of approximately 65% at Day 29 (our unpublished data). Based on the AUC ratios we observed, rHuPH20 increased the bioavailability of bococizumab by approximately 18 to 37% across the dose range studied, with the bococizumab 300 mg + rHuPH20 dosing group displaying a 36.6% increase. This did not translate into an enhanced reduction in LDL‐C, however, with the observed mean percent reduction from baseline in LDL‐C at Day 29 being approximately 50% for both the bococizumab 300 mg and bococizumab 300 mg + rHuPH20 groups. The extent of LDL‐C reduction we observed in the bococizumab 150 mg + rHuPH20 dosing group of the current study was similar to data from other phase 1, single‐dose studies of bococizumab alone.9, 11 A study investigating 3 different injection sites reported LDL‐C reductions from baseline of 30 to 35% at Day 29 following a single subcutaneous dose of bococizumab 150 mg,11 whereas we observed a reduction of 34.3% in our bococizumab 150 mg + rHuPH20 dosing group. A similar LDL‐C reduction of approximately 30% was reported in another single‐dose phase 1 study that included a slightly higher subcutaneous bococizumab dose of 200 mg.9 The dosing group that received bococizumab 450 mg + rHuPH20 also showed increased bioavailability, but the magnitude and duration of the LDL‐C response observed were consistent with what could be expected for this size of dose without rHuPH20 co‐mixture, which indicated that the observed effect on PD parameters was not due to enhanced bioavailability.
The safety profile of bococizumab in the current study was consistent with that seen in previous studies and showed that a co‐mixture of bococizumab and rHuPH20 was generally safe and well tolerated by healthy adults.9, 11, 12, 13, 14, 16, 17 Overall, the most frequently reported AEs were injection‐site erythema, injection‐site bruising, and nasopharyngitis. All injection‐site AEs were considered mild in severity, with their overall incidence being highest in the bococizumab 450 mg + rHuPH20 dosing group. The incidence of ADAs in our study was similar to the rate observed in the phase 1 study of bococizumab that investigated 3 different injection sites: The incidence in this previous study was 30.7% (23/75 subjects),11 while the rate we observed was 21.7%. The incidence of nAbs to bococizumab in the present study was higher than previously observed in the single‐dose study that investigated 3 different injection sites (11.7% vs 5.3%), but it is important to note several differences in methodology: The injection‐site study only included subjects with fasting LDL‐C ≥ 130 mg/dL, only investigated bococizumab administered alone, and only involved a bococizumab dose of 150 mg, which was the dose level with which we observed no incidence of ADA development at all. The dosing groups and the number of subjects who developed ADAs were not of sufficient size to support a meaningful evaluation of the impact of immunogenicity on PK or PD parameters.
Our phase 1 study has some limitations. First, the participants in our study were not hypercholesterolemic and did not receive the study medication against a background of lipid‐lowering therapy. Therefore, the PK/PD behavior of bococizumab in our study may not have fully reflected the situation in patients eligible for treatment with a PCSK9 inhibitor. Second, the participating subjects received only a single dose of study treatment, and this does not reflect the PK, PD, or safety profiles that a chronic dosing paradigm would induce. Third, our results may not be representative for broader patient groups because the study was conducted at a single European center where more than 90% of the participants were of white race. Fourth, while the aim of including the bococizumab 450 mg + rHuPH20 dosing group was to assess the overall tolerability of this size of subcutaneous injection, one that is usually considered too large for clinical practice, the absence of a dosing group that received bococizumab 450 mg alone prevented a direct comparison and limited interpretation of the impact of rHuPH20 at this dose level.
On November 1, 2016, Pfizer Inc. announced the discontinuation of the global clinical development program for bococizumab.25 This decision was based on the totality of clinical information for bococizumab, as well as the evolving treatment and market landscape for lipid‐lowering agents. Pfizer observed an unanticipated attenuation of the LDL‐C–lowering effect over time, as well as a higher level of immunogenicity and higher rate of injection‐site reactions than shown with the other agents in this class.16 Further analysis of data from the SPIRE trials showed that the extent of LDL‐C reduction at weeks 12 to 14, as well as at study completion, in patients with or without familial hypercholesterolemia (FH; n = 1,578 and n = 15,959, respectively) was similar, that the incidence of bococizumab ADAs was slightly higher in patients with FH compared with trial participants without FH (43.2% vs 35.8%, respectively), and that the risk of a patient with FH experiencing a major adverse cardiovascular event was not significantly different from the risk observed in participants without FH.26
5. CONCLUSION
Co‐mixture of bococizumab with rHuPH20 increased the bioavailability of bococizumab following a single subcutaneous dose. Administration of a 300‐mg dose of bococizumab in co‐mixture with rHuPH20 did not result in enhanced LDL‐C reductions at Day 29, with time‐course profiles suggesting that co‐mixture with rHuPH20 reduced the nadir of LDL‐C reduction but prolonged the duration of effect. A larger‐volume, 450‐mg dose of bococizumab co‐mixed with rHuPH20, was found to be tolerable and had a dose‐proportional effect on LDL‐C. The co‐mixture of bococizumab and rHuPH20 was well tolerated and showed a safety profile that was consistent with previous studies of bococizumab.
FUNDING
This study was sponsored by Pfizer.
CONFLICT OF INTEREST
Almasa Bass and David R. Plowchalk were employees of Pfizer when this study was conducted. Anna Plotka, Catherine Sattler, and Albert M. Kim are employees of Pfizer. Khurshid Mridha is an employee of Science Recruitment Group Ltd, who was a paid consultant to Pfizer in connection with the development of this manuscript. These conflicts of interest did not affect the design, conduct, or reporting of the study.
AUTHOR CONTRIBUTIONS
Conceptualization: Albert M. Kim, David R. Plowchalk
Data curation: Anna Plotka
Formal analysis: Anna Plotka
Methodology: Albert M. Kim, David R. Plowchalk, Anna Plotka
Project administration: Almasa Bass
Supervision: Almasa Bass
Visualization: Almasa Bass, Anna Plotka, Khurshid Mridha, Catherine Sattler, Albert M. Kim, David R. Plowchalk
Writing—original draft preparation: Almasa Bass, Anna Plotka, Khurshid Mridha, Catherine Sattler, Albert M. Kim, David R. Plowchalk
Writing—review and editing: Almasa Bass, Anna Plotka, Khurshid Mridha, Catherine Sattler, Albert M. Kim, David R. Plowchalk
Supporting information
Data S1. Supporting Information
ACKNOWLEDGEMENTS
The authors thank the principal investigator of the study, Ekaterina Tankisheva, MD, PhD, for expert supervision at the Pfizer CRU in Brussels, Belgium, and also thank Fred McCush, MA, for expert bioanalytical support during the study. This study was sponsored by Pfizer. Medical writing support was provided by David Wateridge, PhD, of Engage Scientific Solutions (Horsham, UK) and funded by Pfizer.
Bass A, Plotka A, Mridha K, Sattler C, Kim AM, Plowchalk DR. Pharmacokinetics, pharmacodynamics, and safety of bococizumab, a monoclonal antibody against proprotein convertase subtilisin/kexin type 9, in healthy subjects when administered in co‐mixture with recombinant human hyaluronidase: A phase 1 randomized trial. Health Sci Rep. 2018;1:e61 10.1002/hsr2.61
Almasa Bass and David R. Plowchalk had the denoted affiliations during study conduct.
REFERENCES
- 1. World Health Organization . The top 10 causes of death. January 2017. http://www.who.int/mediacentre/factsheets/fs310/en/ [Accessed April 30, 2018].
- 2. Dawber TR, Moore FE, Mann GV. Coronary heart disease in the Framingham study. Am J Public Health Nations Health. 1957;47:4‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rosengren A, Hawken S, Ounpuu S, et al. Association of psychosocial risk factors with risk of acute myocardial infarction in 11119 cases and 13648 controls from 52 countries (the INTERHEART study): case‐control study. Lancet. 2004;364(9438):953‐962. [DOI] [PubMed] [Google Scholar]
- 4. O'Keefe JH Jr, Cordain L, Harris WH, Moe RM, Vogel R. Optimal low‐density lipoprotein is 50 to 70 mg/dl: lower is better and physiologically normal. J Am Coll Cardiol. 2004;43(11):2142‐2146. [DOI] [PubMed] [Google Scholar]
- 5. Mitchell S, Roso S, Samuel M, Pladevall‐Vila M. Unmet need in the hyperlipidaemia population with high risk of cardiovascular disease: a targeted literature review of observational studies. BMC Cardiovasc Disord. 2016;16:74 10.1186/s12872-016-0241-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Banach M, Stulc T, Dent R, Toth PP. Statin non‐adherence and residual cardiovascular risk: there is need for substantial improvement. Int J Cardiol. 2016;225:184‐196. [DOI] [PubMed] [Google Scholar]
- 7. Lloyd‐Jones DM, Morris PB, Ballantyne CM, et al. 2016 ACC Expert Consensus Decision Pathway on the role of non‐statin therapies for LDL‐cholesterol lowering in the management of atherosclerotic cardiovascular disease risk: a report of the American College of Cardiology Task Force on Clinical Expert Consensus Documents. J Am Coll Cardiol. 2016;68(1):92‐125. [DOI] [PubMed] [Google Scholar]
- 8. Liang H, Chaparro‐Riggers J, Strop P, et al. Proprotein convertase subtilisin/kexin type 9 antagonism reduces low‐density lipoprotein cholesterol in statin‐treated hypercholesterolemic nonhuman primates. J Pharmacol Exp Ther. 2012;340(2):228‐236. [DOI] [PubMed] [Google Scholar]
- 9. Gumbiner B, Udata C, Joh T, et al. The effects of single‐ and multiple‐dose administration of bococizumab (RN316/PF‐04950615), a humanized IgG2Δa monoclonal antibody binding proprotein convertase subtilisin/kexin type 9, in hypercholesterolemic subjects treated with and without atorvastatin: results from four phase I studies. Cardiovasc Ther [Epub ahead of print] Published online October 27,. 2017. 10.1111/1755-5922.12309 [Accessed November 20, 2017];36(1). [DOI] [PubMed] [Google Scholar]
- 10. Udata C, Garzone PD, Gumbiner B, et al. A mechanism‐based pharmacokinetic/pharmacodynamic model for bococizumab, a humanized monoclonal antibody against proprotein convertase subtilisin/kexin type 9, and its application in early clinical development. J Clin Pharmacol. 2017;57(7):855‐864. [DOI] [PubMed] [Google Scholar]
- 11. Wang EQ, Plotka A, Salageanu J, Sattler C, Yunis C. Pharmacokinetics and pharmacodynamics of bococizumab, a monoclonal antibody to PCSK9, after single subcutaneous injection at three sites [NCT 02043301]. Cardiovasc Ther. 2017. Oct;35(5). 10.1111/1755-5922.12278 [DOI] [PubMed] [Google Scholar]
- 12. Fazio S, Robertson DG, Joh T, et al. Effects of 12 weeks of treatment with intravenously administered bococizumab, a humanized monoclonal antibody blocking proprotein convertase subtilisin/kexin type 9, in hypercholesterolemic subjects on high‐dose statin. Cardiovasc Ther [Epub ahead of print] Published online October 23. 2017. 10.1111/1755-5922.12308 [Accessed November 20, 2017];36(1). [DOI] [PubMed] [Google Scholar]
- 13. Ballantyne CM, Neutel J, Cropp A, et al. Results of bococizumab, a monoclonal antibody against proprotein convertase subtilisin/kexin type 9, from a randomized, placebo‐controlled, dose‐ranging study in statin‐treated subjects with hypercholesterolemia. Am J Cardiol. 2015;115(9):1212‐1221. [DOI] [PubMed] [Google Scholar]
- 14. Yokote K, Kanada S, Matsuoka O, et al. Efficacy and safety of bococizumab (RN316/PF‐04950615), a monoclonal antibody against proprotein convertase subtilisin/kexin type 9, in hypercholesterolemic Japanese subjects who are receiving a stable dose of atorvastatin or are treatment‐naive: results from a randomized, Placebo‐controlled, dose‐ranging study. Circ J. 2017;81(10):1496‐1505. [DOI] [PubMed] [Google Scholar]
- 15. Ridker PM, Amarenco P, Brunell R, et al. Evaluating bococizumab, a monoclonal antibody to PCSK9, on lipid levels and clinical events in broad patient groups with and without prior cardiovascular events: Rationale and design of the Studies of PCSK9 Inhibition and the Reduction of vascular Events (SPIRE) Lipid Lowering and SPIRE Cardiovascular Outcomes Trials. Am Heart J. 2016;178:135‐144. [DOI] [PubMed] [Google Scholar]
- 16. Ridker PM, Tardif JC, Amarenco P, et al. Lipid‐reduction variability and antidrug‐antibody formation with bococizumab. N Engl J Med. 2017;376(16):1517‐1526. [DOI] [PubMed] [Google Scholar]
- 17. Ridker PM, Revkin J, Amarenco P, et al. Cardiovascular efficacy and safety of bococizumab in high‐risk patients. N Engl J Med. 2017;376(16):1527‐1539. [DOI] [PubMed] [Google Scholar]
- 18. Seidah NG, Benjannet S, Wickham L, et al. The secretory proprotein convertase neural apoptosis‐regulated convertase 1 (NARC‐1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci U S A. 2003;100(3):928‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maxwell KN, Breslow JL. Adenoviral‐mediated expression of Pcsk9 in mice results in a low‐density lipoprotein receptor knockout phenotype. Proc Natl Acad Sci U S A. 2004;101(18):7100‐7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nassoury N, Blasiole DA, Tebon Oler A, et al. The cellular trafficking of the secretory proprotein convertase PCSK9 and its dependence on the LDLR. Traffic. 2007;8(6):718‐732. [DOI] [PubMed] [Google Scholar]
- 21. Vaughn DE, Yocum RC, Muchmore DB, et al. Accelerated pharmacokinetics and glucodynamics of prandial insulins injected with recombinant human hyaluronidase. Diabetes Technol Ther. 2009;11(6):345‐352. [DOI] [PubMed] [Google Scholar]
- 22. Dychter SS, Harrigan R, Bahn JD, et al. Tolerability and pharmacokinetic properties of ondansetron administered subcutaneously with recombinant human hyaluronidase in minipigs and healthy volunteers. Clin Ther. 2014;36(2):211‐224. [DOI] [PubMed] [Google Scholar]
- 23. Rosengren S, Dychter SS, Printz MA, et al. Clinical immunogenicity of rHuPH20, a hyaluronidase enabling subcutaneous drug administration. AAPS J. 2015;17(5):1144‐1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dias CS, Shaywitz AJ, Wasserman SM, et al. Effects of AMG 145 on low‐density lipoprotein cholesterol levels: results from 2 randomized, double‐blind, placebo‐controlled, ascending‐dose phase 1 studies in healthy volunteers and hypercholesterolemic subjects on statins. J Am Coll Cardiol. 2012;60(19):1888‐1898. [DOI] [PubMed] [Google Scholar]
- 25. Ferri N, Corsini A, Sirtori CR, Ruscica M. Bococizumab for the treatment of hypercholesterolaemia. Expert Opin Biol Ther. 2017;17(2):237‐243. [DOI] [PubMed] [Google Scholar]
- 26. Ridker PM, Rose LM, Kastelein JJP, et al. Cardiovascular event reduction with PCSK9 inhibition among 1578 patients with familial hypercholesterolemia: Results from the SPIRE randomized trials of bococizumab. J Clin Lipidol. 2018. Apr 3. pii: S1933–2874(18)30193–4. 10.1016/j.jacl.2018.03.088. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information