

Abstract
Background
The basic helix-loop-helix transcription factors play important roles in diverse cellular and molecular processes. Comparative functional genomics can provide powerful approaches to draw inferences about gene function and evolution among species. The comprehensive comparison of bHLH gene family in different gramineous plants has not yet been reported.
Results
In this study, a total of 183, 231 and 571 bHLHs were identified in rice, maize and wheat genomes respectively, and 1154 bHLH genes from the three species and Arabidopsis were classified into 36 subfamilies. Of the identified genes, 110 OsbHLHs, 188 ZmbHLHs and 209 TabHLHs with relatively high mRNA abundances were detected in one or more tissues during development, and some of them exhibited tissue-specific expression such as TabHLH454–459, ZmbHLH099–101 and OsbHLH037 in root, TabHLH559–562, − 046, − 047 and ZmbHLH010, − 072, − 226 in leaf, TabHLH216–221, − 333, − 335, − 340 and OsbHLH005, − 141 in inflorescence, TabHLH081, ZmbHLH139 and OsbHLH144 in seed. Forty five, twenty nine and thirty one differentially expressed bHLHs were respectively detected in wheat, maize and rice under drought stresses using RNA-seq technology. Among them, the expressions of TabHLH046, − 047, ZmbHLH097, − 098, OsbHLH006 and − 185 were strongly induced, whereas TabHLH303, − 562, ZmbHLH155, − 154, OsbHLH152 and − 113 showed significant down-regulation. Twenty two TabHLHs were induced after stripe rust infection at 24 h and nine of them were suppressed at 72 hpi, whereas 28 and 6 TabHLHs exhibited obviously down- and up-regulation after powdery mildew attack respectively. Forty one ZmbHLHs were differentially expressed in response to F. verticillioides infection. Twenty two co-expression modules were identified by the WGCNA, some of which were associated with particular tissue types. And GO enrichment analysis for the modules showed that some TabHLHs were involved in the control of several biological processes, such as tapetal PCD, lipid metabolism, iron absorption, stress responses and signal regulation.
Conclusion
The present study identifies the bHLH family in rice, maize and wheat genomes, and detailedly discusses the evolutionary relationships, expression and function of bHLHs. This study provides some novel and detail information about bHLHs, and may facilitate understanding the molecular basis of the plant growth, development and stress physiology.
Electronic supplementary material
The online version of this article (10.1186/s12870-018-1529-5) contains supplementary material, which is available to authorized users.
Keywords: bHLHs, Gramineous crops, Expression regulation, Growth and development, Stress responses
Background
The basic helix-loop-helix (bHLH) transcription factors constitute one of the largest transcription factor families in plants and are involved in a wide and diverse array of biological processes. A series of evidences showed that bHLHs participated in the regulation of plant growth and development including morphogenesis [1–3], iron homeostasis [4], root vascular cell proliferation [5], shoot branching [6], stomatal initiation [7], flowering time [8], pollen, gynoecium and fruit development [9, 10], and grain yield [11]. Previous studies revealed that bHLHs played very important roles in response of plants to abiotic stresses such as drought, salt and cold. AtbHLH068 and OsbHLH148 overexpressing in transgenic Arabidopsis and rice respectively conferred plant tolerance to drought stress via ABA- and JA-mediated signaling pathway [12, 13]. OsbHLH062, OsJAZ9 and OsNINJA formed a transcriptional regulation complex to fine tune the expression of JA-responsive genes involved in salt stress tolerance in rice, such as OsHAK21 [14]. AtICE1, AtICE2, ZmmICE1, TaICE41 and TaICE87, the five MYC-like bHLHs, functioned as key regulators at the upstream of CBF (C-repeat binding factor) transcriptional cascade controlling cold tolerance [15–17]. TabHLH1 can mediate tobacco adaptation to osmotic stress via ABA-dependent pathway [18], and improve tolerance to Pi and N deprivation through transcriptional regulation of phosphate transporter, nitrate transporter and antioxidant enzyme encoding genes [19]. In rice, OsPTF1 overexpression resulted in significantly higher total root length and surface area in response to Pi starvation [20]. And repression of OsIRO2 led to lower mugineic acid family phytosiderophores (MAs) secretion and hypersensitivity to Fe deficiency [21]. Also, plant bHLHs involve in pathogen stress adaptation and resistance development. OsDPF was induced in rice leaves by blast infection, and DPF overexpressing and DPF knockdown rice led to remarkably increased and decreased accumulation of momilactones and phytocassanes, respectively [22]. And wheat bHLH060 overexpression negatively regulated plant resistance to Pseudomonas syringae through jasmonic acid (JA) and ethylene (ET) signaling in transgenic Arabidopsis [23]. Although some functions of bHLHs have been characterized, the biological functions of most plant bHLHs remain unclear, especially in gramineous crops such as rice, maize and wheat.
The bHLHs are characterized by the signature domain which consists of two functionally distinctive regions, the basic and helix-loop-helix (HLH) regions. The basic region located at the N-terminus contains approximately 17 residues, which is typically rich in basic amino acids, and the region with at least five basic amino acids is expected to recognize and bind specific DNA sequence. In the region, Glu-13 and Arg-16 are essential in E-box-binding recognition, and two additional residues His/Lys-9 and Arg-17 provide DNA-binding specificity for G-box, a specific type of E-box [24]. According to the sequence information in the region, plant bHLHs can be divided into two categories: DNA- and non DNA-binders. The HLH region includes two amphipathic α-helices separated by a loop of variable length and sequence, allowing the formation of homodimers or heterodimers. Additionally, the bHLH domain is composed of around 60 amino acids, of which 25 are conserved residues, including five in the basic region, six in the first helix, two in the loop, and 12 in the second helix [25]. Based on phylogenetic analysis, 638 bHLHs, including 167 from Arabidopsis, 177 from rice and the rest from poplar, moss and algae, were classified into 32 subfamilies [25]. The more species genomes had been sequenced, the more studies about bHLH gene family were reported, such as 230 genes organized into 24 subfamilies in the Chinese cabbage [26], 152, 159 genes separated into 21 or 26 subfamilies in tomato [27, 28], 117 genes assigned to 23 subfamilies in Nelumbo nucifera [29], 127 genes grouped into 25 subfamilies in Salvia miltiorrhiza [30], 155 genes clustered into 21 subfamilies in common bean [31], and 197 genes divided into 24 subfamilies in maize [32]. Also, conservative motifs outside the domain region were identified, and most of them were conserved within a subfamily.
Rice, maize and wheat are the three leading food crops in the world, and the grain yield is severely affected by adverse environmental conditions. Uncovering the molecular mechanism underlying the roles of bHLHs in plant growth, development and stress responses may contribute to genetics and molecular breeding. It’s indispensable to perform whole-genome identification and expression analysis for wheat bHLH gene family referencing new hexaploid bread wheat (Triticum aestivum) genome. The rice pseudomolecules were reconstructed and the new annotations were released in 2011, and therefore, it is necessary to perform a genome-scale analysis for rice bHLHs based on new genome assembly and expression data. Similarly, the identification of atypical bHLHs and expression analysis of bHLHs were not reported in maize. In this study, we aim to build connections between genetic variation and phenotypic evolution for Arabidopsis, rice, maize and wheat. For this purpose, full-genome identification and comparative evolutionary analysis of bHLH family in Arabidopsis thaliana (TAIRv10), Oryza sativa (MSUv7.0), Zea mays (AGPv3) and Triticum aestivum genome (TGACv1, updated in September 2016) were performed. The expression patterns and functions of TabHLHs, ZmbHLHs and OsbHLHs during plant life cycle and under biotic and abiotic stresses were systematically investigated. Then, WGCNA (weighted gene co-expression network analysis) and the Gene Ontology (GO) enrichment analysis were conducted to identify wheat tissue-specific and stress-responsive genes.
Results
Identification and prediction of DNA-binding ability for bHLH proteins
A total of 571, 183 and 231 bHLH genes were identified in wheat, rice and maize, respectively (Additional file 1: Table S1). Of the 571 TabHLHs, 180 genes may be the same as those previously reported by Xiao-Jiang Guo et al. [33], as listed in Additional file 2: Table S2. Other 45 out of 225 bHLHs identified in the previous study did not match any of the 571 TabHLHs in the current study. Obviously, there are some great differences between the old and new genome assembly versions. It may be resulted from complicated genome assembly of wheat with large genomes, polyploidy and a high proportion of repetitive elements. For maize and rice, 36 and 10 bHLHs were novel. All TabHLHs and ZmbHLHs and 10 novel OsbHLHs (OsbHLH179–188) were renamed. For multi-transcript genes, a putative transcript with fewer mismatches from bHLH consensus motif and longest sequence length was chosen to represent each of them. As shown in Additional file 1: Table S1, bHLHs account for approximately 0.55, 0.47, 0.59, and 0.61% of the wheat (103,539), rice (39,045), maize (39,469) and Arabidopsis protein-coding genes (27,655), respectively, constituting one of the largest transcription factor families in the species. The TabHLH domains with the highest number (361, 63.22%) are composed of 60 amino acids, and those with the second (87, 15.23%) and third highest numbers (52, 9.11%) consist of 61, 62 residues, respectively. The loop regions within most of TabHLH domains (364, 63.75%) are six residues long but divergent in terms of amino acid composition. Based on information of gene annotation, 176, 193 and 183 TabHLHs are non-randomly distributed in the A, B and D sub-genomes respectively, while 19 TabHLHs are located on scaffolds. The minimum of 12, 13 and 14 TabHLHs are localized on chromosome 1A, 1B and 1D, while the maximum of 42, 42 and 35 on chromosome 5A, 4B and 4D, respectively (Additional file 3: Table S3).
Using the criteria proposed by Xiaoxing Li et al. [34], all the 571 TabHLHs and 231 ZmbHLHs were respectively divided into two major groups based on sequence information of the basic region within bHLH domains Table 1 (i) a large group of 460 TabHLHs or 180 ZmbHLHs containing five to eleven basic residues within their basic region were expected to bind DNA, and (ii) a smaller group of 111 TabHLHs or 51 ZmbHLHs with low basic region were tentatively predicted to be non DNA binders. The DNA-binding bHLHs were further classified into two subgroups: E-box binders (376 TabHLHs or 142 ZmbHLHs) and non E-box binders (84 TabHLHs or 38 ZmbHLHs), depending on whether both Glu-13 and Arg-16 are present in the basic region. Of E-box binder subgroup, 291 TabHLHs and 117 ZmbHLHs were predicted to bind G-boxes, which contain additional residues His/Lys-9 and Arg-17 at the basic region. Additionally, according to the criteria suggested by Carretero-Paulet et al. [25], a subset of non DNA binders containing the essential residues in E-box- (eight TabHLHs and five ZmbHLHs) and G-box-binding (three TabHLHs and two ZmbHLHs) recognition motifs were potential DNA-binding bHLHs. Our prediction results of the DNA-binding ability of four AtbHLHs (AtbHLH026, AtbHLH047, AtbHLH109 and AtbHLH142) and 20 OsbHLHs (OsbHLH006, OsbHLH007, OsbHLH012, OsbHLH042 and so on) were different from that reported by Carretero-Paulet et al., as can be seen in Additional file 1: Table S1.
Table 1.
Predicted DNA-binding categories based on the bHLH domain
| > = 5 basic amino acids | < 5 basic amino acids | Total | |||||
|---|---|---|---|---|---|---|---|
| G binder | E non G | non E binder | E-box | G-box | non DNA binder | ||
| Arabidopsis | 88 | 20 | 38 | 4 | 1 | 18 | 169 |
| Rice | 89 | 18 | 30 | 4 | 2 | 40 | 183 |
| Wheat | 291 | 85 | 84 | 8 | 3 | 100 | 571 |
| Maize | 117 | 25 | 38 | 5 | 2 | 44 | 231 |
| Total | 585 | 148 | 190 | 21 | 8 | 202 | 1154 |
Multiple sequence alignments and phylogenetic tree construction of bHLHs
As the flanking sequences of the bHLH proteins from independent subfamilies are generally too divergent to be reliably aligned, the bHLH domain was used for this analysis. From the alignment, we identified 31 residues that are conserved in at least 50% of the 1154 bHLH domains from Arabidopsis, rice, maize and wheat (Additional file 4: Figure S1, indicated at the bottom of the alignment). An unrooted NJ phylogenetic tree was constructed using the alignment of the bHLH domain sequences with bootstrap analysis (1000 replicated) to observe the evolutionary relationship of bHLHs in four species (Fig. 1 and Additional file 5: Figure S2). A total of 1136 bHLHs were grouped into 36 subfamilies according to the clades with at least 50% support, topology of the tree and the classification of the Arabidopsis and rice [34–36]. The internal nodes have low support and therefore the evolutionary relationships between different bHLH subfamilies could not be inferred. The remaining 18 bHLHs were considered as orphans, likely representing highly diverged lineage-specific genes. Among the 36 subfamilies, 26 subfamilies are consistent with previously defined groups [36]. And 10 new subfamilies (10 and 26–34) are formed by 73 bHLHs which are from monocotyledons except for AtbHLH151 (previously considered member of subfamily IVd by Nuno Pires and Liam Dolan [36]), AtbHLH147, AtbHLH148, AtbHLH149 and AtbHLH150 (previously classified as orphans). As shown in Additional file 6: Table S4, 28 subfamilies are common to the four species, while the remaining 8 subfamilies are from wheat and maize and/or rice, indicating that these bHLHs might have formed after the divergence of the monocotyledon and dicotyledon, and be required for monocotyledon-specific traits. Interestingly, we noted 29 TabHLHs along with 42 PIFs and PIF-likes (PILs) identified in Arabidopsis (15), rice (13) and maize (14) clustered in subfamily VII(a + b) [32, 37, 38]. Twenty four, fifteen, and twelve TabHLHs were respectively clustered into three subfamilies III(d + e), IIIf and XIII, which MYB-interacting-region (MIR) containing proteins AtTT8 (AtbHLH042), AtEGL3 and AtLHW belong to. TabHLH183 and − 184 share high sequence similarities with AtMYC2 at amino acid level. While only 19 ZmbHLHs are found in the three subfamilies (ten in subfamily III(d + e), two in subfamily IIIf and seven in subfamily XIII), of which ZmbHLH103 and − 104 are homologous to OsMYC2 (OsbHLH009).
Fig. 1.
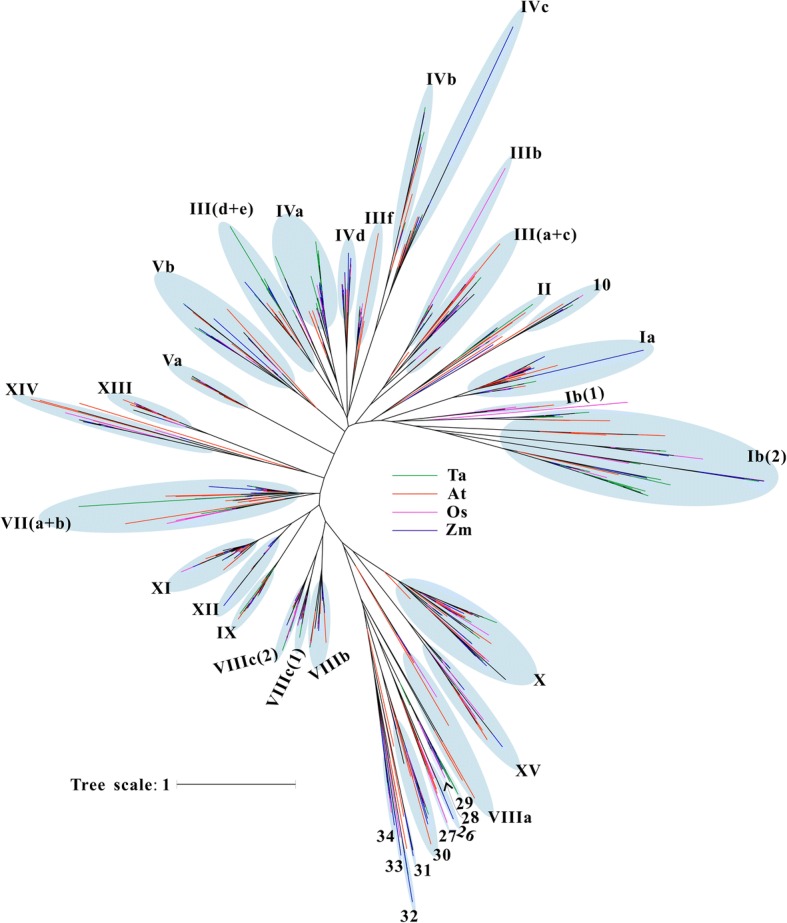
Phylogenetic relationship among Arabidopsis, rice, maize, and wheat bHLH proteins. An unrooted cladogram shows the phylogenetic relationships among 1154 bHLHs from Arabidopsis thaliana (At), Oryza sativa (Os), Zea mays (Zm) and Triticum aestivum (Ta). The blue balloons delineate the 36 subfamilies of bHLH proteins. Colored lines symbolize the species to which the proteins belong (red: Arabidopsis thaliana; purple: Oryza sativa; blue: Zea mays; green: Triticum aestivum). A full tree with protein names, proportional branch lengths, and clade support values is given in Additional file 5: Figure S2
Intron distribution pattern within the bHLH domain
To better understand the gene structural features of TabHLHs and ZmbHLHs, their intron/exon organization and splicing phase were analyzed based on their phylogenetic relationships (Additional files 7 and 8: Figures S3 and S4). In full-length genes, the exon number of TabHLHs varies from 1 to 12; and 64 genes are intronless. Contrastingly, ZmbHLHs contain up to 14 exons; and 45 genes are intronless. It was observed that the structures of genes within a subfamily show high similarity. To name a few, all members of subfamilies XIV, 26, 27, 32 and 34 are intronless, and most genes in subfamilies VIIIb, Vb, IVc contain one, two and five exons, respectively. Additionally, a great number of exons are symmetric with phase zero which is likely to facilitate gene assembly via exon shuffling and recombination [39].
Furthermore, we analyzed the intron distribution, relative position and phase within the coding sequence of bHLH domains for each gene, and found 22 different intron patterns designed as A to V (Additional file 1: Table S1). Six patterns B, D, F, I, J and Q were not found in rice and Arabidopsis, and M, O, R-V were found only in wheat. The intron number varies from zero to three and their lengths are quite different even at the same position (Additional files 7 and 8: Figures S3 and S4). As can be seen in Fig. 2, 93 TabHLHs, 50 ZmbHLHs, 32 OsbHLHs and 37 AtbHLHs do not contain intron within their bHLH domain encoding regions, forming the third common pattern P. Pattern A containing three phase-zero introns at three highly conserved positions (indicated by red “丫”) is the second common pattern in the four species. The most common pattern N has only one phase-zero intron at loop region in the species, which widely distributed across 18 subfamilies (Additional file 6: Table S4). Intron pattern distribution within most subfamilies was almost absolutely conserved, which provide a reliable support to our phylogenetic analysis. For example, pattern A was observed in 93.55% (87 out of 93) and 91.76% (78 out of 85) of XII and X subfamily members, respectively. Pattern P is presented in each member of subfamilies XIV, VIIIb, 26–28, 30, and 32–34.
Fig. 2.

Intron distribution within the bHLH domain of the AtbHLH, OsbHLH, ZmbHLH, TabHLH proteins. Scheme of intron distribution patterns (designated A to V) within the bHLH domains. Position of introns is indicated by “丫” based on the bHLH region of TabHLH136, which is shown at the top, and the number corresponds to the intron phase. The count and percentage of bHLHs displaying each pattern in wheat (Ta), Arabidopsis (At), rice (Os) and maize (Zm) are given in the table at right
Conserved motifs in most bHLH subfamilies
We searched for amino-acid sequence patterns in our data set of bHLH proteins according to their evolutionary relationships, and then each motif was characterized and named as motif 1 to 38 (Additional file 9: Table S5). The relative position of most motifs is conserved, as shown in the Additional file 10: Figure S5. bHLH domain includes motif 1 (21 amino acids in length) covering partial DNA-binding and complete helix 1 regions and motif 2 (21 amino acids) identified as a part of the loop and helix 2 regions. Outside the domain, subfamily-specific motifs were found, and some of them have been characterized as defining additional functional properties. Motifs 6, 5, and 10 observed in the majority members of subfamilies Ia, IVa and II have been reported to form a high conserved C-terminal domain (the SMF domain) of AtSPCH, AtMUTE and AtFAMA [7]. Additionally, motifs 6 and 5 were also detected in a great many proteins in subfamilies IIIf and III(d + e), such as TabHLH239, AtMYC2, TabHLH184 and ZmbHLH103, and we found they were significantly matched with an ACT domain that contributed to the recruitment of the C1 R2R3-MYB factor to the C1 binding sites located in the promoters of flavonoid biosynthetic genes [40]. Motif 7, observed in all members of subfamilies IVb and IVc, was unequivocally characterized as a ZIP dimerization domain. Motifs 13, 27, 11, 32 and 15, conserved among most members of subfamilies IIIf and III(d + e), overlap with the MIR and MYC_N domain which can interact with JAZs [41]. Interestingly, motifs 13, 27, 33, 32 and 15 form the N-terminal region of subfamily XIII proteins, which was likely responsible for activating transcription [42]. Then we discussed a detailed analysis of the structure of N-terminal fragment of proteins in the three subfamilies (IIIf, III(d + e) and XIII) through mapping secondary structure elements of N-termini of MYC3 (PDB code: 4RRU) onto the sequence alignment, as illustrated in Additional file 11: Figure S6. The five motifs (13, 27, 11, 32 and 15) respectively correspond to distinct regions: α2-helix to β1-sheet, β2, α4 to α5, α7 to β5 and β6 to α8, and the last one overlaps the transcription activation domain (TAD). Motif 12 shared by members (except OsbHLH188) of subfamily XIII has been characterized in AtLHW as necessary for homodimerization [42]. Motif 38 is present in several proteins of subfamily VII(a + b) and overlaps with the N-terminal of active phytochrome binding (APB) motif [43]. Besides, some other motifs demonstrate subfamily-specificity, yet their functions are still unclear. For instance, motif 4 is observed in almost all members of subfamily XI (except ZmbHLH048) and X (except TabHLH360 and OsbHLH179). bHLHs in subfamilies XV and XII have a motif 8 immediately C-terminal to the second helix. Motif 9 is adjacent to the N-terminal of motif 1 in subfamily Ia.
Cis-regulatory elements analysis
The cis- regulatory elements (CREs) are essential for gene expression, which participate in the control of plant growth, development and stress responses. In order to investigate the possible biological functions and regulation network of TabHLHs involved in, CREs in the 1500 bp nucleotide sequences upstream of the 5′-UTR of these genes were identified. A total of 128 distinct CREs were found in that region of TabHLHs (Additional file 12: Table S6). Among them, light response elements, such as G-box, ACE, AE-box, ATCT-motif and Box I, were abundantly presented in the promoter region of TabHLHs. A great number of CREs required for the regulation of particular tissue development were detected, such as AACA, GCN4 and skn-1 motifs and prolamin-box for endosperm, RY-element for seed, as1 for root, and HD-Zip 1 and 2 for leaf. Fifteen types of stress responsive element were found, e.g. box E, C-repeat/DRE, W-box, JERE and LTR. At least one hormone-responsive element, particularly ABRE (ABA-responsive), TCA-element (SA-responsive), CGTCA- and TGACG-motifs (JA-responsive), is presented in the promoter region of most genes. Seventy one genes contain at least one cell cycle-related regulatory motif, including E2Fa, E2Fb and MSA-like. Twenty eight genes have MBSI and/or MBSII, which are the MYB binding sites involved in flavonoid biosynthetic genes regulation. The different types of CREs presenting in TabHLHs indicate functional diversity and complexity of the regulatory networks.
Expression profiles of bHLHs in different tissues and developmental stages
To investigate the gene expression alterations in the development of bread wheat, deep transcriptome sequencing was performed in duplicates in 15 samples corresponding to five different organs (root, stem, leaf, spike and grain) at three development stages each [44]. After removing reads with low-quality, a total of about 2.82 billion paired-end reads were generated, with average of 93.56 million filtered reads for each library, as indicated in Additional file 13: Table S7. Eventually, approximately 96.08% of the reads were mapped onto the bread wheat genome, of which 79.46% were mapped uniquely in each library. In our study, 209 TabHLHs with expression values greater than 10 TPM (transcripts per million) in one or more tissues were selected for expression analysis (Additional file 14: Table S8). A total of 188 ZmbHLHs and 110 OsbHLHs were used for comparative analysis, which were expressed at medium and high levels (FPKM ≥5) in one or more tissues (Additional files 15 and 16: Tables S9 and S10). These wheat, maize and rice bHLHs were respectively grouped into five (A to E), four (F to I) and five (J to N) clusters according to the hierarchical clustering of gene expression data (Fig. 3a, Additional files 17 and 18: Figures S7 and S8). The 51 TabHLHs in cluster A were expressed with intermediate levels in stem at elongation stages, inflorescence at each stage and grain at early formation stage, particularly, TabHLH273, − 094, − 084 and − 121. And some genes with very low expression values were found in stem and leaf at different reproductive stages. Cluster B consists of 13 members, of which eight had the highest expression levels in grain at ripening stage, such as TabHLH142, − 081 and − 143. Cluster C with 41 genes can be further divided into three subclusters, of which nine in cluster C1 exhibited inflorescence-specific expression. In cluster C2, TabHLH553, − 554, − 244 and − 246 presented a relatively high expression levels in leaf at cotyledon emergence stage, while four genes TabHLH479, − 480, − 156, − 157 were highly expressed in grain at early formation stage. And TabHLH337 was specifically expressed in the grains at filling stages, which may be a homologue of endosperm-specific ZHOUPIs of Arabidopsis and maize. Several ZmbHLHs in cluster G were intensely expressed in embryo (ZmbHLH090 and − 161), endosperm and seed (ZmbHLH139 (ZmZHOUPI), − 093 (ZmmICE1) and − 094). OsbHLH144 and − 001 respectively homologous to ZmZHOUPI and ZmmICE1 were highly expressed in seed, which belong to cluster M where several genes showed higher transcript levels in panicle (OsbHLH170, − 091, − 157 and − 095) and seed (OsbHLH177, − 002 and − 147). However, OsbHLH160–162 and − 166 sharing high sequence similarities to ZmbHLH161 showed root-specific expression. Members in cluster C3 showed higher transcript abundance in leaf than in other organs, including eight predicted PIFs/PILs (TabHLH061–063, − 070, − 072 and − 076-078) in subfamily VII(a + b), five (TabHLH552 and − 559-562) in subfamily Ib(2), four (TabHLH299, − 301, − 304 and − 305) in subfamily III(a + c) and two (TabHLH047 and − 046) in subfamily VIIIb. OsBU1/ILI4 (OsbHLH172) was reported to function in controlling rice lamina inclination [45], while mainly expressed in callues in our analysis, which belongs to cluster K that comprises of callus-specific genes, such as OsbHLH035, − 042 and − 047. Genes in cluster D were expressed with high levels in root, especially for TabHLH454–459 and − 311–313. ZmHLH155 and ZmbHLH101, showing high sequence similarities to TabHLH455 and TabHLH312 respectively, belong to cluster I which contained a large number of tissue-preferentially expressed genes, such as ZmHLH057 and − 058 in germinating seed and embryo, ZmbHLH096, − 110, − 111, − 121, − 130, − 145, − 146, − 149, − 150 and − 206 in leaf, ZmbHLH138 in seed and ZmbHLH081, − 099, − 100, − 117, − 120, − 123, − 125, − 180, − 188, − 208, − 209 and − 222 in root. Whereas, OsIRO2 (OsbHLH056), the rice homologue of TabHLH454–459, was strongly expressed not only in root but also in shoot. All genes in cluster E exhibited relatively high transcript abundance in each organ, with different expression patterns during development stages. Most TabHLHs within the subfamilies IVb and IVc were found in subcluster E1. In contrast, each ZmbHLH (except ZmbHLH169 in cluster H) and OsbHLH in the two subfamilies respectively fell into clusters F and J in which genes were widely expressed in most tissues. As a member of subfamily IVb, OsIRO3 (OsbHLH063) was expressed at higher levels in shoot and root than in other tissues. Interestingly, 17 genes in subcluster E2 were expressed at very low levels in grain at ripening stage, e.g., TabHLH295, − 296 and − 485-487. Most of ZmbHLHs in cluster H were expressed at higher levels in vegetative organs than in reproductive organs.
Fig. 3.

Expression profiles of TabHLHs in five organs at three developmental stages. a Hierarchical cluster analysis of TabHLH gene expression in 15 tissues. b Expression profiles of five clusters in five organs (R, root; S, steam; L, leaf; I, inflorescence; F, fruit) are shown. The gray, light-green and light-blue lines represent the expression changes of genes, and the red lines represent the mean expression trend of bHLHs belonging to each cluster. c Proposed model for the roles of several TabHLHs in Fe uptake. X, xylem; P, phloem; EX, exodermis; EP, epidermis; RH, rhizosphere. d The co-expression subnetwork constructed using TabHLH046–048 as guide genes in modules “Lightsteelblue1” and “Greenyellow” with edge weight > 0.25. Other genes (except for TaNCED1) in the subnetwork are labeled according to their homologous genes in rice. GH, glycosyl hydrolase; GSTF, glutathione S-transferase; OPR, 12-oxophytodienoate reductase; SALP stress associated little protein; ZOS10–11, zinc-finger transcription factor; ABIL, A type 2C protein phosphatase; HOX, Homeobox-leucine zipper protein; MT3A, type 3 metallolthionein isoform; PUB, TPR and U-box domain containing protein. e Proposed model for the roles of TabHLHs in anther development. V, vascular bundle; T, tapetum layer; ML, middle layer; En, endothecium; E, epidermis; C, cuticular wax and cutin; ER, endoplasmic reticulum
Expression profiles of TabHLHs, ZmbHLHs and OsbHLHs under drought stress
In this study, we focused on 80 TabHLHs that were expressed at levels above 10 TPM in one or more conditions, out of which 45 were identified as differentially expressed genes (DEGs) with fold change ≥2 and adjusted P values ≤0.05 in at least one condition compared to control (Fig. 4a, and Additional file 19: Table S11). Thirty two (including 11 for D1h, and 31 for D6h) of the 45 DEGs in wheat were significantly down-regulated, and the rest (including 6 for D1h, and 12 for D6h) were up-regulated under drought stress, as shown in Fig. 4b. While 78 ZmbHLHs expressed at levels ≥5 FPKM in at least one sample were selected for downstream analysis, of which eight and 20 were significantly affected by ≥2 folds down-regulation and up-regulation, respectively (Additional file 20: Table S12). In rice, 59 bHLHs were chosen for subsequent analysis due to their expression levels greater than 5 RPKM, of which 31 were considered as DEGs (Additional file 21: Table S13) including 11 down-regulated and 20 up-regulated genes. The transcript abundance of TabHLH562, − 552, − 304 and − 303 were relatively high in control group and gradually decreased along with the enhancement of drought degree. Intriguingly, ZmbHLH205 and − 206 that are likely to be homologs of TabHLH562 and − 552 were up-regulated under drought stress. ZmbHLH097, − 098 and OsbHLH006 (OsRERJ) that belong to the same subfamily III(a + c) along with TabHLH304 and − 303 were up-regulated also. Several members of subfamily VII(a + b), TabHLH063 and − 069-078, were significantly down-regulated, only TabHLH065 was up-regulated. Analogously, ZmbHLH054 and OsbHLH109 orthologous to TabHLH065 were up-regulated whereas others (especially ZmbHLH051, − 059, and OsbHLH104, − 103, − 102, − 113 and − 152) in this subfamily were down-regulated. TabHLH047, − 046 and − 048 were strongly up-regulated after drought stress treatment, and their rice homolog OsqRT9 (OsbHLH120) showed increased expression as well. It’s worth noting that MYC-like genes ZmbHLH103, − 104 and OsMYC2 (OsbHLH009) were up-regulated, which are different from TabHLH183 and − 184 showing relatively stable expression. Additionally, ZmbHLH155 and − 154 expressions were inhibited, and levels of ZmbHLH156 and its rice homologues OsbHLH148 and − 185 were significantly increased.
Fig. 4.

Expression profiles of TabHLHs under drought stress. a Heatmap shows expression profile of TabHLHs in drought tress and irrigation (c) conditions. b Heatmap presents statistically significant fold changes (log2-tranformed) calculated between each drought stress and irrigation condition. c qPCR analysis of selected genes in drought tress and irrigation conditions. d Proposed model for the role of several TabHLHs in response to drought
The expression patterns of several selected TabHLHs, OsbHLHs and ZmbHLHs were validated by qPCR analysis. The primers used in this analysis were listed in Additional file 22: Table S14. Figure 4c showed that TabHLH046, − 047, − 048, − 414 and − 562 were rapidly up regulated within 1 h of drought stress, and TabHLH562, − 552, − 303, − 304, − 072 and − 560 were down regulated after 6 h of drought treatment. TabHLHs expression patterns tested in this assay consistent with that found in RNA-seq data. OsbHLH006, − 185, − 152 and − 065 were significantly down-regulated after 6 h of drought treatment, while OsbHLH113 did not show significant decrease in mRNA levels. Lower transcription abundances of ZmbHLH205, − 097 and − 156 were detected 12 h after stopping the daily watering, and ZmbHLH154 were significantly up-regulated.
Expression profiles of TabHLHs and ZmbHLHs during fungal infection
Stripe rust (Puccinia striiformis f. sp. tritici; Pst) and powdery mildew (Blumeria graminis f. sp. tritici; Bgt) are devastating diseases of wheat (Triticum aestivum). An RNA-Seq experiment with three biological replicates in each of seven conditions was performed, and Pst and Bgt fungus-inoculated wheat leaves were collected at 0, 24, 48, and 72 h post-inoculation (hpi) [46]. Fusarium verticillioides causes ear rot in maize and accumulation of mycotoxins affecting human and animal health. A deep sequencing data was generated for F. verticillioides inoculated and uninoculated resistant CO441 and susceptible CO354 maize genotypes at 72 hpi to study transcriptional changes [47]. To explore the roles of maize and wheat bHLHs in the stress responses of the fungal pathogens, the raw sequencing data was processed as described in method. A total of 85 TabHLHs were singled out for detailed analysis, which were expressed with levels ≥10 TPM in one or more conditions (Fig. 5a and Additional file 23: Table S15). And 223 RefGen_v3 IDs of ZmbHLHs were converted into RefGen_v4 IDs by the Maize Inflorescence Project (http://www.maizeinflorescence.org/v4/convert/index.php), of which 147 genes with expression levels ≥5 RPKM in one or more conditions were selected for further analysis (Additional files 24 and 25: Tables S16 and S17). As illustrated in Fig. 5a and b, our count-based differential expression analysis showed that 53 TabHLHs and 41 ZmbHLHs were significantly differentially expressed. Twenty two TabHLHs were up-regulated under Pst attack at 24 h, whereas nine of them decreased their expression levels at 72 hpi. To name but a few, the transcript abundance of TabHLH492 and − 493 showed nearly 5.0- and 3.0-fold increase at 24 hpi, respectively, and then all decreased to less than the normal levels at 48 and 72 hpi. Under Bgt-induced stress, six genes were up-expressed, and 28 were down-expressed. Interestingly, TabHLH144, − 145 and − 147 were up-expressed under stripe rust stress and down-expressed under powdery mildew stress, whereas TabHLH161 exhibited opposite expression pattern. Four genes (TabHLH304, − 299, − 061 and − 302) were down-regulated and two (TabHLH317 and − 318) were up-regulated in response to both Bgt- and Pst-induced stress. The transcript levels of ZmbHLH144, − 145, − 147, − 148, − 150 and − 151 homologous to TabHLH317 and − 318 increased up to 4.39- to 86.74-fold in two genotypes. Of another 23 up-regulated ZmbHLHs, 12 genes were significantly induced in resistant genotype, such as, ZmbHLH096, − 155, − 054 and − 059, and three (ZmbHLH037, − 169 and − 088) were found in susceptible line. The remaining 13 genes were down-regulated, of which three (ZmbHLH225, − 159 and − 122) were found in the susceptible. In addition, TabHLH071–078 belonging to PIF-like subfamily were significantly induced by Pst infection at 24 hpi and repressed at 48 hpi, while gradually up-regulated in response to Bgt infection, particularly TabHLH077. Similarly, ZmbHLH058, − 059 and − 054 in the subfamily were induced remarkably in the resistant line.
Fig. 5.

Expression profiles of TabHLHs and ZmbHLHs under different fungal stresses. a Heatmap depicts statistically significant fold changes (log2-tranformed) of expression after Bgt and Pst inoculation in wheat. b Heatmap illustrates statistically significant fold changes (log2-tranformed) of expression after F. verticillioides infection in resistant and susceptible maize genotypes. RI and SI represent the resistant and susceptible infection groups respectively; RC and SC represent the resistant and susceptible control groups, respectively. c Proposed model for the role of several TabHLHs in JA, SA, ABA and GA signaling pathways in response to pathogen stress
Gene co-expression module generation and functional enrichment analysis
Recently, co-expression network analysis became an effective approach for gene functional annotations [48]. In this study, we performed WGCNA which introduces a soft-threshold method. According to approximate scale free-topology criterion, a suitable soft-threshold value of 12 was employed to construct gene co-expression modules (Additional file 26: Figure S9). A total of 36,258 genes were parsed into 22 gene modules ranging from 55 (darkorange2) to 9396 (darkorange) genes and represented by color classifiers (Additional file 27: Figure S10). As shown in Fig. 6a, some modules share high-positive correlation, such as cyan and darkorange, greenyellow and lightsteelblue1, darkgreen and turquoise. Considering high volume data, GO enrichment analysis were used to investigate the functions of co-expressed genes within a module (Additional file 28: Table S18). Cyan module contains anther-specific terms associated with pollen wall assembly, pollen exine formation, sporopollenin biosynthetic and fatty acid metabolic processes (GO:0010208, GO:0010584, GO:0080110 and GO:0006631). Lightgreen module reflects gene functions related with nicotianamine metabolic, nicotianamine biosynthetic and tricarboxylic acid biosynthetic processes (GO:0030417, GO:0030418 and GO:0072351). For lightcyan module, significant terms were enriched in phosphorus metabolic process, protein modification and phosphorylation process (GO:0006793, GO:0036211 and GO:0016310). Then the module-tissue correlation analysis was conducted (Additional file 27: Figure S10). Cyan module is significantly correlated with inflorescence at the maximum stem length reached stage (ISE.99). The lightgreen module show higher correlation to the development of root than that of others. The correlation coefficients between turquoise module and each tissue (expect FR, fruit at whole plant fruit ripening stage) are significantly high, ranging from 0.39 to 0.53. Further GO enrichment analysis showed the genes in this module are implicated not only in the response to abiotic and biotic stresses (GO:0009628 and GO:0006952) but also in various biological functions. As is evident from the Nightingale Rose Diagrams, darkorange and black modules contain the largest numbers of DEGs with different expression patterns during Pst and Bgt infection and drought stress (Fig. 6b-d). However, the percentages of DEGs within darkorange2, ivory and greenyellow modules were higher than those in others. Both tryptophan metabolic and indolalkylamine metabolic processes (GO:0006568 and GO:0006586), which contribute to the capacity for chemical defense against microbes [49], are the significantly enriched GO terms for genes in ivory module. Greenyellow module showed significant enrichment of water, inorganic substance, oxygen-containing compound and abiotic stimulus responses (GO:0009415, GO:0010035, GO:1901700, and GO:0009628). To further understand the biological roles of TabHLHs, ten subnetworks composed of guide genes (bHLHs) along with their first-degree neighbors with edge weight ≥ 0.4 were extracted (Fig. 6e), which are associated with each other possibly due to a common biological process. The general function of these co-expressed modules was given according to the most enriched GO term and P value (Additional file 29: Table S19).
Fig. 6.

Weighted gene co-expression network analysis of wheat genes. a Heatmap of eigengene adjacencies. Blue represents negative correlation and red represents a positive correlation. The colored box on the left side of the heatmap illustrates the corresponding module and gene number. b-d Nightingale rose diagrams show the distribution of DEGs of each module under stripe rust, powdery mildew and drought stresses. The sectors with different colors indicate the distinct expression patterns over time after stress treatment. The larger the radius of a sector, the greater number of DEGs is included. “-”, non-significant; “d”, significant down-regulation; “u”, significant up-regulation. The deep-blue number indicates the percentage of differentially expressed genes in corresponding module. e Ten co-expression subnetworks constructed using several TabHLHs as guide genes. The red notes represent TabHLHs. The numbers in bracket correspond to genes annotated for the top GO term and genes in the module
Discussion
Comparative evolutionary analysis of bHLHs
The gene and protein characteristics, expression patterns and function of some bHLHs have already been unfolded in many plants, whereas the relative research has still not been performed in maize and wheat, two of the most important crops worldwide. The genome sequencing projects in wheat are still in infancy compared to other plants, and currently available wheat genome sequences are relatively fragmented [50], which makes it difficult to study tandem and segmental gene duplication events during the evolution of a gene family. In this study, based on the latest draft genome sequence of hexaploid bread wheat, we systematically identified the bHLH gene family including 571 members (Additional file 1: Table S1). The percentage of the number of bHLH genes in wheat protein-coding genes was only higher than that in rice. Comparative evolutionary analyses among four species, including phylogenetic tree reconstruction, gene structure investigation and conserved motif detection, served as the first step in a comprehensive functional characterization of bHLH transcription factors.
Previous phylogenetic analyses of plant bHLH gene family provided a helpful phylogenetic framework for the classification of bHLHs, but it varied between different studies, which probably due to different methods and sequences adopted [25, 26]. Based on the phylogenetic tree generated, bHLHs in Arabidopsis, rice, maize and wheat can be classified into 36 subfamilies, of which 28 subfamilies include genes from the four species and eight subfamilies (40 genes) are likely to be monocotyledon-specific. Some subfamilies clustered together with a high bootstrap probability (> 0.5) in the tree constructed with the sequences of bHLH domains from four species among the 36 subfamilies were not assigned to a subgroup, such as IVb and IVc, XI and XII, VIIIc(1) and VIIIc(2) due to their distinct gene structures and protein motifs in full-length sequences (Additional files 7, 8 and 10: Figures S3-S5). In the phylogenetic tree of maize bHLH genes, subfamilies 26, 28 and 30–34 clustered with a high bootstrap probability, while in that of wheat only 26–28 and 31 subfamilies clustered together. And some “orphan” fell into one subfamily in phylogenetic tree constructed for single species, such as TabHLH505, ZmbHLH140, ZmbHLH228 and ZmbHLH230. From the results, we speculated that the high degree of sequence divergence of bHLHs is presented in the four species probably due to species-specific specializations. And a lot of small clusters having exactly one representative member from each subgenome were regarded as homologous triplets (Additional file 5: Figure S2).
The members with conserved non-bHLH motifs might play a similar role. The MIR in the N-terminal region of MYC-like bHLHs is responsible for interaction with R2R3-MYB proteins [51]. By interacting with MYB proteins, three MIR containing proteins AtGL3, AtEGL3 and AtTT8 acted in a partially redundant manner to specify root and leaf epidermal cell fates and mediate JA-induced anthocyanin accumulation [52–54]. In addition, AtMYC2, AtMYC3, and AtMYC4 can interact directly with GS-related MYBs to promote glucosinolate (GS) biosynthesis gene expression in response to JA-related defense [55]. Several MYC-like bHLHs have been reported in Arabidopsis, rice, and maize, but none was studied in wheat. Here were 33 TabHLHs in subfamilies III(d + e), IIIf and XIII considered as MYC-like bHLHs based on protein sequence analyses. Heterodimer formed by AtbHLH156/LHW in subfamily XIII and AtbHLH032/TOM5 or AtbHLH030/T5 L1 or SACLs (AtbHLH142–145) coupling with auxin and cytokinin signaling pathways participates in the regulation of Arabidopsis root vascular initial population [56]. bHLHs have multiple functions due to the complex structural characteristics, and future experimental evidence should help to determine whether they can interact with MYBs.
Spatio-temporal expression dynamics of bHLHs during development
In this study, the expression patterns of bHLHs in different tissues and developmental stages were systematically investigated to understand their potential function during the life cycle of rice, maize and wheat. As seen in Fig. 3b, Additional files 17 and 18: Figures S7 and S8, most of genes were expressed in specific organ. Several genes in cluster D were selectively expressed at high levels in root. Among them, TabHLH454–459 have the high sequence similarity with OsIRO2 which positively controlled the expression of key genes involved in Fe absorption, such as OsNAS1, OsNAS2, OsNAAT1 and OsYSL15 [21]. The relatively high-abundance transcript levels of ZmbHLH155 homologous to OsIRO2 were observed in root, stem and leaf. The above mentioned bHLHs display high degree of sequence similarity to four AtbHLH genes which encode AtbHLH038 (ORG2), AtbHLH039 (ORG3), AtbHLH100 and AtbHLH101 that functioned downstream of AtbHLH034-AtbHLH104-AtbHLH105 complex and separately interacted with FIT1 (AtbHLH029) to constitutively regulate the expression of IRT1 (high-affinity ferrous iron transporter) and FRO2 (ferric–chelate reductase) [57]. TabHLH311–313 and ZmbHLH099–101, homologs of FIT1, were expressed with high levels in root as well, whereas twelve TabHLHs (TabHLH401–412) and six ZmbHLHs (ZmbHLH173–178), homologs of AtbHLH105 (ILR3), were widely expressed in various organs including root. Moreover, TabHLH454–459 and − 311–313 were in lightgreen module that contains a number of genes involved in nicotianamine (NA) biosynthesis. NA constitutes the biosynthetic precursor of MAs that efficiently bind Fe(III) in rhizosphere, contributing to metal transport in graminaceous plant species. We identified 31 NA synthase encoding genes (NASs), of which 18 were discovered in wheat genome assembly IWGSC1.0 by Julien Bonneau et al. [58], and 24 out of 31 were presented in the subnetwork constructed using these bHLHs (TabHLH171, − 311–313, and − 454-459) as guide genes (Additional file 30: Table S20). Six putative nicotianamine aminotransferase (NAAT) and three iron(III)-deoxymugineic acid transporter (YSL) encoding genes were also found in the subnetwork. In IVb subfamily, seven TabHLHs (TabHLH414–420) and four ZmbHLHs (ZmbHLH169–172) have higher sequence homologies to AtPYE (AtbHLH047) and OsIRO3 that can negatively regulate the transcription of several metal absorption-related genes [59, 60], and are expressed in all organs, and only TabHLH415 and − 416 were found in lightgreen module. These results suggested that some bHLHs might participate in the regulation of metal absorption and homeostasis in root and other organs (Fig. 3c). Additionally, ZmbHLH180, TabHLH471 and − 473 specifically expressed in root show homologies with AtPRE1/TMO7, OsILI3 and OsILI7 which may be involved in the regulation of cell elongation in response to BR, GA and auxin signals [61, 62]. Both TabHLH047 and − 046 exhibiting significant sequence similarities to AtHECs (AtbHLH088, − 037 and − 043) and OsqRT9 were relatively high expressed in leaf, and our co-expression network analysis showed that the former might be directly connected to the expression of TaNCED1 homologous to OsNCED3 involved in ABA biosyntheses, shaping leaf morphology and vascular bundle development (Fig. 3d) [63]. ZmbHLH064 and − 065, putative maize homologues of AtHECs, were expressed in leaf and primary root. Earlier researches found that AtHECs can dimerize with SPT (AtbHLH024, belonging to subfamily VII(a + b)) to coordinately regulate gynoecium development and control stem cell fate by repressing the stem cell regulators WUS and CLAVATA3 (CLV3) [64, 65]. A recent study reported that AtHECs can not only interact with PIFs to fine-tune photomorphogenesis but also positively regulate cholorophyll and carotenoid biosynthesis [66]. Although eight putative TaPIFs/PILs were expressed with relatively high levels in leaf, they were not presented in lightsteelblue1 module.
TabHLH216–221, orthologues of OsTDR1 (OsbHLH005) that is a key regulator of tapetum development and degeneration by promoting the transcription of OsCP1 and OsC6 [67], showed inflorescence-specific expression. It was reported that OsEAT1 (OsbHLH141) acted downstream of OsTDR1 and regulated the expressions of OsAP25 and OsAP37 which encode aspartic proteases participating in tapetal programmed cell death (PCD) [68]. TabHLH333 and − 335, sharing sequence similarity to OsEAT1, were also specifically expressed in inflorescence at maximum stem length reached stage. Additionally, TabHLH340, a homologue of OsbHLH142 encoding a protein that can interact with OsTDR1 to regulate the expression of OsEAT1 and be directly connected to the expressions of OsCYP703A3, OsCYP704B2, OsMS2 and OsC6 [9], was specifically expressed in inflorescence. From homology-based analysis of downstream-regulated genes OsCP1, OsC6, OsAP25, OsAP37, OsCYP703A3, OsCYP704B2, OsMS2, OsACOS12 and OsABCG15 which are required for regulating tapetal PCD and biosynthesis of sporopollenin and cutin precursors, we found that all of them have orthologs in wheat. Most of them showed inflorescence-specific expression, and were presented in cyan subnetwork (Additional file 30: Table S20). Hence, we speculate that TabHLH216–221, − 333, − 335 and − 340 may play essential roles in anther development (Fig. 3e). TabHLH479, − 480, − 156 and − 157 were expressed with significant levels not only in inflorescence but also in grain at early formation stage. The first two genes show homologies with OsBU1/ILI4 (OsbHLH172) and OsPGL2/ILI5 (OsbHLH170) that controlled bending of the lamina joint and regulated the grain length and weight [45, 69], and their maize homolog ZmbHLH184 was highly expressed in immature leaf and developing seed. Our gene co-expression network analysis showed that TabHLH479 and − 480 highly expressed in fruit formation stage (FF1) were directly connected to alpha-amylase- (TaAMY3.1–4), starch synthase- (TaSSIIa1–3 and TaGBSS1–3) and aldose 1-epimerase- (TaA1E1–4) encoding genes and so forth. TabHLH142, − 081, − 052-054 and − 080 were expressed significantly during grain ripening stage and directly connected to lots of genes encoding organic cation/carnitine transporters (TaOCT1–5), sugar transporters (TaGMST1–2 and TaSWEET1) and late embryogenesis abundant proteins (TaLEA1–26) in turquoise module. Each of them contains Skn-1 motif required for endosperm expression in their promoter regions, and RY-element involved in seed-specific regulation is present in TabHLH081 and − 080. Outstandingly, ZmbHLH058, an orthologue of TabHLH081, − 080, AtPIF4 and AtPIF5 were expressed with extremely high levels in embryo and germinating seed. A series of experiments revealed that PIF4 can facilitate hypocotyl elongation through activating auxin biosynthesis gene YUCCA8 and signaling gene IAA29 in Arabidopsis [70, 71].
Functional divergence of bHLHs in response to drought stress
The crops are often exposed drought conditions which drastically affect the growth and development through distressing various biochemical and physiological processes. Accumulating evidences suggested that bHLHs were involved in drought stress response [13, 72, 73], however the roles of TabHLHs and ZmbHLHs have scarcely been reported in response to water deficiency. In our study, 45 TabHLHs, 29 ZmbHLHs and 31 OsbHLHs were found to be differentially expressed under drought treatment. Drought responses are coordinated by complex signaling networks, and ABA acts as a global regulator. Recent study suggested that TabHLH136 (TC307165) overexpression improved the tobacco growth and osmotic stress-associated traits through modulating ABA signaling [18], and overexpression of its homologue OsPTF1 (OsbHLH096) in rice may increase root length and surface area resulting in higher instantaneous Pi uptake rate [20]. Putative homeologous triplet TabHLH136–138 were up-regulated at 1 h after 20% PEG-6000 treatment, however only TabHLH136 with ABRE in the promoter returned to normal levels at 6 h. And transcripts of ZmbHLH047 orthologous to TabHLH136–138 and OsPTF1 were significantly increased by approximately 3.0-fold under drought treatment. The activation of AKS1/CFLAP1/FBH3 (AtbHLH122), AKS2 (AtbHLH128) and AKS3/FBH4 (AtbHLH130) was suppressed by ABA-induced phosphorylation, decreasing K+ channel (KAT1) expression in guard cells leading to stomatal closure [74]. TabHLH145, − 147 and OsbHLH143 homologous to AtAKSs were significantly down-regulated. Interestingly, there was a significant decrease in the expression levels of some of the genes after 6 h when compared to 1 h but still remained higher than the non stressed controls, such as TabHLH048, − 303, − 304 and − 560. While qPCR assays showed that mRNA accumulation of TabHLH047 rather than TabHLH048 was significantly reduced at 6 h. Coexpression analysis indicated that TabHLH047 was directly linked to several stress-responsive genes encoding TaNCED1, homeobox-leucine zipper proteins (TaHOX22_A, TaHOX22_B and TaHOX22_D), glutathione S-transferases (TaGSTF1–2), glycosyl hydrolases (TaGH1–3) and 12-oxophytodienoate reductase (TaOPR1) and so on (Fig. 3e, Additional file 30: Table S20). TabHLH190 and ZmbHLH108, the homologues of AtJAM2 (AtbHLH013) which functioned mostly antagonistically to AtMYC2 in JA signaling pathway [75], were differentially up-regulated. ZmbHLH103 and − 104 homologous to AtMYC2 were significantly up-regulated, whereas the expressions of wheat homologs (TabHLH183 and − 184) remained relatively constant across different sampling times. In contrast, TabHLH241 having high sequence similarity to AtEGL1/EGL3 which participated in JA-mediated anthocyanin accumulation [54], was up-regulated at 6 h, while its maize homologs ZmbHLH124 and − 125 were expressed at very low levels. According to the above expression analysis combined with the data listed in Additional file 1: Table S1, we speculated that TabHLH190, − 241, ZmbHLH103 and − 104 might bind G-box element of downstream drought-responsive genes and regulate their transcriptions. The expression levels of TabHLH414, − 415 and ZmbHLH171 were increased under drought stress, whose rice ortholog OsbHLH062 encodes a interactor of JAZ9 to regulate the transcriptional activation of JA-, salt stress-responsive genes including ion transporter genes [14]. The higher OsRERJ mRNA levels was detected 1 h after drought treatment and the abundance was decreased rapidly in rice seedlings thereafter, as previously reported [76]. Similarly, the expression patterns of its homologous ZmbHLH097, TabHLH303 and − 304 were observed under drought stress during seedling period. We speculated that their mRNA accumulations peaked less than 1 h after 20% PEG treatment and 12 h drought condition. However, high ZmbHLH097 and − 098 mRNA levels were detected in fertilized ovary and basal leaf meristem tissue under drought stress, about 3 days after irrigation withheld. In addition, our qPCR assay verified that ZmbHLH154 was strongly up-regulated, which is different from its expression profile at reproductive stage. And several putative wheat and maize PIFs/PILs were down-regulated, with the exception of TabHLH065 and ZmbHLH054 that are homologues of OsPIL16/APG which have been reported to be a negative regulator of the grain length and weight [77], suggesting a role in regulation of the balance of growth and drought stress response. This analysis indicated some bHLHs may be associated with drought stress response through diverse regulation processes in different tissues. And it was a major challenge to analyse the mechanism of genes transcriptional regulation in response to water deficiency at different development stages.
Involvement in immune responses against pathogens
In the expression analyses, we observed a differential response of TabHLHs for Pst and Bgt pathogens though both of them are biotrophic fungi (Fig. 5). Salicylic acid (SA) play a central role in the disease resistance against biotrophic pathogens, while JA is critical for activation of defense against necrotrophic pathogens [78]. SA- and JA-responsive signaling are interdependent, and SA antagonize JA signaling downstream of COI1, degrading ORA59 and then suppressing PDF1.2 [79]. When SA levels are intermediate, NPR1 accumulates and interacts with TGA transcription factor, acting as a co-activator to activate SA responsive genes including pathogen-related protein (PR) genes [80]. One earlier study reported that TabHLH407 (GenBank accession: ADC33137) overexpression resulted in the suppression of transcription levels of PR1, PR2 and PR5, and the enhancement of the expression of ORA59 (AP2/ERF domain transcription factor) and PDF1.2 (PLANT DEFENSIN1.2) in transgenic Arabidopsis [23]. In our analysis, putatively homeologous triplet TabHLH407, TabHLH408 and − 409 were suppressed by Bgt and Pst attack. The transcripts of several TaPR1s (TaPR1_1–4, TaPR1_14, TaPR1_18 and TaPR1_20) were highly abundant at 24 hpi and then gradually decreased following Bgt inoculation, and TaPR1_1–4 had similar expression patterns after Pst infection (Additional file 30: Table S20). We identified 50 wheat homologues of AtORA59, of which five (TaERF9, − 16, − 25, − 33 and − 48) were obviously induced by two pathogens and one (TaERF31) was differentially activated by Bgt. Of our identified 140 wheat defensin-like genes (DEFLs) using AtPDF1s and AtPDF2s sequences as queries for local BLAST search, three (TaDEFL18, − 19 and − 59) were up-regulated after Pst incubation at 24 hpi, and one (TaDEFL33) was significantly induced by Bgt infection at 72 hpi (Additional file 31: Figure S11). ZmbHLH174 sharing high sequence similarity with TabHLH407 was down-regulated under F. verticillioides infection in resistant genotype, and ZmPDF1 (LOC100280744) and ZmPDF2 (LOC100280572) were intensely expressed in both genotypes. In comparison, normal mRNA expression levels in susceptible line are higher than those in resistant line. Conversely, higher fold changes were observed in resistant one. Interestingly, the mRNA levels of another five TaDEFLs (TaDEFL57, − 59 and − 80-82) accumulated substantially in grain at ripening stage. As a transcriptional coactivator of TGAs in SA-mediated pathway, TaNPR1_A1, TaNPR1_D1 and TaNPR1_D2 were induced by the Bgt, which were not observed during Pst attack. MYC branch is another branch of JA signaling pathway, in which AtMYC2 acts as the master regulator [81]. TabHLH183 and − 184 with ABRE-, CGTCA- and TGACG-motifs in promoter regions, the homologs of OsMYC2 and AtMYC2, were slightly up-regulated. For their homologous genes in maize, ZmbHLH103 and − 104 expressions were increased by F. verticillioides infection. A series of evidences have substantiated that AtANAC019 and − 055 acted downstream of AtMYC2 and enhanced the expressions of AtVSP1, AtLOX2 and AtBSMT1 but suppressed AtICS1 [82, 83]. Herein, five wheat homologues (TaNAC1–5) of AtANAC019 and − 055 were induced by Pst infection at 24 hpi and then decreased to very low levels, whereas the mRNA levels remained relatively stable after Bst invasion. Besides, TaLOX3.1–3 were significantly induced by both pathogen, particularly TaLOX3.3 at 24 hpi. JAZ and DELLA proteins mediate crosstalk between JA-dependent defenses and GA-triggered elongation growth. Under biotic stress, JA concentrations increase to high levels, resulting in the degradation of JAZs and the release of MYCs and DELLAs to activate JA-responsive genes and to bind PIFs, respectively. In this study, three TaPIFs/PILs (TabHLH071, − 073 and − 075) were up-expressed at 24 hpi after Pst infection and down-expressed at 72 hpi, with opposite expression patterns observed in Bgt infection. In addition, some ZmPIFs/PILs were up-regulated, especially ZmbHLH058, − 059 and ZmPIL1 (ZmbHLH054). OsGAI encoding DELLA protein was reported to integrate and amplify SA- and JA-dependent defense signaling [84]. Its homologous genes TaRHT1A-D were significantly up-regulated after Pst inoculation, especially at 24 hpi, but down-regulated after Bgt infection. Of our identified TaJAZs (Additional files 30 and 32: Table S20 and Figure S12), six of 61 genes were induced under pathogen stress, including TaJAZ14B, −15D, −16A, -17B, -9B and -2B homologous to OsJAZ10, − 11, − 6, − 8, − 4 and OsTIFY1a respectively. In addition, strongly up-regulated ZmbHLH144, − 145 and TabHLH317–319 are homologous with OsDPF (OsbHLH025) which is implicated in the biosynthesis of diterpenoid phytoalexins by positively regulating CPS2 and CYP99A2 [22]. And TabHLH324 significantly induced after Bgt infection was closely connected to some genes listed in Additional file 30: Table S20, which encode CYPs, UDP-glucosyl transferases (UGTs), 2-oxoglutarate/Fe(II)-dependent dioxygenases (2-ODDs) and Tryptophan synthases (TSAs) and may be involved in the biosynthesis of DIMBOA and tryptophan. Our results indicated that bHLHs are crucial components of a complex regulation circuit involved in the plant disease resistance. Due to the lack of genome-scale identification of PR, AP2/ERF, PDF, bZIP, NPR, and other gene families for the updated wheat genome assembly, the current study has a few limitations to study the signaling pathway in response to pathogen stresses.
Conclusions
The updated genome assemblies are expected to serve as a platform for our comparative functional genomics studies. In this study, 183 OsbHLHs, 231 ZmbHLHs and 571 TabHLHs were identified. The comparative evolutionary analysis showed that all bHLHs from Arabidopsis, rice, maize and wheat can be divided into 36 subfamilies. The exon/intron structure and motif compositions were conserved within one subfamily. Some bHLHs were specifically expressed in endosperm, grain, inflorescence, leaf, root, or shoot. Combining with the WGCNA and GO function enrichment analysis, we speculated that TabHLH454–459 and − 311–313 may be related with iron uptake, and TabHLH216–221, − 333, − 335, and − 340 may be involved in anther development. Some differentially expressed bHLHs may be required for enhancing the tolerance of crop plants to drought and pathogen stresses. PIFs/PILs, MYCs and TabHLH407–409 and ZmbHLH174 may be involved in the crosstalk between GA-mediated growth and JA-, ABA-, SA-dependent defenses. And some bHLHs participate in the regulation of secondary metabolism in response to environmental stimulus. Comparative spatiotemporal expression analysis of lots of putative homeologous triplets showed that there was apparent expression divergence among the homeologs, but it was nonradical alterations, suggesting that the subfunctionalization rather than neo-functionalization occurred among three subgenomes. Our analysis will facilitate the future functional analysis of bHLHs in Gramineae species and contribute to molecular breeding for improving yield, stress tolerance and grain quality.
Methods
Database mining and identification of bHLH genes
Rice, maize and bread wheat genome sequences, annotation files in GTF format, and protein sequences were downloaded from TIGR (release 7), Ensembl Plants (release 31, http://plants.ensembl.org/index.html) and Ensembl Plants (relase 32), respectively. Three local protein databases were established with these protein sequences. As per the previous researches, predicted AtbHLHs, OsbHLHs and ZmbHLHs were retrieved from TAIR (release 10), TIGR and Ensembl Plants, respectively [25, 32, 35, 36]. As no typical bHLH domain was found in At1t22380 (AtbHLH152), Os07t11020 (OsbHLH017), Os01t65080 (OsbHLH033), Os01t18290 (OsbHLH105), Os04t35000 (OsbHLH145), GRMZM5G899865_T01 and GRMZM2G137426_T01, they were excluded from the study. All bHLH domain sequences from three species were aligned with MEGA 6.0 software (http://megasoftware.net/) and the obtained multiple alignments were used to construct Hidden Markov Model (HMM) profiles with hmmbuild tool implemented in HMMER3.1 (http://hmmer.org/). The profiles were used to search the local protein databases using hmmsearch tool. Then the bHLH HMM profile (accession number: PF00010) in the Pfam database (http://pfam.xfam.org/) was also applied to search the proteins. All non-redundant sequences were received a conserved domain check using Pfam tool and SMART web server (http://smart.embl-heidelberg.de/). To confirm the amino acid sequences as bHLHs, we examined them by counting the number of matches at each region of the predicted bHLH domain according to a low stringent criterion with allowing 12 mismatches from the HLH consensus.
Phylogenetic analysis
Multiple alignments of identified bHLH domain sequences from 571 TabHLHs, 231 ZmbHLHs, and 183 OsbHLHs were created respectively using MAFFT v7 (http://mafft.cbrc.jp/alignment/software/) with --auto, −-reorder options. The bHLH sequences used for our analysis were renamed as TabHLHXXX, ZmbHLHXXX and OsbHLHXXX, and the number designation of them was based on the order of the multiple sequence alignment. To investigate the evolutionary relationships among Arabidopsis, rice, maize and wheat bHLHs, multiple sequence alignment of 1154 bHLH domains was generated using MAFFT. Phylogenetic tree was constructed by the neighbor-joining (NJ) method with a bootstrap test (1000 replicates) using MEGA v6.0. The alignments were visualized using ESPript v3.0 (http://espript.ibcp.fr/ESPript/ESPript/), and the tree image was generated using iTOL (http://itol.embl.de/).
Gene structure analysis and conserved motif detection
The exon/intron organization and splicing phase of the predicted TabHLHs and ZmbHLHs were investigated based on the GTF annotation files of Triticum aestivum and Zea mays genomes, and then graphically displayed by the Gene Structure Display Server (GSDS, http://gsds.cbi.pku.edu.cn/) [85]. For OsbHLHs, the gene structures were determined by comparing predicted gene coding sequences (CDS) with their corresponding genomic sequences using GSDS. Furthermore, to discover the intron distribution pattern within the coding sequence of the bHLH domain, the different regions of bHLH domain were assigned to different colors. To analyze other conserved motifs in complete amino acid sequence of bHLHs, the MEME tool in Galaxy web-based platform (https://usegalaxy.org/) was used. And each motif was individually checked using FIMO tool in the platform also.
Cis-regulatory elements analysis
The upstream 1500 bp genomic DNA sequences of predicted TabHLH genes were extracted from Triticum aestivum genome using TBtools (http://cj-chen.github.io/tbtools/), and then submitted to PlantCARE database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) for scanning cis- regulatory elements.
Read alignment and expression analysis
The raw sequencing data in FASTQ or SAM formats generated from wheat (cv. Chinese Spring, N9134 and TAM107) of different developmental stages and under different treatment conditions are available from ArrayExpress (http://www.ebi.ac.uk/arrayexpress/) with accession numbers E-MTAB-4484 and E-MTAB-4289, and NCBI Sequence Read Archive (SRA) database (https://www.ncbi.nlm.nih.gov/sra) with accession number SRP045409. With the TGACv1 wheat genome assembly and annotations generated in Dec. 2015, to which 99% genes annotated on the IWGSC1 + popseq assembly can be mapped, more coding genes can be identified. Therefore, we re-processed the raw data according to the following steps: The raw pair-end reads were quality-trimmed with multi-perspective approach via SeqPrep (https://github.com/jstjohn/SeqPrep), Sickle (https://github.com/najoshi/sickle) and Fastx-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Clean data of pair-end reads from each sample were aligned to the TGACv1 wheat genome assembly by HISAT2 software [86]. The raw count of unique mapped reads to each gene was aggregated using featureCounts and used to calculated TPM value utilizing a custom-made Perl script [87]. The DEseq2 package in R software (v3.3.1) (http://www.bioconductor.org/packages/devel/bioc/html/DESeq2.html) was used to perform differential gene expression analysis, and fold change cutoff of 2 and adjusted P value ≤0.05 were taken as statistically significant threshold. The log2-transformed (TPM + 1) values were used for heatmap generation by gplots package.
To analyze the expression patterns of maize and rice bHLHs during development, drought and pathogen stresses, the processed expression data sets were downloaded from NCBI SRA and ArrayExpress databases with accession numbers SRP010680, E-GEOD-40070, E-MTAB-4219, E-MTAB-2037 and E-GEOD-65022. The differential expression analysis of maize genes after Fusarium verticillioides infection was performed using DEseq2 package. The log2-transformed (FPKM+ 1) and log2-transformed fold change values were used to generate heatmaps.
Co-expression network construction and GO enrichment analysis
We extended the expression analysis by carrying out a weighted gene co-expression network nalysis (WGCNA) with package WGCNA in R [88]. As it is believed that low expressed and non-changing genes provide limited information in a co-expression network building, 35,702 wheat genes were selected based on their expression values ≥10 TPM in one or more samples and coefficient of variation (CV) ≥ 0.6. And other 563 genes of interest were added into the analysis. To check that there are no outliers, 57 samples were clustered using the function hclust. Based on the scale free topology analysis by pickSoftThreshold function, the lowest soft-thresholding power value 12 was selected to compute an adjacency matrix (AM). Topological overlap matrix (TOM) was computed from AM, and in turn converted into a dissimilarity TOM. Genes were then clustered using hierarchical clustering and distinct modules were identified using Dynamic Tree Cut approach. Modules whose eigengenes were highly correlated were merged with a mergeCutHeight of 0.25. In order to study the relationships among modules, module similarity was quantified based on the correlation of module eigengenes (MEs). To select potential biologically interesting modules for downstream analysis, the correlation between modules and tissues type were estimated using MEs.
Using BioMart data mining tool supported by Ensembl Plants, Gene Ontology (GO) annotation was obtained to generate a broad overview of groups of genes. GO enrichment analysis was performed for each module using the OmicShare tools (www.omicshare.com/tools). As TFs are crucial in regulating gene expression and triggering multiple aspects of biological process, bHLHs were considered as the “guide-gene” to construct subnetworks. The node and edge information of subnetworks was visualized and analyzed using Cytoscape (http://www.cytoscape.org/).
Plant materials and qPCR validation
Seeds of Nipponbare and a leading wheat variety TAM107 were surface-sterilized in 1% sodium hypochlorite for 20 min, rinsed in distilled water for six times, and soaked in dark overnight at room temperature. The germinated seeds were cultured in water and grew in a growth chamber with 22 °C/18 °C (day/night), 16 h/8 h (light/dark), and 50% humidity. Then 1 week old seedlings were subjected to dehydration stress, replacing water with 20% (m/V) PEG-6000 solution for 1 h, 6 h and 24 h. Maize inbred line B73 were soaked at room temperature and germinated seeds grew in soil in the growth chamber, keeping 90% soil humidity. Then 1 week old seedlings were subjected to drought stress 12 h and 24 h after soil humidity reduced to 50%. Total RNA were isolated from fresh seedling samples, and the quantitative real time PCR (qPCR) reaction was performed on CFX96 qPCR detection system. UBQ (rice: LOC_Os06g46770, maize: GRMZM2G409726) and wheat actin (TRIAE_CS42_1AL_TGACv1_001447_AA0030680) were used as internal reference genes to normalize Ct values of each reaction [89–91].
Additional files
Table S1. List of 1154 bHLHs from Arabidopsis, rice, maize and wheat and their related information. (XLS 509 kb)
Table S2. A table shows 180 new names of wheat bHLHs side by side with previous ones. (XLS 73 kb)
Table S3. The distribution of TabHLHs on wheat chromosomes. (XLS 24 kb)
Figure S1. Amino acid sequence alignment of 1154 Arabidopsis, rice, wheat and maize bHLH domains. The red triangles at the bottom indicate the 31 conserved residues. The loop region has been shortened to better visualize the alignment results. (PDF 358 kb)
Figure S2. The unrooted NJ tree of bHLH domains from Arabidopsis, rice, wheat and maize. The blue, red and green rhombuses indicate the TabHLH in A, B and D sub-genomes respectively. (PDF 510 kb)
Table S4. Comparison of the number of genes and intron distribution patterns within the bHLH domain in each bHLH subfamily between wheat and other species. (XLS 84 kb)
Figure S3. Phylogenetic relationships and gene structure features of TabHLHs. The phylogenetic tree was constructed with MEGA 6.0 based on a multiple alignment of 571 wheat bHLH domain sequences. For better visualization and comparison, all introns were displayed in the same length. (PDF 5746 kb)
Figure S4. Phylogenetic relationships and gene structural features of ZmbHLHs. The phylogenetic tree was constructed with MEGA 6.0 based on a multiple alignment of 231 amino acid sequences of maize bHLH domain. For better visualization and comparison, all introns were displayed in the same length. (PDF 1617 kb)
Table S5. Motif with best possible match and the number in each subfamilies and species. (XLS 38 kb)
Figure S5. Architecture of protein conserved motifs. Motifs are graphically represented as colored boxes drawn to scale for bHLH proteins. (PDF 19037 kb)
Figure S6. Arrangement of secondary structure elements in AtMYC3 (5–242). Secondary structure elements overlaid on the sequence alignment of bHLH N-terminal proteins belonging to three subfamilies III(d + e), XIII and IIIf. (PDF 517 kb)
Table S6. The distribution of cis-regulatory elements in TabHLH promoters. “Y” means that the element is present in the promoter region. (XLS 648 kb)
Table S7. Summary of RNA-seq data generated and mapping statistics. (XLS 35 kb)
Table S8. The TPM values of 209 TabHLHs in 15 tissues. (XLS 85 kb)
Table S9. The FPKM values of 188 ZmbHLHs in 18 tissues. (XLS 78 kb)
Table S10. The FPKM values of 110 OsbHLHs in seven tissues. (XLS 35 kb)
Figure S7. Heatmap showing the transcriptional abundance of ZmbHLHs in 18 tissues. (PDF 448 kb)
Figure S8. Heatmap showing log2-transformed (FPKM+1) values for OsbHLHs in 7 tissues. (PDF 850 kb)
Table S11. The TPM values of 80 TabHLHs under drought stress. (XLS 39 kb)
Table S12. The FPKM values of 78 ZmbHLHs under drought stress. (XLS 71 kb)
Table S13. The RPKM values of 59 OsbHLHs under drought stress. (XLS 29 kb)
Table S14. List of primers used in this study for qPCR. (XLS 27 kb)
Table S15. The TPM values of 88 TabHLHs under stripe rust (S) and powdery mildew (P) stresses. (XLS 41 kb)
Table S16. Convertion of maize AGPv3 gene IDs to maize AGPv4 gene IDs. (XLS 63 kb)
Table S17. The RPKM values for 147 ZmbHLHs after F. verticillioides infection. (XLS 42 kb)
Figure S9. Analysis of network topology for various soft-thresholding powers. a. The scale-free fit index (y-axis) as a function of the soft-thresholding power (x-axis). b. The mean connectivity (degree, y-axis) as a function of the soft-thresholding power (x-axis). (PDF 469 kb)
Figure S10. Module-tissue association. Each row corresponds to a module and each column corresponds to a tissue. The top and bottom number in each cell indicate the correlation coefficient between the module and tissue and p-value of the test, respectively. (PDF 3815 kb)
Table S18. Summary of the significantly enriched GO terms (q-value < 0.001) of genes in each co-expression module. BP, biological process; MF, molecule function; CC, cellular component. (XLS 279 kb)
Table S19. List of the “guide genes” of nine subnetwork, and their related information. (XLS 37 kb)
Table S20. The expression profiles of some genes from other families discussed in this study. (XLS 181 kb)
Figure S11. Plyhogenetic analysis of 140 TaDEFLs, 7 AtPDF1s and 6 AtPDF2s. The phylogenetic tree was constructed using MEGA by the NJ method with 1000 bootstrap replications, and the bootstrap values greater than 700 were displayed on the branches. (PDF 411 kb)
Figure S12. Phylogenetic relationship and motif compositions of TaJAZs. a. The phylogenetic tree of TaJAZs constructed using MEGA by the NJ method with 100 bootstrap replicates. b. Domain distribution of TaJAZs was investigated using the MEME web server. c. The consensus sequence of TIFY and Jas motif from TaJAZs. (PDF 414 kb)
Acknowledgments
We are grateful to the providers who submitted the RNA-seq data to the public expression databases which can be applied freely. We sincerely thank C Yin (Shanghai Majorbio Bio-pharm Technology Co., Ltd.) for her great contribution to the RNA-seq data analysis. And we thank YG Lab for providing reagent and qPCR detection system. We also thank YX Li and W Zhang in our laboratory for useful discussions.
Funding
The project was supported by the Science and Technology Cooperation Project of Fujian Province, China (Grant No.2015I0006). The funding institutions had no direct role in study design, sample collection, analysis, and interpretation of date, nor in manuscript writing. Annual reports were submitted to the funding institutions tracking the progress of the projects.
Availability of data and materials
All of the datasets supporting the results of this article are included within the article and its Additional files.
Abbreviations
- bHLH
Basic helix-loop-helix
- CRE
Cis-acting regulatory
- GO
Gene Ontology
- hpi
Hours post-inoculation
- JA
Jasmonic acid
- PIFs
Phytochrome-Interacting factor
- SA
Salicylic acid
- WGCNA
Weighted gene co-expression network analysis
Authors’ contributions
KW conceptualized, designed the project. HC carried out computational analysis, and RNA-seq expression profiling. HC and KW performed the experiments. HC and KW wrote the manuscript. These authors have contributed equally to this work. Both authors read and approved the final manuscript.
Ethics approval and consent to participate
TAM107 and maize inbred line B73 seeds used in our study were kindly provided by Institute of Crop Sciences, Chinese Academy of Agricultural Sciences and seedlings grew in plant growth chamber in Tsinghua University YG Lab. Rice seeds (Oryza sativa, Nipponbare) were collected by Dr. MX in Anhui Agricultural University. KW. The authors declared the compliance with institutional, national and international guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Kaifa Wei, Email: kaifa-wei@163.com.
Huiqin Chen, Email: chq18@mails.tsinghua.edu.cn.
References
- 1.Miguel DL, Salomé P. PIFs get BRright: PHYTOCHROME INTERACTING FACTORs as integrators of light and hormonal signals. New Phytol. 2014;202(4):1126–1141. doi: 10.1111/nph.12725. [DOI] [PubMed] [Google Scholar]
- 2.Gangappa SN, Kumar SV. DET1 and HY5 control PIF4-mediated Thermosensory elongation growth through distinct mechanisms. Cell Rep. 2017;18(2):344–351. doi: 10.1016/j.celrep.2016.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li K, Yu R, Fan LM, Wei N, Chen H, Deng XW. DELLA-mediated PIF degradation contributes to coordination of light and gibberellin signalling in Arabidopsis. Nat Commun. 2016;7:11868. doi: 10.1038/ncomms11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li X, Zhang H, Ai Q, Liang G, Yu D. Two bHLH transcription factors, bHLH34 and bHLH104, regulate Iron homeostasis in Arabidopsis thaliana. Plant Physiol. 2016;170(4):2478–2493. doi: 10.1104/pp.15.01827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohashi-Ito K, Fukuda H. Functional mechanism of bHLH complexes during early vascular development. Curr Opin Plant Biol. 2016;33:42–47. doi: 10.1016/j.pbi.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 6.Komatsu K, Maekawa M, Ujiie S, Satake Y, Furutani I, Okamoto H, Shimamoto K, Kyozuka J. LAX and SPA: major regulators of shoot branching in rice. Proc Natl Acad Sci U S A. 2003;100(20):11765–11770. doi: 10.1073/pnas.1932414100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raissig MT, Abrash E, Bettadapur A, Vogel JP, Bergmann DC. Grasses use an alternatively wired bHLH transcription factor network to establish stomatal identity. Proc Natl Acad Sci U S A. 2016;113(29):8326–8331. doi: 10.1073/pnas.1606728113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ito S, Song YH, Josephson-Day AR, Miller RJ, Breton G, Olmstead RG, Imaizumi T. FLOWERING BHLH transcriptional activators control expression of the photoperiodic flowering regulator CONSTANS in Arabidopsis. Proc Natl Acad Sci U S A. 2012;109(9):3582–3587. doi: 10.1073/pnas.1118876109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ko SS, Li MJ, Sun-Ben Ku M, Ho YC, Lin YJ, Chuang MH, Hsing HX, Lien YC, Yang HT, Chang HC, et al. The bHLH142 transcription factor coordinates with TDR1 to modulate the expression of EAT1 and regulate pollen development in Rice. Plant Cell. 2014;26(6):2486–2504. doi: 10.1105/tpc.114.126292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Groszmann M, Paicu T, Alvarez JP, Swain SM, Smyth DR. SPATULA and ALCATRAZ, are partially redundant, functionally diverging bHLH genes required for Arabidopsis gynoecium and fruit development. Plant J. 2011;68(5):816–829. doi: 10.1111/j.1365-313X.2011.04732.x. [DOI] [PubMed] [Google Scholar]
- 11.Luo J, Liu H, Zhou T, Gu B, Huang X, Shangguan Y, Zhu J, Li Y, Zhao Y, Wang Y. An-1 encodes a basic Helix-loop-Helix protein that regulates awn development, grain size, and grain number in Rice. Plant Cell. 2013;25(9):3360–3376. doi: 10.1105/tpc.113.113589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le Hir R, Castelain M, Chakraborti D, Moritz T, Dinant S, Bellini C. AtbHLH68 transcription factor contributes to the regulation of ABA homeostasis and drought stress tolerance in Arabidopsis thaliana. Physiol Plant. 2017;160(3):312–327. doi: 10.1111/ppl.12549. [DOI] [PubMed] [Google Scholar]
- 13.Seo JS, Joo J, Kim MJ, Kim YK, Nahm BH, Song SI, Cheong JJ, Lee JS, Kim JK, Choi YD. OsbHLH148, a basic helix-loop-helix protein, interacts with OsJAZ proteins in a jasmonate signaling pathway leading to drought tolerance in rice. Plant J. 2011;65(6):907–921. doi: 10.1111/j.1365-313X.2010.04477.x. [DOI] [PubMed] [Google Scholar]
- 14.Wu H, Ye H, Yao R, Zhang T, Xiong L. OsJAZ9 acts as a transcriptional regulator in jasmonate signaling and modulates salt stress tolerance in rice. Plant Sci. 2015;232:1–12. doi: 10.1016/j.plantsci.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 15.Xu W, Jiao Y, Li R, Zhang N, Xiao D, Ding X, Wang Z. Chinese wild-growing Vitis amurensis ICE1 and ICE2 encode MYC-type bHLH transcription activators that regulate cold tolerance in Arabidopsis. PLoS One. 2014;9(7):e102303. doi: 10.1371/journal.pone.0102303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu X, Yang L, Yu M, Lai J, Wang C, McNeil D, Zhou M, Yang C. A novel Zea mays ssp. mexicana L. MYC-type ICE-like transcription factor gene ZmmICE1, enhances freezing tolerance in transgenic Arabidopsis thaliana. Plant Physiol Biochem. 2017;113:78–88. doi: 10.1016/j.plaphy.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Badawi M, Reddy YV, Agharbaoui Z, Tominaga Y, Danyluk J, Sarhan F, Houde M. Structure and functional analysis of wheat ICE (inducer of CBF expression) genes. Plant Cell Physiol. 2008;49(8):1237–1249. doi: 10.1093/pcp/pcn100. [DOI] [PubMed] [Google Scholar]
- 18.Yang T, Yao S, Hao L, Zhao Y, Lu W, Xiao K. Wheat bHLH-type transcription factor gene TabHLH1 is crucial in mediating osmotic stresses tolerance through modulating largely the ABA-associated pathway. Plant Cell Rep. 2016;35(11):2309–2323. doi: 10.1007/s00299-016-2036-5. [DOI] [PubMed] [Google Scholar]
- 19.Yang T, Hao L, Yao S, Zhao Y, Lu W, Xiao K. TabHLH1, a bHLH-type transcription factor gene in wheat, improves plant tolerance to pi and N deprivation via regulation of nutrient transporter gene transcription and ROS homeostasis. Plant Physiol Biochem. 2016;104:99–113. doi: 10.1016/j.plaphy.2016.03.023. [DOI] [PubMed] [Google Scholar]
- 20.Yi K, Wu Z, Zhou J, Du L, Guo L, Wu Y, Wu P. OsPTF1, a novel transcription factor involved in tolerance to phosphate starvation in rice. Plant Physiol. 2005;138(4):2087–2096. doi: 10.1104/pp.105.063115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ogo Y, Itai RN, Nakanishi H, Kobayashi T, Takahashi M, Mori S, Nishizawa NK. The rice bHLH protein OsIRO2 is an essential regulator of the genes involved in Fe uptake under Fe-deficient conditions. Plant J. 2010;51(3):366–377. doi: 10.1111/j.1365-313X.2007.03149.x. [DOI] [PubMed] [Google Scholar]
- 22.Yamamura C, Mizutani E, Okada K, Nakagawa H, Fukushima S, Tanaka A, Maeda S, Kamakura T, Yamane H, Takatsuji H, et al. Diterpenoid phytoalexin factor, a bHLH transcription factor, plays a central role in the biosynthesis of diterpenoid phytoalexins in rice. Plant J. 2015;84(6):1100–1113. doi: 10.1111/tpj.13065. [DOI] [PubMed] [Google Scholar]
- 23.Wang F, Lin R, Feng J, Qiu D, Chen W, Xu S. Wheat bHLH transcription factor gene, TabHLH060 , enhances susceptibility of transgenic Arabidopsis thaliana to Pseudomonas syringae ☆. Physiol Mol Plant Pathol. 2015;90:123–130. doi: 10.1016/j.pmpp.2015.04.007. [DOI] [Google Scholar]
- 24.Ellenberger T, Fass D, Arnaud M, Harrison SC. Crystal structure of transcription factor E47: E-box recognition by a basic region helix-loop-helix dimer. Genes Dev. 1994;8(8):970–980. doi: 10.1101/gad.8.8.970. [DOI] [PubMed] [Google Scholar]
- 25.Carretero-Paulet L, Galstyan A, Roig-Villanova I, Martinez-Garcia JF, Bilbao-Castro JR, Robertson DL. Genome-wide classification and evolutionary analysis of the bHLH family of transcription factors in Arabidopsis, poplar, rice, moss, and algae. Plant Physiol. 2010;153(3):1398–1412. doi: 10.1104/pp.110.153593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song XM, Huang ZN, Duan WK, Ren J, Liu TK, Li Y, Hou XL. Genome-wide analysis of the bHLH transcription factor family in Chinese cabbage (Brassica rapa ssp. pekinensis) Mol Genet Genomics. 2014;289(1):77–91. doi: 10.1007/s00438-013-0791-3. [DOI] [PubMed] [Google Scholar]
- 27.Sun H, Fan HJ, Ling HQ. Genome-wide identification and characterization of the bHLH gene family in tomato. BMC Genomics. 2015;16:9. doi: 10.1186/s12864-014-1209-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Hu Z, Zhao T, Yang Y, Chen T, Yang M, Yu W, Zhang B. Genome-wide analysis of bHLH transcription factor and involvement in the infection by yellow leaf curl virus in tomato (Solanum lycopersicum) BMC Genomics. 2015;16:39. doi: 10.1186/s12864-015-1249-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hudson KA, Hudson ME. The basic Helix-loop-Helix transcription factor family in the sacred Lotus, Nelumbo Nucifera. Trop Plant Biol. 2014;7(2):65–70. doi: 10.1007/s12042-014-9138-4. [DOI] [Google Scholar]
- 30.Zhang X, Luo H, Xu Z, Zhu Y, Ji A, Song J, Chen S. Genome-wide characterisation and analysis of bHLH transcription factors related to tanshinone biosynthesis in Salvia miltiorrhiza. Sci Rep. 2015;5:11244. doi: 10.1038/srep11244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kavas M, Baloglu MC, Atabay ES, Ziplar UT, Dasgan HY, Unver T. Genome-wide characterization and expression analysis of common bean bHLH transcription factors in response to excess salt concentration. Mol Genet Genomics. 2016;291(1):129–143. doi: 10.1007/s00438-015-1095-6. [DOI] [PubMed] [Google Scholar]
- 32.Kumar I, Swaminathan K, Hudson K, Hudson ME. Evolutionary divergence of phytochrome protein function in Zea mays PIF3 signaling. J Exp Bot. 2016;67(14):4231–4240. doi: 10.1093/jxb/erw217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo XJ, Wang JR. Global identification, structural analysis and expression characterization of bHLH transcription factors in wheat. BMC Plant Biol. 2017;17(1):90. doi: 10.1186/s12870-017-1038-y.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X, Duan X, Jiang H, Sun Y, Tang Y, Yuan Z, Guo J, Liang W, Chen L, Yin J, et al. Genome-wide analysis of basic/helix-loop-helix transcription factor family in rice and Arabidopsis. Plant Physiol. 2006;141(4):1167–1184. doi: 10.1104/pp.106.080580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toledo-Ortiz G, Huq E, Quail PH. The Arabidopsis basic/helix-loop-helix transcription factor family. Plant Cell. 2003;15(8):1749–1770. doi: 10.1105/tpc.013839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pires N, Dolan L. Origin and diversification of basic-helix-loop-helix proteins in plants. Mol Biol Evol. 2010;27(4):862–874. doi: 10.1093/molbev/msp288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leivar P, Quail PH. PIFs: pivotal components in a cellular signaling hub. Trends Plant Sci. 2011;16(1):19–28. doi: 10.1016/j.tplants.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakamura Y, Kato T, Yamashino T, Murakami M, Mizuno T. Characterization of a set of phytochrome-interacting factor-like bHLH proteins in Oryza sativa. Biosci Biotechnol Biochem. 2007;71(5):1183–1191. doi: 10.1271/bbb.60643. [DOI] [PubMed] [Google Scholar]
- 39.Roy SW. Recent evidence for the exon theory of genes. Genetica. 2003;118(2–3):251–266. doi: 10.1023/A:1024190617462. [DOI] [PubMed] [Google Scholar]
- 40.Feller A, Hernandez JM, Grotewold E. An ACT-like domain participates in the dimerization of several plant basic-helix-loop-helix transcription factors. J Biol Chem. 2006;281(39):28964–28974. doi: 10.1074/jbc.M603262200. [DOI] [PubMed] [Google Scholar]
- 41.Zhang F, Yao J, Ke J, Zhang L, Lam VQ, Xin XF, Zhou XE, Chen J, Brunzelle J, Griffin PR. Structural basis of JAZ repression of MYC transcription factors in jasmonate signaling. Nature. 2015;525(7568):269–273. doi: 10.1038/nature14661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohashiito K, Bergmann DC. Regulation of the Arabidopsis root vascular initial populationby LONESOME HIGHWAY. Development. 2007;134(16):2959–2968. doi: 10.1242/dev.006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khanna R, Huq EKikis EA, Al-Sady B, Lanzatella C, Quail PH. A novel molecular recognition motif necessary for targeting photoactivated phytochrome signaling to specific basic helix-loop-helix transcription factors. Plant Cell. 2004;16(11):3033–3044. doi: 10.1105/tpc.104.025643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pingault L, Choulet F, Alberti A, Glover N, Wincker P, Feuillet C, Paux E. Deep transcriptome sequencing provides new insights into the structural and functional organization of the wheat genome. Genome Biol. 2015;16(1):29. doi: 10.1186/s13059-015-0601-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanaka A, Nakagawa H, Tomita C, Shimatani Z, Ohtake M, Nomura T, Jiang CJ, Dubouzet JG, Kikuchi S, Sekimoto H. BRASSINOSTEROID UPREGULATED1, encoding a Helix-loop-Helix protein, is a novel gene involved in Brassinosteroid signaling and controls bending of the Lamina joint in Rice. Plant Physiol. 2009;151(2):669–680. doi: 10.1104/pp.109.140806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang H, Yang Y, Wang C, Liu M, Li H, Fu Y, Wang Y, Nie Y, Liu X, Ji W. Large-scale transcriptome comparison reveals distinct gene activations in wheat responding to stripe rust and powdery mildew. BMC Genomics. 2014;15(1):898. doi: 10.1186/1471-2164-15-898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lanubile A, Ferrarini A, Maschietto V, Delledonne M, Marocco A, Bellin D. Functional genomic analysis of constitutive and inducible defense responses to fusarium verticillioides infection in maize genotypes with contrasting ear rot resistance. BMC Genomics. 2014;15(1):710. doi: 10.1186/1471-2164-15-710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sircar S, Parekh N. Functional characterization of drought-responsive modules and genes in Oryza sativa: a network-based approach. Front Genet. 2015;6:256. doi: 10.3389/fgene.2015.00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bednarek P, Piślewska-Bednarek M, Ver Loren van Themaat E, Maddula RK, Svatoš A, et al. Conservation and clade-specific diversification of pathogen-inducible tryptophan and indole glucosinolate metabolism in Arabidopsis thaliana relatives. New Phytol. 2011;192(3):713–726. doi: 10.1111/j.1469-8137.2011.03824.x. [DOI] [PubMed] [Google Scholar]
- 50.Mayer KFX, Rogers J, Doležel J, Pozniak C, Eversole K, Feuillet C, Gill B, Friebe B, Lukaszewski AJ, Sourdille P. A chromsome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 2014;345(6194):1251788. doi: 10.1126/science.1251788. [DOI] [PubMed] [Google Scholar]
- 51.Feller A, Machemer K, Braun EL, Grotewold E. Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 2011;66(1):94–116. doi: 10.1111/j.1365-313X.2010.04459.x. [DOI] [PubMed] [Google Scholar]
- 52.Bernhardt C, Lee MM, Gonzalez A, Zhang F, Lloyd A, Schiefelbein J. The bHLH genes GLABRA3 (GL3) and ENHANCER OF GLABRA3 (EGL3) specify epidermal cell fate in the Arabidopsis root. Development. 2003;130(26):6431–6439. doi: 10.1242/dev.00880. [DOI] [PubMed] [Google Scholar]
- 53.Morohashi K, Zhao M, Yang M, Read B, Lloyd A, Lamb R, Grotewold E. Participation of the Arabidopsis bHLH factor GL3 in Trichome initiation regulatory events. Plant Physiol. 2007;145(3):736–746. doi: 10.1104/pp.107.104521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qi T, Song S, Ren Q, Wu D, Huang H, Chen Y, Fan M, Peng W, Ren C, Xie D. The Jasmonate-ZIM-domain proteins interact with the WD-repeat/bHLH/MYB complexes to regulate Jasmonate-mediated anthocyanin accumulation and Trichome initiation in Arabidopsis thaliana. Plant Cell. 2011;23(5):1795–1814. doi: 10.1105/tpc.111.083261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schweizer F, Fernã n-CP, Zander M, Diez-Diaz M, Fonseca S, Glauser G, Lewsey MG, Ecker JR, Solano R, Reymond P. Arabidopsis basic helix-loop-helix transcription factors MYC2, MYC3, and MYC4 regulate glucosinolate biosynthesis, insect performance, and feeding behavior. Plant Cell. 2013;25(8):3117–3132. doi: 10.1105/tpc.113.115139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vera-Sirera F, Derybel B, Úrbez C, Kouklas E, Pesquera M, Álvarez-Mahecha JC, Minguet E, Tuominen H, Carbonell J, Borst JW. A bHLH-based feedback loop restricts vascular cell proliferation in plants. Dev Cell. 2015;35(4):432–443. doi: 10.1016/j.devcel.2015.10.022. [DOI] [PubMed] [Google Scholar]
- 57.Wang N, Cui Y, Liu Y, Fan H, Du J, Huang Z, Yuan Y, Wu H, Ling HQ. Requirement and functional redundancy of Ib subgroup bHLH proteins for iron deficiency responses and uptake in Arabidopsis thaliana. Mol Plant. 2013;6(2):503–513. doi: 10.1093/mp/sss089. [DOI] [PubMed] [Google Scholar]
- 58.Bonneau J, Baumann U, Beasley J, Li Y, Johnson AA. Identification and molecular characterization of the nicotianamine synthase gene family in bread wheat. Plant Biotechnol J. 2016;14(12):2228–2239. doi: 10.1111/pbi.12577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Long TA, Tsukagoshi H, Busch W, Lahner B, Salt DE, Benfey PN. The bHLH transcription factor POPEYE regulates response to iron deficiency in Arabidopsis roots. Plant Cell. 2010;22(7):2219–2236. doi: 10.1105/tpc.110.074096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zheng L, Ying Y, Wang L, Wang F, Whelan J, Shou H. Identification of a novel iron regulated basic helix-loop-helix protein involved in Fe homeostasis in Oryza sativa. BMC Plant Biol. 2010;10(1):166. doi: 10.1186/1471-2229-10-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schlereth A, Möller B, Liu W, Kientz M, Flipse J, Rademacher EH, Schmid M, Jürgens G, Weijers D. MONOPTEROS controls embryonic root initiation by regulating a mobile transcription factor. Nature. 2010;464(7290):913–916. doi: 10.1038/nature08836. [DOI] [PubMed] [Google Scholar]
- 62.Zhang LY, Bai MY, Wu J, Zhu JY, Wang H, Zhang Z, Wang W, Sun Y, Zhao J, Sun X. Antagonistic HLH/bHLH transcription factors mediate Brassinosteroid regulation of cell elongation and plant development in Rice and Arabidopsis. Plant Cell. 2009;21(12):3767–3780. doi: 10.1105/tpc.109.070441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sangwang H, Chen HC, Huang WY, Chu YC, Choutou S, Cheng WH. Ectopic expression of rice OsNCED3 in Arabidopsis increases ABA level and alters leaf morphology. Plant Sci. 2010;178(1):12–22. doi: 10.1016/j.plantsci.2009.09.014. [DOI] [Google Scholar]
- 64.Gremski K, Ditta G, Yanofsky MF. The HECATE genes regulate female reproductive tract development in Arabidopsis thaliana. Development. 2007;134(20):3593–3601. doi: 10.1242/dev.011510. [DOI] [PubMed] [Google Scholar]
- 65.Schuster C, Gaillochet C, Medzihradszky A, Busch W, Daum G, Krebs M, Kehle A, Lohmann JU. A regulatory framework for shoot stem cell control integrating metabolic, transcriptional, and phytohormone signals. Dev Cell. 2014;28(4):438–449. doi: 10.1016/j.devcel.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 66.Zhu L, Xin R, Bu Q, Shen H, Dang J, Huq E. A Negative Feedback Loop between PHYTOCHROME INTERACTING FACTORs and HECATE proteins fine-tunes Photomorphogenesis in Arabidopsis. Plant Cell. 2016;28(4):855–874. doi: 10.1105/tpc.16.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li N, Zhang DS, Liu HS, Yin CS, Li XX, Liang WQ, Yuan Z, Xu B, Chu HW, Wang J. The rice tapetum degeneration retardation gene is required for tapetum degradation and anther development. Plant Cell. 2006;18(11):2999–3014. doi: 10.1105/tpc.106.044107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Niu N, Liang W, Yang X, Jin W, Wilson ZA, Hu J, Zhang D. EAT1 promotes tapetal cell death by regulating aspartic proteases during male reproductive development in rice. Nat Commun. 2013;4(4):1445. doi: 10.1038/ncomms2396.. [DOI] [PubMed] [Google Scholar]
- 69.Heang D, Sassa H. An atypical bHLH protein encoded by POSITIVE REGULATOR OF GRAIN LENGTH 2 is involved in controlling grain length and weight of rice through interaction with a typical bHLH protein APG. Breed Sci. 2012;62(2):133–141. doi: 10.1270/jsbbs.62.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang L, Wu LM, Greaves IK, Zhu A, Dennis ES, Peacock WJ. PIF4-controlled auxin pathway contributes to hybrid vigor in Arabidopsis thaliana. Proc Natl Acad Sci U S A. 2017;114(17):E3555. doi: 10.1073/pnas.1703179114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim JH, Lee HJ, Jung JH, Lee S, Park CM. HOS1 facilitates the Phytochrome B-mediated inhibition of PIF4 function during hypocotyl growth in Arabidopsis. Mol Plant. 2017;10(2):274–284. doi: 10.1016/j.molp.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 72.Wang F, Zhu H, Chen D, Li Z, Peng R, Yao Q. A grape bHLH transcription factor gene, VvbHLH1 , increases the accumulation of flavonoids and enhances salt and drought tolerance in transgenic Arabidopsis thaliana. Plant Cell Tiss Org Cult. 2016;125(2):387–398. doi: 10.1007/s11240-016-0953-1. [DOI] [Google Scholar]
- 73.Liu Y, Ji X, Nie X, Qu M, Zheng L, Tan Z, Zhao H, Huo L, Liu S, Zhang B. Arabidopsis AtbHLH112 regulates the expression of genes involved in abiotic stress tolerance by binding to their E-box and GCG-box motifs. New Phytol. 2015;207(3):692–709. doi: 10.1111/nph.13387. [DOI] [PubMed] [Google Scholar]
- 74.Takahashi Y, Ebisu Y, Kinoshita T, Doi M, Okuma E, Murata Y, Shimazaki K. bHLH transcription factors that facilitate K+ uptake during stomatal opening are repressed by abscisic acid through phosphorylation. Sci Signal. 2013;6(280):ra48. doi: 10.1126/scisignal.2003760. [DOI] [PubMed] [Google Scholar]
- 75.Sasakisekimoto Y, Jikumaru Y, Obayashi T, Saito H, Masuda S, Kamiya Y, Ohta H, Shirasu K. Basic Helix-loop-Helix transcription factors JASMONATE-ASSOCIATED MYC2-LIKE1 (JAM1), JAM2, and JAM3 are negative regulators of Jasmonate responses in Arabidopsis. Plant Physiol. 2013;163(1):291–304. doi: 10.1104/pp.113.220129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shimizu T, Nojiri H, Yamane H, Okada K, Mochizuki S. Stress-induced expression of the transcription factor RERJ1 is tightly; regulated in response to jasmonic acid accumulation in rice. Protoplasma. 2013;250(1):241–249. doi: 10.1007/s00709-012-0400-z. [DOI] [PubMed] [Google Scholar]
- 77.Heang D, Sassa H. Antagonistic actions of HLH/bHLH proteins are involved in grain length and weight in Rice. PLoS One. 2012;7(2):e31325. doi: 10.1371/journal.pone.0031325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wasternack C, Hause B. Jasmonates: biosynthesis, perception, signal transduction and action in plant stress response, growth and development. An update to the 2007 review in annals of botany. Ann Bot. 2013;111(6):1021–1058. doi: 10.1093/aob/mct067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Van dDD, Leon-Reyes A, Koornneef A, Van Verk MC, Rodenburg N, Pauwels L, Goossens A, Körbes AP, Memelink J, Ritsema T. Salicylic acid suppresses jasmonic acid signaling downstream of SCFCOI1-JAZ by targeting GCC promoter motifs via transcription factor ORA59. Plant Cell. 2013;25(2):744–761. doi: 10.1105/tpc.112.108548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Caarls L, Pieterse CMJ, Wees SCMV. How salicylic acid takes transcriptional control over jasmonic acid signaling. Front Plant Sci. 2015;6(170):170. doi: 10.3389/fpls.2015.00170.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vos I, Verhage A, Schuurink R, Watt L, Pieterse C, Van Wees S. Onset of herbivore-induced resistance in systemic tissue primed for jasmonate-dependent defenses is activated by abscisic acid. Front Plant Sci. 2013;4(539). 10.3389/fpls.2013.00539. [DOI] [PMC free article] [PubMed]
- 82.Bu Q, Jiang H, Li CB, Zhai Q, Zhang J, Wu X, Sun J, Xie Q, Li C. Role of the Arabidopsis thaliana NAC transcription factors ANAC019 and ANAC055 in regulating jasmonic acid-signaled defense responses. Cell Res. 2009;18(5):756–767. doi: 10.1038/cr.2008.53. [DOI] [PubMed] [Google Scholar]
- 83.Zheng X, Spivey NW, Zeng W, Liu PP, Fu ZQ, Klessig DF, He SY, Dong X. Coronatine promotes Pseudomonas syringae virulence in plants by activating a signaling cascade that inhibits salicylic acid accumulation. Cell Host Microbe. 2012;11(6):587–596. doi: 10.1016/j.chom.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.De VD, Seifi S, Filipe O, Haeck A, Nguyen HS, Demeestere K, Höfte MM. The DELLA protein SLR1 integrates and amplifies salicylic acid- and jasmonic acid-dependent innate immunity in rice. Plant Physiol. 2016;170(3):1831–1847. doi: 10.1104/pp.15.01515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wei K, Wang Y, Xie D. Identification and expression profile analysis of the protein kinase gene superfamily in maize development. Mol Breed. 2014;33(1):155–172. doi: 10.1007/s11032-013-9941-x. [DOI] [Google Scholar]
- 86.Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11(9):1650–1667. doi: 10.1038/nprot.2016.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 88.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9(1):559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gutjahr C, Banba M, Croset V, An K, Miyao A, An G, Hirochika H, Imaizumi-Anraku H, Paszkowski U. Arbuscular mycorrhiza: specific signaling in Rice transcends the common Symbiosis signaling pathway. Plant Cell. 2008;20(11):2989–3005. doi: 10.1105/tpc.108.062414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cui Z, Luo J, Qi C, Ruan Y, Li J, Zhang A, Yang X, He Y. Genome-wide association study (GWAS) reveals the genetic architecture of four husk traits in maize. BMC Genomics. 2016;17(1):946. doi: 10.1186/s12864-016-3229-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu W, Zhao X, Peng Z, Mar TT, Yan L, Zhang Z, Han C, Wang X. A one step real-time RT-PCR assay for the quantitation of wheat yellow mosaic virus (WYMV) Virol J. 2013;10(1):173. doi: 10.1186/1743-422X-10-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of 1154 bHLHs from Arabidopsis, rice, maize and wheat and their related information. (XLS 509 kb)
Table S2. A table shows 180 new names of wheat bHLHs side by side with previous ones. (XLS 73 kb)
Table S3. The distribution of TabHLHs on wheat chromosomes. (XLS 24 kb)
Figure S1. Amino acid sequence alignment of 1154 Arabidopsis, rice, wheat and maize bHLH domains. The red triangles at the bottom indicate the 31 conserved residues. The loop region has been shortened to better visualize the alignment results. (PDF 358 kb)
Figure S2. The unrooted NJ tree of bHLH domains from Arabidopsis, rice, wheat and maize. The blue, red and green rhombuses indicate the TabHLH in A, B and D sub-genomes respectively. (PDF 510 kb)
Table S4. Comparison of the number of genes and intron distribution patterns within the bHLH domain in each bHLH subfamily between wheat and other species. (XLS 84 kb)
Figure S3. Phylogenetic relationships and gene structure features of TabHLHs. The phylogenetic tree was constructed with MEGA 6.0 based on a multiple alignment of 571 wheat bHLH domain sequences. For better visualization and comparison, all introns were displayed in the same length. (PDF 5746 kb)
Figure S4. Phylogenetic relationships and gene structural features of ZmbHLHs. The phylogenetic tree was constructed with MEGA 6.0 based on a multiple alignment of 231 amino acid sequences of maize bHLH domain. For better visualization and comparison, all introns were displayed in the same length. (PDF 1617 kb)
Table S5. Motif with best possible match and the number in each subfamilies and species. (XLS 38 kb)
Figure S5. Architecture of protein conserved motifs. Motifs are graphically represented as colored boxes drawn to scale for bHLH proteins. (PDF 19037 kb)
Figure S6. Arrangement of secondary structure elements in AtMYC3 (5–242). Secondary structure elements overlaid on the sequence alignment of bHLH N-terminal proteins belonging to three subfamilies III(d + e), XIII and IIIf. (PDF 517 kb)
Table S6. The distribution of cis-regulatory elements in TabHLH promoters. “Y” means that the element is present in the promoter region. (XLS 648 kb)
Table S7. Summary of RNA-seq data generated and mapping statistics. (XLS 35 kb)
Table S8. The TPM values of 209 TabHLHs in 15 tissues. (XLS 85 kb)
Table S9. The FPKM values of 188 ZmbHLHs in 18 tissues. (XLS 78 kb)
Table S10. The FPKM values of 110 OsbHLHs in seven tissues. (XLS 35 kb)
Figure S7. Heatmap showing the transcriptional abundance of ZmbHLHs in 18 tissues. (PDF 448 kb)
Figure S8. Heatmap showing log2-transformed (FPKM+1) values for OsbHLHs in 7 tissues. (PDF 850 kb)
Table S11. The TPM values of 80 TabHLHs under drought stress. (XLS 39 kb)
Table S12. The FPKM values of 78 ZmbHLHs under drought stress. (XLS 71 kb)
Table S13. The RPKM values of 59 OsbHLHs under drought stress. (XLS 29 kb)
Table S14. List of primers used in this study for qPCR. (XLS 27 kb)
Table S15. The TPM values of 88 TabHLHs under stripe rust (S) and powdery mildew (P) stresses. (XLS 41 kb)
Table S16. Convertion of maize AGPv3 gene IDs to maize AGPv4 gene IDs. (XLS 63 kb)
Table S17. The RPKM values for 147 ZmbHLHs after F. verticillioides infection. (XLS 42 kb)
Figure S9. Analysis of network topology for various soft-thresholding powers. a. The scale-free fit index (y-axis) as a function of the soft-thresholding power (x-axis). b. The mean connectivity (degree, y-axis) as a function of the soft-thresholding power (x-axis). (PDF 469 kb)
Figure S10. Module-tissue association. Each row corresponds to a module and each column corresponds to a tissue. The top and bottom number in each cell indicate the correlation coefficient between the module and tissue and p-value of the test, respectively. (PDF 3815 kb)
Table S18. Summary of the significantly enriched GO terms (q-value < 0.001) of genes in each co-expression module. BP, biological process; MF, molecule function; CC, cellular component. (XLS 279 kb)
Table S19. List of the “guide genes” of nine subnetwork, and their related information. (XLS 37 kb)
Table S20. The expression profiles of some genes from other families discussed in this study. (XLS 181 kb)
Figure S11. Plyhogenetic analysis of 140 TaDEFLs, 7 AtPDF1s and 6 AtPDF2s. The phylogenetic tree was constructed using MEGA by the NJ method with 1000 bootstrap replications, and the bootstrap values greater than 700 were displayed on the branches. (PDF 411 kb)
Figure S12. Phylogenetic relationship and motif compositions of TaJAZs. a. The phylogenetic tree of TaJAZs constructed using MEGA by the NJ method with 100 bootstrap replicates. b. Domain distribution of TaJAZs was investigated using the MEME web server. c. The consensus sequence of TIFY and Jas motif from TaJAZs. (PDF 414 kb)
Data Availability Statement
All of the datasets supporting the results of this article are included within the article and its Additional files.