

To the Editor:
Among patients with a clinical and imaging-based diagnosis of autosomal dominant polycystic kidney disease (ADPKD), a negative family history for ADPKD (FH−) has been reported in 10%1 and 14.2%2 (this proportion is 27.8% in a broader PKD population3). Clinical reasons for an FH− are unavailable information about the biological parents and undiagnosed mild or variable ADPKD associated with de novo mutations.1–3 The genetic causes for clinically variable ADPKD include locus heterogeneity (PKD1, PKD2, and GANAB), allelic heterogeneity (truncating and non-truncating mutations), hypomorphic alleles, mosaicism, modifier genes, and rare combinations of these.4–7 In 5% to 10% of patients with apparent ADPKD, no mutation is detected (NMD).2,5,6 Seventeen patients in the Early HALT-PKD Trial had both FH− and NMD2; we hypothesized that they would have a different disease course.
A total of 512 patients with ADPKD (418 families) in the Early HALT-PKD Trial2 (part a, Item S1) were analyzed in 4 groups based on a positive family history (FH+) versus FH− and genetic classification (PKD1/PKD2 mutation or NMD): 414 patients with FH+/PKD1/PKD2, 29 with FH+/NMD, 52 with FH−/PKD1/PKD2, and 17 with FH−/NMD (table a, Item S1). Baseline characteristics (part b, table b, Item S1) and disease progression during 60 months were determined for the 4 groups using estimated change from baseline in total kidney volume (TKV), height-adjusted TKV (htTKV), estimated GFR (eGFR), and 24-hour urine albumin excretion (UAE).2 In FH− patients “a diagnosis of ADPKD was based on the presence of at least 20 kidney cysts bilaterally with features consistent with ADPKD.”8(pp. 103–104) Detailed statistical and genetic methods are in parts c and d of Item SI.
Of FH− and FH+ patients, 24.6% and 6.5%, respectively, had NMD (P < 0.001; table a, Item S1). Estimated change from baseline in TKV (Fig 1; Table 1) and htTKV (table c, Item S1) differed significantly across the 4 groups over 60 months (P = 0.03 for both TKV and htTKV) and was lowest in the FH−/NMD group. Annual percentage increases in TKV and htTKV were also significantly different across groups: 3.30% and 3.30%, respectively, in FH−/NMD compared with 5.82% and 5.83% in FH+/PKD1/PKD2, 4.65% and 4.62% in FH+/NMD, and 6.00% and 6.08% in FH−/PKD1/PKD2 (P = 0.03 for both TKV and htTKV; Table 1 and table c of Item S1, respectively). There was no significant difference across the 4 groups in annual change in eGFR (fig a, Item S1, P = 0.09), which was −1.6 mL/min/1.73 m2 in FH−/NMD patients, −2.9 mL/min/1.73 m2 in FH+/PKD1/PKD2, −2.4 mL/min/1.73 m2 in FH+/NMD, and −3.3 mL/min/1.73 m2 in FH−/PKD1/PKD2. Acute and chronic eGFR changes (part e, Item S1) and changes in UAE (fig b, Item S1; P = 0.1) were not significantly different. There were also no significant differences in baseline height-adjusted liver volumes (table b, Item S1). The older patient age at the time of ADPKD diagnosis in FH−/NMD patients suggested milder disease (table b, Item S1).
Figure 1.
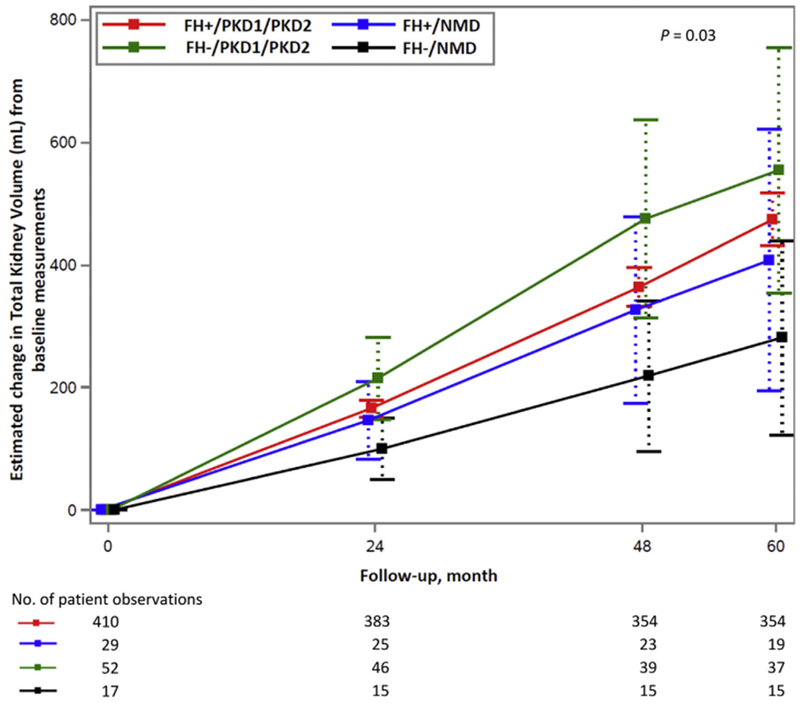
Estimated change in TKV from baseline measurements in 4 patient groups during 60 months of follow-up with 95% confidence intervals. Across patient groups there was a significant difference in the increase in TKV (P = 0.03), with FH−/NMD patients lowest.
Table 1.
Estimated Mean Change in TKV From Baseline Measurements in 4 Patient Groups During 60 Months of Follow-up With 95% Confidence Intervals
| Model Estimated Value at Baseline, mL | Change From Baseline, mL | |||
|---|---|---|---|---|
| 24 mo | 48 mo | 60 mo | ||
| FH+/PKD1/PKD2 | 1,210.68 | 165.32 | 364.01 | 474.11 |
| Lower 95% CI | 1,105.73 | 151.65 | 332.11 | 431.08 |
| Upper 95% CI | 1,331.21 | 179.00 | 395.91 | 517.14 |
| FH+/NMD | 1,093.69 | 146.10 | 326.25 | 407.43 |
| Lower 95% CI | 1,000.32 | 79.45 | 166.09 | 180.44 |
| Upper 95% CI | 1,195.82 | 212.76 | 486.41 | 634.42 |
| FH−/PKD1/PKD2 | 1,353.09 | 214.70 | 474.83 | 554.60 |
| Lower 95% CI | 1,239.09 | 145.86 | 308.45 | 348.58 |
| Upper 95% CI | 1,477.62 | 283.54 | 641.21 | 760.61 |
| FH−/NMD | 1,061.59 | 99.67 | 218.62 | 280.71 |
| Lower 95% CI | 972.22 | 44.53 | 84.67 | 107.79 |
| Upper 95% CI | 1,159.19 | 154.82 | 352.57 | 453.63 |
Note: P = 0.03. See part c, Item S1 for statistical methods and for the formula used to convert per-month slopes into annual percentage changes.
Assessment of FH− relies on each patient’s personal and family history, renal imaging, and genetic analyses. Unlike Reed et al1 and Iliuta et al,3 we were unable to examine the parents of our trial participants. In a recent study, after extensive investigation of the parents of 209 patients with PKD, there were 58 FH− patients: 32 (15.3%) had a de novo mutation, 22 (10.5%) still had indeterminate family history, and 4 (1.9%) had an FH+ in retrospect.3 Uniquely, our study provides longitudinal data on the course of FH−/NMD patients, showing that they have evidence of slower disease progression. These results developed in a controlled clinical trial that eliminated a confounding effect from uncontrolled hypertension (part a, Item S1).2 Because our criteria focused on ADPKD,8 we believe that we minimized the inclusion of atypical cases of PKD with FH−2 that were less likely to be ADPKD.3 However, the onset of ADPKD in FH−/NMD patients could appear delayed when compared to its onset in FH−/PKD1/PKD2 patients if the latter group had predominantly de novo PKD1 mutations. Among our 52 FH−/PKD1/PKD2 patients, 48 (92.3%) had a presumed de novo PKD1 mutation, similar to results of others who reported that PKD1 mutations were more common in de novo disease.1,3 The de novo PKD1 mutations in FH−/PKD1/PKD2 patients could have been responsible for more rapid disease progression and an earlier ADPKD diagnosis despite being FH−.
In summary, 17 FH−/NMD patients with clinically documented ADPKD had a lower annual increase in TKV and htTKV over 60 months, consistent with slower disease progression. This subset of ADPKD patients and their biological parents might be a rewarding group for study with advanced genetic techniques capable of determining with next-generation sequencing if there were undetected mutations at known loci, a new ADPKD locus, a de novo mutation, and/or mosaicism.5,9
Supplementary Material
Acknowledgements:
We thank the hundreds of patients who took part in the HALT-PKD study; the dedicated study coordinators who guided them through the years of participation; the research program coordinators and program managers at WUSTL and the University of Pittsburgh; the staff at the Image Analysis Center; Diane Comer at the Center for Research on Health Data Center for expert assistance with statistical analyses; and Ms Nikki Williams for expert manuscript preparation.
Support: This work was supported by grants from NIDDK (DK62402 to RWS, DK62411 to RDP, DK62410 to VET, DK082230 to CGM, DK62408 to ABC, and DK62401 to Washington University in St. Louis [WUSTL]) and the National Center for Research Resources General Clinical Research Centers (RR000039 to Emory University, RR000585 to the Mayo Clinic, RR000054 to Tufts Medical Center, RR000051 to the University of Colorado, RR023940 to the University of Kansas Medical Center, and RR001032 to Beth Israel Deaconess Medical Center) and by National Center for Advancing Translational Sciences Clinical and Translational Science Awards (RR025008, TR000454 to Emory University; RR024150, TR00135 to the Mayo Clinic; RR025752, TR001064 to Tufts University; RR025780, TR001082 to the University of Colorado; RR025758, TR001102 to Beth Israel Deaconess Medical Center; RR033179, TR000001 to the University of Kansas Medical Center; RR024989, TR000439 to Cleveland Clinic), by funding from the Zell Family Foundation (to the University of Colorado), and by a grant from the PKD Foundation. Study drugs were donated by Boehringer Ingelheim Pharmaceuticals Inc (telmisartan and matched placebo) and Merck & Co Inc (lisinopril).
Footnotes
Supplementary Material
Item S1: Detailed methods and additional results.
Financial Disclosure: RWS reports receiving fees for serving on advisory boards from Otsuka Pharmaceuticals, Janssen Pharmaceuticals, and Ikaria; RDP, consulting fees from Sanofi-Genzyme and Vertex Pharmaceuticals and consulting fees and grant support through his institution from Otsuka; VET and PCH, grant support from Otsuka; WEB, prior grant support from Wyeth-Pfizer; TIS, grant support from Otsuka and Kadmon; MCH, grant support from Novartis and Otsuka; FFR-O, fees for serving on advisory boards from Otsuka; KTB, consulting fees from Otsuka; and ABC, consulting fees from Kadmon, Otsuka, and Pfizer and grant support from Otsuka. The other authors declare that they have no relevant financial interests. JJG died before this manuscript was submitted. WEB affirms that to the best of his knowledge JJG had no relevant financial interests.
Peer Review: Received June 7, 2017. Evaluated by 2 external peer reviewers, with editorial input from a Statistics/Methods Editor, an Associate Editor, and the Editor-in-Chief. Accepted in revised form September 7, 2017.
References
- 1.Reed B, McFann K, Kimberling WJ, et al. Presence of de novo mutations in autosomal dominant polycystic kidney disease patients without family history. Am J Kidney Dis. 2008;52(6): 1042–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schrier RW, Abebe KZ, Perrone RD, et al. ; for the HALT-PKD Trial Investigators. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med. 2014;371(24): 2255–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iliuta I-A, Kalatharan V, Wang K, et al. Polycystic kidney disease without any apparent family history. J Am Soc Nephrol. 2017;28(9):2768–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schrier RW, Brosnahan G, Cadnapaphornchai MA, et al. Predictors of autosomal dominant polycystic kidney disease progression. J Am Soc Nephrol. 2014;25(11):2399–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Porath B, Gainullin VG, Cornec-LeGall E, et al. Mutations in GANAB, encoding the glucosidase 11a subunit, cause autosomal-dominant polycystic kidney and liver disease. Am J Hum Genet. 2016;98(6):1193–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cornec-Le Gall E, Audrezet MP, Chen JM, et al. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol. 2013;24(6):1006–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heyer C, Sundsbak J, Abebe K, et al. Classification of predicted mutation strength of non-truncating PKD1 mutations aids genotype/phenotype correlations in ADPKD. J Am Soc Nephrol. 2016;27(9):2872–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chapman AB, Torres VE, Perrone RD, et al. The HALT polycystic kidney disease trials: design and implementation. Clin J Am Soc Nephrol. 2010;5(1):102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu JT, Campeau PM, Lee BH. Genotype-phenotype correlation — promiscuity in the era of next-generation sequencing. N Engl J Med. 2014;371(7):593–596. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.