

Abstract
Contact guidance or bidirectional migration along aligned fibers modulates many physiological and pathological processes such as wound healing and cancer invasion. Aligned 2D collagen fibrils epitaxially grown on mica substrates replicate many features of contact guidance seen in aligned 3D collagen fiber networks. However, these 2D collagen self-assembled substrates are difficult to image through, do not have known or tunable mechanical properties and cells degrade and mechanically detach collagen fibrils from the surface, leading to an inability to assess contact guidance over long times. Here, we describe the transfer of aligned collagen fibrils from mica substrates to three different functionalized target substrates: glass, polydimethylsiloxane (PDMS) and polyacrylamide (PA). Aligned collagen fibrils can be efficiently transferred to all three substrates. This transfer resulted in substrates that were to varying degrees resistant to cell-mediated collagen fibril deformation that resulted in detachment of the collagen fibril field, allowing for contact guidance to be observed over longer time periods. On these transferred substrates, cell speed is lowest on softer contact guidance cues for both MDA-MB-231 and MTLn3 cells. Intermediate stiffness resulted in the fastest migration. MTLn3 cell directionality was low on soft contact guidance cues, whereas MDA-MB-231 cell directionality marginally increased. It appears that the stiffness of the contact guidance cue regulates contact guidance differently between cell types. The development of this collagen fibril transfer method allows for the attachment of aligned collagen fibrils on substrates, particularly flexible substrates, that do not normally promote aligned collagen fibril growth, increasing the utility of this collagen self-assembly system for the fundamental examination of mechanical regulation of contact guidance.
Keywords: Collagen fibrils, gelatin, polydimethylsiloxane, polyacrylamide, traction force microscopy, tension, stiffness, elastic modulus
Graphical Abstract:
Introduction:
Cells sense several different characteristics of the extracellular matrix (ECM) including composition1, mechanical properties2 and topography3. These properties in turn regulate migration, proliferation, differentiation and other cellular behaviors. One structural feature that drives many physiological and pathological processes is ECM fiber alignment. Both pancreatic as well as breast cancers assemble a tumor microenvironment (TME) composed of aligned collagen fibers that are oriented perpendicular to the tumor margin 4, 5, leading to invasion, metastasis and poor prognosis. These aligned collagen fibers alter force transmission throughout the TME and regulate directional migration of cancer cells away from as well as stromal and immune cell towards the tumor in a process called contact guidance. Directional migration allows for more efficient cell migration towards blood vessels, lymph vessels and along nerve fibers. Contact guidance is driven by preferential focal adhesion strengthening6, contractile actomyosin cytoskeletal alignment7 and protrusion along fibers or fiber like structures8, 9. Cancer, stromal and immune cells in the TME regulate the molecular properties of the contact guidance cue such as collagen density, fiber thickness and fiber crosslinking. Changes in these chemical and biophysical properties result in alteration of the mechanical properties of the TME, including the elastic modulus. It has been shown that human tumors are stiffer than normal tissue10, leading to a more invasive phenotype11. The elastic modulus of normal breast tissue is ~400 Pa, while that of tumor tissue rises to more than 5 kPa10. ECM stiffness regulates the degree of cell-ECM adhesion, the size and turnover of focal adhesions as well as the formation of contractile actomyosin networks or fibers, thus affecting cell motility12. These changes in stiffness of the TME may enhance or dampen the directional response to aligned collagen fibers13. Controlling the mechanical properties independently of the degree of alignment of a fiber network is needed to parse the role of multiple signals in the TME in guiding cell migration. Numerous studies have shown that tumor cell invasion and migration is increased on stiffer ECM. However, how matrix stiffness affects contact guidance behavior is less well understood.
Numerous approaches to fabricate contact guidance cues have been used including fabrication of grooves or gratings6 and microcontact printing14. Contact guidance cues can also be made through electrospinning15 or fiber drawing16. These approaches present non-natural aligned fibers or fiber-like structures. Naturally assembled collagen fibers can be aligned by cellular contraction17, 18, through flow2, 19, 20, magnetic fields21, 22, mechanical rotation or strain23–25, freeze-drying26, confinement27 or self-assembly on mica28–30. We have chosen to focus our work over the past several years on examining contact guidance on collagen fibrils self-assembled on mica. The mica substrate appears to induce an epitaxial growth, leading to aligned collagen fibrils oriented in one direction over a very large area28. While this system is a 2D contact guidance system, it holds several advantages. First, the technique is relatively easy and only requires mica as a substrate and collagen. Second, microscopy in 3D contact guidance environments can be challenging, particularly when examining subcellular processes31, but cells attached to and migrating on epitaxially grown and aligned collagen fibrils are easily imaged7, 28. Third, epitaxially grown and aligned collagen fibrils allows for the tuning of the degree of alignment, collagen density and fibril size characteristics through changes in the collagen concentration, incubation time, pH, ionic composition and strength29. Finally, while this does represent a 2D system, it appears to replicate contact guidance behavior seen in 3D. In 3D contact guidance we observe that MDA-MB-231 cells have high contact guidance fidelity, while MTLn3 cells have low contact guidance fidelity, corresponding to higher invasion of MDA-MB-231 cells shown in vivo32. This behavior is also seen on epitaxially grown and aligned collagen fibrils, while it is not observed on other 2D contact guidance cues28. While there have been cell type differences seen on other 2D contact guidance cues6, 33, most 2D contact guidance cues are quite potent and induce contact guidance in all cell types. Unfortunately, the mechanical properties of the collagen fibril on the surface of the mica is unknown and not tunable, eliminating the possibility of testing how stiffness affects the cells ability to sense contact guidance cues.
Previously, we used epitaxially grown collagen fibrils on mica to show that MDA-MB-231 cells robustly sense aligned collagen fibrils7, 28. Contact guidance fidelity on these substrates differs between cells and is modulated by Rho-kinase mediated contractility7, 28. While collagen can be aligned in different ways, mica is the only substrate that is known to cause the epitaxial growth and alignment of collagen fibrils, but as mentioned above the mechanical properties of the collagen fibril on the surface of the mica is unknown and not tunable. Polydimethylsiloxane (PDMS)34 and polyacrylamide (PA)35 are two of the most commonly used tunable flexible substrates by which to assess the role of the stiffness of the substrate on cell function. Here, we developed an approach to transfer aligned collagen fibrils from mica substrates to other functionalized substrates with different stiffness (glass, polydimethylsiloxane (PDMS), and polyacrylamide (PA)). Attachment of fibers to flexible materials allows for their mechanical characteristics to be probed36. Transfer of materials in general or fibers specifically can be commonly carried out using PDMS stamps14, 37–40, direct contact41–43 or transfer via thermal polymers like poly (N-isopropylacrylamide) (PNIPAAm)44. In this paper we used gelatin as the transfer agent. Gelatin is soluble in aqueous solution, attaches well to the collagen and can form thermally reversible gels around temperatures that do not damage ECM45. We utilized second harmonic generation (SHG)46 and atomic force microscopy (AFM) to quantify the transfer efficiency from mica to target substrates and the maintenance of structural alignment of collagen fibrils on targeted substrates. Furthermore, we quantified collagen fibril deformation that results in cell detachment, alignment and migration on aligned collagen fibrils that have been transferred to both stiff and soft substrates. This transfer method allows for the assessment of the role of mechanical properties in controlling contact guidance. It also opens the possibility of transferring these aligned collagen fibrils to a variety of target substrates with properties different from mica.
Experimental:
Cell Culture
A human mammary basal/claudin low carcinoma cell line (MDA-MB-231, ATCC, Manassas, VA, USA) was cultured in Dulbecco’s Modified Eagles Medium (DMEM) (Sigma Aldrich, St. Louis, MO, USA) containing 10% fetal bovine serum (FBS) (Gibco, Grand Island, New York, USA) and 1% penicillin-streptomycin (pen-strep) (Gibco) at 37°C in 5% CO2. A rat mammary basal adenocarcinoma cell line (MTLn3, Jeffrey E. Segall, Albert Einstein College of Medicine) was authenticated using IDEXX BioResearch (Westbrook, Maine, USA) and cultured in MEMα (Gibco) supplemented with 5% FBS (Gibco) and 1% pen-strep (Gibco) at 37°C in 5% CO2. Imaging media for MDA-MB-231 and MTLn3 cells was the same as the subculturing media, with the exception that no phenol red was included and that 12 mM HEPES (Sigma Aldrich) was included.
Assembling Aligned Collagen Fibril Substrates
Collagen fibrils were epitaxially grown on 15 mm x 15 mm pieces of muscovite mica (highest grade VI, Ted Pella, Redding, CA, USA) that were freshly cleaved using tape. 28 Collagen type I was diluted (10 µg/ml) in the buffer solution consisted of 50 mM Tris-HCl (Fisher Scientific) and 200 mM KCl (Fisher Scientific) at pH 9.2. After incubation of 6–18 h the collagen solution was washed with deionized water, the mica was laid against the edge of a tissue culture dish and the mica was allowed to dry overnight and was used the next day.
Functionalization of Substrates
Glass Coverslips Preparation
Coverslips (glass) were cleaned and functionalized with 1% aminopropyltriethylsilane (Fisher Scientific, Hampton, New Hampshire, USA) in 10 mM acetic acid (Alfa Aesar, Ward Hill, MA, USA) and 6% glutaraldehyde (Electron Microscopy Sciences, Hatfield, PA, USA) in phosphate buffered saline (PBS) without calcium and magnesium (Gibco).
Polydimethylsiloxane (PDMS) Preparation
Flexible PDMS substrates were made by mixing 184 Silicone Elastomer Base (Dow Corning, Midland, MI, USA) with its curing agent in a 20:1 weight ratio. Mixed PDMS was either poured or spin coated onto a plain 22 mm × 22 mm no. 1.5 coverslip, forming a 400–500 µm or 30–40 μm thick substrate, respectively. PDMS was exposed to a vacuum to remove any air bubbles and then cured for 1 h at 60°C. PDMS substrates then were oxidized by oxygen plasma treatment, functionalized with 1% aminopropyltriethylsilane in 10 mM acetic acid and 6% glutaraldehyde in PBS without calcium and magnesium.
Polyacrylamide (PA) Preparation
Flexible PA substrates were generated as described previously. 47 Briefly, 22 mm ×22 mm no. 1.5 coverslips were activated by serial treatments of 0.1 M NaOH, 5% 3-aminopropyltrimethyloxysilane, and 0.5% glutaraldehyde, and each treatment was followed by extensive distilled H2O washing. Activated coverslips were inverted onto a 75µl of 0.03%/0.1% bis and 3% /4% acrylamide, tetramethylethylenediamine, and ammonium persulfate, producing 100–200 µm thick substrates with moduli of 200 Pa and 2000 Pa, respectively. Some of polyacrylamide samples contain 0.2 µm far-red microspheres (Molecular Probes). Coverslips with attached gels were washed in water and spun dry using a custom-made coverslip spinner. Gels were activated by coupling with 2 mg/ml Sulfo-SANPAH with two 8-min UV exposures 1 inch from two 10 W 254 nm UV bulbs (UVP, San Gabriel, CA, USA).
Transferring Fibrils to Different Functionalized Substrates
A gelatin solution (15% w/v) was made and melted completely in 37 °C water bath or incubator. The solution was put on top of collagen fibrils assembled on mica substrates (see above) for 1 h (Figure 1A). After gelatin solution gelled, it was peeled from the surface and transferred to functionalized glass, PDMS and PA substrates for another 1 h. The transferred substrates were submerged in PBS and incubated at 37°C until the gel was completely melted and washed with fresh PBS in 37°C incubator for several hours, making sure no gelatin residue was left. Glass and PDMS substrates were blown dry with an air gun and PA substrates were stored in PBS until use.
Figure 1: Transferring assembled collagen fibrils onto alternative substrates.
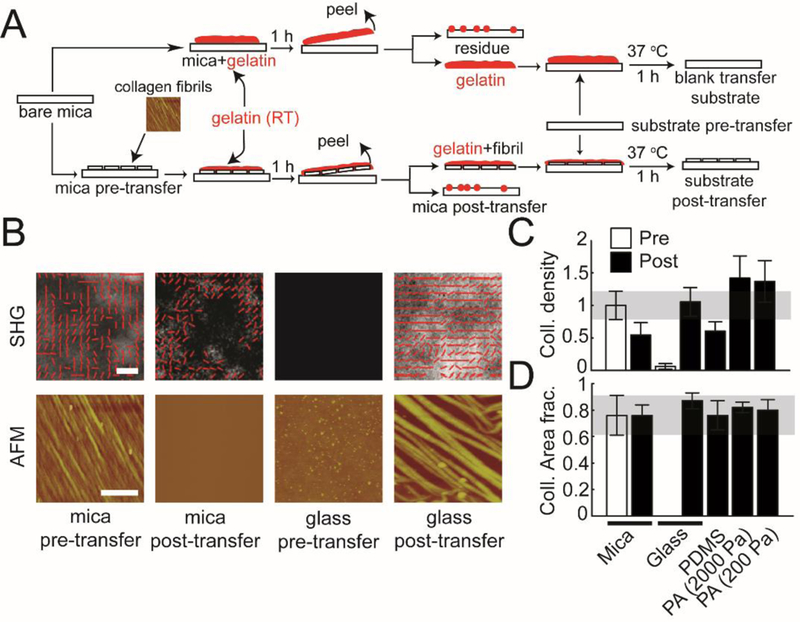
(A) The schematic shows the side view of the steps involved in transferring collagen fibrils from mica to functionalized substrates with different stiffness (glass, PDMS, and PA (2000 and 200 Pa)) using the gelatin protocol described in the Experimental section. (B) Representative AFM and SHG images of aligned collagen fibrils transferred from mica substrates to functionalized glass substrates. ‘mica pre-transfer’ images indicate collagen fibrils on mica substrates before transferring. ‘mica post-transfer’ images show collagen fibrils on mica substrates after transferring. ‘glass pre-transfer’ indicate lack of collagen fibrils on glass substrates before transferring. ‘glass post-transfer’ show collagen fibrils on glass substrates after transferring. (C) Normalized collagen density and (D) collagen area fraction on mica substrates (pre- and post-transfer), glass substrates (pre- and post-transfer), PDMS, and PA (2000Pa and 200Pa). Error bars represent 95% confidence intervals. Calibration bar length for AFM images is 2 μm. Calibration bar length for SHG images is 20 μm.
Imaging Fixed Cells
MDA-MB-231 cells were plated at 40,000–60,000 cells/ml in 2 ml of media for 4, 12 and 24 h on aligned type I collagen fibrils. Cells that were incubated for 12 and 24 h on aligned type I collagen fibrils transferred from mica to target substrates (glass, PDMS, and PA (both 2000 Pa and 200 Pa)). Cells were fixed in 4% paraformaldehyde w/v, dissolved in cytoskeleton buffer. The cytoskeleton buffer included 10 mM MES pH 6.1, 3 mM MgCl, 138 mM KCl, and 2 mM EGTA. Cell samples were placed on heating plates while fixed to maintain cell shape. Cells were then treated with a 100 mM solution of glycine (to react with unreacted aldehydes) and a 0.5% triton X (to permeabolize the cell membrane) for 10 minutes each. Both the glycine and triton X solutions were diluted in cytoskeleton buffer. Cells were then washed with Tris-buffered saline (TBS) containing 150 mM NaCl and 20 mM Tris-Cl pH 7.4 3x for 5 min each. Samples were removed from their last TBS wash and flipped face side down onto a droplet of blocking buffer placed on parafilm for 1 h. The blocking buffer was made with TBS, 0.1% v/v tween-20, and 2% w/v BSA and contained a 1:40 dilution of alexa 488-phalloidin (Molecular Probes). After the hour of blocking and staining, samples were mounted using ProLong Gold mounting media onto microscope slides face down for imaging sealed with nail polish. Fixed and stained cells were imaged using a 20x objective (NA = 0.50, Nikon, Melville, NY, USA) with a charge-coupled device (CoolSNAP HQ2, Photometrics, Tucson, AZ, USA) attached to an inverted microscope (Eclipse Ti, Nikon).
Assessing Cell Alignment
A 4 × 4 matrix of images was taken for each sample for a particular condition. After the images were taken, ImageJ was used to analyze cell orientation angle and aspect ratio. The fluorescence cell image was background subtracted with a 50 pixel rolling ball algorithm, thresholded and converted to a binary image. The binary image was eroded, dilated, opened, closed, eroded, and finally dilated. The cell ROIs were then analyzed for any ROIs greater than 500 pixels. The analyzed outlines were compared to the original phase contrast image to ensure the size and shape was accurate. Overlapping cells were outlined manually to ensure an accurate cell count and area. Only single cells were used in analysis of orientation and aspect ratio. ImageJ was used to analyze mean greyscale value, area, and ellipse properties for each ROI. The ellipse fit in ImageJ gives the major and minor axes lengths as well as the angle of the major axis with respect to a horizontal vector. The aspect ratio of the ellipse was used to determine whether the orientation angle of the cell was used. Cells with an aspect ratio greater than 2 were considered oriented and included in the orientation analysis. Distributions of orientation angles were generated. These distributions were fit to the sum of three von Mises distributions offset by 60° each. The equation used is
| (1) |
where ai is the fraction of cells in peak i, κ determines the breadth of the distribution where large κ results in narrower peaks, μ is the location of each peak (offset by 60°), I0 is the modified Bessel function of the first kind.
The angle distribution for a particular sample was determined to have a peak if the fit to the von Mises distribution resulting in at least one peak amplitude with a 95% confidence interval that did not include zero. Samples with peaks were reoriented so the major peak was at 30° and the second peak was at 90°. The non-aligned samples were not reoriented. Fractional distributions were then averaged across samples for a given condition. These averaged distributions were then fitted to Equation 1. The aligned cell fraction was the area underneath the most significant peak in each condition’s average von Mises fit. In addition, the κ was calculated for each condition based on the fit.
Imaging Microsphere Deformations on Aligned Collagen Fibrils
Microspheres were added to substrates to mark deformations. Carboxylate-modified microspheres (580 nm excitation max, 605 nm emission max, 0.04 μm, ThermoFisher Scientific, Waltham, MA USA) were sonicated to fully suspend microspheres and disrupt aggregates. Then microspheres were diluted in PBS and added to collagen fibrils assembled on mica for 45 mins. Collagen fibrils with attached microspheres were washed 3×, the last time with distilled water and dried before use in deformation assays. Cells were plated at 30,000 cells/ml with microsphere-labeled collagen fibrils. Imaging media used for live-cell experiments was conditioned in a dish containing 960,000 cells/ml of MDA-MB-231 cells the day prior to use. Conditioned media was centrifuged to clarify any cells or cell debris. MDA-MB-231 cells were incubated in the conditioned imaging medium for 2–3 h, respectively, before imaging. Substrates with attached cells were inverted onto two strips of double sided tape attached to a microscope slide to generate a flow chamber. The chamber was filled with conditioned imaging media and sealed with a 1:1:1 mixture of Vaseline, lanolin and paraffin wax. Chambers were imaged by phase contrast microscopy on a heated stage at 37°C every 30 s for 6 h. Phase contrast images were captured at 20x (NA = 0.50, Nikon) with a charge-coupled device (CoolSNAP HQ2, Photometrics) attached to an inverted microscope (Eclipse Ti, Nikon). Fluorescent images were captured at 20x (NA = 0.50, Nikon) with the microscope connected to a fluorescence illumination system (Lumen200PRO, Prior) and an excitation filter 555 and an emission filter 605 was used for microspheres on fibrils.
Matlab was used to detect microspheres positions in each image and then we applied an intensity threshold to find each microsphere centroid and track each from image sequences using a standard particle-tracking algorithm (http://site.physics.georgetown.edu/matlab/tutorial.html) 48 and measure the mean displacement of microspheres. The average displacement of microsphere at any time point, i, was defined as:
| (2) |
where xi is an x-axis position of a single microsphere at time point, i, yi is a y-axis position of a single microsphere at time point, i, and and are average x and y positions over the entire timelapse of a single microsphere. In addition to the displacement, the distance from the cell outline was calculated at each time point, allowing one to bin displacements as function of distance from the cell.
Imaging Collagen Fibrils
Atomic Force Microscope (AFM) Imaging.
Different substrates were imaged using a Dimension 3100 scanning probe microscope with Nanoscope IV controller (Veeco Metrology, LLC, Santa Barbara, CA, USA) was utilized to obtain images of collagen fibrils on mica and transferred coverslips.
Secondary Harmonic Generation (SHG) Microscopy Imaging
The fixed cell samples on the different substrates (mica, transferred glass, PDMS, and PA (2000 Pa) and PA (200 Pa)) were imaged using a mode-locked Ti:Sapphire laser (100 fs pulse width, 1 kHz repetition rate, Libra, Coherent, Santa Clara, CA) that produces an 800 nm fundamental beam. The average power at the sample image plane was controlled using a combination of a half-wave plate and a Glan-Thompson polarizer (Thorlabs, Newton, NJ, USA). For this setup, an inverted microscope (AmScope, Irvine, CA, USA) and Nikon Plan Fluorite objective (20x, 0.50 NA, 2.1 mm WD) was used to focus the beam and the SHG transmission was collected with a Nikon water immersion objective (40x, 0.8 NA, 3.5 mm WD, Nikon). The transmitted SHG signal was reflected by a dichroic mirror (DMLP425T, Thorlabs) and separated from the fundamental beam with two short pass filters, < 450 nm (FGB37M, Thorlabs) and 808 nm notch filter (NF-808.0-E-25.0M, Melles Griot, Rochester, NY, USA), before detection by an intensified charge coupled device (CCD) (iCCD, iStar 334T, Andor, Belfast, UK). Polarized SHG imaging was conducted using a Glan-Thompson polarizer and a half-wave plate mounted on a motor driven rotational stage (Thorlabs) to achieve linear polarization. Images of the samples were collected every 10° from 0° to 350°. The second harmonic signal from the samples on different substrates were imaged in transmission mode in order to quantify the collagen amount. Differences in transmission efficiency were accounted for by measuring the power of the laser signal before and after the sample plane. Additionally, we measured the signal for blank transfer substrates (Figure 1A). This signal was subtracted from that observed for post-transfer substrates (glass, PDMS and the two polyacrylamide samples) in order to calculate the amount of collagen present. A minimum of three images for each experimental condition was taken. From this collection of images, regions of interest (ROIs) were fit using the following equation:
| (3) |
where χzzz/χzxx and χxzx/χzxx are second-order susceptibility tensor element ratios, θe and θo are incident polarization angle and collagen fibril angle, respectively, and c is a normalization constant. The orientation angle of collagen in each ROI was calculated and a histogram was generated. Collagen organization was observed by fitting the orientation angle histogram with a von Mises equation, over a 180° profile. The different ROIs were classified as ordered, non-ordered and non-collagen, to evaluate the micro-scale ordering of the collagen fibrils in the samples. The non-ordered ROIs were included in the 180° bin and were determined by subtracting the model von Mises distribution from the experimentally observed distribution for that bin. The non-collagen ROIs were identified as those ROIs that did not properly fit Equation 3. As the total number of ROIs were kept constant throughout the analysis, the remaining ROIs were calculated as the ordered area fraction.
Statistics
The number of experiments and cells were stated in all the figure legends. At least three independent experiments were conducted. In general, means were calculated with error bars representing 95% confidence intervals.
Results
Collagen Transfer Efficiency
In order to transfer the aligned collagen fibrils from mica to other substrates, a gelatin solution is poured onto the collagen fibrils assembled on mica substrates. After gelation, the gelatin is peeled off and transferred to functionalized substrates: glass (~10 GPa49), PDMS (PDMS, ~300 kPa50) and PA (PA, ~2000 Pa and ~200 Pa) (Figure 1A)35. Glass and PDMS substrates were aldehyde-functionalized and PA substrates were functionalized using Sulfo-SANPAH. To assess the transfer efficiency of fibrillar collagen, both SHG and AFM imaging were conducted (Figure 1B). Both SHG and AFM showed that collagen fibrils were highly aligned and densely packed on mica substrates (mica pre-transfer). After fibrils were transferred to functionalized glass, images of the mica substrates were taken again (mica post-transfer). AFM showed an absence of collagen fibrils, while SHG showed many fewer fibrils on the surface. The fibrils that remained were primarily unaligned, showing no alignment peak (Figure 1B, Supplementary Figure 1). Due to the larger imaging region, the SHG images allowed for a better global view of collagen structure than AFM (125 μm×125 μm vs. 6 μm×6 μm). Functionalized glass substrates before transfer (glass pre-transfer) showed no collagen fibrils on the surface, whereas functionalized glass substrates after transfer (glass post-transfer) showed highly aligned collagen fibrils, albeit the density was sparser. To quantify the transfer efficiency on functionalized substrates, we measured normalized collagen density and collagen area fraction from SHG images. Compared to mica substrates, collagen fibril density on functionalized glass and PA substrates was not significantly different from that of the mica substrates (Figure 1C). However, we did see a somewhat lower collagen fibril density on PDMS substrates (Figure 1C). Collagen area fraction showed no significant difference between mica substrates and all transferred substrates (Figure 1D). Given the good transfer efficiency of collagen fibrils from mica substrates onto different functionalized substrates, we were interested in how MDA-MB-231 cells responded to aligned collagen fibrils transferred from mica substrates.
Fibril Transfer Blocks Cell Detachment
MDA-MB-231 cells sense contact guidance cues robustly over the first 12 h7, 28. However, when cells migrated for a longer time (12–24 h), they began to detach the collagen fibrils. Consequently, MDA-MB-231 cells were fixed, stained and imaged for F-actin at 12 and 24 h after plating. Cell density was normalized to the predicted initial cell density. On mica substrates, normalized cell density decreased over 24 h. The area fraction that the cells occupy increased from 0 to 12 h, due to cell spreading, but then decreased from 12 to 24 h as would be expected for cells detaching from the substrate (Figure 2A&B). Cell detachment was blocked to similar extents when collagen fibrils were transferred to all functionalized substrates (Figure 2A). The decrease in area fraction that the cells occupy is also blocked when collagen fibrils are transferred to all functionalized substrates. However, different substrates block this decrease to different extents. PDMS enhances cell area fraction, indicating that the cells are spreading to a greater extent. Glass and PA substrates block the decrease in area fraction between 12 and 24 h, but do not facilitate an increase in area fraction. Given that aligned collagen fibrils transferred to functionalized substrates appear to block cell detachment, we were interested in whether cell alignment still occurs.
Figure 2: MDA-MB-231 cell detachment on collagen fibrils transferred from mica to alternative substrates.
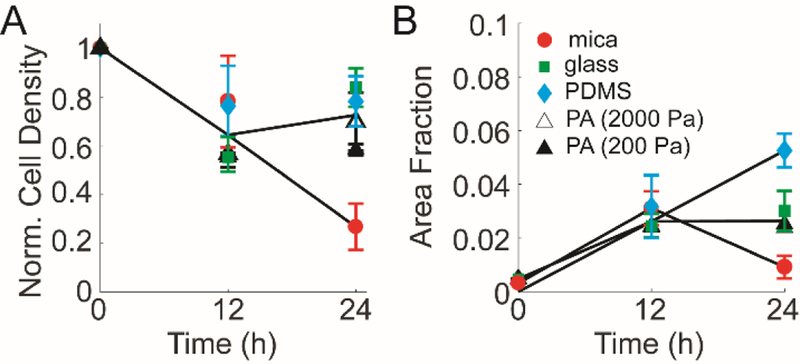
(A) Normalized cell density and (B) area fraction occupied by MDA-MB-231 cells change with time on aligned collagen fibrils assembled on mica (circles). The decrease seen at 24 h is blocked when aligned collagen fibrils are transferred to glass (closed square), PDMS (closed diamond), and PA (2000Pa (open triangle) and 200 Pa (closed triangle)) as compared to aligned collagen fibrils assembled on mica (closed circle). Error bars represent 95% confidence intervals.
Fibril Transfer Facilitates Cell Alignment
We assessed cell alignment on the mica and functionalized substrates, where collagen fibrils have been transferred at various time points by examining the orientation distribution of cells as we have done previously28. Images of fixed MDA-MB-231 cells were taken at 12 h on mica, glass, PDMS and PA (2000 Pa and 200 Pa) substrates. Representative images at 12 h were shown (Figure 3A–E). Cells on mica substrates were not frequently elongated and aligned, showing minimal directional orientation. This differs from previous studies where we observed high alignment and directional migration7, 28, however, in those studies we examined early time points between 1–12 h, well before cell and collagen fibril detachment were occurring. Cells on other flexible substrates presented spindle-like morphology and showed high alignment. Cell angle distributions were calculated from hundreds of cells and fit with von Mises distributions as described in the Materials and Methods section (Figure 3F–H). The glass substrate resulted in the highest cell alignment (narrowest peak). Mica substrates showed low cell alignment. The first peak in the von Mises distribution is the dominant cell alignment direction. One measure of cell alignment is the fraction of cells that fall under the significant peak. This is referred to as the aligned cell fraction. This was calculated for all substrates at 12 h (Figure 3I). Glass substrates appear to be more aligned than mica and other flexible substrates at 12 h. Because the area fraction taken up by cells changes over time (Figure 2B), we calculated the product of the area fraction and the aligned cell fraction, which yields the cell alignment area fraction (Figure 3J). This rewards substrates for both resisting cell detachment and showing cell alignment. Cell alignment area fraction on transferred substrates were all significantly higher than that on mica substrates at 12 h (Figure 3J). In addition to probing cell alignment at 12 h, we also assessed cell alignment at 24 h.
Figure 3: MDA-MB-231 cell alignment on collagen fibrils transferred from mica to alternative substrates.
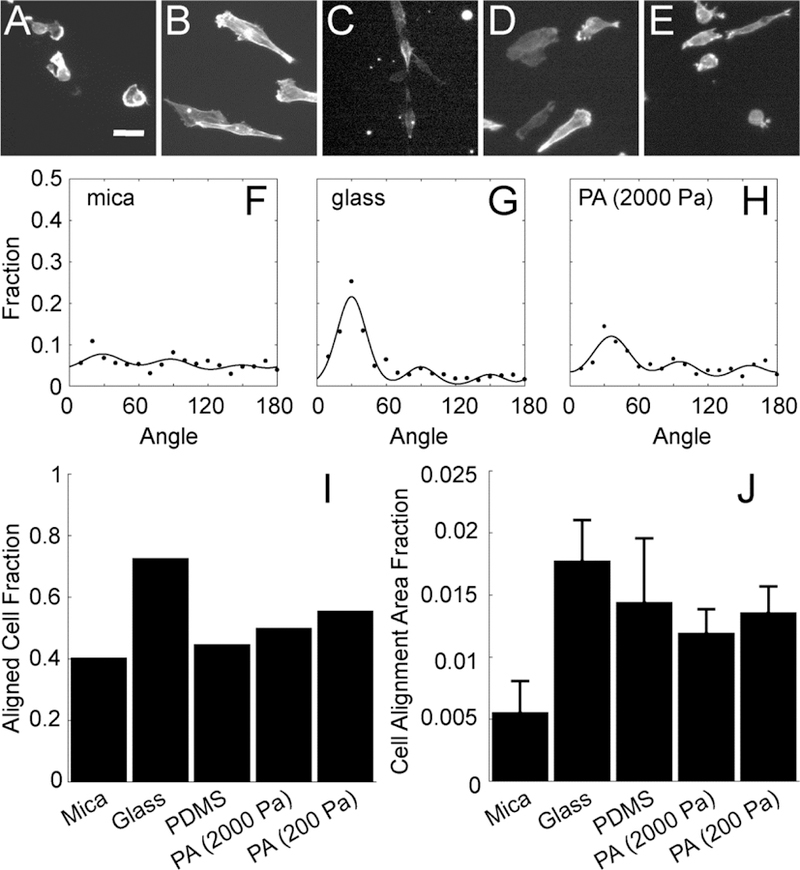
Images of F-actin in cells plated on aligned collagen fibrils assembled on (A) mica or transferred to (B) glass, (C) PDMS, (D) PA (2000 Pa) or (E) PA (200 Pa). (F-H) The angle distribution of cells 12 h after plating on aligned collagen fibrils assembled on mica, transferred glass and PA (2000 Pa). The sum of three von Mises distributions separated by 60 degrees is used to fit the data. (I) Aligned cell fraction and (J) cell alignment area fraction of cells adhered to aligned collagen fibrils assmbled on mica or transferred to glass, PDMS or PA (2000 Pa and 200 Pa). Error bars represent 95% confidence intervals. Calibration bar length is 30 μm.
We assessed aligned cell fraction and cell alignment area fraction on mica and flexible substrates from 12 to 24 h. Aligned cell fraction increased on mica substrates modestly (Figure 4A), while cell alignment area fraction remained relatively low (Figure 4B). On functionalized substrates, aligned cell fraction decreased slightly or remained relatively constant (Figure 4A). However, cell alignment area fraction on PDMS appears to increase, whereas glass remains constant and PA marginally decreases between 12 and 24 h (Figure 4B). Cell alignment area fraction on all functionalized substrates is higher than that on mica substrates at 24 h, indicating a blocking of the cell detachment phenotype, but retention of the cell alignment phenotype. Another measure of alignment is the width of the von Mises distribution, characterized by κ (Equation 1) calculated from the fit of the cell orientation distribution (Figure 4C). The κcell for mica substrates increased from 12 to 24 h, agreeing with the measurement of the aligned cell fraction. The κcell did not change dramatically over time for functionalized substrates, indicating that contact guidance was not decreasing and alignment was remaining constant. Given that transferring collagen fibrils from mica to functionalized substrates block cell detachment while retaining cell alignment, we wanted to measure if there was a quantifiable change in collagen fibril deformation under transfer conditions.
Figure 4: MDA-MB-231 cell alignment on collagen fibrils transferred from mica to alternative substrates at different times.
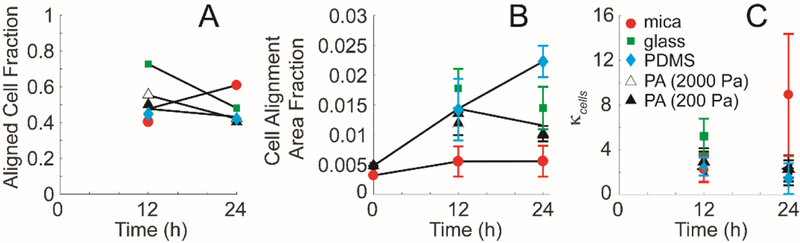
(A) Aligned cell fraction, (B) cell alignment area fraction and (C) distribution spread parameter, κcells as a function of time. Cells adhered to aligned collagen fibrils assmbled on mica (closed circle) or transferred to glass (closed square), PDMS (closed diamond) or PA (2000 Pa (open triangle) and 200 Pa (closed triangle)). Error bars represent 95% confidence intervals.
Fibril Transfer Modulates Cell-mediated Collagen Fibril Deformations
In order to assess the cells’ abilities to deform, remodel and potentially detach collagen fibrils on substrates, fluorescently labeled carboxylate-modified microspheres were adsorbed to the collagen fibrils assembled on the mica substrates or assembled and then transferred to functionalized glass and PA substrates. Microspheres appear to more tightly adsorb to the substrate when collagen fibrils are present indicating a preference for binding collagen fibrils over mica, glass or PA substrates (Figure 5G). Indeed, others have shown carboxylate microspheres bind collagen well51. Microsphere positions were tracked over time and displacements were calculated. Displacement was calculated as the distance between a single microsphere at a given time point, t and the same microsphere at the initial time, t0 (Equation 2). In addition, the distance at any time point t between the cell edge and the microsphere can be calculated. Average displacements can be the result of two mechanisms: reversible collagen fibril deformation and irreversible collagen fibril degradation. Three different colors were used to represent large average displacements (>2 μm, blue), intermediate average displacements (600 nm – 2 μm, green) and small average displacements of collagen fibrils (< 600 nm, red). MDA-MB-231 cells on mica substrates exerted larger collagen fibril displacements than cells plated on collagen fibrils transferred to glass or PA (Figure 5A–C). Microspheres that were close to the cell edge or underneath the MDA-MB-231 cells on mica substrates responded with large displacements, indicating a tearing of the collagen fibrils from the surface in a concerted fashion (Figure 5A). When collagen fibrils were transferred to glass or PA substrates, the magnitudes of the deformations at the same positions are much smaller and no tearing of the collagen fibrils from the surface is detected (Figure 5B&C). The displacement of these substrates was sorted according to distance between the cell edge and a single microsphere. The distribution of displacements was then plotted for collagen fibrils assembled on mica substrates and also assembled on mica substrates and transferred to glass or PA substrates. Displacements should be larger for small distances between the cell edge and microsphere (0–10 µm), than for large distances between the cell edge and microsphere (180–190 μm). Indeed, mica substrates showed a smaller fraction of small displacements as compared to either glass or PA substrates (Figure 5D). In addition, the mica substrate showed a much larger fraction of large (> 6 μm) displacements than either the glass or PA substrates (Figure 5E). The microsphere displacement at which the distribution of microsphere displacements for glass substrates crosses over that for mica substrates is lower than for PA substrates, indicating that PA substrates allow for intermediate displacements. This is reasonable given that they are more flexible than the glass substrates. The distribution of microsphere displacements is different for microspheres far away from the cell edge. In general, the microsphere displacements are much lower as compared to areas closer to the cell edge. However, the flexible substrates still show larger microsphere displacements as compared to either mica or glass, indicating that the cell may be able to transmit forces over relatively large distances on these substrates (Figure 5F). Given that transferring collagen fibrils from mica to functionalized substrates appears to block irreversible collagen fibril deformations leading to cell detachment, we examined the collagen fibril orientation directly using SHG.
Figure 5: Trajectories of fluorescent microspheres adsorbed on aligned collagen fibrils.
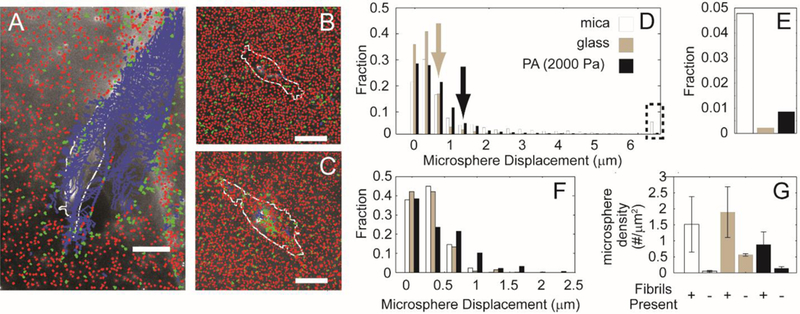
Collagen fibrils (A) assembled on mica substrates, (B) transferred to glass substrates or (C) transferred to 2000 Pa PA substrates and labeled with the fluorescent microspheres. Microsphere trajectories are colored differently depending on their average displacement. Blue indicates mean displacements larger than 2 μm, green indicates average displacements between 600 nm and 2 μm and red indicates average displacements smaller than 600 nm. The cell outline from one time point is shown in white. (D) Fraction of microsphere displacements when the distances between the cell edge and the microsphere is very small (0–10 μm). Arrows indicate the microsphere at which the transferred substrate crosses the mica substrate (E) Fraction of large displacements (> 6 μm). (F) Fraction of microsphere displacements when the distances between the cell edge and the microsphere is very large (180–190 μm). (G) Microsphere number per μm2 on different substrates with and without fibrils. Calibration bar length is 30 μm.
Collagen Alignment is Retained after Fibril Transfer
The collagen fibrils formed on mica substrates and transferred to functionalized substrates were imaged using SHG imaging and the distribution of collagen fibril orientations was determined and fitted to the same distribution as used above for cell alignment. On mica substrates, collagen orientation was not significantly different between substrates on which no cells had been plated and substrates on which cells had been plated for 12 h (Figure 6A&B). However, collagen fibril alignment decreased dramatically between 12 and 24 h on mica substrates (Figure 6C). Aligned cell fraction on these substrates, however, increased from 12 to 24h, indicating that of the cells that remained on the collagen fibrils, their alignment was higher (Figure 4A). The collagen fibrils on functionalized substrates at 0 h were more aligned as characterized by a narrower angle distribution when compared to the mica substrates (Figure 6D&G&J&M). All functionalized substrates appeared to inhibit the decrease in collagen alignment through 24 h when compared to mica substrates (Figure 6F&I&L&O). This result was consistent with the trend observed for the cell alignment area fraction on the functionalized substrates at 24 h compared to those on mica (Figure 4B). Since this distribution indicates orientation and not density, we quantified the collagen density under the same conditions by examining the intensity of the SHG images. Aligned collagen on all functionalized substrates in the presence of cells showed a decrease in normalized collagen density over time (Figure 7A). However, decreases in cell area fraction were only observed on the mica samples, suggesting that these decreases did not dramatically alter cell adhesion (Figure 2B). Cell density did not show any significant differences for the transferred substrates. In addition, normalized collagen density on PDMS substrates was smaller than mica and other functionalized substrates. In parallel to normalized collagen density, we then determined what fraction of the substrate area was occupied by collagen on mica and functionalized substrates (Figure 7B). Collagen area fraction on mica, glass and PDMS substrates decreased dramatically over 24 h. All three substrates showed different trends when comparing their cell area fractions. Cells on mica had area fractions that had the same trend as the collagen area fraction. The opposite trend was seen for the PDMS substrates where the cell area fraction actually increased over time. In the case of glass substrates, there were no significant changes for cell area fraction. Collagen area fraction on PA substrates (2000 Pa and 200 Pa) did not change (Figure 7B). In line with this, the cell area fraction also did not change over the same time period (Figure 2B). There were no significant differences in the non-ordered collagen fractions across all samples (Figure 7C). The aligned collagen fractions were also evaluated and decreased marginally over 24 h with mica substrates being significantly lower at 24 h (Figure 7D). A similar trend was observed for the aligned cell fraction over the same time period (Figure 2A). The fitting parameter κcollagen, which describes the width of the von Mises distribution showed significant differences between mica and functionalized substrates at 0 h, but showed no significant difference across the substrates at 24 h. Given that collagen density and alignment decreases were blocked after transferring collagen fibrils to functionalized substrates, we wanted to examine whether cell migration differs on mica and transferred substrates.
Figure 6: Angle distribution of collagen on the mica substrates, and transferred substrates (glass, PDMS, and 2000Pa and 200Pa polyacrylamide) seeded with MDA-MB-231 cells.
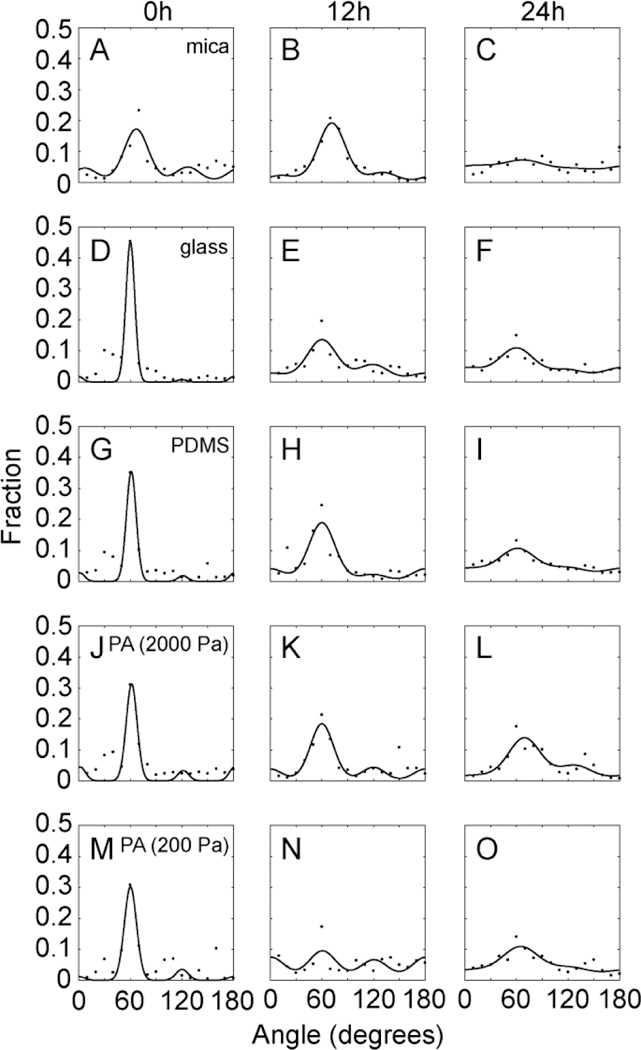
(A-C) The angle distribution of collagen at 0, 12, and 24 h on mica substrates, (D-F) The angle distribution of collagen at 0, 12, and 24 h after transfer to glass substrates, (G-I) The angle distribution of collagen a 0, 12, and 24 h after transfer to PDMS substrates (J-L) The angle distribution of collagen at 0, 12, and 24 h after transfer to 2000 Pa PA substrates (M-O) The angular distribution of collagen at 0, 12, and 24 h after transfer to 200 Pa PA substrates. Points represent data and the line represents the von Mises fit.
Figure 7: Collagen fibril alignment on mica substrates and transferred substrates (glass, PDMS, and 2000Pa and 200Pa polyacrylamide) seeded with MDA-MB-231 cells.
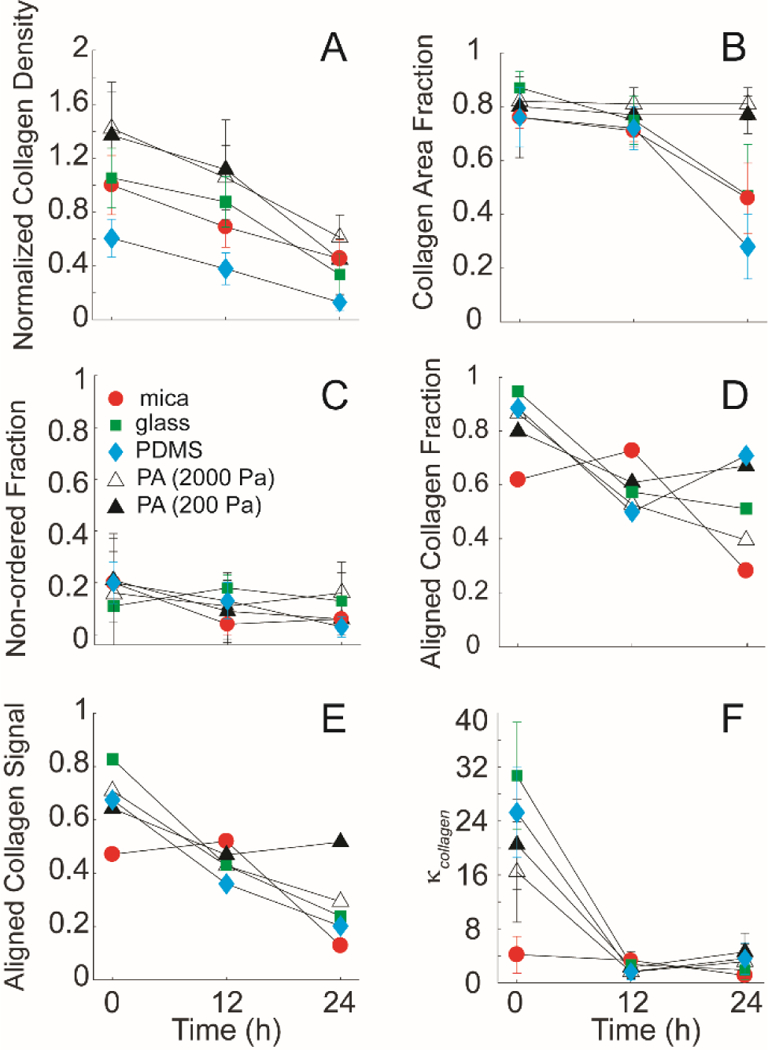
(A) The relative intensity of the collagen fibrils on the mica-based substrates over 24 hours, normalized to the initial intensity of the control at 0 hours. Collagen fibrils on all substrates showed rapid degradation of the collagen signal over time. (B) The collagen area fraction representing the sum of both ordered and non-ordered fractions of the collagen signal on the substrates showed significant difference over 24 hours on mica, glass and PDMS substrates. (C) The non-ordered collagen fractions of the collagen signal from the different substrates showed no significant differences (D) Collagen alignment fraction, (E) Aligned collagen signal fraction, as a product of the aligned collagen fraction and the collagen area fraction and (F) distribution spread parameter, κcollagen as a function of time on function. Cells plated on aligned collagen fibrils assembled on mica (circle), on transferred glass (closed squares), PDMS (closed diamond), and 2000Pa (open triangle) and 200Pa (closed triangle). Error bars represent 95% confidence intervals.
We tested two different breast cancer cell lines (MDA-MB-231 cells and MTLn3 cells) in vitro on mica and transferred substrates. They were plated on aligned collagen fibrils assembled on mica substrates and aligned collagen fibrils transferred to glass substrates (approximately 10 GPa49), thin PDMS substrates (approximately 280 kPa50), thick PDMS substrates (approximately 280 kPa50), and 2000 Pa and 200 Pa PA substrates. Migration speed and directionality were calculated. The thin and thick PDMS substrates have the same stiffness because the ratio of base and curing agent is the same, but the different thicknesses result in a different sensed stiffness. The order in stiffness is presented in Figure 8 from stiff (glass substrates) to soft (200 Pa PA substrates). The speed of MTLn3 cells increased from glass substrates to thick PDMS substrates and then decreased dramatically on 2000 Pa and 200 Pa PA substrates (Figure 8A). The fastest speed of MTLn3 cells is on the thick PDMS substrates. MTLn3 cells directionality was not significantly different between mica and flexible substrates (Figure 8B). The speed of MDA-MB-231 cells increased from glass substrates to 2000 Pa PA substrates and then decreased dramatically on 200 Pa PA substrates (Figure 8C). MDA-MB-231 cell directionality on PA substrate was slightly higher than that on mica substrates and MDA-MB-231 cell directionality had little significant difference among transferred substrates, indicating that the mechanical properties of the substrate only modestly governs directionality, but has a much more dramatic effect on cell migration speed (Figure 8D).
Figure 8: Migration behavior of MDA-MB-231 and MTLn3 cells on mica substrates and transferred substrates (glass, PDMS, and 2000 Pa and 200 Pa polyacrylamide).
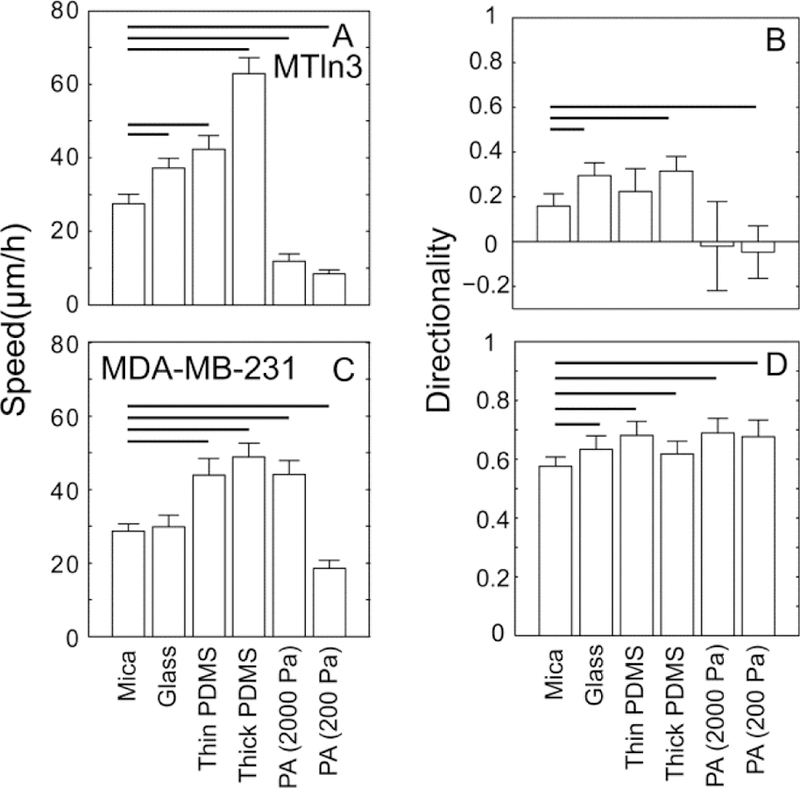
(A) speed and (B) directionality of MTLn3 cells. (C) speed and (D) directionality of MDA-MB-231 cells. Error bars are 95% confidence intervals. Solid black lines indicate that the means are statistically significant from the mica substrates (p < 0.05 using two-tailed student’s t-test).
Discussion
Highly aligned collagen fibers in the ECM of the TME correlates with cancer cell invasion52 and cancer cells in environments with higher collagen alignment migrate faster53 and with high persistence54. The TME not only contains aligned collagen fibers, but has also been shown to be stiffer than normal tissue, enhancing the invasive characteristic of cancer cells55, 56. The tumor is much stiffer than normal tissues. In vivo, normal breast tissue is predominantly around 400 Pa, while the mean stiffness of invasive tissue elevates to as much as 5 kPa.57 In this paper we used a method to transfer epitaxially grown collagen fibrils from mica substrates to flexible substrates and studied how stiffness of these flexible substrates affect contact guidance. Although collagen fibrils assembled on mica substrates are very similar to native fibril structures in vivo, the stiffness of mica substrates is likely different from the stiffness of tumor tissues in vivo. While epitaxial growth of aligned collagen fibrils represents a reasonably physiological contact guidance cue, the unknown and uncontrollable mechanical properties represent challenges. Epitaxially grown aligned collagen fibrils can only be assembled because of mica’s special surface properties58 and this assembly leads to collagen assembly into fibrils, regulated by collagen−collagen and collagen−mica electrostatic interactions.58, 59 The fibril bundles resist deformation in the direction parallel to the alignment direction while the fibril bundles are more readily deformed in the direction perpendicular to the alignment indicating that epitaxially collagen fibrils are loosely attached on mica substrates and can be transferred to flexible substrates with different stiffness. Therefore, in this paper we are presenting a novel way to transfer epitaxially grown aligned collagen fibrils from mica substrates to other substrates.
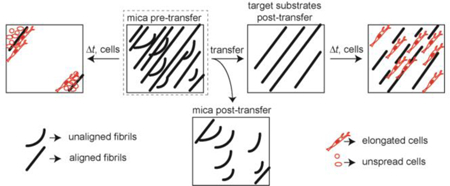
Collagen fibril transfer efficiency seemed to vary significantly among the target substrates. The amount of collagen transferred to glass and PA substrates was about the same. However, the amount of collagen transferred to PDMS substrates was smaller than that transferred to either glass or PA substrates. In addition, there was no difference in transfer efficiency between 200 Pa and 2000 Pa PA substrates. This would suggest that transfer efficiency is not influenced by mechanical rigidity, even though substrate stiffness as modulated through thickness and elastic modulus have been shown to affect material transfer.60, 61 In addition, the stiffest target substrate is glass, followed by PDMS and finally PA, also suggesting no correlation between transfer efficiency and stiffness. However, different functionalization chemistry is used, complicating this comparison. PA substrates uses sulfo-SANPAH, whereas glass and PDMS substrates use aldehyde functionalized silanes to attach the collagen to the surface. When only considering the aldehyde functionalized substrates, glass has a higher transfer efficiency than PDMS. This differs from that seen with globular proteins and heterotrimeric collagen, where PDMS appears to bind protein with higher affinity than glass62, 63. An alternative hypothesis is that mechanical properties do affect transfer, but only for relatively stiff substrates, much stiffer than the 200 Pa and 2000 Pa PA substrates. Interestingly, collagen area fraction of all target substrates after collagen transfer is about the same, even though the collagen density is different. This could be caused by two different mechanisms. The first explanation is a thinning of the epitaxially grown collagen fibrils, however the collagen fibrils are already quite thin and on the order of ~ 1–4 nm28, 29. This is on the order of the diameter of a collagen heterotrimer, making it unlikely that only part of the height is transferred. The second explanation is that only some of the collagen within each ROI is transferred. This causes a decrease in the collagen density as measured by intensity, however, the ROI is still flagged as having collagen. The ROI is on the order of 6 μm x 6 μm. Each collagen fibril is approximately 150 nm in width28 (Figure 1B), which corresponds to approximately 40–60 collagen fibril widths depending on the orientation angle of the fibrils with respect to the ROI. It is possible that only a fraction of these are transferred. Not only did collagen density and area fraction differ among target substrate, but the degree of collagen alignment immediately after collagen fibril transfer also differed. The fibrils appeared more aligned on all target substrates (Figure 7D-F). Although not statistically significant, glass appeared to show the most ordering, followed by PDMS and finally the PA. This does not map to what is seen for transfer efficiency, where the order was PA, glass and PDMS. Consequently, the transfer of ordered collagen must be somewhat different than that of nonordered collagen (Figure 9). Bolstering this fact is the observation that the collagen left behind after transfer was primarily disordered (Supplemental Figure 1). This could have contributed to the fact that highly directional cells like MDA-MB-231 cells align better on the transferred substrates (Figure 3I&J and Figure 8D).
Figure 9: Schematic of fibrils transfer and cell behavior from mica substrates to target substrates.
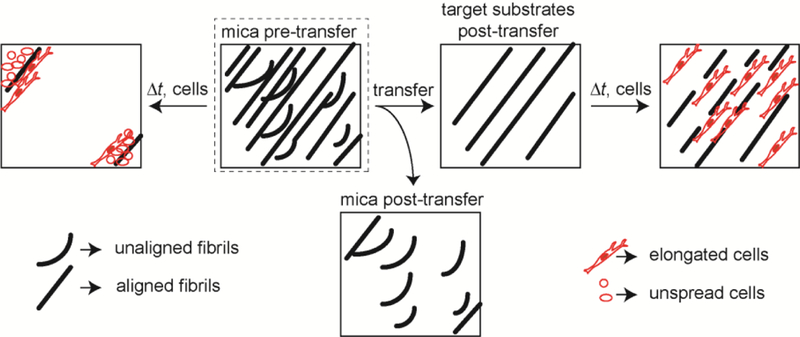
After we transferred aligned collagen fibrils from mica substrates to flexible substrates, we wanted to understand how stiffness of flexible substrates affects cell’s contact guidance behavior. When MDA-MB-231 cells were plated on mica substrates for longer time (24 h), both cell density and cell alignment area fractions were much smaller than that on transferred substrates. This occurs because self-assembled collagen fibrils were loosely attached and cells were able to remodel fibrils easily and distort them from their original alignment. Therefore, after MDA-MB-231 cells were plated on mica substrates for 24 h, cells form large aggregates and most of fibrils detached from the substrate (Figure 9). When MDA-MB-231 cells were plated on the flexible substrates, the cross-linking reaction covalently links amine groups within the collagen to the surface exposed aldehyde. Thus, collagen fibrils attached to flexible substrates are much stronger than those on mica substrates, and block in part ECM remodeling. While cells are still able to somewhat alter collagen alignment on flexible substrates, cells did not aggregate and detach from surfaces as was seen on collagen fibrils assembled on mica (Figure 9), which is shown by measuring cell density and cell alignment area fraction (Figure 4A&B). This illustrates that this substrate could be used for longer term migration assays in situations where contact guidance cue detachment is undesired. Also, by transferring fibrils to glass substrates imaging approaches like total internal reflection microscopy are now available to be used in examining contact guidance on these collagen fibrils.
Our initial analysis was geared toward understanding how mechanical properties of the substrate regulate contact guidance. Mechanical properties of the contact guidance cue likely have impact, given the overlapping signaling pathways between migration in response to stiffness gradients and aligned fiber or fiber-like structures3, 64, 65. We compared MDA-MB-231 and MTLn3 cells on epitaxially grown aligned collagen fibrils on mica substrates. We showed previously that MDA-MB-231 cells sense contact guidance cues, however MTLn3 cells do not.7, 28 MTLn3 cells, exhibit an amoeboid mode of cell migration, have small adhesive force and are less depend on cell-ECM interactions, while MDA-MB-231 cells, exhibit a mesenchymal mode of cell migration, have large adhesive force and are more depend on cell-ECM interactions.66–68 The cell migration speed of MTLn3 and MDA-MB-231 cells showed a biphasic response to stiffness as has been shown previously69. It is less clear how stiffness affects directionality. There has been some indication that contact guidance and durotaxis share overlapping intracellular signaling pathways3. Since durotactic index or the directional fidelity during durotaxis appears to be in part related to the average stiffness of the substrate, it is likely that contact guidance operates similarly. We have shown that 3D collagen environments made stiffer through collagen glycation result in lower directionality in MDA-MB-231 cells, which agrees with the work presented here23. There is likely some feedback that enhances local collagen alignment that is diminished when the ECM is stiff. However, MTLn3 cells appear to show a different trend. This cell line tends to respond poorly to contact guidance and contact guidance is not enhanced when the cue is softer. This suggesting that only cells that are able to robustly remodel the ECM can amplify and consequently respond to changes in the stiffness of the contact guidance cue. Even though these flexible substrates show no dramatic change in directional fidelity, they do represent potential substrates on which to study force transmission during contact guidance.
In addition to contact guidance, applications where structured fibers could be coated or transferred to implantable biomaterials offer another application for this transfer process. Much work has established minimal ECM peptides as potential approaches for better implant grafting70. Attachment of cells to the surfaces of implants for bone regeneration or dental applications is important. Structuring the ECM on the interface of these implants may improve mechanical integration and implant acceptance. This is particularly important for dental implants, where attachment of the periodontal ligament is mediated through aligned collagen71. A flexible approach in which to transfer aligned collagen fibrils to these surfaces will expand the ability to engineer these biomaterials interfaces for enhanced biological function.
Conclusions:
In this paper, we examine an approach to transfer self-assembled aligned collagen fibrils from mica substrates onto functionalized substrates. Using this approach, most of the aligned collagen fibrils on mica substrates are transferred to functionalized substrates, leaving primarily unaligned collagen fibrils. Furthermore, cell and collagen alignment on functionalized substrates to which aligned collagen fibrils have been transferred are similar to mica substrates. In addition, transfer of aligned collagen fibrils to functionalized substrates blocks the cells’ ability to remodel aligned collagen fibrils and pull them from the surface. Finally, MTLn3 and MDA-MB-231 cells respond similarly to stiffness, migrating faster on intermediate stiffness substrates and slower when stiffness is tuned up or down. However, tuning the stiffness of the contact guidance cue appears to have only marginal impact on directionality.
Supplementary Material
Insight Box:
Understanding how cells sense and respond to contact guidance cues will lend insight into such diverse processes as wound healing and cancer cell invasion. Self-assembly of aligned collagen fibrils on mica substrates provides an excellent model system in which to examine contact guidance. However, the stiffness is unknown and uncontrollable, cells detach these collagen fibrils over time and mica is only marginally transparent. We have developed and characterized a novel method by which to transfer self-assembled collagen fibrils from mica, a relatively limiting substrate to a variety of substrates with tunable mechanical properties. This method allows for the generation of flexible aligned collagen fiber substrates to examine the role of mechanics in governing contact guidance.
Acknowledgements:
ICS acknowledges support from the National Institutes of Health/National Cancer Institute [R03CA184575] for general project funding. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
References:
- 1.Pickup MW, Mouw JK and Weaver VM, EMBO Rep, 2014, 15, 1243–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saeidi N, Sander EA and Ruberti JW, Biomaterials, 2009, 30, 6581–6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Janson IA and Putnam AJ, J. Biomed. Mater. Res. Part A, 2015, 103, 1246–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG and Keely PJ, BMC Med, 2006, 4, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drifka CR, Loeffler AG, Mathewson K, Keikhosravi A, Eickhoff JC, Liu YM, Weber SM, Kao WJ and Eliceiri KW, Oncotarget, 2016, 7, 76197–76213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ray A, Lee O, Win Z, Edwards RM, Alford PW, Kim DH and Provenzano PP, Nat. Commun, 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang J and Schneider IC, Biomaterials, 2017, 120, 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kubow KE, Shuklis VD, Sales DJ and Horwitz AR, Sci Rep, 2017, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramirez-San Juan GR, Oakes PW and Gardel ML, Mol. Biol. Cell, 2017, 28, 1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, Chen YY, Liphardt J, Hwang ES and Weaver VM, Integr. Biol, 2015, 7, 1120–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bordeleau F, Mason BN, Lollis EM, Mazzola M, Zanotelli MR, Somasegar S, Califano JP, Montague C, LaValley DJ, Huynh J, Mencia-Trinchant N, Abril YLN, Hassane DC, Bonassar LJ, Butcher JT, Weiss RS and Reinhart-King CA, Proc. Natl. Acad. Sci. U. S. A, 2017, 114, 492–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gardel ML, Schneider IC, Aratyn-Schaus Y and Waterman CM, in Annual Review of Cell and Developmental Biology, Vol 26, eds. Schekman R, Goldstein L and Lehmann R, Annual Reviews, Palo Alto, 2010, vol. 26, pp. 315–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Helvert S, Storm C and Friedl P, Nat. Cell Biol, 2018, 20, 8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Romsey NR, Hou Y, Rodriguez LL and Schneider IC, Cellular and Molecular Bioengineering, 2014, 7, 122–135. [Google Scholar]
- 15.Xue JJ, Xie JW, Liu WY and Xia YN, Accounts Chem. Res, 2017, 50, 1976–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nain AS, Phillippi JA, Sitti M, MacKrell J, Campbell PG and Amon C, Small, 2008, 4, 1153–1159. [DOI] [PubMed] [Google Scholar]
- 17.Ray A, Slama ZM, Morford RK, Madden SA and Provenzano PP, Biophys. J, 2017, 112, 1023–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barocas VH and Tranquillo RT, J. Biomech. Eng.-Trans. ASME, 1997, 119, 137–145. [DOI] [PubMed] [Google Scholar]
- 19.Koster S, Leach JB, Struth B, Pfohl T and Wong JY, Langmuir, 2007, 23, 357–359. [DOI] [PubMed] [Google Scholar]
- 20.Lee P, Lin R, Moon J and Lee LP, Biomed. Microdevices, 2006, 8, 35–41. [DOI] [PubMed] [Google Scholar]
- 21.Antman-Passig M and Shefi O, Nano Letters, 2016, 16, 2567–2573. [DOI] [PubMed] [Google Scholar]
- 22.Guo C and Kaufman LJ, Biomaterials, 2007, 28, 1105–1114. [DOI] [PubMed] [Google Scholar]
- 23.Nuhn JAM, Perez AM and Schneider IC, Acta Biomater, 2018, 66, 248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Julias M, Edgar LT, Buettner HM and Shreiber DI, Biomed. Eng. Online, 2008, 7. [DOI] [PMC free article] [PubMed]
- 25.Antman-Passig M, Levy S, Gartenberg C, Schori H and Shefi O, Tissue Eng. Part A, 2017, 23, 403–414. [DOI] [PubMed] [Google Scholar]
- 26.Lowe CJ, Reucroft IM, Grota MC and Shreiber DI, ACS Biomater. Sci. Eng, 2016, 2, 643–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saeidi N, Sander EA, Zareian R and Ruberti JW, Acta Biomater, 2011, 7, 2437–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Petefish JW, Hillier AC and Schneider IC, Langmuir, 2015, 31, 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang FZ, Horber H, Howard J and Muller DJ, J. Struct. Biol, 2004, 148, 268–278. [DOI] [PubMed] [Google Scholar]
- 30.Poole K, Khairy K, Friedrichs J, Franz C, Cisneros DA, Howard J and Mueller D, J. Mol. Biol, 2005, 349, 380–386. [DOI] [PubMed] [Google Scholar]
- 31.Yamada KM and Cukierman E, Cell, 2007, 130, 601–610. [DOI] [PubMed] [Google Scholar]
- 32.Zhou ZN, Sharma VP, Beaty BT, Roh-Johnson M, Peterson EA, Van Rooijen N, Kenny PA, Wiley HS, Condeelis JS and Segall JE, Oncogene, 2014, 33, 3784–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clark P, Connolly P and Moores GR, J. Cell Sci, 1992, 103, 287–292. [DOI] [PubMed] [Google Scholar]
- 34.Trappmann B, Gautrot JE, Connelly JT, Strange DGT, Li Y, Oyen ML, Stuart MAC, Boehm H, Li BJ, Vogel V, Spatz JP, Watt FM and Huck WTS, Nat. Mater, 2012, 11, 642–649. [DOI] [PubMed] [Google Scholar]
- 35.Tse JR and Engler AJ, PLoS One, 2011, 6, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szymanski JM, Zhang KR and Feinberg AW, Sci Rep, 2017, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szymanski JM, Sevcik EN, Zhang KR and Feinberg AW, Biomater. Sci, 2017, 5, 1629–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qin D, Xia YN and Whitesides GM, Nat. Protoc, 2010, 5, 491–502. [DOI] [PubMed] [Google Scholar]
- 39.Shi J, Wang L and Chen Y, Langmuir, 2009, 25, 6015–6018. [DOI] [PubMed] [Google Scholar]
- 40.Hu T, Li QT, Dong H, Xiao WW, Li L and Cao XD, Small, 2017, 13. [DOI] [PubMed] [Google Scholar]
- 41.Duan YQ, Huang YA and Yin ZP, Thin Solid Films, 2013, 544, 152–156. [Google Scholar]
- 42.Doan VA, Nobukawa S and Yamaguchi M, Compos. Pt. B-Eng, 2012, 43, 1218–1223. [Google Scholar]
- 43.Liu J, Shi JA, Secret E, Wang L, Zhang HL and Chen Y, Microelectron. Eng, 2010, 87, 2513–2517. [Google Scholar]
- 44.Sun Y, Jallerat Q, Szymanski JM and Feinberg AW, Nat. Methods, 2015, 12, 134–U168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karim AA and Bhat R, Food Hydrocolloids, 2009, 23, 563–576. [Google Scholar]
- 46.Chen XY, Nadiarynkh O, Plotnikov S and Campagnola PJ, Nat. Protoc, 2012, 7, 654–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pelham RJ and Wang YL, Proc. Natl. Acad. Sci. U. S. A, 1997, 94, 13661–13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crocker JC and Grier DG, J. Colloid Interface Sci, 1996, 179, 298–310. [Google Scholar]
- 49.Palchesko RN, Zhang L, Sun Y and Feinberg AW, PLoS One, 2012, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park JY, Yoo SJ, Lee EJ, Lee DH, Kim JY and Lee SH, BioChip J, 2010, 4, 230–236. [Google Scholar]
- 51.Bhide VM, Laschinger CA, Arora PD, Lee W, Hakkinen L, Larjava H, Sodek J and McCulloch CA, J. Biol. Chem, 2005, 280, 23103–23113. [DOI] [PubMed] [Google Scholar]
- 52.Provenzano PP, Eliceiri KW, Yan L, Ada-Nguema A, Conklin MW, Inman DR and Keely PJ, Microsc. microanal, 2008, 14, 532–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fraley SI, Wu PH, He LJ, Feng YF, Krisnamurthy R, Longmore GD and Wirtz D, Sci Rep, 2015, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Riching KM, Cox BL, Salick MR, Pehlke C, Riching AS, Ponik SM, Bass BR, Crone WC, Jiang Y, Weaver AM, Eliceiri KW and Keely PJ, Biophys. J, 2014, 107, 2546–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stowers RS, Allen SC, Sanchez K, Davis CL, Ebelt ND, Van Den Berg C and Suggs LJ, Cellular and Molecular Bioengineering, 2017, 10, 114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA and Weaver VM, Cancer Cell, 2005, 8, 241–254. [DOI] [PubMed] [Google Scholar]
- 57.Shoulders MD and Raines RT, in Annual Review of Biochemistry, Annual Reviews, Palo Alto, 2009, vol. 78, pp. 929–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leow WW and Hwang W, Langmuir, 2011, 27, 10907–10913. [DOI] [PubMed] [Google Scholar]
- 59.Narayanan B, Gilmer GH, Tao JH, De Yoreo JJ and Ciobanu CV, Langmuir, 2014, 30, 1343–1350. [DOI] [PubMed] [Google Scholar]
- 60.Bartlett MD and Crosby AJ, Mater. Horizons, 2014, 1, 507–512. [Google Scholar]
- 61.Eisenhaure J and Kim S, Micromachines, 2017, 8. [Google Scholar]
- 62.Xiao DQ, Zhang H and Wirth M, Langmuir, 2002, 18, 9971–9976. [Google Scholar]
- 63.Li C, Ding YC, Kuddannaya S, Zhang YL and Yang L, J. Mater. Sci, 2017, 52, 9963–9978. [Google Scholar]
- 64.Hadden WJ, Young JL, Holle AW, McFetridge ML, Kim DY, Wijesinghe P, Taylor-Weiner H, Wen JH, Lee AR, Bieback K, Vo BN, Sampson DD, Kennedy BF, Spatz JP, Engler AJ and Choi YS, Proc. Natl. Acad. Sci. U. S. A, 2017, 114, 5647–5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hartman CD, Isenberg BC, Chua SG and Wong JY, Proc. Natl. Acad. Sci. U. S. A, 2016, 113, 11190–11195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kraning-Rush CM, Carey SP, Califano JP, Smith BN and Reinhart-King CA, Phys. Biol, 2011, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mierke CT, Rosel D, Fabry B and Brabek J, Eur. J. Cell Biol, 2008, 87, 669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wyckoff JB, Pinner SE, Gschmeissner S, Condeelis JS and Sahai E, Curr. Biol, 2006, 16, 1515–1523. [DOI] [PubMed] [Google Scholar]
- 69.Peyton SR and Putnam AJ, J. Cell. Physiol, 2005, 204, 198–209. [DOI] [PubMed] [Google Scholar]
- 70.Agarwal R and Garcia AJ, Adv. Drug Deliv. Rev, 2015, 94, 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim SG, Kim SG, Viechnicki B, Kim S and Nah HD, J. Clin. Periodontol, 2011, 38, 1130–1136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.