

Abstract
Background
Semaglutide, a glucagon-like peptide-1 (GLP-1) analogue, has been co-formulated with the absorption enhancer sodium N-(8-[2-hydroxybenzoyl] amino) caprylate (SNAC) as a tablet for oral administration. This trial (NCT02014259) investigated the pharmacokinetics, safety and tolerability of oral semaglutide in subjects with and without renal impairment.
Methods
Subjects were categorised as having normal renal function (n = 24), mild (n = 12), moderate (n = 12) or severe (n = 12) renal impairment, or end-stage renal disease (ESRD) requiring haemodialysis (n = 11) and received once-daily oral semaglutide (5 mg for 5 days followed by 10 mg for 5 days) in the fasting state, followed by 30 min fasting after dosing. Semaglutide plasma concentrations were measured during dosing and for up to 21 days after the last dose.
Results
Semaglutide exposure (area under the plasma concentration–time curve from time zero to 24 h after the tenth dose and maximum concentration after the tenth dose) did not vary in a consistent pattern across the renal function groups. Similarly, there was no apparent effect of renal impairment on the semaglutide half-life (geometric mean range 152–165 h). Except for one subject in the ESRD group, semaglutide was not detected in urine. Haemodialysis did not affect the pharmacokinetics of semaglutide. Adverse events were in line with those observed for other GLP-1 receptor agonists and no safety concerns were identified.
Conclusion
There was no apparent effect of renal impairment or haemodialysis on the pharmacokinetics of oral semaglutide. Based on this trial, renal impairment should not affect dose recommendations for oral semaglutide.
Key Points
| Renal impairment did not appear to impact the pharmacokinetic properties of oral semaglutide following ten consecutive once-daily doses. |
| Oral semaglutide was well-tolerated in subjects with varying degrees of renal impairment. |
| These data suggest that renal impairment should not affect the dosing recommendations of oral semaglutide. |
Introduction
Renal impairment is a frequent co-morbidity in patients with type 2 diabetes mellitus and can affect the metabolism and excretion of antidiabetic medications [1]. Altered pharmacokinetic properties of antidiabetic drugs in subjects with renal impairment may lead to an increased risk of adverse effects [2]. Hence, some antidiabetic medications are contraindicated or should be used with caution in this population, leading to more limited treatment options in renally impaired patients with type 2 diabetes.
Semaglutide is a glucagon-like peptide-1 (GLP-1) analogue in late-stage clinical development for the treatment of type 2 diabetes. Semaglutide has 94% sequence homology to native human GLP-1 [3]. Structural differences between native GLP-1 and semaglutide include amino acid substitutions at position 8 (alanine to α-aminoisobutyric acid) and position 34 (lysine to arginine), and acylation of the lysine in position 26 with a spacer and C-18 fatty diacid chain [4]. The substitution at position 8 renders semaglutide less susceptible to degradation by dipeptidyl peptidase-4 (DPP-4), while the lysine acylation improves binding to albumin [4]. In phase III trials in patients with type 2 diabetes, once-weekly subcutaneous administration of semaglutide has been shown to significantly improve glycaemic control and reduce body weight compared with placebo when given as monotherapy, and compared with sitagliptin, extended-release exenatide or insulin glargine when given as add-on therapy to oral antidiabetic drugs [5–8]. Treatment with once-weekly subcutaneous semaglutide has also been associated with a reduced incidence of cardiovascular events in patients with type 2 diabetes at high cardiovascular risk [9].
An oral formulation of semaglutide may lead to earlier initiation of treatment and may improve acceptance and adherence for some patients. However, peptide-based drugs, including GLP-1 receptor agonists, typically have very low bioavailability when orally administered due to extensive degradation by proteolytic enzymes and poor absorption across the gastrointestinal mucosa [10, 11]. To achieve adequate bioavailability after oral administration, co-formulation with an absorption enhancer is necessary. Oral semaglutide is being developed as a co-formulation with the absorption enhancer sodium N-(8-[2-hydroxybenzoyl] amino) caprylate (SNAC). The mode of action of SNAC involves a localised increase in pH that protects semaglutide against proteolytic degradation and facilitates the absorption of semaglutide across the gastric epithelium [12, 13]. Once-daily oral semaglutide resulted in better glycaemic control and greater reductions in body weight than placebo in a 26-week phase II dose–response study in patients with type 2 diabetes [14].
Semaglutide is metabolised by proteolytic cleavage of the peptide backbone and sequential β-oxidation of the fatty acid side chain. As semaglutide is not cleared by a specific organ, the impact of reduced renal function on its pharmacokinetics is expected to be limited, as supported by the previous observation that the pharmacokinetics and safety of subcutaneous semaglutide was not affected by renal impairment [15]. However, these data may not predict the impact of potentially altered gastrointestinal physiology due to renal impairment on the pharmacokinetics, safety and tolerability of oral semaglutide. Renal impairment could affect pathways of gut drug metabolism and can be associated with other changes, such as changes in absorption, plasma protein binding, transport and tissue distribution. Further, the effect of renal impairment on the pharmacokinetics of SNAC is unknown. The aim of this trial was to assess the pharmacokinetics, safety and tolerability of oral semaglutide in subjects with renal impairment compared to those with normal renal function.
Patients and Methods
Male or female subjects were eligible for inclusion in the trial if they were aged 18–85 years with body mass index 18.5–40.0 kg/m2. Subjects with renal impairment were categorised according to their estimated creatinine clearance at screening. Clearance of creatinine was estimated by the Cockcroft-Gault formula [16] and was based on age, weight, serum creatinine and sex, and adjusted to body surface area calculated by the Dubois and Dubois formula [17]: mild impairment (60–89 mL/min/1.73 m2), moderate impairment (30–59 mL/min/1.73 m2) and severe impairment (15–29 mL/min/1.73 m2). The end-stage renal disease (ESRD) group included subjects with renal disease requiring haemodialysis. Subjects with normal renal function were judged to be of general good health by the investigator based on medical history, physical examination, vital signs and laboratory safety tests. In line with US Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidelines for studies in subjects with renal impairment [18, 19], the aim was to recruit normal and impaired renal function groups which were comparable with respect to sex, age and body weight as far as was possible.
Key exclusion criteria for all subjects included previous renal transplantation; peritoneal dialysis; Crohn’s disease, ulcerative colitis or other inflammatory bowel disease; chronic pancreatitis or idiopathic acute pancreatitis; personal or family history of medullary thyroid carcinoma or type 2 multiple endocrine neoplasia syndrome; serious cardiac disease (New York Heart Association heart failure functional class III or IV; myocardial infarction within 3 months; unstable angina pectoris); uncontrolled hypertension (diastolic blood pressure ≥ 100 mmHg or systolic blood pressure ≥ 180 mmHg); current hepatic dysfunction or severe hepatic disease; and chronic malabsorption. Subjects with renal impairment using agents known to alter tubular secretion of creatinine (e.g. cimetidine, trimethoprim) were also excluded. Concomitant use of lipid-lowering agents, anti-hypertensive agents and platelet aggregation inhibitors, such as aspirin, was permitted. Subjects with diabetes, other than those being treated with GLP-1 receptor agonists or DPP-4 inhibitors, could be enrolled in the renally impaired groups.
All subjects provided written, informed consent. Relevant Ethics Committees approved the protocol and the trial was conducted in accordance with Good Clinical Practice, the Declaration of Helsinki and FDA/EMA guidelines for studies in subjects with renal impairment [18, 19].
Trial Design
This was a multicentre, open-label, multiple-dose, parallel-group trial (NCT02014259). All subjects were treated with oral semaglutide (Novo Nordisk A/S, Søborg, Denmark) for 10 consecutive days, with dose escalation to mitigate the risk of gastrointestinal adverse effects: 5 days of semaglutide 5 mg followed by 5 days of semaglutide 10 mg (Fig. 1). Oral semaglutide tablets included 300 mg of SNAC. The 10-day dosing regimen was chosen to avoid subjects with all measured concentrations below the lower limit of quantification (LLOQ) and to reduce the variability observed in exposure after a single dose [20]. Subjects received a single tablet of oral semaglutide with 120 mL of water in the morning after overnight fasting (≥ 6 h), with no fluid intake for ≥ 2 h before dosing. Subjects had no food or liquid intake for 30 min after dosing, after which a standardised breakfast was started. Administration of oral concomitant medication was avoided within ± 2 h of dosing. During the dosing period, haemodialysis sessions in subjects with ESRD (three times per week) were started immediately after semaglutide administration. Haemodialysis was performed at two trial sites; the haemodialyser used was either a Fresenius Medical Care (Bad Homburg, Germany) machine or B Braun (Hessen, Germany) Dialog® machine using Xevonta low flux membrane (Lo 15) or high flux membrane (Hi 15 or Hi 18). The blood flow rate was 500–600 mL/min and the ultrafiltration rate was 0–23 mL/min.
Fig. 1.
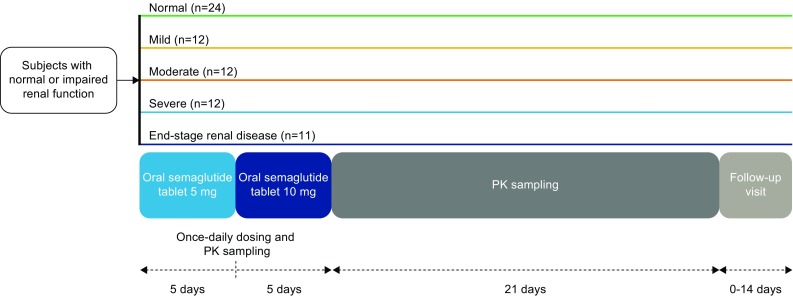
Trial design. PK pharmacokinetics
Blood samples for determination of semaglutide concentration in plasma were drawn pre-dose and at frequent timepoints until 504 h (21 days) after the last dosing. Blood samples for analysis of plasma concentrations of SNAC were collected at additional timepoints within the first 24 h after the last dosing to account for the shorter half-life of SNAC. Additional samples pre- and post-dialysis were collected to determine semaglutide and SNAC concentrations in subjects with ESRD; samples were also collected to determine SNAC concentrations during dialysis on day 9.
Semaglutide Plasma Bioanalysis
Venous blood samples were drawn in K3EDTA tubes and stored at − 20 °C until analysed. A liquid chromatography–tandem mass spectroscopy (LC–MS/MS) assay was used following precipitation of the plasma proteins (Celerion Switzerland AG, Fehraltorf, Switzerland). The LC–MS/MS assay was validated according to current guidelines for bioanalysis of plasma samples in the concentration range 0.729–60.8 nmol/L (3.00–250 ng/mL). A fivefold dilution of each sample was validated to extend the assay range above 60.8 nmol/L. A stable isotope-labelled analogue of semaglutide was used as an internal standard (IS). The analysis was carried out using an AB Sciex API QTrap® 5500 mass spectrometer (Sciex, Framingham, MA, USA). Positive ions were monitored in the multiple reaction-monitoring (MRM) mode with mass transitions m/z 1029.1 → 136.0 Da (semaglutide) and m/z 1033.2 → 136.0 Da (IS). The LC system was a Waters Acquity UPLC® system and the LC column an Acquity UPLC® BEH300 C18, 2.1 × 50 mm (Waters, Elstree, UK). Quantification was performed by peak area ratios of semaglutide versus IS. The calibration curve fitting was done by weighted linear regression (1/concentration2). The lower LLOQ for semaglutide was 0.729 nmol/L.
Sodium N-(8-[2-Hydroxybenzoyl] Amino) Caprylate (SNAC) Plasma Bioanalysis
Venous blood samples were drawn in K3EDTA tubes and stored at − 20 °C until analysed. A LC–MS/MS assay was used following in-line solid-phase sample preparation (Celerion Switzerland AG, Fehraltorf, Switzerland). The LC–MS/MS assay was validated according to current guidelines for bioanalysis of plasma samples in the concentration range 5.00–2000 ng/mL. A fivefold dilution of each sample was validated to extend the assay range above 2000 ng/mL. A structural analogue of SNAC was used as an IS. The analysis was carried out using an AB SCIEX API 4000™ Triple Quadrupole Mass Spectrometer (Sciex, Framingham, MA, USA). Negative ions were monitored in the MRM mode with mass transitions m/z 278.1 → 118.0 Da (SNAC) and m/z 249.0 → 135.0 Da (IS). The LC system was a Cohesive Turbulent Flow system with LC loading column TurboFlowTM Cyclone-P, 50 × 0.5 mm (Thermo Fisher Scientific, Waltham, MA, USA) and analytical column OnyxTM Monolithic C18, 50 × 2.0 mm (Phenomenex, Torrance, CA, USA). Quantification was performed using the peak area ratios of SNAC versus IS. The calibration curve fitting was done by weighted linear regression (1/concentration2). The LLOQ for SNAC was 5.00 ng/mL.
Urine samples were collected from subjects producing urine pre-dose on day 1 and on days 10–11 with fractionated urine collection in predefined intervals after dosing.
Semaglutide and SNAC Urine Bioanalysis
Urine samples were stored at − 20 °C until analysed. To avoid non-specific binding of semaglutide to urine collection containers, 1% Triton™ X-100 (Sigma-Aldrich, Saint Louis, MO, USA) was added to the urine samples in a ratio of 1 part per 9 parts urine. LC–MS/MS assays identical to the above for plasma were used for analysing urine samples for semaglutide and SNAC. The assay ranges and the LLOQ were identical to those in plasma but due to dilution with Triton X-100, the assay ranges were corrected for dilution (correction factor 1.111).
SNAC metabolites were also analysed (data not reported).
Trial Endpoints and Statistical Analysis
Sample size was calculated based on the precision of the ratio of the area under the semaglutide plasma concentration–time curve (AUC) from time zero to 24 h after the tenth dose (AUC24,Day10) between the group of subjects with normal renal function and any one of the groups of subjects with renal impairment. The between-subject standard deviation of log(AUC24,Day10) used in the sample size calculation was 0.60. Twenty-two subjects with evaluable pharmacokinetic profiles in the normal renal function group and 11 in each of the renal impairment groups provided at least 80% probability of achieving a two-sided 90% confidence interval (CI) for the true ratio (R) of AUC24,Day10 between the normal renal function group and a specific renal impairment group, which was contained within the interval [0.66*; 1.51*] where represented the estimated ratio that would be achieved in the present trial.
The primary endpoint was the AUC24,Day10. Secondary endpoints included maximum observed semaglutide plasma concentration 0–24 h after the tenth dose (Cmax,Day10), time to Cmax,Day10 (tmax,Day10), renal clearance of semaglutide (based on 0–36 h after the tenth dose) (CLR) and terminal half-life of semaglutide after the tenth dose (t½,Day10). Analyses of pharmacokinetic endpoints were based on the full analysis set, which consisted of all subjects who were exposed to at least one dose of trial product.
AUC24,Day10 was calculated using the linear trapezoidal method based on observed values and actual sampling times. Cmax,Day10 and tmax,Day10 were derived from the observed pharmacokinetic profiles. t½,Day 10 was calculated as ln(2)/λz, where the terminal elimination rate constant (λz) was estimated by log-linear regression on the terminal part of the pharmacokinetic profiles. AUC24,Day10 and Cmax,Day10 were compared between the normal renal function group and each of the four groups of subjects with renal impairment using a linear normal model with log-transformed endpoint as dependent variable and log (weight) and age as continuous covariates, and sex and renal function group as categorical fixed effects. The model included data from all four renal impairment groups as well as the group with normal renal function and allowed for different variations in each of these five groups. Estimated differences in log-transformed values between the group with normal renal function and each of the four groups with renal impairment were back-transformed to the original scale. For each parameter, the results were presented as the estimated ratios with the corresponding 90% CIs. This was done with an equivalence approach in mind; however, the CIs were not expected to lie within any specific no-effect boundaries due to the large variability seen with oral semaglutide.
In subjects with ESRD, the ratio of the observed AUC of semaglutide from 144 to 148 h during haemodialysis (day 16) over the predicted AUC of semaglutide from 144 to 148 h (AUC144–148 h,dial/AUC144–148 h,pred) was calculated using the terminal elimination rate (λz) (as estimated using the semaglutide plasma concentrations between two haemodialysis sessions at 76, 96, 120 and 144 h) after the tenth dosing.
Pharmacokinetic endpoints were also calculated based on estimated glomerular filtration rate (eGFR) using the Modification of Diet in Renal Disease (MDRD) equation [21].
Endpoints derived for SNAC included AUC24,Day10, Cmax,Day10, tmax,Day10 and CLR. In addition, in subjects with ESRD, the area under the SNAC plasma concentration–time curve from time zero to 4 h (AUC4) after dosing was calculated during haemodialysis on day 9 and with no haemodialysis on day 10 and these were compared using a linear normal model with log-transformed endpoint as dependent variable and haemodialysis (yes/no) and subject as fixed factors. All statistical analyses were performed using SAS® version 9.3 or 9.4 (SAS Institute, Cary, NC, USA).
Safety and tolerability were assessed through reported adverse events (AEs), hypoglycaemic episodes (defined as confirmed if the episode was severe according to the American Diabetes Association [ADA], i.e. requiring third-party assistance [22], or verified by a plasma glucose level of < 3.1 mmol/L [≤ 56 mg/dL]), laboratory safety variables, physical examination, vital signs and electrocardiogram.
Results
Subjects
A total of 71 subjects received oral semaglutide, all of whom completed the trial and were included in the full analysis and safety analysis sets. Of those entering the trial, 24 subjects had normal renal function, 12 subjects each had mild, moderate or severe renal impairment based on the Cockcroft-Gault formula, and 11 had ESRD. Twelve subjects had type 2 diabetes and two had type 1 diabetes; of these, eight were being treated with insulin analogues and six with oral antidiabetic drugs. Baseline demographic and clinical characteristics are summarised in Table 1.
Table 1.
Baseline demographic and clinical characteristics
| Parameters | Renal function group (full analysis set) | ||||
|---|---|---|---|---|---|
| Normal (n = 24) | Mild (n = 12) | Moderate (n = 12) | Severe (n = 12) | ESRD (n = 11) | |
| Age (years) [mean (SD)] | 52 (8) | 57 (13) | 59 (11) | 57 (12) | 54 (13) |
| Sex, male [n (%)] | 15 (62.5) | 5 (41.7) | 9 (75.0) | 9 (75.0) | 7 (63.6) |
| Weight (kg) [mean (SD)] | 84.9 (12.9) | 83.4 (19.5) | 87.2 (15.8) | 85.5 (12.2) | 75.0 (15.3) |
| Body mass index (kg/m2) [mean (SD)] | 28.4 (3.9) | 29.0 (5.5) | 30.1 (5.2) | 28.5 (3.9) | 26.9 (5.3) |
| HbA1c (%) [mean (SD)] | 5.5 (0.3) | 5.9 (0.7) | 6.0 (0.8) | 6.1 (0.6) | 6.2 (1.3) |
| Creatinine clearance (mL/min/1.73 m2) [mean (range)] | 107 (90–132) | 71 (60–89) | 47 (34–56) | 18 (15–27) | 11 (9–14) |
| Subjects with diabetes [n (%)] | 0 | 2 (16.7) | 3 (25.0) | 5 (41.7) | 4 (36.4) |
ESRD end-stage renal disease, HbA1c glycosylated haemoglobin, SD standard deviation
Pharmacokinetics
Geometric mean concentration–time profiles of semaglutide after the tenth dose by renal function group are shown in Fig. 2. There was no consistent pattern of increase or decrease in semaglutide exposure (AUC24,Day10 and Cmax,Day10) by renal function group on day 10 (Table 2 and Fig. 3). Compared to the group with normal renal function, the mean exposure of semaglutide appeared to be higher in the group with mild renal impairment (estimated ratio: AUC24,Day10 1.37 [90% CI 0.91–2.06] and Cmax,Day10 1.39 [0.93–2.06]), whereas lower exposure was observed in the group with severe renal impairment (estimated ratio: AUC24,Day10 0.61 [90% CI 0.42–0.88] and Cmax,Day10 0.61 [0.42–0.87]). Semaglutide exposure was similar for the groups with moderate renal impairment and normal renal function. No consistent or clinically relevant pattern of increase or decrease in semaglutide exposure was observed when subjects were categorised into renal function groups by eGFR based on MDRD, with similar exposure in all groups with the exception of higher apparent mean exposure in the moderately renally impaired group (data not shown).
Fig. 2.

Geometric mean concentration–time profiles of semaglutide after the tenth dose by renal function. Full analysis set
Table 2.
Pharmacokinetic endpoints for semaglutide after the tenth dosing
| Parameters | Renal function group (full analysis set) | ||||
|---|---|---|---|---|---|
| Normal (n = 24) | Mild (n = 12) | Moderate (n = 12) | Severe (n = 12) | ESRD (n = 11) | |
| AUC24,Day10 (nmol·h/L) | 283.7 (53.3) | 378.2 (78.9) | 298.5 (107.3) | 163.5 (65.6) | 287.7 (128.7) |
| Cmax,Day10 (nmol/L) | 14.9 (53.2) | 20.2 (75.9) | 16.6 (102.0) | 8.6 (62.9) | 15.7 (128.3) |
| tmax,Day10 (h) | 1.0 (0.5, 4.0) | 1.0 (0.5, 2.5) | 1.0 (0.5, 4.0) | 1.5 (0.5, 4.0) | 1.0 (0.5, 2.0) |
| t½ (h) | 151.7 (9.1) | 159.3 (12.0) | 162.8 (11.2) | 164.9 (8.9) | 152.8 (49.0) |
Data are geometric means (coefficient of variation) except for tmax,Day10 where median (minimum, maximum) values are presented
AUC24,Day10 area under the plasma concentration–time curve from time zero to 24 h after the tenth dose, Cmax,Day10 maximum plasma concentration 0–24 h after the tenth dose, ESRD end-stage renal disease, tmax,Day10 time to reach Cmax,Day10, t½ terminal half-life
Fig. 3.
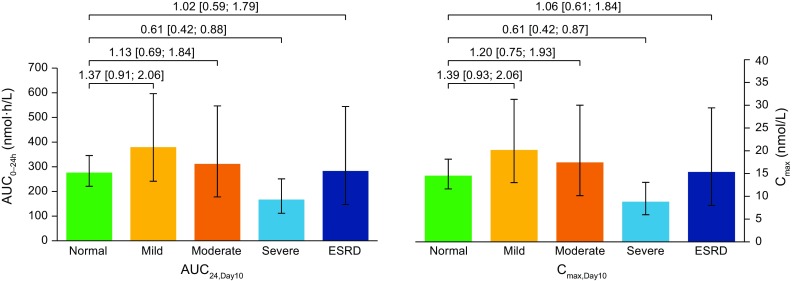
AUC24,Day10 and Cmax,Day10 for semaglutide after the tenth dose by renal function. Full analysis set. Bars are estimated means and 95% confidence intervals. Treatment comparisons show estimated treatment ratio and 90% confidence interval. AUC24,Day10 area under the plasma concentration–time curve from time 0 to 24 h after the tenth dose, CLR renal clearance, Cmax,Day10 maximum plasma concentration 0–24 h after the tenth dose, ESRD end-stage renal disease
The median tmax,Day10 for semaglutide was similar across all groups and ranged from 1.0 to 1.5 h (Table 2). The geometric mean for t½,Day10 appeared to increase slightly from 152 h in subjects with normal renal function to 165 h in those with severe renal impairment, but was similar in the ESRD group and normal renal function group (Table 2). Semaglutide was not generally detected in urine samples except in one subject in the ESRD group (who had a very low urine volume) and therefore CLR was not assessed.
Mean concentration–time profiles of SNAC after the tenth dose by renal function group are shown in Fig. 4 with pharmacokinetic parameters presented in Table 3. The AUC24,Day 10 of SNAC increased with increasing degree of renal impairment, except for in the ESRD group. The estimated ratio of the AUC24,Day10 in each of the renal impairment groups to that in the group with normal renal function was 1.19 (90% CI 1.01–1.42) for subjects with mild renal impairment, 1.30 (90% CI 1.08–1.57) with moderate renal impairment, 1.49 (90% CI 1.26–1.75) with severe renal impairment and 1.32 (90% CI 1.13–1.55) with ESRD. The Cmax,Day10 of SNAC showed no consistent pattern with increasing renal impairment: the estimated ratio compared with normal renal function was 0.94 (90% CI 0.63–1.41) in subjects with mild renal impairment, 0.70 (90% CI 0.45–1.10) with moderate renal impairment, 1.06 (90% CI 0.64–1.75) with severe renal impairment and 0.79 (90% CI 0.51–1.22) with ESRD. The median time to reach the maximum concentration (tmax) after the tenth dosing was similar across the renal function groups and ranged from 0.58 to 0.83 h, while a clear terminal phase was not observed for SNAC and the half-life (t½) could not be estimated. The CLR of SNAC based on 0–36 h after the tenth dosing decreased with severe renal impairment.
Fig. 4.

Mean concentration–time profiles of SNAC after the tenth dose by renal function. Full analysis set. ESRD end-stage renal disease, SNAC sodium N-(8-[2-hydroxybenzoyl] amino) caprylate
Table 3.
Pharmacokinetic endpoints for sodium N-(8-[2-hydroxybenzoyl] amino) caprylate (SNAC) after the tenth dosing
| Parameters | Renal function group (full analysis set) | ||||
|---|---|---|---|---|---|
| Normal (n = 24) | Mild (n = 12) | Moderate (n = 12) | Severe (n = 12) | ESRD (n = 11) | |
| AUC24,Day10 (nmol·h/L) | 1028.8 (29.2) | 1311.6 (31.5) | 1335.9 (34.6) | 1522.8 (31.8) | 1471.4 (27.0) |
| Cmax,Day10 (nmol/L) | 971.4 (89.4) | 1086.5 (99.0) | 734.7 (70.4) | 1074.4 (95.0) | 936.3 (88.5) |
| tmax,Day10 (h) | 0.75 (0.33, 3.00) | 0.58 (0.33, 2.50) | 0.67 (0.33, 6.00) | 0.67 (0.17, 1.00) | 0.83 (0.17, 5.00) |
| CLR (L/h) | 0.06 (104.33) | 0.08 (87.88) | 0.09 (92.63) | 0.04 (98.22) | 0.01 (508.51) |
Data are geometric means (coefficient of variation) except for tmax,Day10 where median (minimum, maximum) values are presented
AUC24,Day10 area under the plasma concentration–time curve from time zero to 24 h after the tenth dose, CLR renal clearance, Cmax,Day10 maximum plasma concentration 0–24 h after the tenth dose, ESRD end-stage renal disease, tmax,Day10 time to reach Cmax,Day10, t½ terminal half-life
Regarding pharmacokinetic endpoints in subjects with ESRD, haemodialysis did not appear to affect exposure of semaglutide, since AUC144–148 h,dial/AUC144–148 h,pred was close to 1 (estimated ratio: 0.97, 95% CI 0.92–1.01). For SNAC, exposure was compared between a dialysis and non-dialysis day. The estimated ratio of the AUC4 of SNAC on day 9 during haemodialysis to the AUC4 of SNAC on day 10 was 0.95 (95% CI 0.74–1.23). These data suggest that haemodialysis did not affect either semaglutide or SNAC exposure.
Safety and Tolerability
Across all renal function groups, 25 subjects (35.2%) reported a total of 53 AEs (Table 4). The proportion of subjects with AEs was higher in the groups with renal impairment (25.0–58.3%) than in the group with normal renal function (20.8%); however, the overall occurrence of AEs did not increase with increasing renal impairment. The majority of AEs were mild (48 events in 23 subjects); four subjects reported five moderate AEs, while none were severe. There were no serious AEs, no deaths and no AEs leading to trial withdrawal. The most frequently reported AEs were gastrointestinal disorders (abdominal distension, vomiting, nausea and abdominal discomfort).
Table 4.
Adverse events and hypoglycaemic episodes
| AEs | Renal function group [N (%), E] | |||||
|---|---|---|---|---|---|---|
| Normal (n = 24) | Mild (n = 12) | Moderate (n = 12) | Severe (n = 12) | ESRD (n = 11) | Total (n = 71) | |
| Overall AEs | 5 (20.8), 5 | 7 (58.3), 19 | 7 (58.3), 16 | 3 (25.0), 7 | 3 (27.3), 6 | 25 (35.2), 53 |
| AEs occurring in > 3% of subjects overall | ||||||
| Abdominal distension | 1 (4.2), 1 | 2 (16.7), 2 | 2 (16.7), 7 | 1 (8.3), 1 | 0 (0), 0 | 6 (8.5), 11 |
| Vomiting | 0 (0), 0 | 1 (8.3), 1 | 1 (8.3), 1 | 2 (16.7), 3 | 2 (18.2), 5 | 6 (8.5), 10 |
| Headache | 2 (8.3), 2 | 2 (16.7), 6 | 0 (0), 0 | 1 (8.3), 1 | 0 (0), 0 | 5 (7.0), 9 |
| Nausea | 1 (4.2), 1 | 0 (0), 0 | 2 (16.7), 2 | 1 (8.3), 1 | 0 (0), 0 | 4 (5.6), 4 |
| Abdominal discomfort | 0 (0), 0 | 3 (25.0), 3 | 0 (0), 0 | 0 (0), 0 | 0 (0), 0 | 3 (4.2), 3 |
| Hypoglycaemiaa | ||||||
| Severe | 0 (0), 0 | 0 (0), 0 | 0 (0), 0 | 0 (0), 0 | 0 (0), 0 | 0 (0), 0 |
| Documented symptomatic | 0 (0), 0 | 0 (0), 0 | 3 (25.0), 5 | 2 (16.7), 2 | 0 (0), 0 | 5 (7.0), 7 |
| Confirmed | 0 (0), 0 | 0 (0), 0 | 1 (8.3), 1 | 0 (0), 0 | 1 (9.1), 1 | 2 (2.8), 2 |
AE adverse event, E number of AEs, ESRD end-stage renal disease, N number of subjects with AE
aHypoglycaemic events were categorised based on American Diabetes Association (ADA) definitions [22]. Severe hypoglycaemia was defined as an event requiring assistance of another person to actively administer carbohydrates or glucagon or take other corrective actions. Documented symptomatic hypoglycaemia was defined as an event during which typical symptoms of hypoglycaemia were accompanied by a measured plasma glucose concentration ≤ 3.9 mmol/L (≤ 70 mg/dL). Confirmed hypoglycaemia was defined as severe according to the ADA classification and/or biochemically confirmed by a plasma glucose value of < 3.1 mmol/L (≤ 56 mg/dL), with or without symptoms consistent with hypoglycaemia
Hypoglycaemia confirmed by plasma glucose concentration of < 3.1 mmol/L (< 56 mg/dL) was reported by two subjects (one episode each), both of whom had diabetes; one in the moderate renal impairment group treated with sulphonylureas and one in the ESRD group treated with insulin (Table 4). Documented symptomatic hypoglycaemia (according to ADA classification) [22] was also low (three subjects [five events] in the moderate renal impairment group and two subjects [two events] in the severe renal impairment group). No severe hypoglycaemic episodes were reported.
There was an increase in the mean pulse rate at the end of treatment in all of the renal function groups but there was no trend towards a greater increase with increasing degree of renal impairment. There were no clinically relevant changes in systolic or diastolic blood pressure, physical examination or electrocardiogram. There were minor increases in mean lipase values in all renal function groups but mean values did not increase with decreasing renal function.
Discussion
Overall, no consistent or clinically relevant pattern of increase or decrease in semaglutide exposure (AUC from time zero to 24 h [AUC24] and maximum concentration [Cmax]) between subjects with varying degrees of renal impairment and normal renal function was observed after 10 consecutive days of once-daily oral administration. This is largely consistent with previous studies that have suggested that impaired renal function has minimal effect on the pharmacokinetics and pharmacodynamics of other GLP-1 receptor agonists [23–27]. Importantly, this trial is also in line with the corresponding study for subcutaneous semaglutide that showed no clinically relevant effect on the pharmacokinetics of semaglutide when administered by subcutaneous injection in 56 subjects [15]. This indicates that the administration form for semaglutide does not affect the pharmacokinetics in subjects with renal impairment.
The median tmax for semaglutide was also similar across renal function groups, while t½ appeared to increase slightly with decreasing renal function (with the exception of the ESRD group), although this was not considered clinically relevant. These data indicate that absorption of oral semaglutide across the gastric epithelium is unaffected by renal impairment and any associated changes in gastrointestinal physiology. There was limited or no excretion of semaglutide via urine, with semaglutide only detected in the urine of one subject in the ESRD group. This is in line with data after subcutaneous administration which showed that intact semaglutide in urine accounted for 3.1% of the administered dose in humans, less than 1% in rats and was not detected in the urine in monkeys [28].
Consistent with observations in subjects with mild-to-severe renal impairment, there appeared to be no effect on semaglutide exposure in subjects requiring haemodialysis. The lack of an effect of ESRD on the pharmacokinetics of GLP-1 analogues is in line with previous results with subcutaneous semaglutide in healthy subjects [15], as well as liraglutide in patients with type 2 diabetes and ESRD [29].
Regarding the impact of renal impairment on the pharmacokinetics of SNAC, AUC24,Day10 increased with increasing impairment, except for in the ESRD group, while Cmax,Day10 revealed no consistent pattern in relation to renal function. As SNAC is an absorption enhancer with no anticipated systemic pharmacodynamic effects, the increased exposure observed (for AUC24,Day10) was not considered clinically relevant. However, there are no published data on the long-term safety of SNAC in subjects with renal impairment. A decrease in CLR was observed with increasing renal impairment; however, no accumulation of SNAC was seen. Furthermore, the estimated ratio of the AUC4 for SNAC during haemodialysis to not during haemodialysis was close to 1.
Previous phase I studies have reported that GLP-1 receptor agonist therapy is well-tolerated in patients with renal impairment [15, 23]. Similarly, in the current trial, oral semaglutide was well-tolerated with no trend towards an increase in the proportion of subjects with AEs with increasing degree of renal impairment, which is in line with the lack of a consistent effect of renal dysfunction on semaglutide exposure. The most frequently reported AEs in all groups were gastrointestinal disorders, which are considered a class effect of GLP-1 receptor agonists. Most gastrointestinal AEs were mild in severity. No severe hypoglycaemic episodes were reported and confirmed hypoglycaemia occurred rarely, consistent with the glucose-dependent mode of action of GLP-1 receptor agonists [30], with no apparent increase with decreasing renal function. These findings are consistent with results from larger trials with GLP-1 receptor agonists [31, 32]. For instance, in the 26-week LIRA-RENAL study of 279 patients with moderate renal impairment, liraglutide did not affect renal function; withdrawals due to gastrointestinal AEs were higher than with placebo but with no increase in hypoglycaemia risk [32]. Minor increases in lipase levels were observed across all renal function groups, which is consistent with increases observed in patients with type 2 diabetes treated with liraglutide [33].
A limitation of the trial is the short duration, which does not permit conclusions on long-term safety. However, no unexpected safety findings were reported in a 26-week phase II trial, in which the safety profile of oral semaglutide was comparable with subcutaneous semaglutide [14]. Previous pharmacokinetic and longer-term phase III studies with subcutaneous semaglutide have indicated a similar safety profile to that of other GLP-1 receptor agonists [5–8, 15, 34]. Further long-term safety data on oral semaglutide will be provided by the ongoing PIONEER phase III clinical trial programme in a large and broad population of subjects with type 2 diabetes, which includes the PIONEER 5 trial in subjects with type 2 diabetes and moderate renal impairment (NCT02827708). A strength of the trial is that the effect of renal impairment on pharmacokinetics was assessed after clinic-based multiple dosing of semaglutide. Effects of renal impairment on pharmacokinetics at steady state were not characterised, as steady state is predicted to be reached after 4–5 weeks. The sample size may appear limited at 11–12 subjects per renally impaired group. However, the number of subjects was more than double that in a comparable trial with liraglutide [29] and also had more patients with normal renal function than in the subcutaneous semaglutide trial [15]. This was to compensate for the loss in statistical power due to the higher inherent variability in semaglutide exposure when orally administered. The inconsistent pattern in semaglutide exposure across renal function groups can be attributed to a lack of effect of renal impairment on semaglutide exposure, as was previously observed with subcutaneous semaglutide [15], together with the higher intrinsic variability of orally administered semaglutide. In addition, the trial was conducted according to regulatory standards and comprised four renal impairment groups (including a group with ESRD) with more than ten subjects in each group and included a reference group with normal renal function that was comparable with respect to baseline characteristics [18, 19].
Conclusion
The pharmacokinetics of oral semaglutide did not appear to be affected by renal impairment, including in subjects with ESRD undergoing haemodialysis. Oral semaglutide was also well-tolerated with no increase in AEs with increasing renal dysfunction. These results indicate that adjusted dosing for oral semaglutide may not be required for patients with impaired renal function.
Acknowledgements
Medical writing and editorial assistance was provided by Andy Bond and Emma Marshman of Spirit Medical Communications Ltd, and was supported by Novo Nordisk A/S.
The authors would like to thank all subjects who participated in this trial. We also acknowledge the trial investigators: Ágnes Réthy (Péterfy Sándor utcai Kórház, Budapest, Hungary), István Kiss and Szent Imre Egyetemi Oktatókórház, Budapest, Hungary), Petr Šrámek (Pharmaceutical Research Associates CZ, s.r.o. Prague, Czech Republic) and Sylvia Dusilová-Sulková (Hemodialyzační středisko, Hradec Králové-Nový Hradec Králové, Czech Republic); Michael Pilgaard Andersen (Novo Nordisk A/S) for support with the semaglutide and SNAC bioanalyses; and Morten Donsmark and Salvatore Calanna (Novo Nordisk A/S) for their review of the manuscript.
Funding
This trial was sponsored by Novo Nordisk A/S, Søborg, Denmark.
Author Contributions
All authors participated in the trial design, were involved in data analysis and interpretation, and participated in writing the manuscript together with medical writing services provided by the sponsor. All authors have approved the submitted manuscript.
Conflict of interest
CG, FLS, MT and TWA are employees of, and own stock in, Novo Nordisk A/S.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Consent to participate
Informed consent was obtained from all individual participants included in the trial.
References
- 1.Davies M, Chatterjee S, Khunti K. The treatment of type 2 diabetes in the presence of renal impairment: what we should know about newer therapies. Clin Pharmacol. 2016;8:61–81. doi: 10.2147/CPAA.S82008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arnouts P, Bolignano D, Nistor I, et al. Glucose-lowering drugs in patients with chronic kidney disease: a narrative review on pharmacokinetic properties. Nephrol Dial Transpl. 2014;29(7):1284–1300. doi: 10.1093/ndt/gft462. [DOI] [PubMed] [Google Scholar]
- 3.Kapitza C, Nosek L, Jensen L, Hartvig H, Jensen CB, Flint A. Semaglutide, a once-weekly human GLP-1 analog, does not reduce the bioavailability of the combined oral contraceptive, ethinylestradiol/levonorgestrel. J Clin Pharmacol. 2015;55(5):497–504. doi: 10.1002/jcph.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lau J, Bloch P, Schäffer L, et al. Discovery of the once-weekly glucagon-like peptide-1 (GLP-1) analogue semaglutide. J Med Chem. 2015;58(18):7370–7380. doi: 10.1021/acs.jmedchem.5b00726. [DOI] [PubMed] [Google Scholar]
- 5.Sorli C, Harashima SI, Tsoukas GM, et al. Efficacy and safety of once-weekly semaglutide monotherapy versus placebo in patients with type 2 diabetes (SUSTAIN 1): a double-blind, randomised, placebo-controlled, parallel-group, multinational, multicentre phase 3a trial. Lancet Diabetes Endocrinol. 2017;5(4):251–260. doi: 10.1016/S2213-8587(17)30013-X. [DOI] [PubMed] [Google Scholar]
- 6.Ahrén B, Masmiquel L, Kumar H, et al. Efficacy and safety of once-weekly semaglutide versus once-daily sitagliptin as an add-on to metformin, thiazolidinediones, or both, in patients with type 2 diabetes (SUSTAIN 2): a 56-week, double-blind, phase 3a, randomised trial. Lancet Diabetes Endocrinol. 2017;5(5):341–354. doi: 10.1016/S2213-8587(17)30092-X. [DOI] [PubMed] [Google Scholar]
- 7.Aroda VR, Bain SC, Cariou B, et al. Efficacy and safety of once-weekly semaglutide versus once-daily insulin glargine as add-on to metformin (with or without sulfonylureas) in insulin-naive patients with type 2 diabetes (SUSTAIN 4): a randomised, open-label, parallel-group, multicentre, multinational, phase 3a trial. Lancet Diabetes Endocrinol. 2017;5(5):355–366. doi: 10.1016/S2213-8587(17)30085-2. [DOI] [PubMed] [Google Scholar]
- 8.Ahmann AJ, Capehorn M, Charpentier G, et al. Efficacy and safety of once-weekly semaglutide versus exenatide ER in subjects with type 2 diabetes (SUSTAIN 3): a 56-week, open-label, randomized clinical trial. Diabetes Care. 2018;41(2):258–266. doi: 10.2337/dc17-0417. [DOI] [PubMed] [Google Scholar]
- 9.Marso SP, Bain SC, Consoli A, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375(19):1834–1844. doi: 10.1056/NEJMoa1607141. [DOI] [PubMed] [Google Scholar]
- 10.Mahato RI, Narang AS, Thoma L, Miller DD. Emerging trends in oral delivery of peptide and protein drugs. Crit Rev Ther Drug Carrier Syst. 2003;20(2–3):153–214. doi: 10.1615/CritRevTherDrugCarrierSyst.v20.i23.30. [DOI] [PubMed] [Google Scholar]
- 11.Graaf CD, Donnelly D, Wootten D, et al. Glucagon-like peptide-1 and its class B G protein-coupled receptors: a long march to therapeutic successes. Pharmacol Rev. 2016;68(4):954–1013. doi: 10.1124/pr.115.011395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buckley ST, Schéele SG, Kirk RK, Knudsen LB. Mechanism of absorption mediated by SNAC in an oral formulation of semaglutide [poster] Diabetes. 2017;66(Suppl 1):1206-P. [Google Scholar]
- 13.Connor A, Borregaard J, Buckley ST, et al. Site of absorption of an oral formulation of semaglutide [poster] Diabetes. 2017;66(Suppl 1):1180-P. [Google Scholar]
- 14.Davies M, Pieber TR, Hartoft-Nielsen M-L, Hansen OKH, Jabbour S, Rosenstock J. Effect of oral semaglutide compared with placebo and subcutaneous semaglutide on glycemic control in patients with type 2 diabetes: a randomized clinical trial. JAMA. 2017;318(15):1460–1470. doi: 10.1001/jama.2017.14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marbury TC, Flint A, Jacobsen JB, Derving Karsbøl J, Lasseter K. Pharmacokinetics and tolerability of a single dose of semaglutide, a human glucagon-like peptide-1 analog, in subjects with and without renal impairment. Clin Pharmacokinet. 2017;56(11):1381–1390. doi: 10.1007/s40262-017-0528-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 17.DuBois D, DuBois EF. A formula to estimate the approximate surface area if height and weight be known. Arch Intern Med. 1916;17:863–871. doi: 10.1001/archinte.1916.00080130010002. [DOI] [Google Scholar]
- 18.European Medicines Agency. Note for guidance on the evaluation of the pharmacokinetics of medicinal products in patients with impaired renal function. 2004. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003123.pdf. Accessed Nov 2017.
- 19.US Food and Drug Administration. Center for Drug Evaluation and Research. Guidance for industry. Pharmacokinetics in patients with impaired renal function—study design, data analysis, and impact on dosing and labeling, Draft Guidance. 2010. Revision 1. https://www.fda.gov/downloads/drugs/guidances/ucm204959.pdf. Accessed Nov 2017.
- 20.Bækdal TA, Blicher TM, Donsmark M, Søndergaard FL. Safety, tolerability, and pharmacokinetics of single escalating doses of oral semaglutide in healthy male subjects [poster] Diabetes. 2017;66(Suppl 1):1191-P. [Google Scholar]
- 21.Levey AS, Bosch JP, Lewis JB, et al. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130(6):461–470. doi: 10.7326/0003-4819-130-6-199903160-00002. [DOI] [PubMed] [Google Scholar]
- 22.Seaquist ER, Anderson J, Childs B, et al. Hypoglycemia and diabetes: a report of a workgroup of the American Diabetes Association and the Endocrine Society. Diabetes Care. 2013;36(5):1384–1395. doi: 10.2337/dc12-2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacobsen LV, Hindsberger C, Robson R, Zdravkovic M. Effect of renal impairment on the pharmacokinetics of the GLP-1 analogue liraglutide. Br J Clin Pharmacol. 2009;68(6):898–905. doi: 10.1111/j.1365-2125.2009.03536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davidson JA, Brett J, Falahati A, Scott D. Mild renal impairment and the efficacy and safety of liraglutide. Endocr Pract. 2011;17(3):345–355. doi: 10.4158/EP10215.OR. [DOI] [PubMed] [Google Scholar]
- 25.Loghin C, de la Peña A, Cui X, Zhang X, Geiser JS, Chien JY. Pharmacokinetics of once weekly dulaglutide in special populations [abstract no. 857] Diabetologia. 2014;57(Suppl 1):S349. [Google Scholar]
- 26.Young MA, Wald JA, Matthews JE, Yang F, Reinhardt RR. Effect of renal impairment on the pharmacokinetics, efficacy, and safety of albiglutide. Postgrad Med. 2014;126(3):35–46. doi: 10.3810/pgm.2014.05.2754. [DOI] [PubMed] [Google Scholar]
- 27.Hanefeld M, Arteaga JM, Leiter LA, et al. Efficacy and safety of lixisenatide in patients with type 2 diabetes and renal impairment. Diabetes Obes Metab. 2017;19(11):1594–1601. doi: 10.1111/dom.12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jensen L, Helleberg H, Roffel A, et al. Absorption, metabolism and excretion of the GLP-1 analogue semaglutide in humans and nonclinical species. Eur J Pharm Sci. 2017;104:31–41. doi: 10.1016/j.ejps.2017.03.020. [DOI] [PubMed] [Google Scholar]
- 29.Osonoi T, Saito M, Tamasawa A, et al. Effect of hemodialysis on plasma glucose profile and plasma level of liraglutide in patients with type 2 diabetes mellitus and end-stage renal disease: a pilot study. PLoS One. 2014;9(12):e113468. doi: 10.1371/journal.pone.0113468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nauck M. Incretin therapies: highlighting common features and differences in the modes of action of glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Diabetes Obes Metab. 2016;18(3):203–216. doi: 10.1111/dom.12591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leiter LA, Carr MC, Stewart M, et al. Efficacy and safety of the once-weekly GLP-1 receptor agonist albiglutide versus sitagliptin in patients with type 2 diabetes and renal impairment: a randomized phase III study. Diabetes Care. 2014;37(10):2723–2730. doi: 10.2337/dc13-2855. [DOI] [PubMed] [Google Scholar]
- 32.Davies MJ, Bain SC, Atkin SL, et al. Efficacy and safety of liraglutide versus placebo as add-on to glucose-lowering therapy in patients with type 2 diabetes and moderate renal impairment (LIRA-RENAL): a randomized clinical trial. Diabetes Care. 2016;39(2):222–230. doi: 10.2337/dc14-2883. [DOI] [PubMed] [Google Scholar]
- 33.Steinberg WM, Buse JB, Ghorbani MLM, Ørsted DD, Nauck MA, LEADER Steering Committee; LEADER Trial Investigators Amylase, lipase, and acute pancreatitis in people with type 2 diabetes treated with liraglutide: results from the LEADER randomized trial. Diabetes Care. 2017;40(7):966–972. doi: 10.2337/dc16-2747. [DOI] [PubMed] [Google Scholar]
- 34.Jensen L, Kupcova V, Arold G, et al. Pharmacokinetics and tolerability of semaglutide in people with hepatic impairment. Diabetes Obes Metab Epub. 2017 doi: 10.1111/dom.13186. [DOI] [PMC free article] [PubMed] [Google Scholar]