

Summary
Our understanding of the spatial variation in the chemical and microbial make-up of an entire human organ remains limited, in part due to the size and heterogeneity of human organs and the complexity of the associated metabolome and microbiome. To address this challenge we developed a workflow to enable the cartography of metabolomic and microbiome data onto a three dimensional (3D) organ reconstruction built off radiological images. This enabled the direct visualization of the microbial and chemical makeup of a human lung from a cystic fibrosis patient. We detected host-derived molecules, microbial metabolites, medications, and region specific metabolism of medications and placed it in the context of microbial distributions in the lung. Our tool further created browsable maps of a 3D microbiome/metabolome reconstruction map on a radiological image of a human lung and forms an interactive resource for the scientific community.
eTOC blurb
Garg et al introduces the technology that enables the volume cartography of the molecules and microbes onto a radiological image such as X-ray or CT-scan of human organ. They demonstrate the concept by revealing the chemistry associated with the microbial communities from a Cystic fibrosis patient.
Introduction
Mammalian organs are inherently complex and vary in anatomical structure, in physical conditions such as temperature, and in chemical conditions such as oxygen saturation and pH. This variation within the organ may result in discrete local microbial niches that contain different subpopulations, which in turn may affect the chemical makeup of these regions (Bumann, 2015). Genome sequencing of microbial isolates from different regions of the lung has accordingly shown variation in subpopulation localization (Chung et al., 2017; Jorth et al., 2015; Willner et al., 2012), and 16S rRNA sequencing of human skin has shown distinct microbial taxonomies across anatomically distinct regions of skin (Bouslimani et al., 2015; Grice et al., 2009; Oh et al., 2014), similar to observations in various other body parts (Human Microbiome Project Consortium, 2012). While metabolomics-based regional variation has been demonstrated by surface mapping of human skin (Bouslimani et al., 2015), the metabolomic volumetric mapping of an entire human organ in 3D, spanning tens of centimeters, has not been performed before. The difficulty in procuring human organs and lack of technology to map omics data on an entire organ representation leads to a gap in our ability to inventory and understand the chemical environment, including the penetration of drugs, host immune response, and microbial virulence factors, and limits our ability to correlate this chemical environment to the microbial environment. The availability of such technology could reveal how biogeography influences the production of microbial small-molecule virulence factors, and even how microbes evolve their response to drugs, allowing us to develop new, more useful drugs. To create methodology for volumetric mapping of an entire human organ in 3D, we chose a human lung associated with cystic fibrosis (CF) as our model system. CF is a genetic disease typified by persistent and recurrent microbial infections of the lung that become antibiotic resistant (Hauser et al., 2011; Zhao et al., 2012). Regional variation in inflammation within the lungs has been observed on chest radiographs and is also evident from quantification of regional variation in neutrophils within the lungs (Meyer and Sharma, 1997). Two recent studies also showed that location-specific selection, diversification or evolution takes place in CF pathogens, Pseudomonas (Jorth et al., 2015) and Stenotrophomonas (Chung et al., 2017). These studies were performed by isolating bacteria from anatomically distinct locations within lungs, then sequencing the genomes and characterizing the phenotypes of a large number of these isolates. Such studies provide much-needed advances in our knowledge of microbial evolution by revealing mutations and genome diversification. However, they lack the ability to directly visualize microbial molecules within the lung (for example, the presence of microbial virulence factors involved in processes such as biofilm formation), or how they correlate with exposure to antibiotics and the presence of specific microbes individually or in combination. Thus, lungs associated with CF represent a good model system for investigating spatial variation in the microbes, microbial metabolism, and xenobiotics within a single organ.
Accordingly, we created methodology to visualize spatial diversity of omics data in the human lung in 3D, and applied this methodology to map 16S rRNA sequencing and metabolomics data to this common patient-specific reference space. Because we used an end-stage CF lung, one pathogen, Pseudomonas, dominated the lung, but many other microbes showed spatial heterogeneity within the lung when compared to controls, allowing us to create an atlas of microbes and metabolites throughout the lung. We observed that even though Pseudomonas was abundant throughout the lung, its associated virulence factors, as seen in in vitro studies, were not. The presence of another CF-associated pathogen, Achromobacter, was negatively spatially correlated with the presence of the antibiotic Meropenem. Spatial maps generated in this study allow such correlations to be observed. Visualization of antibiotics revealed that not all antibiotics show uniform penetration within the lung, and that antibiotic metabolism varies spatially.
Results
Generation of 3D lung models and mapping of data
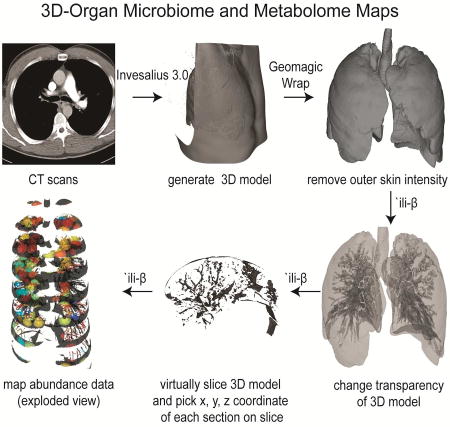
To generate 3D lung models for mapping omics data, we procured computed tomography (CT) scan images of a human lung. These images were processed to generate the 3D model using the software Invesalius version 3.0 (Figure 1). The pixels corresponding to the chest and back were then removed with the software Geomagic Wrap and the model was further cleaned to represent intensity corresponding to the lung (Figure S1). The methodology to map the abundances from omics data onto these models was created by modifying the Google Chrome extension `ili. This application allows one to map data on the surface of a 2D and a 3D model using the physical x, y, and z coordinates of the sampling site. The abundance data corresponding to all sampling sites is plotted on the same model enabling visualization of anatomical/regional spatial variation. Herein, the code was modified to allow us to map the abundance data not only on the surface but also within the model, enabling volumetric mapping of abundance data. This was achieved by incorporating various new features. The new features allow one to change the opacity of the model, section the model that mimics sectioning of the lung at the hospital (enabling picking of x, y, z coordinates of the sampling site) and expansion of the model (exploded view) along either x, y or z axis for visualization purposes (Figure 1). Whenever a 3D model can be generated and x, y, z coordinates of sampling sites are available, one can map and correlate abundances of various data types: metagenomics, transcriptomics, 16S rRNA gene sequencing, 18S rRNA gene sequencing, ITS sequencing, metabolomics, and proteomics. As proof of principle, we used this methodology to map and correlate the 16S rRNA gene sequencing and metabolomics data onto the left-side lung of a patient with CF.
Figure 1.

A. The description of the workflow used to create 3D metabolome and microbiome organ maps from the observation matrix. B. The description of the workflow used for generating the metabolome and microbiome. (See also Figure S1).
Generation and analyses of microbiome and metabolome data
To build a resource for mapping the microbiome and metabolome of a human lung in 3D, the left lung of a patient afflicted with CF was procured in coordination with the patient’s physician and a pathologist who were previously familiarized with the research design. The pathologist assisted in the sectioning of the lung tissue, wherein the entire left lung was processed into sections. During sectioning, each of the sections was swabbed to isolate DNA for 16S rRNA gene (a bacterial and archaeal marker gene) amplicon sequencing and analysis. The tissue sections were extracted to generate mass spectrometry-based (MS) metabolomics data. To gain insight into the small molecules, detected as ions, the untargeted metabolomics data was subjected to molecular networking using the Global Natural Product Social Molecular Networking analysis infrastructure (GNPS) (Wang et al., 2016). Molecular networking is an approach that utilizes spectral alignment to merge all identical MS/MS spectra and performs a spectral similarity search to find spectra/molecules that are similar to each other and to annotate known spectra/molecules that are deposited in spectral libraries (Watrous et al., 2012). The resulting spectral alignment is displayed as a molecular network (Figure S2). In the molecular network generated from data collected on lung tissue extracts, 149,353 spectra were observed (not all of these spectra represent unique compounds as they merge into 7,412 unique spectra or nodes), with 12% of the spectra annotated as known molecules (2.5% of all nodes). The known annotations include drugs administered to the patient and phthalates from the environment, as well small molecules (such as peptides, amino acids and amino acid metabolites, sugars, fatty acids, lipids, and bile acids) from the host and the microbes (virulence factors) and are available at GNPS.
To annotate microbial molecules, the CF-associated microbes were cultured and the MS data was collected on the extracted cultures and compared with the data collected on lung tissue using molecular networking. The small molecule virulence factors detected in the lung tissue include quinolones and rhamnolipids that are produced by Pseudomonas aeruginosa in the cultured isolates (Figure 2 and 3). Among various roles, rhamnolipids are biosurfactants that are involved in maintaining open channels in biofilms formed by Pseudomonas (Abdel-Mawgoud et al., 2010). Quinolones are produced by Pseudomonas, and are involved in processes such as quorum sensing, regulation of gene expression for production of virulence factors and as antimicrobials, thus significantly affecting P. aeruginosa virulence (Heeb et al., 2011). A total of 56 different quinolones have been reported in the primary literature, and the repertoire of quinolones includes 2-alkyl-4-quinolones (AHQ’s) such as 2-heptyl-4-quinolone (HHQ), and 2-nonyl-4-quinolone (NHQ), 2-alkyl-4-quinolone-N-oxide such as 2-heptyl-4-quinolone-N-oxide (HQNO), and 2-nonyl-4-quinolone-N-oxide (NQNO) (Lepine et al., 2004). A third class of quinolones namely, 2-alkyl-3-hydroxy-4-quinolone, with 2-heptyl-3-hydroxy-4-quinolone, also called pseudomonas quinolone signal (PQS) as the most prominent and well-studied member are also produced. The AHQ’s serve as the biosynthetic precursors for this third class of quinolones. PQS, in addition to homoserine lactones, is considered one of the quorum sensing circuits regulating virulence factor gene expression and biofilm formation by P. aeruginosa (Williams and Camara, 2009). Herein, as expected, a larger diversity of quinolones and rhamnolipids was detected in the cultured isolates, represented by cyan nodes in the molecular network (Figure 2 and 3), as opposed to a smaller diversity of quinolones directly detected in the lung tissue, represented by orange nodes. Furthermore, PQS was detected in the cultured isolates, but not in lung tissue. These findings highlight disconnect between the potential role of small molecules in pure culture and the biologically relevant activity in the diseased organ. It is likely that these molecules are either not produced, are degraded, are cleared or modified in the host tissue.
Figure 2.

The molecular network corresponding to quinolones produced by Pseudomonas. The orange nodes represents quinolones detected in the extracts of both the lung tissue and the cultured microbial isolates; the cyan nodes represent quinolones detected only in the extracts of cultured microbial isolates. The distributions of the quinolones are shown as the exploded view of the lung (left) on a jet color scheme where red represents the highest abundance and blue represents the lowest abundance. The mapping has been performed on the left lung. The distribution of other quinolones is shown in Figure S4. Numbers inside the nodes represent the m/z. The video S1 provides visualization of HHQ and the interactive visualization is available at the weblinks: HHQ (64-fold difference between highest and lowest detected abundance (henceforth: fold difference)), NHQ (226-fold difference), NHQ-C9:1db (68-fold difference), NQNO (18-fold difference). There are three steps for interactive visualization: loading of data, mapping of data, and processing (at the end exploded view of the lung with abundances mapped is loaded, which may take tens of seconds to load completely).
Figure 3.

The molecular network corresponding to rhamnolipids produced by Pseudomonas. The orange nodes represents rhamnolipids detected in the extracts of both the lung tissue and the cultured microbial isolates; the cyan nodes represent rhamnolipids detected only in the extracts of cultured microbial isolates. The distribution is shown on a jet color scheme where red represents the highest abundance and blue represents the lowest abundance. The mapping has been performed on the left lung. The interactive visualization is available at the weblinks: Rha-Rha-C10-C10 (17-fold difference), Rha-Rha-C10-C12 (11-fold difference).
The MS collected in this study on the lung tissue and cultured microbes was also aligned with previously deposited metabolomics datasets on the skin, gut, and sputum of CF patients. Because this analyses is performed using MS2-based molecular networking, it provides global insight into the shared metabolome between different sample types that is amenable to MS-based detection and fragmentation. Using the network statistics analyses, by counting the number of nodes that are unique or are shared between sample types, 25% of the metabolome was unique to the lung tissue and 4% was exclusively shared between lung tissue and the closely related sputum samples (Figure S3). The physiological difference between the lung tissue and the sputum likely leads to low overlap between their metabolomes.
To visualize the distribution of xenobiotics, microbial molecules, and host molecules, the 3D metabolome lung maps were generated by relative quantification of the MS1-based abundances of the molecules. The molecules were annotated using MS2-based molecular networking and dereplication as described above. The distribution of select molecules belonging to different structural class and origin (host, microbial, and xenobiotics) is shown in Figure 4 (refer to videoS1) and the navigable atlas of both the microbes (Microbe_maps) and molecules (Molecule_maps) is also provided. Related molecules, such as the two trisaccharides, were identically distributed, in contrast, structurally unrelated molecules were differentially distributed. For example, the distribution pattern of the drug Albuterol, a bronchodilator given via inhalation, is completely different from that of intravenous antibiotics (Figure 5), representing the extensive damage to the airways also reported by hyperpolarized helium-3 magnetic resonance imaging in CF patients (Koumellis et al., 2005). The dioctylphthalate that constitutes a component of our exposome was more abundant at the base of the lung in the lower lobe than in the upper lobe, while microbial quinolones were largely abundant in the upper and middle lobes of the lung (Figure 2 and Figure S4). As expected, different quinolones originating from the same biosynthetic pathway were identically distributed. The microbial rhamnolipids were largely abundant at the top of the lung with poor abundance observed at the middle and lower lobes of the lung (Figure 3). Both Pseudomonas virulence factors -the quinolones and rhamnolipids, which are synthesized from two different biosynthetic pathways were similarly distributed with largest abundances at the lung apex. Thus, the Pseudomonas biofilms showed spatial heterogeneity within the lung based on the detected virulence factors.
Figure 4.

Distributions of selected features identified using molecular networking. The distribution is shown on a jet color scheme where red represents the highest abundance and blue represents the lowest abundance. The mapping has been performed on the left lung. The interactive visualization is available at the weblinks: Glycurodeoxycholic acid (6-fold difference), Trisaccharide sugar 1 (5-fold difference), Trisaccharade sugar 2 (4-fold difference), HHQ (64-fold difference), Dioctylphthalate (5-fold difference), Meropenem (27-fold difference) , Albuterol (4-fold difference), N-Acetyltryptophan (14-fold difference), and LysoPAF (3-fold difference). The navigable atlas for all detected molecules is available at weblink: (Molecule_maps).
Figure 5.

Distributions of antibiotics and their breakdown products throughout the lung. The distribution is shown on a jet color scheme where red represents the highest abundance and blue represents the lowest abundance. The mapping has been performed on the left lung. The interactive visualization is available at the weblinks: Meropenem (27-fold difference), Piperacillin (7-fold difference), Desethyl-piperacillin (3-fold difference), Ciprofloxacin (3-fold difference), and Descladinose-azithromycin (6-fold difference).
Visualization of the penetration of antibiotics administered intravenously to this patient (Figure 5) revealed that the β-lactam drugs Meropenem (administered for 2 days prior to tissue collection), Piperacillin (administered for 5 days prior to tissue collection) and its metabolite desethyl-piperacillin poorly penetrated to the base of the lung, whereas the fluoroquinolone antibiotic Ciprofloxacin (administered for 2 days prior to tissue collection) and descladinose-azithromycin, a metabolite of the macrolide antibiotic Azithromycin (administration stopped one month prior to tissue collection), uniformly penetrated the lung. Thus, different drugs may differentially penetrate the lung, limiting exposure to effective dosage and promoting the development of antibiotic resistance. Our methodology allows direct visualization of antibiotic penetration within an entire diseased human organ. Future studies examining these effects on larger numbers of lungs will assist in building predictive models to explain antibiotic failure.
Among the microbes detected using 16S rRNA gene sequencing and analyses, Pseudomonas dominated throughout the lungs (95.8–99.9%) and was detected in all sections (Figure 6 and Figure 7A). Some of the less abundant microbes, including Achromobacter (0–2.6%), Prevotella (0–0.2%), Haemophilus (0–0.8%), Staphylococcus (0–0.02%), and Stenotrophomonas (0–0.4%) were also detected (Figure 6). These microbes were not present in the DNA sequenced from blank swabs processed at the hospital during tissue collection, and hence represent DNA from bacteria present in the lungs (although the specific experiments performed do not confirm that these microbes were alive in the lung). The microbial spatial maps (Figure 6) along with microbe-microbe correlation network (Figure S5) allowed visualization of microbial co-localization. Among CF pathogens, Pseudomonas was positively correlated with Staphylococcus and negatively correlated with Granulicatella, Streptococcus and Haemophilus. A strong co-occurrence was observed for CF pathogens Haemophilus, Rothia, and Prevotella (Figure 6 and S5). Various other low-abundance microbes were also detected (Data S1).
Figure 6.

The distributions of various genera. The maps reveal relative percent of 16S reads in each sample and may not reflect absolute changes in bacterial load. The mapping has been performed on the left lung.
Figure 7.

A) The distribution of Pseudomonas and Pseudomonas-derived molecules were not spatially correlated. B) Achromobacter distributions were anti-correlated with the drug Meropenem. The mapping has been performed on the left lung. The navigable atlas for all detected molecules and microbes is available at weblinks: Molecule_maps and Microbe_maps.
Correlation between microbial and molecular penetration
We compared the spatial maps of microbial abundances to the spatial maps of molecular abundances, specifically the virulence factors and the antibiotics administered. All the quinolones and rhamnolipids were strongly spatially correlated; i.e., the structurally different quinolones and rhamnolipids colocalized (Figures 2 and 3 and Figure S4). Similar to spatial maps, the metabolite-metabolite correlation network also showed statistically significant clustering of different quinolones (Figure S6). However, the presence of quinolones or the rhamnolipids was not strongly correlated with the microbe Pseudomonas, which was largely abundant through the lung and produces these molecules (Figure 7A). The metabolite-metabolite correlation network (Figure S6) also showed clustering of the parent molecule Piperacillin with its metabolite desethyl-piperacillin, supporting the observation from 3D maps that the parent drug and metabolite show strong spatial correlation. Unlike quinolones and rhamnolipids, the different antibiotics were not strongly spatially correlated, but a strong anti-correlation was observed for the distribution of Meropenem and Achromobacter (Figure 7B). This observation suggests that bacteria can persist in regions of the lung where antibiotic penetration is sub-therapeutic, posing an important clinical concern from the standpoint of the development of antibiotic resistance. It has been suggested that Achromobacter can persist in the lung for long periods of time by developing antibiotic resistance (Ormerod et al., 2015). Our data suggests that Achromobacter may survive due to the lower exposure of the antibiotic deeper into the lung. In support of this hypothesis we tested the antibiotic susceptibility of Achromobacter. Achromobacter was susceptible to Meropenem with a minimal inhibitory concentration of 0.5 µg/mL and a sub-MIC exposure resulted in significant change in the Achromobacter metabolome (Figure S7). The antibiotics do not penetrate the entire lung, likely making it easier to develop resistance that can then spread to other regions of the lung. This provides an example of how spatial mapping and microbe-metabolome correlation analyses facilitates the construction of testable hypotheses by interrogating direct spatial correlations difficult to observe and understand via other methods.
Discussion
The last decade has brought a significant advance in our understanding of the role of the human microbiome in health and disease. Numerous diseases, including diabetes, inflammatory bowel disease, irritable bowel syndrome, obesity, autism spectrum disorders, and Parkinson’s disease have been linked to our microbiome (Gilbert et al., 2016). The challenges associated with deciphering causation versus correlation in the involvement of our microbiome in health and disease hampers methodical development of treatments that would be directed towards the microbes in question. Therefore, we must develop methodologies and resources that allow us to build testable hypothesis directly from the diseased samples when available to explain treatment failure, and to build predictive models that will guide treatment of new cases. In infectious diseases, spatial variation in the microbiota may be associated with disease severity and may result in variation in the chemical make-up. Thus, visualization and cross-correlation between the microbiome via 16S rRNA amplicon or metagenome sequencing and the molecular profile via metabolomics will allow us to investigate possible mechanisms by which microbes interact with each other, the host, and the treatments administered. In this study, we created methodology to construct and analyze 3D spatial metabolome and microbiome maps on radiology imaging of an entire human lung (n=1) at the late stage of disease. Using mass spectrometry, we made annotations corresponding to microbial molecules, host-associated molecules, and the administered drugs. These molecular annotations, along with 16S rRNA gene amplicon sequences, were mapped onto a 3D image of the human lung. We created browsable maps of 16,379 molecules and 56 microbes that serve as a resource for scientists researching CF and other lung-associated diseases. These maps revealed that the drug penetration and metabolism varies spatially within the lung and may influence the local bacterial community structure as suggested by the presence of Achromobacter at the base of lung where lower penetration of Meropenem was observed. When determining the correct dosage of a new drug, the organ is considered as a whole, and spatial variation of drug availability under the disease scenario is not tested, which has implications for the development of drug resistance. Our observations suggest that it is important to test spatial variation in drug availability at various disease stages. Furthermore, we detected the drug metabolite descladinose-azithromycin a month after the parent drug, Azithromycin, was administered. It is known that Azithromycin is taken up and retained intracellularly (Bosnar et al., 2005), but the persistence of Azithromycin metabolites for many weeks had not previously been described. It is important to understand how long such metabolites last, as the longevity of active metabolites (or parent drug) has direct implications in the development of resistance to the drug. When such active molecules last in the lung, chances of developing resistance increase, a problem often encountered in CF patients (Conway et al., 2003; Ormerod et al., 2015; Sherrard et al., 2014).
As expected, the microbiome maps showed domination by one pathogen, Pseudomonas aeruginosa, because the lung under investigation was associated with end stage CF disease. Nevertheless, the distribution of Pseudomonas-specific molecules involved in biofilm formation was not similar to the distribution of the microbe itself, demonstrating spatial heterogeneity in microbial biofilms within the lung. The spatial variation in quinolones and rhamnolipids may stem from the variation in genetic mutations in quinolone biosynthetic pathways and physical separation of genetically different variants of Pseudomonas, and co-occurrence of other micro-organisms and exposure to different levels of antibiotics. Previously, different isolates cultured from one CF patient at a given time and between patients over time had accumulated different mutations and varied in genetic potential for production of quinolones (Feltner et al., 2016; Markussen et al., 2014; Smith et al., 2006; Wilder et al., 2009). Our maps showed that antibiotic penetration or presence of a specific local microbial community did not result in the spatial variation observed for quinolones and rhamnolipids and hence is most likely accounted for by the genetic variation in the Pseudomonas subpopulation and local environmental variation. Spatial variation of transcripts from metatranscriptomics data or flow-sorting viable cells with a suitable stain is required to provide additional valuable information with regards to viable component of the microbial community observed through 16S rRNA amplicon sequencing. Although there exists significant variability between CF patients, the atlas of distribution of microbes and molecules in the lung generated in this study will serve as a resource for the scientific community studying lung-associated diseases to cross correlate this data with their observations. As the future unravels specific microbiota-derived molecules (Donia and Fischbach, 2015; Garg et al., 2017), their spatial and temporal visualization will provide a means to infer their biological significance.
Our methodology gives global insights into metabolome and microbiome of an entire organ which has not been possible previously. Our maps reveal that local environments within the lung vary in terms of pathogen abundance, characteristics of biofilm at the molecular level, pharmaceutical and environmental exposure directly from the diseased tissue. As previously suggested from studies with microbial isolates from spatially distinct anatomical lung locations (Chung et al., 2017; Jorth et al., 2015), such variation may be the underlying cause for variation of disease severity within the lung and clinical episodes of exacerbations. Furthermore, the methodology developed herein can be extended to any human organ (notably, organs with tumors, which are known to be associated with their own unique microbiomes (Cavarretta et al., 2017; Cummins and Tangney, 2013; Flemer et al., 2017) and to map and correlate various other types of omics data such as metagenomes and transcriptomes. The method also opens the possibility to detect and visualize the environmental chemicals that a human is exposed to. This methodology can be modified to vary the scale of the sample collection and applied to different disease states and even whole animal models to investigate metabolome changes in response to a disease or exposure to drugs such as antibiotics.
STAR methods
CONTACT FOR REAGENT AND RESOURCE SHARING
For clinical aspects of this study, including clinical isolates: dconrad@ucsd.edu
Regarding ‘ili: theodore.alexandrov@embl.de
The sequencing and DNA availability: rknight@ucsd.edu
The mass spectrometry, 3D visualization, the project and interpretation: pdorrestein@ucsd.edu
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Pieter C. Dorrestein (pdorrestein@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subject
The subject is a male, 41 years of age. The antibiotic Meropenem was administered for 2 days prior to tissue collection, Piperacillin was administered for 5 days prior to tissue collection and Ciprofloxacin was administered for 2 days prior to tissue collection. Azithromycin administration was stopped one month prior to tissue collection. This study was approved by the University of California Institutional Review Board (project #081500).
Microbial isolates
The cultures were routinely collected from this patient and analyzed at the Center of Advanced Laboratory Medicine using a MALDI Biotyper. The cultures analyzed at the Center of Advanced laboratory Medicine prior to tissue collection for this patient were identified by the Center using a MALDI-BIOTYPER. All bacterial isolates were cultured on sheep blood agar and MacConkey agar for 24 h prior to extraction for metabolomics analysis.
METHOD DETAILS
Tissue collection and processing
The tissue sectioning was carried out in accordance with the approved protocol and informed consent was obtained from patient before scheduled lung replacement surgery but passed and then agreed upon by next of kin post-mortem. The tissue sectioning was performed at Hillcrest hospital in San Diego, California immediately after the post mortem lungs became available (10 h after the patient passed away and was kept in the cold room). The lungs were first sliced horizontally. To avoid contamination, the knife was cleaned with 70% ethanol after each slice. The anatomical orientation of each slice was recorded. Every alternate slice starting from apex of the lung was further sub-sectioned into small sections 1–2 cm3 in size maintaining the recorded orientation resulting in 86 sections. Each of the sub-sectioned tissue pieces were swabbed with sterile soft foam swabs moistened with Tris-EDTA, pH 7.4. The swabs were stored in 96-well bead plate provided in the MO BIO PowerSoil®-htp 96 Well DNA Isolation Kit placed on dry ice. The individual tissue sections were stored in pre-cooled glass jars placed on dry ice and the jars containing the tissue section were kept on dry ice during the processing of entire tissue. These samples were kept frozen at −80 °C until further processing. Bacterial DNA was isolated from the swabs using the PowerSoil®-htp 96 Well Soil DNA Isolation Kit following the manufacturer’s instructions and was subjected to prokaryotic ribosomal 16S rRNA gene amplicon sequencing using the standardized Earth Microbiome Protocol (http://www.earthmicrobiome.org/emp-standard-protocols/). Amplicons were cleaned and pooled and then sequenced on an Illumina MiSeq. The sequencing run was performed at the BioFrontiers Next-Generation Genomics Facility at the University of Colorado Boulder. The microbial isolates were obtained from the Center of Advanced Clinical Medicine, UC San Diego where the isolates were cultured from tissue swabs and sputum on sheep blood agar and MacConkey agar in CO2 incubator at 30 °C for 24h. The cultures were routinely collected from this patient and analyzed at the Center of Advanced Laboratory Medicine using a MALDI Biotyper. The cultures analyzed at the Center of Advanced laboratory Medicine prior to tissue collection for this patient were identified as Pseudomonas. Tissue sections were weighed and extracted with 1 mL/g of tissue with a 1:1:1 mixture of chloroform, methanol and water followed by a second extraction with 2:2:1 mixture of ethyl acetate, methanol and water. Equal volumes of both extraction conditions were combined and an aliquot of 150 µL of the extract was dried for each tissue section and analyzed by mass spectrometry.
MS acquisition
The tissue extracts and extracts of bacterial isolates from CF subjects cultured on sheep blood agar and MacConkey agar for 24 h were resuspended in 80% methanol containing 1µM sulfadimethoxine and analyzed with UltiMate 3000 UPLC system (Thermo Scientific) using a Kinetex™ 1.7 µm C18 reversed phase UHPLC column (50 × 2.1 mm) and Maxis Q-TOF mass spectrometer (Bruker Daltonics) equipped with ESI source. The column was equilibrated with 2% solvent B (98% acetonitrile, 0.1% formic acid in LC-MS grade water with solvent A as 0.1% formic acid in water) for 1 min, followed by a linear gradient from 2% B to 100% B in 10 min, held at 100% B for 2.5 min. A small wash segment was employed to wash the column (100% B for 0.5 min, 100%-10 % B in 0.5 min) following which the column was kept at 2% B for 0.5 min at a flow rate of 0.5 mL/min throughout the run. MS spectra were acquired in positive ion mode in the range of 50–2000 m/z. A mixture of 10 µg/mL of each sulfamethazine, sulfamethizole, sulfachloropyridazine sulfadimethoxine, amitriptyline, and coumarin-314 was run after every eight injections for quality control. An external calibration with ESI-L Low Concentration Tuning Mix (Agilent technologies) was performed prior to data collection and internal calibrant Hexakis(1H,1H,3H-tertrafluoropropoxy)phosphazene was used throughout the runs. The capillary voltage of 4500 V, nebulizer gas pressure (nitrogen) of 2 bar, ion source temperature of 200 °C, dry gas flow of 9 L/min sour ce temperature, spectral rate of 3 Hz for MS1 and 10 Hz for MS2 was used. For acquiring MS/MS fragmentation, 10 most intense ions per MS1 were selected. Basic stepping function was used to fragment ions at 50% and 125% of the CID with timing of 50% for each step. Similarly, basic stepping of collision RF of 550 and 800 Vpp with a timing of 50% for each step and transfer time stepping of 57 and 90 µs with a timing of 50% for each step was employed. MS/MS active exclusion parameter was set to 3 and released after 30 seconds. The mass of internal calibrant was excluded from the MS/MS list using a mass range of m/z 921.5–924.5. The data was deposited in the online repository namely MassIVE through GNPS and is available under the id MSV000079652. The new annotations of the spectral features made by the host laboratory and the scientific community are continuously available through a GNPS subscription to the data set (Wang et al., 2016).
Molecular networking
The molecular network was created using the online workflow at GNPS platform. The data was then clustered with MS-Cluster with a parent mass tolerance of 0.1 Da and a MS/MS fragment ion tolerance of 0.1 Da to create consensus spectra. Further, consensus spectra that contained less than 3 spectra were discarded. A network was then created where edges were filtered to have a cosine score above 0.65 and more than 4 matched peaks. The edges between two nodes were kept in the network if and only if each of the nodes appeared in each other's respective top 10 most similar nodes. The spectra in the network were then searched against GNPS spectral libraries. All matches kept between network spectra and library spectra were required to have a score above 0.65 and at least 4 matched peaks. The molecular networks and the parameters used are available at the links below:
The molecular network and parameters for the patient data are available at: http://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=63e8945df5e94828a25e8c71e26a1f6chttp://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=07d7ee88c5f240c296468813929e2c8a
The molecular network and parameters for the patient data and data acquired on cultured microbial isolates are available at: http://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=62239894791e45258ad23e6d82db9d8bhttp://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=7f6ce726d8df4ad6975bcb0c24a76e1d
The molecular network and parameters for comparison between the patient data, and public data acquired on sputum, gut and skin under the relevant mass spectrometry conditions are available at http://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=f0488dde74204aac826a305a339c6513
The annotations made using the dereplication workflow are available at http://gnps.ucsd.edu/ProteoSAFe/result.jsp?task=071cb47acc8c4a9a89003fca749f5dcf&view=view_all_annotations_DB
MS1 feature detection
The metabolite features were detected and integrated using the Optimus workflow (https://github.com/MolecularCartography/Optimus). The data analysis was conducted using the following parameter values: m/z tolerance: 10 ppm, noise threshold: 2500, RT tolerance: 60 s, intensity factor as compared to blanks: 3.0. The workflow produced a table with intensities of ions detected across all LC-MS runs. The x, y, z coordinates of each tissue section were added to this table for visualization (Data S2).
16S rRNA gene analysis
The sequences were processed and analyzed using QIIME v1.9 (Caporaso et al., 2010). First, sequences were quality trimmed and filtered using QIIME default parameters (split_libraries_fastq.py). Next, sequences were clustered into operational taxonomic units (OTUs) using the closed reference OTU picking method at 97% sequence similarity. UCLUST (Edgar, 2010) was the underlying clustering algorithm and Greengenes (August 2013 release) was the reference database used. OTU tables were filtered so that OTUs with a number of sequences <0.005% of the total number of sequences were discarded (Navas-Molina et al., 2013). The resulting OTU table was then rarefied to 3,369 sequences per sample (the number in the sample that yielded the fewest sequences) and used in downstream analyses, including the creation of taxonomy summaries and in the calculation of the UniFrac distance (unweighted) between samples.
3D lung model generation and visualization
The CT- scan images in the dicom format were obtained from the radiology department at the Hillcrest hospital in San Diego. These images were imported in the publically available software InVesalius 3.0. Herein, the images were combined to create a 3D lung model and the model was exported in the .stl format. The exported lung model is embedded in the tissue density arising from chest and back. The pixels corresponding to the chest and back were selected and manually deleted using the 3D modelling software, Geomagic Wrap. The underlying code for a previously developed Google Chrome extension, namely `ili (https://github.com/MetaboliteCartography/ili) was modified and adapted to plot abundance of microbes and molecules in three dimensions on the surface as well as inside of the lung. The modified code is deposited at GitHub and is available at https://github.com/mwang87/ili. The models were virtually sliced using this in-house developed web-based tool following the orientation that was used to slice the lung at the hospital. These virtual slices were used to obtain the physical x, y, and z coordinates of each of the tissue section that was cut out from the slices using the point coordinates feature of Geomagic Wrap. A radius of 20 mm was assigned to each coordinates and the abundance of microbes and molecules were plotted using the in-house developed web based tool available at http://mwang87.github.io/ili. The excel file containing the abundance of microbes and molecules is provided as supplementary material as Data S1 and S2, the script with the features utilized for mapping the abundances is available at https://raw.githubusercontent.com/mwang87/ili/master/offline_scripts/orientation_for_paper/patient1/orientation.js
Generation of heat map and pie chart for antibiotic exposure experiment
The minimal inhibitory concentration (MIC) of Achromobacter against Meropenem was determined on solid agar using disk diffusion assay. The concentration above which complete cessation of growth was observed was recorded as MIC and was found to be 0.5 µg/mL. The Achromobacter was cultured using Winogradsky-based culture system in artificial sputum media as described previously (Quinn et al., 2015) containing 0, 0.0625, 0.125, 0.25 µg/mL of Meropenem. The cultures were grown for 24 h and extracted and mass spectrometry data was acquired under the same conditions as described for the extraction of lung tissue. The metabolite features were detected and integrated using the Optimus workflow (https://github.com/MolecularCartography/Optimus). The feature table was used to create the heat map. The columns (molecules) were ordered by hierarchical clustering using the canberra dissimilarity measure and Ward grouping method. In order to detect which metabolic features were changing according to antibiotic concentration, the Kruskal-Wallis rank sum non-parametric test was applied. After the test, only features significantly changing in at least one antibiotic concentration were retained. The features were then summarized in pie charts on the following classes: Increasing/decreasing in all antibiotic concentrations (higher than zero); Increasing/decreasing only on two highest concentrations (0.125, 0.25 µg/mL) and Increasing/decreasing only on the highest concentration (0.25 µg/mL). The R package pheatmap (https://CRAN.R-project.org/package=pheatmap) was used to draw the heat map and the package plotly (https://CRAN.R-project.org/package=plotly) was used to draw the pie charts.
QUANTIFICATION AND STATISTICAL ANALYSIS
Microbe-Microbe and Metabolite-Metabolite correlation networks
The OTU tables generated as described above were used to create the correlation networks using the SparCC method (Friedman and Alm, 2012), using as parameters 100 iterations and 100 simulated datasets for p-value calculation. The resulting network was filtered by significant two sided p-values (< 0.05) and absolute value of correlation score (> 0.05). The correlation network was subsequently visualized in Cytoscape.
The metabolite feature table was used to generate the Pearson correlation network. Pairwise correlations were filtered by absolute correlations scores (r > 0.97). The node size was represented proportionally to the normalized feature average intensity along the samples. The networks were further overlaid with GNPS spectral networks, coloring node MS1 masses of green if a MS2 spectrum was recorded within a close m/z (10 ppm window) and retention time (20 s window) precursor ion. The edges were represented as a thick line if a correlation and a spectral similarity were found by the same node pair. The calculations were performed using R statistical environment and the network visualized using Cytoscape.
DATA AND SOFTWARE AVAILABILITY
Mass spectrometry data, software and analysis
The mass spectrometry data is deposited in GNPS-MassIVE at http://gnps.ucsd.edu under accession number MSV000079652 and the molecular networks were also created within this infrastructure (Wang et al 2016). The molecular network and parameters for the patient data are available at: http://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=63e8945df5e94828a25e8c71e26a1f6chttp://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=07d7ee88c5f240c296468813929e2c8a
The molecular network and parameters for the patient data and data acquired on cultured microbial isolates are available at: http://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=62239894791e45258ad23e6d82db9d8bhttp://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=7f6ce726d8df4ad6975bcb0c24a76e1d
The molecular network and parameters for comparison between the patient data, and public data acquired on sputum, gut and skin under the relevant mass spectrometry conditions are available at http://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=f0488dde74204aac826a305a339c6513
The annotations made using the dereplication workflow are available at http://gnps.ucsd.edu/ProteoSAFe/result.jsp?task=071cb47acc8c4a9a89003fca749f5dcf&view=view_all_annotations_DB
Sequencing data: EBI accession number is ERP051557 and it is study 11335 in Qiita
ADDITIONAL RESOURCES
3D cartography software
The analysis tool ‘ili for 3D volume cartography is available at http://mingwangbeta.ucsd.edu/public/ili/ or the original versions for surface 3D cartography as google extension https://chrome.google.com/webstore/detail/%60ili/nhannoeblkmkmljpddfhcfpjlnanfmkc?hl=en-US
Supplementary Material
Abundance of all detected microbes. Related to Figure 6.
Abundance of all detected molecules. Related to Figure STAR methods: MS1 feature detection”.
Video S1 provides visualization of HHQ. Related to Figure 1 and 2.
Highlights.
CT scan images used to generate 3D models of human lungs for mapping omics data
Microbiome and metabolome data obtained from a CF lung
Omics data plotted on 3D lung model to visualize spatial interactions and variations
Regiospecific metabolism of medications placed in the context of microbial distribution
Acknowledgments
We thank the Center of Advanced Clinical Medicine, UC San Diego and Vanessa Phelan, Assistant Professor, Department of Pharmaceutical Sciences, University of Colorado for providing the CF-associated Achromobacter isolate. We thank the following financial support: National Institute of Health (NIH) Grants 3R01GM095384-03S1 subcontracted to FLR and PCD and UCSD Clinical and Translational Research Institute Pilot award UL1 RR031980 written by Dr. Katrine Whiteson at San Diego State University at the time and now at University of California Irvine. We also would like to acknowledge the Bruker Therapeutic Discovery Mass Spectrometry Center at UCSD Skaggs School of Pharmacy and Pharmaceutical Sciences. MW and NB were partially supported by the US National Institutes of Health Grant 2 P41 GM103484–06A1 from the National Institute of General Medical Sciences; NB is an Alfred P. Sloan Research Fellow. IP and TA acknowledge funding from the European Union’s Horizon2020 program under the grant agreement 634402.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
NG, DC, FR, PCD created the idea for the work
NG, PCD wrote the paper
NG, RW, AB, YWL, GYL, DJC collected lung tissue
NG, TS, SSB created 3D model
NG processed the model
NG, AM picked coordinates on lung model
NG, WC performed Meropenem exposure experiments
RS performed correlative statistical analysis
EH, NG, GH, JG, GA, RK performed 16S rRNA sequencing and analysis
MW, NB modified the tool `ili created by IP, TA
IP, TA created the tool Optimus
NG extracted lung tissue for acquisition of mass spectrometry data
NG acquired mass spectrometry data
NG, IP, PCD analyzed mass spectrometry data
Competing financial interests
NB is a co-founder, has an equity interest and receives income from Digital Proteomics, LLC. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. Digital Proteomics was not involved in the research presented here.
References
- Abdel-Mawgoud AM, Lepine F, Deziel E. Rhamnolipids: diversity of structures, microbial origins and roles. Appl Microbiol Biotechnol. 2010;86:1323–1336. doi: 10.1007/s00253-010-2498-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosnar M, Kelneric Z, Munic V, Erakovic V, Parnham MJ. Cellular uptake and efflux of azithromycin, erythromycin, clarithromycin, telithromycin, and cethromycin. Antimicrob Agents Chemother. 2005;49:2372–2377. doi: 10.1128/AAC.49.6.2372-2377.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouslimani A, Porto C, Rath CM, Wang M, Guo Y, Gonzalez A, Berg-Lyon D, Ackermann G, Moeller Christensen GJ, Nakatsuji T, et al. Molecular cartography of the human skin surface in 3D. Proc Natl Acad Sci U S A. 2015;112:E2120–2129. doi: 10.1073/pnas.1424409112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumann D. Heterogeneous host-pathogen encounters: act locally, think globally. Cell Host Microbe. 2015;17:13–19. doi: 10.1016/j.chom.2014.12.006. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavarretta I, Ferrarese R, Cazzaniga W, Saita D, Luciano R, Ceresola ER, Locatelli I, Visconti L, Lavorgna G, Briganti A, et al. The Microbiome of the Prostate Tumor Microenvironment. Eur Urol. 2017 doi: 10.1016/j.eururo.2017.03.029. [DOI] [PubMed] [Google Scholar]
- Chung H, Lieberman TD, Vargas SO, Flett KB, McAdam AJ, Priebe GP, Kishony R. Global and local selection acting on the pathogen Stenotrophomonas maltophilia in the human lung. Nat Commun. 2017;8:14078. doi: 10.1038/ncomms14078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway SP, Brownlee KG, Denton M, Peckham DG. Antibiotic treatment of multidrug-resistant organisms in cystic fibrosis. Am J Respir Med. 2003;2:321–332. doi: 10.1007/BF03256660. [DOI] [PubMed] [Google Scholar]
- Cummins J, Tangney M. Bacteria and tumours: causative agents or opportunistic inhabitants? Infect Agent Cancer. 2013;8:11. doi: 10.1186/1750-9378-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donia MS, Fischbach MA. HUMAN MICROBIOTA. Small molecules from the human microbiota. Science. 2015;349:1254766. doi: 10.1126/science.1254766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Feltner JB, Wolter DJ, Pope CE, Groleau MC, Smalley NE, Greenberg EP, Mayer-Hamblett N, Burns J, Deziel E, Hoffman LR, et al. LasR Variant Cystic Fibrosis Isolates Reveal an Adaptable Quorum-Sensing Hierarchy in Pseudomonas aeruginosa. MBio. 2016;7 doi: 10.1128/mBio.01513-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemer B, Lynch DB, Brown JM, Jeffery IB, Ryan FJ, Claesson MJ, O'Riordain M, Shanahan F, O'Toole PW. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut. 2017;66:633–643. doi: 10.1136/gutjnl-2015-309595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, Alm EJ. Inferring correlation networks from genomic survey data. PLoS Comput Biol. 2012;8:e1002687. doi: 10.1371/journal.pcbi.1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg N, Luzzatto-Knaan T, Melnik AV, Caraballo-Rodriguez AM, Floros DJ, Petras D, Gregor R, Dorrestein PC, Phelan VV. Natural products as mediators of disease. Nat Prod Rep. 2017;34:194–219. doi: 10.1039/c6np00063k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert JA, Quinn RA, Debelius J, Xu ZZ, Morton J, Garg N, Jansson JK, Dorrestein PC, Knight R. Microbiome-wide association studies link dynamic microbial consortia to disease. Nature. 2016;535:94–103. doi: 10.1038/nature18850. [DOI] [PubMed] [Google Scholar]
- Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, Bouffard GG, Blakesley RW, Murray PR, Green ED, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser AR, Jain M, Bar-Meir M, McColley SA. Clinical significance of microbial infection and adaptation in cystic fibrosis. Clin Microbiol Rev. 2011;24:29–70. doi: 10.1128/CMR.00036-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeb S, Fletcher MP, Chhabra SR, Diggle SP, Williams P, Camara M. Quinolones: from antibiotics to autoinducers. FEMS Microbiol Rev. 2011;35:247–274. doi: 10.1111/j.1574-6976.2010.00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorth P, Staudinger BJ, Wu X, Hisert KB, Hayden H, Garudathri J, Harding CL, Radey MC, Rezayat A, Bautista G, et al. Regional Isolation Drives Bacterial Diversification within Cystic Fibrosis Lungs. Cell Host Microbe. 2015;18:307–319. doi: 10.1016/j.chom.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koumellis P, van Beek EJ, Woodhouse N, Fichele S, Swift AJ, Paley MN, Hill C, Taylor CJ, Wild JM. Quantitative analysis of regional airways obstruction using dynamic hyperpolarized 3He MRI-preliminary results in children with cystic fibrosis. J Magn Reson Imaging. 2005;22:420–426. doi: 10.1002/jmri.20402. [DOI] [PubMed] [Google Scholar]
- Lepine F, Milot S, Deziel E, He J, Rahme LG. Electrospray/mass spectrometric identification and analysis of 4-hydroxy-2-alkylquinolines (HAQs) produced by Pseudomonas aeruginosa. J Am Soc Mass Spectrom. 2004;15:862–869. doi: 10.1016/j.jasms.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Markussen T, Marvig RL, Gomez-Lozano M, Aanaes K, Burleigh AE, Hoiby N, Johansen HK, Molin S, Jelsbak L. Environmental heterogeneity drives within-host diversification and evolution of Pseudomonas aeruginosa. MBio. 2014;5 doi: 10.1128/mBio.01592-14. e01592-01514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KC, Sharma A. Regional variability of lung inflammation in cystic fibrosis. Am J Respir Crit Care Med. 1997;156:1536–1540. doi: 10.1164/ajrccm.156.5.9701098. [DOI] [PubMed] [Google Scholar]
- Navas-Molina JA, Peralta-Sanchez JM, Gonzalez A, McMurdie PJ, Vazquez-Baeza Y, Xu Z, Ursell LK, Lauber C, Zhou H, Song SJ, et al. Advancing our understanding of the human microbiome using QIIME. Methods Enzymol. 2013;531:371–444. doi: 10.1016/B978-0-12-407863-5.00019-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Byrd AL, Deming C, Conlan S, Kong HH, Segre JA. Biogeography and individuality shape function in the human skin metagenome. Nature. 2014;514:59–64. doi: 10.1038/nature13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormerod KL, George NM, Fraser JA, Wainwright C, Hugenholtz P. Comparative genomics of non-pseudomonal bacterial species colonising paediatric cystic fibrosis patients. PeerJ. 2015;3:e1223. doi: 10.7717/peerj.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn RA, Whiteson K, Lim YW, Salamon P, Bailey B, Mienardi S, Sanchez SE, Blake D, Conrad D, Rohwer F. A Winogradsky-based culture system shows an association between microbial fermentation and cystic fibrosis exacerbation. Isme J. 2015;9:1052. doi: 10.1038/ismej.2014.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrard LJ, Tunney MM, Elborn JS. Antimicrobial resistance in the respiratory microbiota of people with cystic fibrosis. Lancet. 2014;384:703–713. doi: 10.1016/S0140-6736(14)61137-5. [DOI] [PubMed] [Google Scholar]
- Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D'Argenio DA, Miller SI, Ramsey BW, Speert DP, Moskowitz SM, et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A. 2006;103:8487–8492. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Carver JJ, Phelan VV, Sanchez LM, Garg N, Peng Y, Nguyen DD, Watrous J, Kapono CA, Luzzatto-Knaan T, et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat Biotechnol. 2016;34:828–837. doi: 10.1038/nbt.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watrous J, Roach P, Alexandrov T, Heath BS, Yang JY, Kersten RD, van der Voort M, Pogliano K, Gross H, Raaijmakers JM, et al. Mass spectral molecular networking of living microbial colonies. Proc Natl Acad Sci U S A. 2012;109:E1743–1752. doi: 10.1073/pnas.1203689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilder CN, Allada G, Schuster M. Instantaneous within-patient diversity of Pseudomonas aeruginosa quorum-sensing populations from cystic fibrosis lung infections. Infect Immun. 2009;77:5631–5639. doi: 10.1128/IAI.00755-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams P, Camara M. Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: a tale of regulatory networks and multifunctional signal molecules. Curr Opin Microbiol. 2009;12:182–191. doi: 10.1016/j.mib.2009.01.005. [DOI] [PubMed] [Google Scholar]
- Willner D, Haynes MR, Furlan M, Schmieder R, Lim YW, Rainey PB, Rohwer F, Conrad D. Spatial distribution of microbial communities in the cystic fibrosis lung. Isme J. 2012;6:471–474. doi: 10.1038/ismej.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Schloss PD, Kalikin LM, Carmody LA, Foster BK, Petrosino JF, Cavalcoli JD, VanDevanter DR, Murray S, Li JZ, et al. Decade-long bacterial community dynamics in cystic fibrosis airways. Proc Natl Acad Sci U S A. 2012;109:5809–5814. doi: 10.1073/pnas.1120577109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Abundance of all detected microbes. Related to Figure 6.
Abundance of all detected molecules. Related to Figure STAR methods: MS1 feature detection”.
Video S1 provides visualization of HHQ. Related to Figure 1 and 2.