

Abstract
The endoplasmic reticulum is the port of entry for proteins into the secretory pathway and the site of synthesis for several important lipids, including cholesterol, triacylglycerol, and phospholipids. Protein production within the endoplasmic reticulum is tightly regulated by a cohort of resident machinery that coordinates the folding, modification, and deployment of secreted and integral membrane proteins. Proteins failing to attain their native conformation are degraded through the endoplasmic reticulum–associated degradation (ERAD) pathway via a series of tightly coupled steps: substrate recognition, dislocation, and ubiquitin-dependent proteasomal destruction. The same ERAD machinery also controls the flux through various metabolic pathways by coupling the turnover of metabolic enzymes to the levels of key metabolites. We review the current understanding and biological significance of ERAD-mediated regulation of lipid metabolism in mammalian cells.
Keywords: ERAD, quality control, ubiquitin, cholesterol, triacylglycerol, lipid droplet
INTRODUCTION
The endoplasmic reticulum (ER) is a dynamic, membrane-bound organelle that is contiguous with the nuclear envelope and that extends throughout the cytoplasm as a netlike array of connected sacs and branching tubules. The ER hosts metabolic pathways that synthesize a variety of lipids, including cholesterol, phospholipids, and neutral lipids [e.g., steryl esters and triacylglycerol (TAG)] (93). Lipids synthesized in the ER are distributed throughout the cell to their respective target membranes via both vesicular and nonvesicular mechanisms (93). The extensive reticular structure of the ER enables the formation of membrane contact sites between the ER and a myriad of organelles (e.g., mitochondria, plasma membrane, endosomes, and others), providing sites for the rapid interorganelle transfer of lipids and signaling molecules (29, 140). The ER also mediates the biogenesis of lipid droplets (LDs), which are organelles that specialize in the storage and mobilization of neutral lipids for membrane biogenesis, energy production, and signaling pathways (139, 189).
As a central hub of lipid metabolism, the ER must sense and respond to fluctuations in lipid levels and the demand for lipids to maintain cellular homeostasis. Multiple mechanisms are employed to fine-tune the flux through metabolic pathways, including transcriptional programs that control the expression of genes involved in lipid biosynthesis, allosteric regulators of enzyme activity, and posttranslational modifications (e.g., phosphorylation) that regulate enzyme activity. Cells also control the levels of lipid biosynthetic enzymes by targeting them for degradation by the ubiquitin-proteasome system (UPS). In the ER, this process is termed ER-associated degradation (ERAD) (20, 132, 197). ERAD is a sophisticated, multistep process that recognizes, extracts, and ubiquitinates ER proteins for clearance by the cytosolic 26S proteasome (20, 132, 197).
In this review we focus on our current understanding of the ERAD mechanism and the role of ERAD in regulating lipid metabolism in mammalian cells.
THE ENDOPLASMIC RETICULUM: THE GATEWAY INTO THE SECRETORY PATHWAY
The ER is a major protein-folding compartment that mediates the modification, structural maturation, and targeted deployment of approximately one-third of the cellular proteome (Figure 1a) (7, 156). Proteins processed within the lumen and membrane of the ER include secreted proteins, as well as proteins that reside in the ER, plasma membrane, Golgi complex, LDs, peroxisomes, and lysosomes. Through its role as the gateway into the secretory pathway, the ER broadly impacts intracellular organelle function and cellular communication with the extracellular environment.
Figure 1.
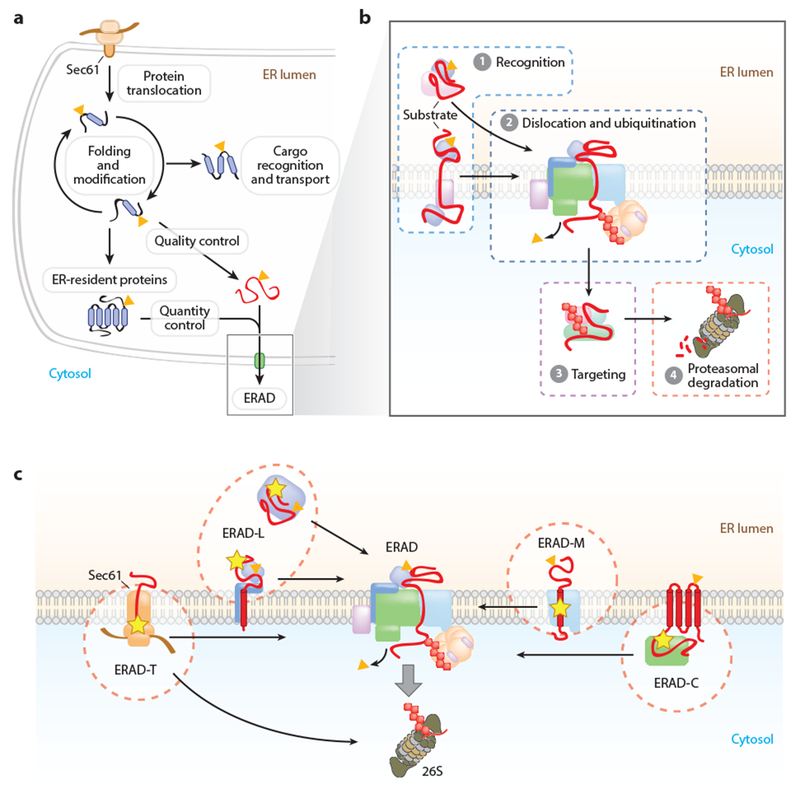
Mechanisms of protein quality and quantity control in the endoplasmic reticulum (ER). (a) Protein folding and maturation in the ER. Proteins inserted through the Sec61 translocon are subject to cotranslational and posttranslational folding. Proteins that have achieved their native conformation are recognized and transported to their final cellular destination. Proteins failing to achieve their proper conformation are triaged for ER-associated degradation (ERAD) (quality control). The levels of some properly folded proteins are also controlled by regulated degradation by ERAD (quantity control). (b) ERAD mediates the delivery of proteins from the ER to the cytosolic 26S proteasome through a series of coupled steps: ① substrate recognition, ② dislocation and ubiquitin conjugation, and ③ targeting of the ubiquitinated proteins to the proteasome for ④ proteolysis. (c) The ERAD system comprises multiple pathways that mediate the recognition and degradation of topologically diverse substrates. These pathways are commonly designated based upon the location of the substrate degradation signal: luminal (ERAD-L), cytosolic (ERAD-C), membrane (ERAD-M), or translocon (ERAD-T). (Yellow star indicates degradation signal; orange triangle indicates N-glycan.)
Secretory proteins, in most cases, contain N-terminal signal sequences or transmembrane domains that are recognized by the ribosome-associated signal recognition particle (SRP) immediately upon emergence from the ribosome exit tunnel (2, 156). The binding of SRP to the signal sequence or transmembrane domain shields the hydrophobic stretch of amino acids from the surrounding aqueous cytoplasm and pauses translation, reducing the overall risk of protein aggregation (2, 156). Upon association with the ER-resident SRP receptor, the emerging protein-ribosome complex is transferred to the Sec61 translocon and translation resumes (2, 156). The α-subunit of the heterotrimeric Sec61 complex forms the translocation channel, folding into an hourglass-shaped aqueous pore that constricts in the middle of the lipid bilayer (185, 188). Following termination of translation, the signal sequence is either laterally gated into the membrane as a transmembrane domain or cleaved by signal peptidases (143). In some cases, proteins lack an N-terminal signal sequence and instead contain a C-terminal transmembrane domain. These tail-anchored proteins fold in the cytoplasm and employ a posttranslational insertion pathway termed the guided entry of tail-anchored proteins (GET) pathway (27, 156), remaining mostly cytosolic with a short C-terminal transmembrane tether.
Proteins translocated into the ER lumen fold into functional three-dimensional conformations within the unique oxidative environment of the ER lumen (7, 176). Protein folding occurs both cotranslationally and posttranslationally, and is assisted by an array of ER-resident chaperones, oxidoreductases, and glycosylation enzymes (7, 176). The relatively slow rate of cotranslational insertion, together with the rapid association of ER-resident chaperones, constrains the rate of protein folding and promotes the native conformation by enabling local domain folding, thus protecting the folding intermediates from aggregation and facilitating the establishment of long-distance interdomain interactions (7, 176).
The majority of secretory proteins are modified by N-linked glycosylation (160). The translocon-associated oligosaccharyltransferase (OST) complex mediates the en bloc transfer of a preassembled oligosaccharide, composed of two N-acetylglucosamine (GlcNAc), nine mannose (Man), and three glucose (Glc) residues (GlcNAc2Man9Glc3), from a dolichol pyrophosphate-linked donor to the asparagine residue within a conserved glycosylation consensus sequence (NXS/T, where X is any amino acid except proline) (160). This N-linked glycan increases the solubility of the protein and directly influences the available protein folding pathways (176). The two most terminal glucose residues are rapidly removed by the successive actions of α-glucosidase I and α-glucosidase II, exposing a single glucose that supports association with two important ER-resident lectin chaperones, calnexin (integral membrane) and calreticulin (soluble) (176). The removal of the final glucose residue by α-glucosidase II prevents reassociation with calnexin and calreticulin (176). UDP-glucose glycoprotein glucosyltransferase 1 (UGT1) participates in an early quality control surveillance step by detecting any remaining unfolded regions, reglucosylating the glycan, and permitting the protein to reassociate with calnexin and calreticulin (176). This calnexin-calreticulin folding cycle continues until the protein is fully folded. Secretory proteins that achieve their native conformation—correct three-dimensional structure, modifications, and oligomeric partners—are packaged into COPII (coat protein complex II) vesicles for trafficking to the Golgi complex and downstream compartments (206).
ERAD MECHANISM
ERAD Participates in Protein Quality and Quantity Control
ERAD consists of a series of temporally and spatially coupled activities that mediate the recognition, cytosolic dislocation (also known as retrotranslocation), and ubiquitin-dependent proteasomal degradation of select proteins from the ER (Figure 1a,b) (132, 197). ERAD maintains the quality of the secretory proteome by degrading proteins that fail to achieve their native conformation due to mutations, errors in transcription or translation, or inefficient assembly into their native oligomeric complexes (Figure 1a). ERAD must achieve a precise balance to preserve both the overall quality and function of the secretory proteome. The failure to detect misfolded proteins can have disastrous consequences, such as the intracellular or extracellular accumulation of toxic aggregates that underlie many human diseases (49). For example, the ineffective triage of misfolded transthyretin for ERAD permits the secretion of misfolded monomeric transthyretin that forms extracellular amyloid deposits in peripheral tissues and causes progressive peripheral sensorimotor neuropathy, autonomic neuropathy, cardiomyopathy, and nephropathy (149, 153). However, an overly stringent ERAD system is also not advantageous. The ΔF508 mutant form of the cystic fibrosis transmembrane conductance regulator (CFTR), the most common cause of cystic fibrosis, is retained and degraded efficiently by ERAD despite partial functionality (42, 193). The failure of the protein to mature and traffic to the plasma membrane results in the severe chloride transport defect that manifests as thickened mucus in the lungs, pancreas, and other organs. The finding that the ΔF508 mutant CFTR is still partially functional provides hope that pharmacological agents that selectively impair ERAD and promote the maturation of functional mutant proteins could cure cystic fibrosis as well as other diseases associated with alterations in protein maturation (102). Thus, depending on the disease, pharmacological agents that improve or selectively impair substrate ERAD are required.
ERAD also has an important role in controlling the abundance of properly folded, functional proteins (Figure 1a). These quantity control substrates include a wide variety of ER-resident and secretory proteins, such as lipid biosynthetic enzymes [e.g., 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR)] (24), calcium transporters (e.g., inositol triphosphate receptors) (195), regulators of transcription pathways (e.g., IRE1α) (173), receptors involved in ligand-induced signaling (e.g., epidermal growth factor receptor) (209), and plasma membrane nutrient transporters (e.g., glutamine transporters) (68). Early studies of ERAD were dominated by analyses of severely misfolded mutant proteins. The identification and analysis of endogenous ERAD substrates will help to define the cellular roles of ERAD, the features of substrates detected by different ERAD pathways, and the complex mechanisms that regulate ERAD.
The Architecture of the ERAD System: Multiple Topologically Organized Routes to the Proteasome
ERAD substrates reflect the topological diversity of the secretory system clientele, and can be completely (i.e., luminal substrates) or partially (i.e., integral membrane substrates) segregated from the cytosolic UPS. Thus, ERAD must recognize conformational determinants presented in three very different subcellular compartments: the ER lumen, the ER membrane, and the cytosol (Figure 1c). ERAD pathways are commonly defined by the localization of the misfolded lesion or degradation signal that is presented by the substrate protein: ERAD-L (luminal), ERAD-C (cytosolic), ERAD-M (membrane), and ERAD-T (translocon-associated or preemptive degradation) (Figure 1c). Although these designations can be overly simplistic, they provide a generally useful organizational principle for the ERAD system, intuitively implying that the distinct spatial presentations of the substrate degradation signal require the engagement of different sets of ERAD factors for recognition and processing. ERAD pathways are largely organized around membrane-embedded complexes that physically and functionally scaffold luminal ERAD components, membrane dislocation factors, and cytosolic ubiquitination and extraction machinery (Figure 1b,c) (14, 18, 132). This is best characterized in yeast where the ERAD pathways are mostly segregated between two membrane-embedded E3 ligase complexes, the Doa10p complex (ERAD-C, ERAD-M) and the Hrd1p complex (ERAD-L, ERAD-M, and ERAD-T) (14, 144, 177). A new ERAD pathway in yeast that utilizes the inner nuclear membrane-localized Asi E3 ligase complex has recently been identified, and its substrates appear to include both soluble and membrane proteins (38, 75). In mammalian cells, the number of ERAD components has been significantly expanded, and the various ERAD pathways utilize overlapping sets of machinery that are combinatorially engaged to degrade substrates displaying different features (18, 132). This modular architecture likely enables the functional recruitment of substrate-specific adaptors and processing enzymes to accommodate the more diverse substrates that transit the mammalian secretory pathway.
Quality Control: Generalizable Mechanisms of Substrate Triage
One of the greatest challenges for the ERAD system is discriminating and segregating misfolded proteins from productive folding intermediates, folded proteins that are being trafficked out of the ER, and stable ER-resident proteins. This challenge is exacerbated by the diversity of secretory proteins, which exhibit differences not only in topology (Figure 1c) but also in modification (e.g., disulfide bonds and glycans), size, folding rate, and oligomeric state. In most cases, the mechanism of ERAD substrate recognition is incompletely understood, but clearly, the varied substrate clientele demands a system capable of recognizing general features of protein conformation. The recognition of aberrantly exposed regions of substrates by chaperones provides one such mechanism. The exposure of hydrophobic regions in aqueous environments leads to recognition by either luminal (e.g., BiP) (130) or cytosolic chaperones (e.g., Hsp70) (48, 106), which have dual roles in protein folding and ERAD targeting. Similarly, the failure of integral membrane proteins to properly fold, oligomerize, or both, may expose hydrophilic residues within the hydrophobic interior of the ER lipid bilayer that can be directly recognized by membrane-embedded ERAD components, such as the Hrd1 E3 ligase (146).
The majority of secretory proteins are glycosylated, and the transition from protein folding to targeted degradation is strongly influenced by the structure of substrate-conjugated glycans, which mediate binding to functionally distinct lectins (55, 176, 197). For example, GlcNAc2Man9Gluc1 enables binding to the calnexin-calreticulin chaperones and enhances protein folding (55, 176, 197). Although the removal of the final glucose by α-glucosidase II is important for release from the calnexin-calreticulin folding cycle and for ER exit, it is not a prerequisite for subsequent mannose trimming and targeting towards ERAD (175). Glycan trimming by ER mannosidase I (ERManI) and the ER degradation-enhancing α-mannosidase-like protein (EDEM) family of mannosidases generates the trimmed glycan structures that shunt proteins toward the ERAD pathway (176, 197). EDEM2, and potentially ERManI, mediates the first mannose-trimming event, converting GlcNAc2Man9 to GlcNAc2Man8, through the removal of a single mannose residue from the B chain (127, 197). EDEM1 and EDEM3 mediate further trimming that exposes an α1,6-linked mannose residue on the C branch (127, 150, 197), committing the substrate to ERAD. EDEM1 binds to both nonnative substrate proteins and the luminal domain of the Hrd1 ERAD-L adaptor SEL1L (23), providing a mechanism for substrate delivery to Hrd1 dislocation complexes. EDEM1 also associates with ERdj5, which has reductase activity for cleavage of substrate disulfide bonds and a J domain that binds BiP (52, 182). Thus, EDEM1 complexes are capable of mediating substrate recognition, processing, and delivery to the SEL1L-Hrd1 dislocation complex.
The ER contains two additional lectins that have been implicated in ERAD, OS-9 and XTP3-B. OS-9 contains one mannose-6-phosphate receptor homology (MRH) domain, and XTP3-B contains two MRH domains. Structural analyses of OS-9 revealed selective binding of the Manα1,6Manα1,6Man residues on the processed C branch (59, 150), identifying OS-9 as a receptor for the EDEM-trimmed glycan structure. OS-9 and XTP3-B bind to the luminal face of SEL1L (19), supporting an attractive model in which lectins deliver ERAD substrates bearing the trimmed glycan signal to the Hrd1 dislocation complex. In some cases, OS-9 and XTP3-B appear to have redundant functions, and depletion of both lectins was required to impact the degradation of a set of ERAD-L substrates (5). However, several findings argue against OS-9 and XTP3-B as wholly interchangeable ERAD lectins. For example, OS-9 and XTP3-B interact with distinct sets of ERAD components and display different mechanisms of substrate association (18, 19, 181). In addition, in contrast to OS-9, the C-terminal MRH domain of XTP3-B binds purified GlcNAc2Man9 in vitro and GlcNAc2Man9 conjugated to the null Hong Kong (NHK) mutant variant of α-1 antitrypsin in vivo, not the ERAD-associated trimmed glycan structure (40). This finding has led to speculation that XTP3-B may negatively regulate ERAD by binding and retaining substrates bearing the untrimmed glycan structure (40). Finally, many ERAD components are themselves glycosylated, and mutations in the OS-9, XTP3-B, and EDEM1 lectin domains reduce their association with SEL1L (19, 23). Thus, these results suggest a more complex model than the simple recognition and delivery of proteins bearing trimmed glycans. The data clearly indicate an important role for highly coordinated glycan trimming and binding by ERAD lectins, but the precise temporal ordering of the sugar binding and substrate hand off events remains to be determined.
The mechanism of nonglycosylated substrate degradation is less clear. BiP and ERdj5 act independently of EDEM1 in the recognition and degradation pathway of at least one ERAD-L substrate, a glycosylation-deficient mutant form of NHK (NHK-QQQ) (183). Interestingly, NHK-QQQ is degraded more quickly than the glycosylated NHK (60, 126), supporting a role for the substrate glycan in an additional surveillance checkpoint to prevent premature protein degradation. BiP has also been implicated in the degradation of nonglycosylated ERAD-L substrates through an ERAD pathway involving Herp (homocysteine-induced ER protein), Hrd1, Derlin-1 (degradation in endoplasmic reticulum protein 1), and valosin-containing protein (VCP) (130). Interestingly, glycosylated proteins utilize glycosylation-independent ERAD pathways during ER stress conditions (183) or if the substrate is severely misfolded (126), indicating that the substrate folding state can function as the dominant degradation signal. In rare cases, substrate misfolding exposes a cryptic glycosylation site that enables posttranslational glycosylation by STT3B-containing oligosaccharyltransferase complexes and the engagement of glycan-dependent ERAD mechanisms (148).
Quantity Control: Implementing Substrate-Specific Adaptors
In contrast to quality control, which requires recognition of generalizable degradation signals, regulated quantity control demands strict substrate specificity. For select quantity control substrates, bona fide substrate-specific adaptors have been identified that bridge the substrate and ERAD machinery, functioning as substrate identification and recruitment factors. For example, the viral US2 and US11 proteins deliver major histocompatibility complex class I proteins to, respectively, the E3 ligase Trc8 (translocation in renal carcinoma on chromosome 8 protein) (167, 186) and TMEM129 (184, 186, 187). Other examples include the sterol-regulated recruitment of HMGCRtotheE3 ligase gp78 by Insig (insulin-induced gene protein) 1 and 2 (24, 155, 166) and the activation-dependent recruitment of inositol triphosphate receptors to the E3 ligase RNF170 by Erlin (endoplasmic reticulum lipid raft–associated protein) 1 and 2 (101, 135, 195).
Substrate Dislocation: Gaining Access to the Cytosol
The process of ERAD substrate transport across the ER lipid bilayer is unique, relative to other protein translocation events, which involve the threading of mostly unfolded proteins through a narrow proteinaceous pore (e.g., Sec61) (143). In contrast, ERAD is able to dislocate large bulky cargo, including intact viral particles (41, 66), ligand-stabilized folded proteins (178), oligomeric complexes (136), and glycosylated proteins (6). ERAD dislocation of large cargo is reminiscent of peroxisome protein import, which is known to translocate oligomerized, folded substrates and even 9-nm-sized gold particles (151). Purified peroxisome importer complexes form large, substrate-regulated pores in planar lipid bilayer experiments (111), and it is attractive to speculate that membrane-embedded ERAD complexes may also form similarly dynamic, substrate-induced dislocation pores.
A number of candidates for the ERAD dislocation channel have been proposed, including Sec61 (137), the derlin family of proteins (96, 202), and Hrd1 (15, 169). In yeast, residues within transmembrane domains of the derlin ortholog Der1p (110) and the Hrd1 ortholog Hrd1p (15) cross-link to residues within the ERAD-L substrate CPY*, indicating direct physical association during transit across the membrane. In addition, the overexpression of Hrd1p was sufficient to bypass the requirement for several key upstream ERAD components (Der1p, Hrd3p, and Yos9p) (15), and Hrd1p was sufficient for substrate binding, ubiquitination, and extraction in a reconstituted system using purified proteins (169). The current models suggest that Der1p may function in the initial portion of dislocation, but that Hrd1p itself likely forms the major structural portions of the dislocation channel (15, 110, 169). Not all substrates require Hrd1p, implying that other dislocation channels exist or that some substrates, such as integral membrane substrates, may not require a dislocation channel and are extracted directly from the membrane. Although several integral membrane proteins are dislocated as full-length proteins (54, 92), some integral membrane substrates are proteolytically processed prior to extraction by intramembrane proteases, such as the ubiquitin-dependent rhomboid protease RHBDL4 (36, 47) and signal peptide peptidase (16). This additional processing step may lower the energetic barrier for extraction from the membrane. It is not clear why some membrane substrates, but not others, require this additional proteolytic clipping step for dislocation.
The energy that drives substrate dislocation is derived from adenosine triphosphate (ATP) hydrolysis by the abundant, ubiquitin-dependent hexameric AAA+ ATPase VCP (also known as p97 or CDC48p in yeast) (112). VCP is recruited to ERAD dislocation complexes through direct interactions with several ERAD components that contain a variety of VCP-recruitment domains and motifs [e.g., VCP interacting motif (VIM), SHP, and ubiquitin regulatory X (UBX)] (112). Interestingly, several ERAD components in the same complex often contain VCP-binding motifs (18). It is not clear whether these multiple binding motifs coordinately orient VCP for optimal substrate engagement or whether their interactions with VCP allow ATP hydrolysis to drive conformational changes in the dislocation machinery that promote substrate dislocation. VCP associates with ubiquitinated substrates directly, or indirectly through its heterodimeric cofactors Ufd1 and Npl4, and mediates their physical extraction from the membrane (112). The cytosolic extraction of a subset of substrates may be mediated, or assisted, by the AAA+ ATPase subunits of the 19S regulatory particle of the proteasome (91, 97, 117).
Given the unique properties of ERAD dislocation and the challenge of irrefutably identifying the ERAD dislocation channel, an alternative model has been proposed that couples substrate dislocation to LD biogenesis (138). This model posits that substrates exit the ER either through transient pores formed in the membrane during LD biogenesis (ERAD-L substrates) or through integration into the membrane of budding LDs (ERAD-M and ERAD-C substrates). An analysis of the degradation kinetics of multiple, topologically distinct substrates in yeast lacking LDs was normal (131), arguing against a conserved role for LDs in the dislocation step. However, several ERAD substrates in mammalian cells have been observed in close association with LDs, and their degradation was inhibited by the long chain acyl-CoA synthetase inhibitor triacsin C (54, 70, 78), which blocks LD biogenesis. The LD model of substrate dislocation has not been directly examined in mammalian cells, and it remains possible that a subset of ERAD substrates may be influenced by LDs. It is also possible that LD association for some ERAD substrates, such as HMGCR, may serve as a mechanism of reversible inactivation by sequestration in LDs or LD-associated ER subdomains.
The Ubiquitin System in ERAD: Ligases, Deubiquitinases, and Ubiquitin-Binding Proteins
Ubiquitin is a small 76 amino acid protein that can be covalently attached to substrate lysine residues, the N terminus, and, infrequently, to serine and threonine residues (79). The addition of ubiquitin occurs via sequential enzymatic reactions involving an E1 ubiquitin-activating enzyme, an E2 ubiquitin-conjugating enzyme, and an E3 ubiquitin ligase that mediates the final transfer of ubiquitin to the substrate (79). The covalent addition of ubiquitin to one of the seven lysine residues, or the N terminus, of ubiquitin leads to the construction of a chain of linked ubiquitin moieties (i.e., a polyubiquitin chain) (79). Deubiquitinating enzymes (DUBs) are proteases that counteract this process by removing ubiquitin or disassembling polyubiquitin chains.
ERAD substrate polyubiquitination functions as a binding site that facilitates extraction by VCP and acts as the canonical signal for proteasomal degradation (20). In mammalian cells, more than a dozen E3 ligases have been implicated in ERAD, including orthologs of the yeast E3 ligases Hrd1p [Hrd1 and gp78 (glycoprotein 78)] and Doa10p [MARCH6 (membrane-associated RING finger protein 6)] (20). For many of the mammalian ERAD E3 ligases, the extent of their contributions to ERAD and their substrate specificities remain mostly unknown. ERAD E3 ligases may act alone or cooperate asE3 ligase pairs [e.g., gp78 and Trc8 (71), Hrd1 and gp78 (210), gp78 and RNF5 (116)], possibly functioning sequentially in chain initiation and chain elongation. Substrate polyubiquitin chains can also be further extended or edited by VCP-associated ubiquitination machinery (123, 191). In addition to substrates targeted for degradation, ERAD E3 ligases may autoubiquitinate or ubiquitinate other ERAD components to recruit VCP or, possibly, other ERAD components that contain ubiquitin-binding domains [e.g., UBXD8 (UBX domain–containing protein 8), UBAC2 (UBA domain–containing protein 2), AUP1 (ancient ubiquitous protein 1), gp78] (20, 169). A role for ubiquitin in regulating ERAD machinery is implied by the requirement for ubiquitin in the ERAD of a nonubiquitinated mutant of the α subunit of the T cell antigen receptor-α (204, 205) and the influence of the deubiquitinating enzyme YOD1 in the dislocation of nonubiquitinated cholera toxin A1 (4).
DUBs have been implicated at various steps in the ERAD process, and can promote or prevent substrate degradation. The continuous low-rate removal of ubiquitin moieties likely functions as a checkpoint that reduces spurious degradation of integral membrane proteins and improves detection of bona fide substrates (211). DUBs such as USP13, which is found in complex with gp78, maintain ERAD function by preventing the ubiquitin-dependent inactivation of associated ERAD factors (100). DUBs also act on substrates during and postextraction. Proteasome-associated DUBs remove ubiquitin for recycling to allow the substrate to efficiently pass through the proteasomal pore (107). A similar model for the VCP-associated DUB YOD1 has been hypothesized to allow substrate passage through the narrow VCP pore (31). This model would require two rounds of ubiquitination and deubiquitination for the VCP extraction and proteasomal degradation steps (30). However, existing crystal structures of VCP indicate that a zinc ion occludes the central pore (25) and mutations in most D1 pore residues had little effect on ERAD (26), suggesting that, in the absence of dramatic conformational changes in vivo, substrates do not pass through the VCP pore.
Maintaining Substrate Solubility During Proteasomal Shuttling
The cytosolic dislocation of misfolded and integral membrane proteins with exposed hydrophobic stretches of amino acids presents considerable potential for protein aggregation. Indeed, under conditions of proteasomal impairment, dislocated ERAD substrates accumulate in the cytosol, aggregate, and are actively targeted to ubiquitin-positive inclusion bodies called aggresomes (73). Cells utilize several strategies to overcome this problem. The dislocation of substrates can be directly coupled to proteasomal degradation, eliminating the cytosolic intermediate (64). In the absence of direct proteasomal coupling, cytosolic chaperone complexes can preserve substrate solubility. For example, a chaperone complex composed of SGTA, Bag6, Ubl4A, and Trc35 associates with the gp78 E3 ligase complex and maintains the solubility of dislocated substrates en route to the proteasome (192,198). Association with the surface of LDs has also been suggested as a potential mechanism for combating protein aggregation, either passively through the sequestration of hydrophobic regions of the substrate (128, 174) or actively through the release of detergent-like sterols (115).
ERAD REGULATION OF CHOLESTEROL METABOLISM
Cellular Cholesterol Homeostasis
Cholesterol is essential for mammalian life (105, 161), playing vital roles in maintaining membrane fluidity and integrity (163), facilitating membrane curvature and fusion (21), and serving as a precursor for steroid hormones, bile acids, and oxysterols. It is also thought to be critical for the formation of lipid rafts, which are cholesterol and sphingolipid-enriched membrane microdomains that concentrate proteins in an optimal environment for processes such as signal transduction (162). The synthesis of cholesterol is energetically expensive, requiring more than 20 enzymes. Despite its importance, cholesterol is toxic in excess (32). Thus, excess free cholesterol is sequestered as cholesteryl esters in LDs (139, 189). At the level of the human body, high cholesterol levels are an important risk factor for the development of atherosclerosis and cardiovascular disease (108).
Cellular cholesterol levels are maintained within a narrow range, in part through transcriptional regulation. The proteins involved in cholesterol synthesis and uptake are targets of the sterol regulatory element–binding protein (SREBP) 2 master transcription factor, which preferentially regulates cholesterogenic genes, such as the low-density lipoprotein receptor and the rate-controlling enzymes HMGCR and squalene monooxygenase (57, 58). SREBPs are subject to sterol-regulated proteolysis. SREBP exists as an inactive precursor protein in the ER membrane, which under low-sterol conditions is transported by SREBP cleavage-activating protein (SCAP) to the Golgi complex. In the Golgi complex, SREBP is proteolytically processed to release the active transcription factor, which enters the nucleus and induces the expression of cholesterogenic genes containing sterol-response elements in their promoter regions (45). When cholesterol levels exceed approximately 5% of total ER lipids (141), SCAP undergoes a conformational change (9) that causes it to bind to Insig-1 and Insig-2 (200) via its sterol-sensing domain (201). The association of SCAP and Insig occludes a sequence required for recognition by COPII vesicle assembly components, resulting in retention of the SCAP-SREBP2 complex in the ER (172). The predicted sterol-sensing domain in SCAP consists of five consecutive transmembrane α-helices, but cholesterol appears to bind to an adjacent membrane-associated luminal loop (118). The conformational change can also be made more sensitive to lower levels of cholesterol through increased expression of Insig or via binding of side-chain oxysterols to Insig, which are more potent regulators than cholesterol itself (142). The plasma membrane is cholesterol rich. When the concentration of cholesterol exceeds the complexing capacity of the surrounding lipid acyl chains, the cholesterol enters a pool of chemically active cholesterol that is more susceptible to trafficking by transport proteins (168), allowing delivery to the sterol-sensing machinery in the sterol-poor ER. In some cases, the sterol-sensing machinery may require the conversion of cholesterol to oxysterols in mitochondria (168).
In addition to responding to direct cholesterol binding at a saturable site (118), SCAP can also sense the properties of the bulk membrane environment of the ER. The ability of sterols to induce the conformational change is associated with their ability to affect membrane properties, and nonsteroid cationic amphiphiles that alter membrane structure have a similar effect (1, 9). Similarly, the enantiomer of cholesterol is able to robustly induce conformational change and inhibit SREBP2 processing (80). This mirror image form should not bind to a specific site on a protein, but it exerts identical effects on the physical properties of the ER membrane.
In contrast to SREBP2, SREBP1c chiefly regulates genes involved in fatty acid (FA) synthesis, whereas SREBP1a also has some specificity for genes involved in cholesterol synthesis (58). The activation of SREBP1 does not appear to be controlled by sterol-regulated processing to an appreciable extent, but rather responds strongly to the level of unsaturated FAs, which inhibit proteolytic processing by an unknown mechanism (53,121). Unsaturated FAs also inhibit SREBP1 at the transcriptional level by antagonizing the liver X receptor transcription factor, which targets SREBP1 (53, 134).
ERAD Regulation of Cholesterol Synthesis
The repression of cholesterol synthesis would be a slow process if it depended on transcriptional control alone, as it takes hours for message levels of SREBP2 target genes to fall in response to sterol accumulation, and this would also leave existing biosynthetic enzymes. Consequently, several levels of posttranscriptional regulation have evolved. One of these mechanisms is through regulated ERAD (Figure 2 and Table 1), achieved by accelerated degradation of the rate-controlling enzymes HMGCR and squalene monooxygenase.
Figure 2.
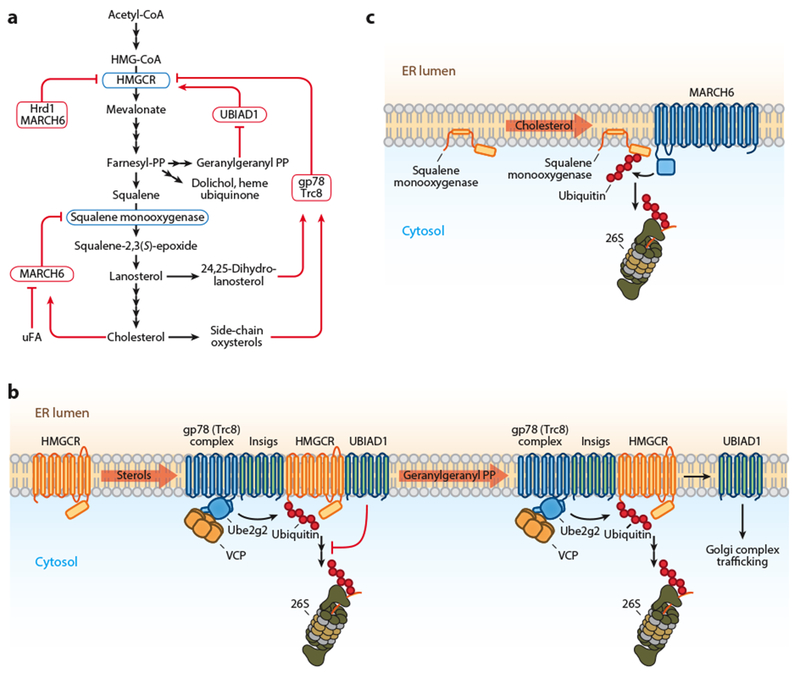
ERAD regulation of cholesterol metabolism. (a) Schematic of the cholesterol synthesis pathway with the points of ERAD-mediated regulation indicated. Regulated enzymes are shown in blue boxes and ERAD machinery in red boxes. (b) In the presence of sterols, Insigs recruit HMGCR to the gp78 and Trc8 complexes for degradation. For simplicity, only gp78 is illustrated. Sterols also stimulate the interaction of HMGCR with UBIAD1, which impairs the dislocation of a portion of HMGCR. Geranylgeranyl-PP binding releases UBIAD1 from the degradation complex, promoting HMGCR dislocation and degradation. The released UBIAD1 traffics to the Golgi complex. (c) Cholesterol induces MARCH6 degradation of squalene monooxygenase, whereas unsaturated FAs stabilize it. Abbreviations: ERAD, endoplasmic reticulum-associated degradation; FA, fatty acid; uFA, unsaturated fatty acid; gp78, glycoprotein 78; HMGCR, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; Insig, insulin-induced gene protein; MARCH6, membrane-associated RING finger protein 6; PP, pyrophosphate; Trc8, translocation in renal carcinoma on chromosome 8 protein; UBIAD1, UbiA prenyltransferase domain–containing protein 1.
Table 1.
Endoplasmic reticulum–associated degradation (ERAD) factors implicated in lipid metabolism
| Protein | Alternative name(s) | ERAD function | Domain(s) | Topology | Associated metabolic process | Regulated substrate | Reference(s) |
|---|---|---|---|---|---|---|---|
| Insig-1 | Substrate-specific adaptor | M, poly | Cholesterol synthesis | HMGCR | 71, 154, 155, 166 | ||
| Insig-2 | Substrate-specific adaptor | M, poly | Cholesterol synthesis | HMGCR | 71,154 | ||
| UBIAD1 | Substrate-specific regulator | Prenyltransferase | M, poly | Cholesterol synthesis | HMGCR | 152 | |
| Hrd1 | Synoviolin | E3 ligase | RING | M, poly | Cholesterol synthesis | HMGCR | 76 |
| gp78 | AMFR, RNF45 | E3 ligase | RING, CUE, G2BR, VIM | M, poly | Cholesterol synthesis, SREBP processing, TAG synthesis, lipoprotein secretion | HMGCR, Insig-1, DGAT2, ApoB100 | 34,71,85,95, 166 |
| Trc8 | RNF138 | E3 ligase | RING, sterol-binding | M, poly | Cholesterol synthesis, SREBP processing | HMGCR, Insig-1 | 71, 88 |
| MARCH6 | Teb4, RNF176 | E3 ligase | RING | M, poly | Cholesterol synthesis | HMGCR, SM | 39,44, 170 |
| Ube2g2 | Ubc7 | E2-conjugating enzyme | C | Cholesterol synthesis | HMGCR | 113 | |
| AUP1 | Ube2g2 recruitment | CUE, G2BR | M, HP | Cholesterol synthesis | HMGCR | 70 | |
| Derlin-1 | Dislocation | Rhomboid, SHP box | M, poly | Lipoprotein secretion | ApoB100 | 174 | |
| Erlin-2 | SPFH2 | Scaffold | SPFH-like | M, SP | Cholesterol synthesis | HMGCR | 72 |
| TMUB1 | DULP | Scaffold | UBL | M, SP | Cholesterol synthesis | HMGCR | 72 |
| UBXD8 | Faf2, ETEA | VCP recruitment | UBA, UAS, UBX | M, HP | TAG synthesis, TAG degradation, SREBP processing, lipoprotein secretion | Insig-1, ApoB100, ATGL | 84, 87, 133, 174 |
| VCP | p97 | AAA+ ATPase | AAA+ | C | Cholesterol synthesis, TAG synthesis and degradation, lipoprotein secretion | HMGCR, Insig-1, ApoB100, SCD1 | 35, 74, 87, 166 |
Abbreviations: AAA+, ATPases associated with diverse cellular activities; ApoB100, apolipoprotein B100; ATGL, adipose triglyceride lipase; AUP1, ancient ubiquitous protein 1; C, cytosolic; CUE, coupling of ubiquitin to endoplasmic reticulum degradation; Derlin-1, degradation in endoplasmic reticulum protein 1; DGAT2, diacylglycerol acyltransferase 2; Erlin-2, endoplasmic reticulum lipid raft–associated protein 2; G2BR, Ube2g2-binding region; HMGCR, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; HP, hairpin; Insig-1, insulin-induced gene 1 protein; Insig-2, insulin-induced gene 2 protein; M, membrane; MARCH6, membrane-associated RING finger protein 6; poly, polytopic; SCD1, stearoyl-CoA desaturase 1; SM, squalene monooxygenase; SP, single pass; SREBP, sterol regulatory element–binding protein; TAG, triacylglycerol; TMUB1, transmembrane and ubiquitin-like domain–containing protein 1; Trc8, translocation in renal carcinoma on chromosome 8 protein; UBA, ubiquitin-associated; Ube2g2, ubiquitin-conjugating enzyme E2 G2; UBIAD1, UbiA prenyltransferase domain–containing protein 1; UBL, ubiquitin-like; UBX, ubiquitin regulatory X; UBXD8, UBX domain–containing protein 8; VCP, valosin-containing protein; VIM, VCP interacting motif.
3-Hydroxy-3-Methylglutaryl-Coenzyme A Reductase
Acute regulation of cholesterol synthesis centers on HMGCR, a rate-limiting enzyme that catalyzes the conversion of HMG-CoA into mevalonate and is the target for the blockbuster statin class of drugs. Mevalonate is a precursor not only for sterols but also for isoprenoids that are essential for cell growth, such as ubiquinone and heme A in the electron transport chain, dolichol for N-linked glycosylation, and the farnesyl, geranylgeranyl, and prenyl groups that anchor proteins to membranes. In addition to transcriptional control by SREBPs, translation of HMGCR messenger RNA is reduced in response to a derivative of mevalonate (122), and HMGCR can be inactivated by reversible phosphorylation (10, 11), likely as a mechanism for conserving energy when ATP levels are low (147).
The accelerated proteasomal degradation of HMGCR is one of the best-studied examples of metabolite-regulated ERAD (Figure 2a,b), in which sterols reduce the protein’s half-life from approximately 12 hours to less than 1 hour (65, 155). Regulated turnover depends on the N-terminal 339 residues of the enzyme (164), which contain 8 transmembrane domains embedded in the ER membrane, including a sterol-sensing domain with sequence homology to that of SCAP. This regulatory domain is distinct from the 548 amino acids comprising the cytosolic, catalytic C terminus (43). Sterols cause Insig proteins to bind to the sterol-sensing domain of HMGCR, leading to HMGCR degradation (89, 154, 155). This association involves interactions that are similar, but not identical, to SCAP binding, suggesting that SCAP and Insigs compete for the same binding site on HMGCR (89, 154, 155). Accordingly, sterols fail to accelerate HMGCR degradation in Insig-deficient cells (90), and overexpression of the sterol-sensing domain of HMGCR or SCAP stabilizes HMGCR unless there is a concomitant increase in Insig-1 (154). Insigs recruit E3 ubiquitin ligases that ubiquitinate HMGCR, marking it for degradation. Sterol-dependent ubiquitination appears to be mediated in part by gp78, which also has binding sites for VCP and the ubiquitin-conjugating enzyme Ube2g2 (ubiquitin-conjugating enzyme E2 G2, a homolog of yeast Ubc7p) (166). After sterol treatment, Insig-1, gp78, and HMGCR can be isolated in a single complex, and HMGCR is stabilized through overexpression of inactive forms of gp78 (166). Small interfering RNA depletion of gp78, or liver-specific deletion of gp78, also prevented sterol-dependent ubiquitination of HMGCR and increased HMGCR protein levels (99, 166). Ufd1, in addition to its established roles in VCP-mediated dislocation, acts as a gp78 cofactor that accelerates HMGCR degradation by stimulating gp78 polyubiquitin chain initiation (13). A second E3 ligase, Trc8, also stimulates the degradation of HMGCR in response to sterols via binding to both Insig-1 and Insig-2, leading to a more complete degradation in concert with gp78 (71). Ube2g2 can act as the cognate E2 for both gp78 and Trc8, and AUP1 increases Ube2g2 recruitment and facilitates HMGCR degradation (70). In some contexts, the gp78 complex that ubiquitinates HMGCR also includes Erlin-2 (also known as SPFH2), which is recruited by TMUB1 (transmembrane and ubiquitin-like domain–containing protein 1) (72). The importance of gp78 for HMGCR degradation has been called into question recently by a report in which small interfering RNA depletion of gp78 and knockout of gp78 in mouse cells did not impact the sterol-dependent degradation of HMGCR (180). This suggests that additional E3 ligases may be involved, even if gp78 is the key E3 ligase in the liver (99). Indeed, Hrd1 and MARCH6 (membrane-associated RING finger protein 6) have been shown to play a role in the turnover of HMGCR, although this degradation event does not appear to be sterol regulated (76, 208). In Drosophila S2 cells, the regulatory domain of mammalian HMGCR is subject to normal sterol-regulated degradation when coexpressed with mammalian Insigs, but in the absence of a direct gp78 homolog, HMGCR is ubiquitinated by fly Hrd1 (124).
The major physiological sterol signal for HMGCR degradation is not cholesterol itself, but rather side-chain oxysterols (81, 165). This appears to be mediated primarily by 27-hydroxycholesterol, which is synthesized from cholesterol in mitochondria, as exogenous cholesterol failed to proteolytically inactivate HMGCR in 27-hydroxylase-deficient cells (81). In contrast, the cholesterol precursor 24,25-dihydrolanosterol (81) and 24(S),25-epoxycholesterol (165) also stimulate degradation of HMGCR, but are likely to be more important as signals for the flux through the endogenous cholesterol synthesis pathway (196), smoothening homeostatic responses by preventing sudden toxic bursts of cholesterol synthesis. The effect of 24,25-dihydrolanosterol has also been proposed to act as an oxygen sensor because hypoxia stimulates the degradation of HMGCR by slowing demethylation of lanosterol and inducing expression of both Insigs (125). Also, it has long been known that the maximum degradation of HMGCR requires an additional derivative of mevalonate. Other than sterol derivatives of mevalonate, the major nonsterol has been shown to be geranylgeraniol, which does not affect ubiquitination but, rather, enhances dislocation (154). It appears that a pool of HMGCR binds to the prenyltransferase UbiA prenyltransferase domain–containing protein 1 (UBIAD1), preventing dislocation of HMGCR from the ER (152). This interaction is stimulated by sterols via Insig, but treatment with geranylgeraniol, which is converted to geranylgeranyl pyrophosphate, causes UBIAD1 to dissociate from HMGCR and traffic from the ER to the Golgi complex (152). The disassociation of UBIAD1 likely allows binding or action of the dislocation machinery required for HMGCR degradation that is otherwise blocked. This mechanism may maintain a low level of protected active HMGCR to allow continual low-level production of essential nonsterol isoprenoids in the presence of an influx of exogenous sterols.
The dislocation of HMGCR appears to involve multiple steps. Sterols first trigger its translocation to a membrane compartment resembling LDs, as sterol or mevalonate treatment causes HMGCR to appear in an LD-enriched fraction (54). This localization seems to be functionally important because inhibition of LD formation with the acyl-CoA synthetase inhibitor triacsin C blunts HMGCR ERAD (54). Interestingly, the transfer of HMGCR to this LD-like compartment does not require ubiquitination (54). In support of a role for ubiquitination in such a compartment, AUP1 recruits Ube2g2 to LDs (70,78), and LD-enriched fractions mediate ubiquitination in vitro (78). In any case, VCP acts to extract ubiquitinated HMGCR from within the membrane to an intermediate state associated with the membrane, akin to a peripheral membrane protein (117). The final extraction from the membrane surface into the cytosol requires ubiquitination of the membrane domain of HMGCR (54) and is performed by ATPases in the 19S regulatory particle of the proteasome (117).
Squalene Monooxygenase
Owing to the importance of HMGCR in controlling cholesterol synthesis, the study of later enzymes in the pathway has been relatively neglected. One such enzyme is squalene monooxygenase (formerly squalene epoxidase), which catalyzes the conversion of squalene to 2,3(S)-monooxidosqualene [squalene-2,3(S)-epoxide], a molecule that is committed to sterol synthesis alone, unlike mevalonate. The enzyme can also act upon its product a second time to generate 2,3(S):22(S),23-dioxidosqualene, leading to synthesis of 24(S),25-epoxycholesterol, a potent oxysterol that is uniquely generated directly through the activity of the pathway in parallel to cholesterol (196).
Squalene monooxygenase constitutes an important, second, rate-limiting step in cholesterol synthesis that is subject to robust cholesterol-dependent degradation by the UPS (Figure 2a,c) (44). This mechanism is distinct from that of HMGCR, as accelerated degradation is stimulated by cholesterol itself, does not require Insigs, and is mediated by the E3 ligase MARCH6 (39, 44, 208). Cholesterol-regulated degradation requires the first 100 amino acids (N100) of squalene monooxygenase (44), which are integrally associated with the ER membrane via a single reentrant loop (61). Thus, N100 fusion proteins are degraded with similar kinetics to the full-length enzyme, with cholesterol reducing their half-lives from approximately 14 to approximately 3 hours (44).
Both direct cholesterol binding and bulk membrane effects appear to control squalene monooxygenase turnover (80). Squalene monooxygenase has been observed to bind a sterol probe in a large-scale screen (63), but the enantiomer of cholesterol can still stimulate degradation, albeit less potently than cholesterol (80). Cholesterol accumulation causes a conformational change in N100, detected by altered derivatization of specific residues by polyethylene glycol maleimide (61). Treatment with the molecular chaperone glycerol, which helps to retain proteins in their native confirmation, stabilizes the levels of N100 fused to green fluorescent protein, suggesting that cholesterol causes squalene monooxygenase to exhibit the properties of a misfolded, quality control substrate (61). The sterol-sensing domain of the HMGCR homolog in yeast, Hmg2p, has previously been observed to behave analogously in response to nonsterol stimuli, as detected by altered proteolysis (157, 158), but this has not yet been observed for the mammalian enzyme. In contrast to mammalian HMGCR, ubiquitination of an internal lysine in the membrane-bound, sterol-responsive N100 region of squalene monooxygenase is not required for degradation (44), which suggests that after recognition, MARCH6 can ubiquitinate the N terminus or any one of the accessible lysines. As the conformational changes observed in response to cholesterol appear to be subtle, it may be that structural changes within the membrane are detected by MARCH6, as has been previously observed for recognition of a transmembrane degron in the Sec61 β-subunit homolog 2 by the yeast homolog of MARCH6, Doa10p (51, 61). Whether additional adaptor proteins are required for squalene monooxygenase recognition remains to be determined, as do the details of the squalene monooxygenase dislocation mechanism. The relative insensitivity of squalene monooxygenase degradation to triacsin C suggests that the dislocation mechanism differs from the mechanism employed by HMGCR (170).
Squalene monooxygenase is stabilized by unsaturated FAs, which inhibit binding and ubiquitination by MARCH6 (170). This might reflect the sensitivity of the squalene monooxygenase folding state to the nature of the local membrane environment, and it could possibly assist in coordinating phospholipid and cholesterol synthesis. Importantly, accelerated degradation of squalene monooxygenase provides the cell with a robust mechanism for regulating sterol production alone, without affecting the synthesis of nonsterol isoprenoids produced by HMGCR. For example, it could allow a higher rate of isoprenoid production in the presence of an acute load of exogenous cholesterol.
ERAD REGULATION OF TRIACYLGLYCEROL METABOLISM
Cellular Triacylglycerol Homeostasis
During periods of nutrient excess, FAs are esterified to glycerol 3-phosphate to form TAG, sequestering potentially toxic lipids and providing an on-demand source of FAs during periods of increased cellular need (22). TAG synthesis begins with the activation of FAs through the ATP-dependent formation of thioester-linked FA CoA conjugates (FA-CoA) (Figure 3a) (22). Activated FAs serve as substrates for a series of acyl transferases that mediate their addition to glycerol 3-phosphate to form lysophosphatidic acid and then phosphatidic acid, which is hydrolyzed to form diacylglycerol (DAG) (22). A final esterification step forms TAG (22).
Figure 3.
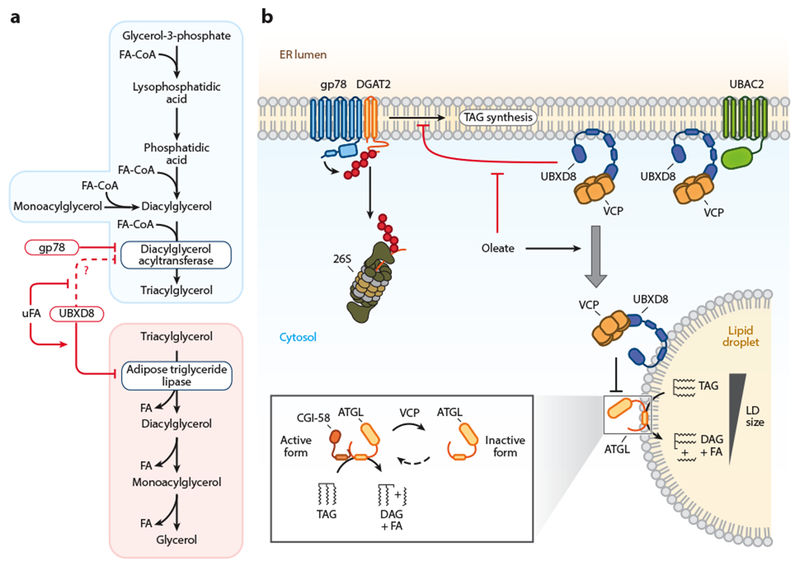
ERAD regulation of triacylglycerol metabolism. (a) Schematic of the triacylglycerol synthesis and degradation pathways with the points of ERAD-mediated regulation indicated. Regulated enzymes are shown in blue boxes and ERAD machinery in red boxes. (b) Constitutive DGAT2 degradation is mediated by gp78. Under low FA conditions, UBXD8 inhibits TAG synthesis. Increases in FA levels release UBXD8 inhibition of TAG synthesis and promote UBXD8 trafficking to LDs. On LDs, UBXD8 impairs lipolysis through the dissociation of ATGL from its cofactor CGI-58. Abbreviations: ATGL, adipose triglyceride lipase; DAG, diacylglycerol; DGAT, diacylglycerol acyltransferase; ERAD, endoplasmic reticulum-associated degradation; FA, fatty acid; uFA, unsaturated fatty acid; FA-CoA, fatty acid coenzyme A; gp78, glycoprotein 78; LD, lipid droplet; TAG, triacylglycerol; UBAC2, UBA domain–containing protein 2; UBXD8, UBX domain–containing protein 8; VCP, valosin-containing protein.
Intracellular TAG is stored in LDs, ubiquitous ER-derived organelles conserved from yeast to humans (139, 189). LD biogenesis is thought to occur through the corralling of TAG deposited between the leaflets of the ER into a lens, which subsequently buds into the cytoplasm through a poorly understood mechanism involving seipin and the fat storage–inducing transmembrane (FIT) family of proteins (139, 189). LDs consist of a neutral lipid core (i.e., TAG and steryl esters) bounded by a phospholipid monolayer decorated with integral and peripheral proteins. The unique LD proteome is gained through the (a) lateral diffusion of proteins from the ER into LDs, (b) direct insertion of proteins into the LD membrane, and (c) recruitment of peripherally associated cytosolic binding partners (139, 189). Many of the LD-associated proteins are involved in local TAG synthesis and TAG breakdown (i.e., lipolysis) (139, 189). During lipolysis, LD-associated lipases mediate the sequential hydrolysis and release of FA for use in energy production, membrane biogenesis, and lipid signaling pathways (Figure 3a) (82).
TAG and cholesterol are also packaged into lipoproteins in enterocytes (i.e., chylomicrons) and hepatocytes [i.e., very-low-density lipoproteins (VLDLs)] for the secretion and transport of lipids throughout the body (159). Lipoproteins are generated in the ER lumen through a two-step process involving microsomal triglyceride transfer protein (MTP). In the first cotranslational step, MTP transfers TAG to apolipoprotein B (ApoB) as it emerges from the Sec61 translocon. In the second posttranslational step, MTP facilitates the association of the nascent ApoB lipoprotein with a preformed, luminal, TAG-rich droplet. A single ApoB molecule (ApoB100 on VLDL and ApoB48 on chylomicrons) encircles the circumference of each lipoprotein, providing structural support and mediating the key protein-protein interactions required for processing. The maturing lipoprotein undergoes further processing and association with additional proteins and lipids during its transport through the secretory system (33). Interestingly, chylomicrons (up to 1,000 nm in diameter) and VLDLs (up to 800 nm in diameter) are too large for classical COPII vesicles (approximately 60–80 nm) and require unique COPII secretory vesicles similar to other large cargos, such as collagen (12, 69, 103).
UBX Domain–Containing Protein 8 and Interorganelle Regulation of Triacylglycerol
UBXD8 is a membrane-embedded VCP recruitment factor that partitions between the ER and LDs (133,174,207). UBXD8 is present in both theHrd1 (119) and gp78 (87) E3 ligase complexes, and it mediates VCP recruitment through its UBX domain to facilitate substrate dislocation. UBXD8 also mediates the association of the cytosolic Bag6 chaperone complex with gp78 (199). Consistent with an important role in ERAD, depletion of UBXD8 or expression of a dominant negative form of UBXD8 impairs the clearance of several ERAD substrates, including US11-induced degradation of class I major histocompatibility complex heavy chains (119), Insig-1 (87), and ApoB100 (174).
Emerging findings have indicated that UBXD8 is a conserved regulator of FA metabolism and TAG storage (Figure 3 and Table 1) (77, 84, 133, 174, 190). In the ER, UBXD8 operates as an FA sensor and regulator of cellular lipids that inhibits the conversion of DAG to TAG by an unknown mechanism (84). Direct binding of unsaturated FAs by the UAS domain of UBXD8 has been proposed to induce UBXD8 oligomerization and release UBXD8 inhibition of TAG synthesis (77, 84). The relationship between UBXD8’s role in ERAD and regulation of TAG synthesis is presently unknown, but one possibility is that UBXD8 is involved in the degradation of a key TAG synthesis enzyme or regulator. In response to increases in FA levels, a portion of UBXD8 traffics from the ER to emerging LDs and recruits VCP to the LD’s surface (133, 174). On LDs, UBXD8 binds and inhibits the rate-limiting enzyme in lipolysis, adipose triglyceride lipase (ATGL), at least in part by stimulating the dissociation of ATGL from its activator CGI-58 (133). These findings support a model in which UBXD8 directly senses FA levels and coordinates their sequestration into TAG in the ER and their lipolytic release from LDs.
The dual localizations and coordinated functions of UBXD8 in lipid metabolism point toward the sorting of UBXD8 between the ER and LDs as a potentially important regulatory step. The hairpin topology of the UBXD8 membrane domain is a shared feature of many LD proteins (189) and it is necessary for UBXD8 membrane association (174), likely enabling the lateral diffusion of UBXD8 from the outer leaflet of the ER into the phospholipid monolayer during LD biogenesis. A recent large-scale proteomic analysis of the ERAD interaction network identified the ER-resident rhomboid pseudoprotease UBAC2 as a UBXD8 binding partner that functions in ERAD (18). UBAC2 also acts as a UBXD8 ER tethering protein that prevents FA-induced trafficking of UBXD8 to LDs (133). Indeed, the loss of endogenous UBAC2 increased the amount of UBXD8 on LDs, and overexpression of UBAC2 blocked UBXD8 trafficking to LDs (133). One function of UBAC2 may be to retain a specific pool of UBXD8 in ER-localized complexes, thereby preserving its ERAD-related functions during fluctuations in cellular FA levels and LD biogenesis. Further studies are necessary to understand the mechanisms that control UBXD8 partitioning between the ER and LDs.
Stearoyl-Coenzyme A Desaturase 1
Stearoyl-CoA desaturase 1 (SCD1) is an ER-resident, four-pass transmembrane Δ9 desaturase that converts saturated FAs into their monounsaturated FA counterparts by introducing a double bond between positions 9 and 10 of the carbon chain. The resulting SCD1 products, primarily palmitoleate and oleate, function as key substrates for the synthesis of TAG, phospholipids, cholesteryl ester, and wax esters (37). SCD1 generation of monounsaturated FAs also facilitates their storage as TAG in LDs and protects against lipotoxicity (98), potentially through substrate channeling facilitated by an association between SCD1 and DGAT2 (diacylglycerol acyltransferase 2) (104). The SCD1 yeast ortholog Ole1p is degraded by ERAD (8), and the mammalian SCD1 is similarly presumed to be degraded via ERAD (74, 120). In support of this possibility, SCD1 is constitutively degraded with a half-life of approximately 3 to 4 hours in cultured cell lines, and inhibition of the proteasome partially stabilizes SCD1 and results in the accumulation of polyubiquitinated SCD1 (74). The N terminus of SCD1 contains multiple PEST sequences, well-characterized UPS degrons, that are necessary for SCD1 degradation and sufficient to promote the degradation of a model ER protein (74, 120). Although SCD1 binds VCP (74), a functional role for VCP in SCD1 degradation has not been tested, and the ERAD components that recognize and ubiquitinate SCD1 remain unknown. The rate of SCD1 degradation is reduced in rats treated with the PPARα agonist clofibric acid (179), suggesting physiologically significant regulation, but it is unclear whether this change reflects regulation of SCD1 ERAD or other proteolytic mechanisms (56).
Diacylglycerol Acyltransferase 2
The terminal step in TAG synthesis is the transfer of an FA from an activated FA-CoA to DAG to form TAG (203). This committed step is catalyzed by the isoenzymes acyl-CoA:diacylglycerol actyltransferase 1 and 2 (DGAT1 and DGAT2) (203). Despite sharing similar enzymatic activities, DGAT1 and DGAT2 are evolutionarily unrelated, exhibiting different protein structures, expression patterns, cellular localizations, and physiological functions (203). DGAT1 is a member of the acyl-CoA:cholesterol acyltransferase family and is an ER-restricted polytopic membrane protein (203). In contrast, DGAT2 is a member of the monoacylglycerol acyltransferase family and has a hairpin topology (109, 171, 203). DGAT2 contains two transmembrane domains joined by a small number of amino acids that are potentially in the ER lumen, and the amino and carboxyl termini are oriented toward the cytosol (109, 171). This hairpin conformation is necessary, but not sufficient, for DGAT2 trafficking from the ER to LDs in response to FA (109, 171). On LDs, DGAT2 cooperates with other TAG synthesis enzymes and contributes to local TAG synthesis and LD growth (194). A recent study demonstrated that DGAT2 (Figure 3), but not DGAT1, is rapidly degraded in cultured HEK293T cells and accumulates in a ubiquitinated form following proteasome inhibition (17). Depletion of the E3 ligase gp78, but not MARCH6, FBXW7, or Hrd1, stabilized DGAT2 and reduced DGAT2 ubiquitination (17). DGAT2-bound VCP and polyubiquitinated DGAT2 accumulated in cells treated with eeyarestatin (17), a small molecule that impairs VCP-associated DUBs (17, 191), suggesting that DGAT2 is extracted by VCP. Given the previous observation that UBXD8 inhibits the conversion of DAG to TAG (84), it will be important to determine if UBXD8 contributes to DGAT2 degradation. In addition, studies are required to determine the physiological significance of DGAT2 ERAD and whether DGAT2 degradation is regulated (e.g., by FA levels). It might be expected that FA treatment would stabilize DGAT2 on LDs because the LD pool of DGAT2 would be sequestered away from the gp78 ERAD pathway.
Apolipoprotein B100
ApoB100 is the principal proteinaceous component of VLDLs secreted from hepatocytes. In the absence of sufficient lipids (28) or during MTP impairment (3), ApoB100 is degraded by ERAD. When lipid amounts are insufficient for VLDL assembly, ApoB100 undergoes translational arrest in the Sec61 translocon (94, 114), adopting an aberrant bitopic topology that may act as a conformational signal to initiate the ERAD-T degradation pathway. Cytosolic (Hsp70 and Hsp90) (50, 212) and luminal (BiP) (145) chaperones are recruited to the translocon-stalled protein, potentially contributing to both the stabilization of partially folded regions of ApoB100 and the eventual targeting of ApoB100 for ERAD. The E3 ligase gp78 polyubiquitinates ApoB100 and facilitates its extraction from the membrane by VCP (34, 35, 95). Strikingly, depletion of gp78, but not VCP, increases VLDL assembly and secretion (34). This result highlights ubiquitination as the commitment step for the degradation fate of ApoB100 and demonstrates that a portion of ApoB100 that is competent for VLDL assembly is constitutively degraded by ERAD. Thus, VLDL assembly is regulated, at least in part, by the competition between ApoB100 lipidation and degradation.
A unique ERAD pathway that involves cytosolic LDs degrades lipidated ApoB100. In cultured Huh7 cells, lipidated ApoB100 accumulates in the ER lumen in association with LDs arrested in the ER membrane (129, 174), possibly reflecting a block in an early stage of LD biogenesis. Following proteasomal impairment, ApoB100 is present in buoyant LD-enriched fractions, is observed on the surface of LDs, and can be cross-linked to the LD protein perilipin 2 (174), indicating that a portion of dislocated cytosolic ApoB100 associates with the LD surface. The presence of ApoB100 in LD-enriched fractions was blocked by MTP inhibitors (174), consistent with LD-associated ApoB100 being in its lipidated state. Derlin-1 was required for ApoB100 cytosolic dislocation and association with the surface of LDs (174). Depletion of UBXD8 resulted in the accumulation of ubiquitinated ApoB100 and a small increase in the amount of LD-associated ApoB100 (174), suggesting that UBXD8 functions downstream of Derlin-1 to facilitate VCP-dependent extraction of ubiquitinated ApoB100 from the LD surface. The E3 ligase responsible for ubiquitination of lipidated ApoB100 in this pathway is unknown. In addition, it is also unclear whether the association with LDs is a requisite intermediate in the ApoB100 degradation pathway. It remains possible that the accumulation of cytosolic, aggregation-prone ApoB100—due to proteasome impairment or UBXD8 depletion—results in an off-pathway association with the LD surface.
ERAD CONTROL OF MASTER REGULATORS OF LIPID-RELATED GENE EXPRESSION
In addition to regulating SREBP activity by acting as an oxysterol sensor and SCAP-binding retention protein, Insig can also form a platform for recruiting E3 ligases, specifically gp78 for Insig-1 and Trc8 for both Insigs (71). This is important in the sterol-regulated degradation not only of HMGCR but also of Insig-1 itself (Figure 4; Table 1). When sterol levels are low, the SCAP-SREBP complex dissociates from Insig-1, allowing binding of gp78, which ubiquitinates Insig-1 and targets it for destruction (46). SREBP2 activity consequently increases the expression of Insig-1, which is an SREBP target gene. When sterol levels are high, SCAP binding displaces gp78, stabilizing Insig-1 and retaining SCAP-SREBP2 in the ER, contributing to a convergent feedback mechanism (46). Early studies in Chinese hamster ovary cells indicated that, in contrast to the rapidly degraded Insig-1 (half-life ~1 h), Insig-2 is a stable protein (half-life >4 h) (83, 86). However, deletion of gp78 in mouse liver resulted in the accumulation of both Insig-1 and Insig-2 (99), indicating that both Insigs are degraded by ERAD in mouse liver. The loss of gp78 in the liver results in important metabolic effects, such as improved insulin sensitivity and reduced lipogenesis by stabilizing Insigs, inhibiting activation of SREBP1 and 2 (99). Insig-1 protein levels are also regulated by the level of unsaturated FAs, leading to stabilization at a postubiquitination step. Unsaturated FAs inhibit binding of UBXD8 to Insig-1, which is required for recruitment of VCP to extract the ubiquitinated protein from the ER to allow proteasomal degradation (87). Thus, unsaturated FAs inhibit SREBP2 activation.
Figure 4.
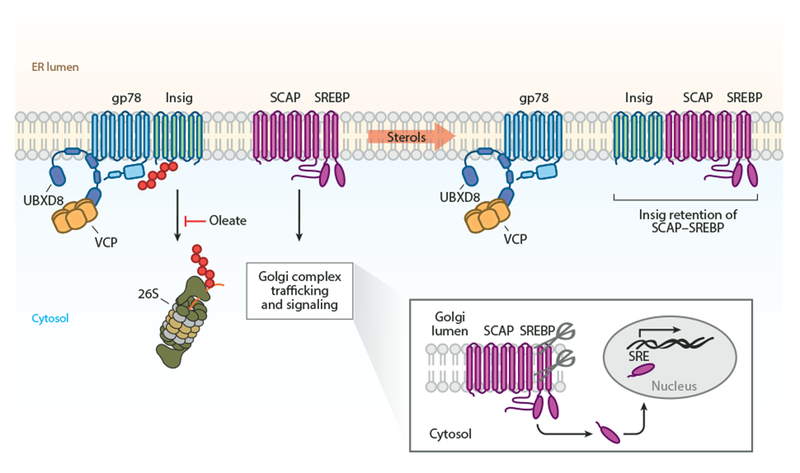
ER-associated degradation control of master transcriptional regulators of lipid metabolism. In the presence of sterols, Insigs mediate the ER retention of the SCAP-SREBP complex. In the absence of sterols, a gp78-UBXD8 complex degrades Insigs, and SCAP-SREBP traffics to the Golgi complex. In the Golgi complex, SREBP is proteolytically processed at two sites, releasing a soluble transcription factor that binds SRE sequences and induces expression of lipid-related genes. Abbreviations: ER, endoplasmic reticulum; gp78, glycoprotein 78; Insig, insulin-induced gene; SCAP, SREBP cleavage-activating protein; SRE, sterol regulatory element; SREBP, sterol regulatory element–binding protein; UBXD8, UBX domain–containing protein 8; VCP, valosin-containing protein.
Independently from its E3 ligase activity, Trc8 inhibits SREBP2 processing by binding to the SCAP-SREBP complex and preventing its packaging into COPII vesicles (67). Trc8 is degraded rapidly in the presence of sterols, but stabilized under low sterol conditions (67), so this mechanism may serve to fine-tune feedback responses when Insig levels are also low. In contrast to liver cells, Trc8 in HEK293 cells mediates the degradation of the SREBP processor (88). Depletion of AUP1 affects levels of SREBP, SCAP, and Insig (70), possibly by enhancing ubiquitination via E2 recruitment to gp78 and Trc8. The Erlins have also been shown to bind cholesterol, and Erlin-1 and Erlin-2 association with the SREBP-SCAP-Insig-1 complex may play a role in preventing SREBP activation (62).
CONCLUDING REMARKS AND FUTURE PERSPECTIVES
The current metabolic disease epidemic highlights the importance of achieving a comprehensive understanding of the regulatory mechanisms governing lipid synthesis, storage, and utilization. The ER, as the primary site of lipid synthesis, LD biogenesis, and lipoprotein assembly, is uniquely positioned to influence lipid physiology. During the past 20 years, ERAD has emerged as a key posttranslational mechanism that controls the flux through the ER-resident metabolic pathways by altering the abundance of lipid biosynthetic enzymes and their regulators. However, despite considerable progress, many outstanding questions remain, such as how multiple metabolic signals are sensed and integrated to influence the appropriate response, what other lipid biosynthetic enzymes and regulators are controlled by ERAD, and how different substrates are selectively recognized and dislocated for degradation. In addition, although our understanding of HMGCR ERAD is quite advanced relative to that of other substrates, the recent discoveries of both new regulators (e.g., UBIAD1 and MARCH6) and new points of downstream regulation (e.g., squalene monooxygenase) in the cholesterol synthesis pathway indicate that the picture is far from complete. It will be particularly interesting to determine how these new regulatory mechanisms are coordinated to influence the flux through a bifurcating metabolic pathway that provides both cholesterol and nonsterol isoprenoids (e.g., vitamins and ubiquinone). In contrast to the relationship of ERAD and cholesterol metabolism, our understanding of the role of ERAD in regulating FA and TAG homeostasis is still in its infancy. Although UBXD8 is clearly an important player, its ER and LD targets remain mostly unknown. Similarly, the degradation pathways for SCD1 and DGAT2, as well as their regulation and physiological significance, remain to be determined. Finally, ERAD does not function in isolation, but rather is integrated into complex cellular responses that enable adaptation to fluctuating environmental conditions and metabolic states. In order to attain a global understanding of lipid homeostasis, it will be important to establish how ERAD regulation of ER-resident metabolic pathways is coordinated with transcriptional programs and the activities of other organelles, such as LDs and mitochondria.
SUMMARY POINTS.
The ER is a central hub of lipid metabolism, functioning as the site of lipid synthesis, LD biogenesis, and lipoprotein assembly.
ERAD plays key roles in maintaining the fidelity of the secretory proteome (quality control) and in mediating the regulated degradation of metabolic enzymes (quantity control).
The regulated degradation of HMGCR and squalene monooxygenase by ERAD enables rapid posttranslational control of the flux through the cholesterol synthesis pathway.
UBXD8 acts as a sensor of unsaturated FA levels that coordinates FA packaging into TAG in the ER and FA mobilization from stored TAG in LDs.
SCD1 and DGAT2 are degraded by ERAD and may provide mechanisms for controlling TAG synthesis.
The competition between ApoB100 lipidation and ERAD influences VLDL assembly and secretion.
ERAD, through its regulation of intracellular lipid synthesis pathways and the assembly and secretion of lipoprotein particles, is capable of influencing both cell-autonomous and noncell-autonomous lipid homeostasis.
ACKNOWLEDGMENTS
We apologize to any authors whose important contributions were not cited in this review due to space limitations. We thank John Christianson, Andrew Nguyen, Milton To, Norbert Volkmar, and Chris Walczak for critical reading of this manuscript. Our research is supported by grants from the National Institutes of Health to J.A.O. (R00DK095921 and R01GM112948).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
The Annual Review of Nutrition is online at nutr.annualreviews.org
LITERATURE CITED
- 1.Adams CM, Goldstein JL, Brown MS. 2003. Cholesterol-induced conformational change in SCAP enhanced by Insig proteins and mimicked by cationic amphiphiles. PNAS 100(19):10647–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akopian D, Shen K, Zhang X, Shan SO. 2013. Signal recognition particle: an essential protein-targeting machine. Annu. Rev. Biochem 82:693–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benoist F, Grand-Perret T. 1997. Co-translational degradation of apolipoprotein B100 by the proteasome is prevented by microsomal triglyceride transfer protein. Synchronized translation studies on HepG2 cells treated with an inhibitor of microsomal triglyceride transfer protein. J. Biol. Chem 272(33):20435–42 [DOI] [PubMed] [Google Scholar]
- 4.Bernardi KM, Williams JM, Inoue T, Schultz A, Tsai B. 2013. A deubiquitinase negatively regulates retro-translocation of non-ubiquitinated substrates. Mol. Biol. Cell 24(22):3545–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernasconi R, Galli C, Calanca V, Nakajima T, Molinari M. 2010. Stringent requirement for HRD1, SEL1L, and OS-9/XTP3-B for disposal of ERAD-Ls substrates. J. Cell Biol 188(2):223–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blom D, Hirsch C, Stern P, Tortorella D, Ploegh HL. 2004. A glycosylated type I membrane protein becomes cytosolic when peptide: N-glycanase is compromised. EMBO J 23(3):650–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braakman I, Bulleid NJ. 2011. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem 80:71–99 [DOI] [PubMed] [Google Scholar]
- 8.Braun S, Matuschewski K, Rape M, Thoms S, Jentsch S. 2002. Role of the ubiquitin-selective CDC48UFD1/NPL4chaperone (segregase) in ERAD of OLE1 and other substrates. EMBO J 21(4):615–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown AJ, Sun L, Feramisco JD, Brown MS, Goldstein JL. 2002. Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol. Cell 10(2):237–45 [DOI] [PubMed] [Google Scholar]
- 10.Brown S, Brunschede Y, Goldstein L. 1975. Inactivation of 3-hydroxy-3-methylglutaryl coenzyme A reductase in vitro: an adenine nucleotide-dependent reaction catalyzed by a factor in human fibroblasts. J. Biol. Chem 250(7):2502–9 [PubMed] [Google Scholar]
- 11.Brown S, Goldstein L, Dietschy M. 1979. Active and inactive forms of 3-hydroxy-3-methylglutaryl coenzyme A reductase in the liver ofthe rat: comparison with the rate ofcholesterol synthesis in different physiological states. J. Biol. Chem 254(12):5144–49 [PubMed] [Google Scholar]
- 12.Butkinaree C, Guo L, Ramkhelawon B, Wanschel A, Brodsky JL, et al. 2014. A regulator of secretory vesicle size, Kelch-like protein 12, facilitates the secretion of apolipoprotein B100 and very-low-density lipoproteins—brief report. Arterioscler. Thromb. Vasc. Biol 34(2):251–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao J, Wang J, Qi W, Miao HH, Wang J, et al. 2007. Ufdl is a cofactor of gp78 and plays a key role in cholesterol metabolism by regulating the stability of HMG-CoA reductase. Cell Metab 6(2):115–28 [DOI] [PubMed] [Google Scholar]
- 14.Carvalho P, Goder V, Rapoport TA. 2006. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 126(2):361–73 [DOI] [PubMed] [Google Scholar]
- 15.Carvalho P, Stanley AM, Rapoport TA. 2010. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell 143(4):579–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen CY, Malchus NS, Hehn B, Stelzer W, Avci D, et al. 2014. Signal peptide peptidase functions in ERAD to cleave the unfolded protein response regulator XBP1u. EMBO J 33(21):2492–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi K, Kim H, Kang H,Lee SY, Lee SJ, et al. 2014. Regulation of diacylglycerol acyltransferase 2 protein stability by gp78-associated endoplasmic-reticulum-associated degradation. FEBS J 281(13):3048–60 [DOI] [PubMed] [Google Scholar]
- 18.Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, et al. 2012. Defining human ERAD networks through an integrative mapping strategy. Nat. Cell Biol 14(1):93–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christianson JC, Shaler TA, Tyler RE, Kopito RR. 2008OS-9 and GRP94 deliver mutant α1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol 10(3):272–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Christianson JC, Ye Y. 2014. Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat. Struct. Mol. Biol 21(4):325–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Churchward MA, Rogasevskaia T, Höfgen J, Bau J, Coorssen JR. 2005. Cholesterol facilitates the native mechanism of Ca2+-triggered membrane fusion. J. Cell Sci 118:4833–48 [DOI] [PubMed] [Google Scholar]
- 22.Coleman RA, Lewin TM, Muoio DM. 2000. Physiological and nutritional regulation of enzymes of triacylglycerol synthesis. Annu. Rev. Nutr 20:77–103 [DOI] [PubMed] [Google Scholar]
- 23.Cormier JH, Tamura T, Sunryd JC, Hebert DN. 2009. EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol. Cell 34(5):627–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DeBose-Boyd RA. 2008. Feedback regulation of cholesterol synthesis: sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res 18(6):609–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeLaBarre B, Brunger AT. 2003. Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat. Struct. Biol 10(10):856–63 [DOI] [PubMed] [Google Scholar]
- 26.DeLaBarre B, Christianson JC, Kopito RR, Brunger AT. 2006. Central pore residues mediate the p97/VCP activity required for ERAD. Mol. Cell 22(4):451–62 [DOI] [PubMed] [Google Scholar]
- 27.Denic V, Dotsch V, Sinning I. 2013. Endoplasmic reticulum targeting and insertion of tail-anchored membrane proteins by the GET pathway. Cold Spring Harb. Perspect. Biol 5(8):a013334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dixon JL, Furukawa S, Ginsberg HN. 1991. Oleate stimulates secretion of apolipoprotein B-containing lipoproteins from Hep G2 cells by inhibiting early intracellular degradation ofapolipoprotein B. J. Biol. Chem 266(8):5080–86 [PubMed] [Google Scholar]
- 29.Elbaz Y, Schuldiner M. 2011. Staying in touch: the molecular era of organelle contact sites. Trends Biochem. Sci 36(11):616–23 [DOI] [PubMed] [Google Scholar]
- 30.Ernst R, Claessen JH, Mueller B, Sanyal S, Spooner E, et al. 2011. Enzymatic blockade of the ubiquitin-proteasome pathway. PLOS Biol 8(3):e1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ernst R, Mueller B, Ploegh HL, Schlieker C. 2009. The otubain YOD1 is a deubiquitinating enzyme that associates with p97 to facilitate protein dislocation from the ER. Mol. Cell 36(1):28–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feng B, Yao PM, Li Y, Devlin CM, Zhang D, et al. 2003. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol 5(9):781–92 [DOI] [PubMed] [Google Scholar]
- 33.Fisher EA, Ginsberg HN. 2002. Complexity in the secretory pathway: the assembly and secretion of apolipoprotein B-containing lipoproteins. J. Biol. Chem 277(20):17377–80 [DOI] [PubMed] [Google Scholar]
- 34.Fisher EA, Khanna NA, McLeod RS. 2011. Ubiquitination regulates the assembly of VLDL in HepG2 cells and is the committing step of the apoB-100 ERAD pathway. J. Lipid Res 52(6):1170–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fisher EA, Lapierre LR, Junkins RD, McLeod RS. 2008. The AAA-ATPase p97 facilitates degradation of apolipoprotein B by the ubiquitin–proteasome pathway. J. Lipid Res 49(10):2149–60 [DOI] [PubMed] [Google Scholar]
- 36.Fleig L, Bergbold N, Sahasrabudhe P, Geiger B, Kaltak L, Lemberg MK. 2012. Ubiquitin-dependent intramembrane rhomboid protease promotes ERAD of membrane proteins. Mol. Cell 47(4):558–69 [DOI] [PubMed] [Google Scholar]
- 37.Flowers MT, Ntambi JM. 2008. Role of stearoyl-coenzyme A desaturase in regulating lipid metabolism. Curr. Opin. Lipidol 19(3):248–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foresti O, Rodriguez-Vaello V, Funaya C, Carvalho P. 2014. Quality control of inner nuclear membrane proteins by the Asi complex. Science 346(6210):751–55 [DOI] [PubMed] [Google Scholar]
- 39.Foresti O, Ruggiano A, Hannibal-Bach HK, Ejsing CS, Carvalho P. 2013. Sterol homeostasis requires regulated degradation of squalene monooxygenase by the ubiquitin ligase Doa10/Teb4. eLife 2:e00953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujimori T, Kamiya Y, Nagata K, Kato K, Hosokawa N. 2013. Endoplasmic reticulum lectin XTP3-B inhibits endoplasmic reticulum-associated degradation of a misfolded α1-antitrypsin variant. FEBS J 280(6):1563–75 [DOI] [PubMed] [Google Scholar]
- 41.Geiger R, Andritschke D, Friebe S, Herzog F, Luisoni S, et al. 2011. Bap31 and Bip are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat. Cell Biol 13(11):1305–14 [DOI] [PubMed] [Google Scholar]
- 42.Gelman MS, Kopito RR. 2002. Rescuing protein conformation: prospects for pharmacological therapy in cystic fibrosis. J. Clin. Investig 110(11):1591–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gil G, Faust JR, Chin DJ, Goldstein JL, Brown MS. 1985. Membrane-bound domain of HMG CoA reductase is required for sterol-enhanced degradation of the enzyme. Cell 41(1):249–58 [DOI] [PubMed] [Google Scholar]
- 44.Gill S, Stevenson J, Kristiana I, Brown AJ. 2011. Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metab 13(3):260–73 [DOI] [PubMed] [Google Scholar]
- 45.Goldstein JL, DeBose-Boyd RA, Brown MS. 2006. Protein sensors for membrane sterols. Cell 124(1):35–46 [DOI] [PubMed] [Google Scholar]
- 46.Gong Y, Lee JN, Lee PC, Goldstein JL, Brown MS, Ye J. 2006. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab 3(1):15–24 [DOI] [PubMed] [Google Scholar]
- 47.Greenblatt EJ, Olzmann JA, Kopito RR. 2012. Making the cut: intramembrane cleavage by a rhomboid protease promotes ERAD. Nat. Struct. Mol. Biol 19(10):979–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grove DE, Fan CY, Ren HY, Cyr DM. 2011. The endoplasmic reticulum–associated Hsp40 DNAJB12 and Hsc70 cooperate to facilitate RMA1 E3-dependent degradation of nascent CFTRAF508. Mol. Biol. Cell 22(3):301–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guerriero CJ, Brodsky JL. 2012. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev 92(2):537–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gusarova V, Caplan AJ, Brodsky JL, Fisher EA. 2001. Apoprotein B degradation is promoted by the molecular chaperones hsp90 and hsp70. J. Biol. Chem 276(27):24891–900 [DOI] [PubMed] [Google Scholar]
- 51.Habeck G, Ebner FA, Shimada-Kreft H, Kreft SG. 2015. The yeast ERAD-C ubiquitin ligase Doa10 recognizes an intramembrane degron. J. Cell Biol 209(2):261–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hagiwara M, Maegawa K, Suzuki M, Ushioda R, Araki K, et al. 2011. Structural basis of an ERAD pathway mediated by the ER-resident protein disulfide reductase ERdj5. Mol. Cell 41(4):432–44 [DOI] [PubMed] [Google Scholar]
- 53.Hannah VC, Ou J, Luong A, Goldstein JL, Brown MS. 2001. Unsaturated fatty acids down-regulate SREBP isoforms 1a and 1c by two mechanisms in HEK-293 cells. J. Biol. Chem 276(6):4365–72 [DOI] [PubMed] [Google Scholar]
- 54.Hartman IZ, Liu P, Zehmer JK, Luby-Phelps K, Jo Y, et al. 2010. Sterol-induced dislocation of 3-hydroxy-3-methylglutaryl coenzyme A reductase from endoplasmic reticulum membranes into the cytosol through a subcellular compartment resembling lipid droplets. J. Biol. Chem 285(25):19288–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hebert DN, Foellmer B, Helenius A. 1995. Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell 81(3):425–33 [DOI] [PubMed] [Google Scholar]
- 56.Heinemann FS, Korza G, Ozols J. 2003. A plasminogen-like protein selectively degrades stearoyl-CoA desaturase in liver microsomes. J. Biol. Chem 278(44):42966–75 [DOI] [PubMed] [Google Scholar]
- 57.Horton JD, Goldstein JL, Brown MS. 2002. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig 109(9):1125–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, et al. 2003. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. PNAS 100(21):12027–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hosokawa N, Kamiya Y, Kamiya D, Kato K, Nagata K. 2009. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J. Biol. Chem 284(25):17061–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hosokawa N, Wada I, Nagasawa K, Moriyama T, Okawa K, Nagata K. 2008. Human XTP3-B forms an endoplasmic reticulum quality control scaffold with the HRD1-SEL1L ubiquitin ligase complex and BiP. J. Biol. Chem 283(30):20914–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Howe V, Chua NK, Stevenson J, Brown AJ. 2015. The regulatory domain of squalene monooxygenase contains a re-entrant loop and senses cholesterol via a conformational change. J. Biol. Chem 290(46):27533–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huber MD, Vesely PW, Datta K, Gerace L. 2013. Erlins restrict SREBP activation in the ER and regulate cellular cholesterol homeostasis. J. Cell Biol 203(3):427–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hulce JJ, Cognetta AB, Niphakis MJ, Tully SE, Cravatt BF. 2013. Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat. Methods 10(3):259–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ikeda Y, Demartino GN, Brown MS, Lee JN, Goldstein JL, Ye J. 2009. Regulated endoplasmic reticulum-associated degradation of a polytopic protein: P97 recruits proteasomes to Insig-1 before extraction from membranes. J. Biol. Chem 284(50):34889–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Inoue S, Bar-Nun S, Roitelman J, Simoni RD. 1991. Inhibition of degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase in vivo by cysteine protease inhibitors. J. Biol. Chem 266(20):13311–17 [PubMed] [Google Scholar]
- 66.Inoue T, Tsai B. 2011. A large and intact viral particle penetrates the endoplasmic reticulum membrane to reach the cytosol. PLOS Pathog 7(5):e1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Irisawa M, Inoue J, Ozawa N, Mori K, Sato R. 2009. The sterol-sensing endoplasmic reticulum (ER) membrane protein TRC8 hampers ER to Golgi transport of sterol regulatory element-binding protein-2 (SREBP-2)/SREBP cleavage-activated protein and reduces SREBP-2 cleavage. J. Biol. Chem 284(42):28995–9004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jeon YJ, Khelifa S, Ratnikov B, Scott DA, Feng Y, et al. 2015. Regulation of glutamine carrier proteins by RNF5 determines breast cancer response to ER stress-inducing chemotherapies. Cancer Cell 27(3):354–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jin L, Pahuja KB, Wickliffe KE, Gorur A, Baumgärtel C, et al. 2012. Ubiquitin-dependent regulation of COPII coat size and function. Nature 482(7386):495–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jo Y, Hartman IZ, DeBose-Boyd RA. 2013. Ancient ubiquitous protein-1 mediates sterol-induced ubiquitination of 3-hydroxy-3-methylglutaryl CoA reductase in lipid droplet-associated endoplasmic reticulum membranes. Mol. Biol. Cell 24(3):169–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jo Y, Lee PC, Sguigna PV, DeBose-Boyd RA. 2011. Sterol-induced degradation of HMG CoA reductase depends on interplay of two Insigs and two ubiquitin ligases, gp78 and Trc8. PNAS 108(51):20503–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jo Y, Sguigna PV, DeBose-Boyd RA. 2011. Membrane-associated ubiquitin ligase complex containing gp78 mediates sterol-accelerated degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J. Biol. Chem 286(17):15022–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Johnston JA, Ward CL, Kopito RR. 1998. Aggresomes: a cellular response to misfolded proteins. J. Cell Biol 143(7):1883–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kato H, Sakaki K, Mihara K. 2006. Ubiquitin-proteasome-dependent degradation of mammalian ER stearoyl-CoA desaturase. J. Cell Sci 119:2342–53 [DOI] [PubMed] [Google Scholar]
- 75.Khmelinskii A, Blaszczak E, Pantazopoulou M, Fischer B, Omnus DJ, et al. 2014. Protein quality control at the inner nuclear membrane. Nature 516(7531):410–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kikkert M, Doolman R, Dai M, Avner R, Hassink G, et al. 2004. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem 279(5):3525–34 [DOI] [PubMed] [Google Scholar]
- 77.Kim H, Zhang H, Meng D, Russell G, Lee JN, Ye J. 2013. UAS domain of Ubxd8 and FAF1 polymerizes upon interaction with long-chain unsaturated fatty acids. J. Lipid Res 54(8):2144–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Klemm EJ, Spooner E, Ploegh HL. 2011. Dual role of ancient ubiquitous protein 1 (AUP1) in lipid droplet accumulation and endoplasmic reticulum (ER) protein quality control. J. Biol. Chem 286(43):37602–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Komander D, Rape M. 2012. The ubiquitin code. Annu. Rev. Biochem 81:203–29 [DOI] [PubMed] [Google Scholar]
- 80.Kristiana I, Luu W, Stevenson J, Cartland S, Jessup W, et al. 2012. Cholesterol through the looking glass: ability of its enantiomer also to elicit homeostatic responses. J. Biol. Chem 287(40):33897–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lange Y, Ory DS, Ye J, Lanier MH, Hsu FF, Steck TL. 2008. Effectors of rapid homeostatic responses of endoplasmic reticulum cholesterol and 3-hydroxy-3-methylglutaryl-CoA reductase. J. Biol. Chem 283(3):1445–55 [DOI] [PubMed] [Google Scholar]
- 82.Lass A, Zimmermann R, Oberer M, Zechner R. 2011. Lipolysis—a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog. Lipid Res 50(1):14–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee JN, Gong Y, Zhang X, Ye J. 2006. Proteasomal degradation of ubiquitinated Insig proteins is determined by serine residues flanking ubiquitinated lysines. PNAS 103(13):4958–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee JN, Kim H, Yao H, Chen Y, Weng K, Ye J. 2010. Identification of Ubxd8 protein as a sensor for unsaturated fatty acids and regulator of triglyceride synthesis. PNAS 107(50):21424–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee JN, Song B, DeBose-Boyd RA, Ye J. 2006. Sterol-regulated degradation ofInsig-1 mediated by the membrane-boundubiquitinligase gp78. J. Biol. Chem 281(51):39308–15 [DOI] [PubMed] [Google Scholar]
- 86.Lee JN, Ye J. 2004. Proteolytic activation of sterol regulatory element-binding protein induced by cellular stress through depletion of Insig-1. J. Biol. Chem 279(43):45257–65 [DOI] [PubMed] [Google Scholar]
- 87.Lee JN, Zhang X, Feramisco JD, Gong Y, Ye J. 2008. Unsaturated fatty acids inhibit proteasomal degradation of Insig-1 at a postubiquitination step. J. Biol. Chem 283(48):33772–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee JP, Brauweiler A, Rudolph M, Hooper JE, Drabkin HA, Gemmill RM. 2010. The Trc8 ubiquitin ligase is sterol regulated and interacts with lipid and protein biosynthetic pathways. Mol. Cancer Res 8(1):93–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee PC, Nguyen AD, Debose-Boyd RA. 2007. Mutations within the membrane domain ofHMG-CoA reductase confer resistance to sterol-accelerated degradation. J. Lipid Res 48(2):318–27 [DOI] [PubMed] [Google Scholar]
- 90.Lee PC, Sever N, Debose-Boyd RA. 2005. Isolation of sterol-resistant Chinese hamster ovary cells with genetic deficiencies in both Insig-1 and Insig-2. J. Biol. Chem 280(26):25242–49 [DOI] [PubMed] [Google Scholar]
- 91.Lee RJ, Liu CW, Harty C, McCracken AA, Latterich M, et al. 2004. Uncoupling retro-translocation and degradation in the ER-associated degradation of a soluble protein. EMBO J 23(11):2206–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Leichner GS, Avner R, Harats D, Roitelman J. 2009. Dislocation of HMG-CoA reductase and Insig-1, two polytopic endoplasmic reticulum proteins, en route to proteasomal degradation. Mol. Biol. Cell 20(14):3330–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lev S 2012. Nonvesicular lipid transfer from the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol 4(10):a013300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liang J, Wu X, Jiang H, Zhou M, Yang H, et al. 1998. Translocation efficiency, susceptibility to proteasomal degradation, and lipid responsiveness of apolipoprotein B are determined by the presence of β sheet domains. J. Biol. Chem 273(52):35216–21 [DOI] [PubMed] [Google Scholar]
- 95.Liang JS, Kim T, Fang S, Yamaguchi J, Weissman AM, et al. 2003. Overexpression of the tumor autocrine motility factor receptor gp78, a ubiquitin protein ligase, results in increased ubiquitinylation and decreased secretion of apolipoprotein B100 in HepG2 cells. J. Biol. Chem 278(26):23984–88 [DOI] [PubMed] [Google Scholar]
- 96.Lilley BN, Ploegh HL. 2004. A membrane protein required for dislocation of misfolded proteins from the ER. Nature 429(6994):834–40 [DOI] [PubMed] [Google Scholar]
- 97.Lipson C, Alalouf G, Bajorek M, Rabinovich E, Atir-Lande A, et al. 2008. A proteasomal ATPase contributes to dislocation of endoplasmic reticulum-associated degradation (ERAD) substrates. J. Biol. Chem 283(11):7166–75 [DOI] [PubMed] [Google Scholar]
- 98.Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, et al. 2003. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. PNAS 100(6):3077–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu TF, Tang JJ, Li PS, Shen Y, Li JG, et al. 2012. Ablation of gp78 in liver improves hyperlipidemia and insulin resistance by inhibiting SREBP to decrease lipid biosynthesis. Cell Metab 16(2):213–25 [DOI] [PubMed] [Google Scholar]
- 100.Liu Y, Soetandyo N, Lee JG, Liu L, Xu Y, et al. 2014. USP13 antagonizes gp78 to maintain functionality of a chaperone in ER-associated degradation. eLife 3:e01369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lu JP, Wang Y, Sliter DA, Pearce MM, Wojcikiewicz RJ. 2011. RNF170 protein, an endoplasmic reticulum membrane ubiquitin ligase, mediates inositol 1,4,5-trisphosphate receptor ubiquitination and degradation. J. Biol. Chem 286(27):24426–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lukacs GL, Verkman AS. 2012. CFTR: folding, misfolding and correcting the ΔF508 conformational defect. Trends Mol. Med 18(2):81–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Malhotra V, Erlmann P. 2011. Protein export at the ER: loading big collagens into COPII carriers. EMBO J 30(17):3475–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Man WC, Miyazaki M, Chu K, Ntambi J. 2006. Colocalization of SCD1 and DGAT2: implying preference for endogenous monounsaturated fatty acids in triglyceride synthesis. J. Lipid Res 47(9):1928–39 [DOI] [PubMed] [Google Scholar]
- 105.Martínez-Botas J 2001. Dose-dependent effects oflovastatin on cell cycle progression: distinct requirement of cholesterol and non-sterol mevalonate derivatives. Biochim. Biophys. Acta 1532(3):185–94 [DOI] [PubMed] [Google Scholar]
- 106.Matsumura Y, Sakai J, Skach WR. 2013. Endoplasmic reticulum protein quality control is determined by cooperative interactions between Hsp/c70 and the CHIP E3 ligase. J. Biol. Chem 288(43):31069–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Matyskiela ME, Martin A. 2013. Design principles of a universal protein degradation machine. J. Mol. Biol 425(2):199–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Maxfield FR, Tabas I. 2005. Role of cholesterol and lipid organization in disease. Nature 438(7068):612–21 [DOI] [PubMed] [Google Scholar]
- 109.McFie PJ, Jin Y, Banman SL, Beauchamp E, Berthiaume LG, Stone SJ. 2014. Characterization of the interaction of diacylglycerol acyltransferase-2 with the endoplasmic reticulum and lipid droplets. Biochim. Biophys. Acta 1841(9):1318–28 [DOI] [PubMed] [Google Scholar]
- 110.Mehnert M, Sommer T, Jarosch E. 2014. Der1 promotes movement of misfolded proteins through the endoplasmic reticulum membrane. Nat. Cell Biol 16(1):77–86 [DOI] [PubMed] [Google Scholar]
- 111.Meinecke M, Cizmowski C, Schliebs W, Krüger V, Beck S, et al. 2010. The peroxisomal importomer constitutes a large and highly dynamic pore. Nat. Cell Biol 12(3):273–77 [DOI] [PubMed] [Google Scholar]
- 112.Meyer H, Bug M, Bremer S. 2012. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol 14(2):117–23 [DOI] [PubMed] [Google Scholar]
- 113.Miao H,Jiang W, Ge L, Li B, Song B. 2010. Tetra-glutamic acid residues adjacent to Lys248 in HMG-CoA reductase are critical for the ubiquitination mediated by gp78 and UBE2G2. Acta Biochim. Biophys. Sin 42(5):303–10 [DOI] [PubMed] [Google Scholar]
- 114.Mitchell DM, Zhou M, Pariyarath R, Wang H, Aitchison JD, et al. 1998. Apoprotein B100 has a prolonged interaction with the translocon during which its lipidation and translocation change from dependence on the microsomal triglyceride transfer protein to independence. PNAS 95(25):14733–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Moldavski O, Amen T, Levin-Zaidman S, Eisenstein M, Rogachev I, et al. 2015. Lipid droplets are essential for efficient clearance of cytosolic inclusion bodies. Dev. Cell 33(5):603–10 [DOI] [PubMed] [Google Scholar]
- 116.Morito D, Hirao K, Oda Y, Hosokawa N, Tokunaga F, et al. 2008. Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTRYF508. Mol. Biol. Cell 19(4):1328–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Morris LL, Hartman IZ, Jun DJ, Seemann J, DeBose-Boyd RA. 2014. Sequential actions of the AAA-ATPase valosin-containing protein (VCP)/p97 and the proteasome 19 S regulatory particle in sterol-accelerated, endoplasmic reticulum (ER)-associated degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J. Biol. Chem 289(27):19053–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Motamed M, Zhang Y, Wang ML, Seemann J, Kwon HJ, et al. 2011. Identification of luminal Loop 1 of Scap protein as the sterol sensor that maintains cholesterol homeostasis. J. Biol. Chem 286(20):18002–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mueller B, Klemm EJ, Spooner E, Claessen JH, Ploegh HL. 2008. SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. PNAS 105(34):12325–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mziaut H, Korza G, Ozols J. 2000. The N terminus of microsomal Δ9 stearoyl-CoA desaturase contains the sequence determinant for its rapid degradation. PNAS 97(16):8883–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nakakuki M, Kawano H, Notsu T, Imada K, Mizuguchi K, Shimano H.2014. A novel processing system of sterol regulatory element-binding protein-1c regulated by polyunsaturated fatty acid. J. Biochem 155(5):301–13 [DOI] [PubMed] [Google Scholar]
- 122.Nakanishi SM, Goldstein L, Brown S. 1988. Multivalent control of3-hydroxy-3-methylglutaryl coenzymeAreductase: Mevalonate-derived product inhibits translation of mRNA and accelerates degradation of enzyme. J. Biol. Chem 263(18):8929–37 [PubMed] [Google Scholar]
- 123.Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL. 2008. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell 132(1):101–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nguyen AD, Lee SH, DeBose-Boyd RA. 2009. Insig-mediated, sterol-accelerated degradation of the membrane domain ofhamster 3-hydroxy-3-methylglutaryl-coenzyme A reductase in insect cells. J. Biol. Chem 284(39):26778–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nguyen AD, McDonald JG, Bruick RK, DeBose-Boyd RA. 2007. Hypoxia stimulates degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase through accumulation of lanosterol and hypoxia-inducible factor-mediated induction ofInsigs. J. Biol. Chem 282(37):27436–46 [DOI] [PubMed] [Google Scholar]
- 126.Ninagawa S, Okada T, Sumitomo Y, Horimoto S, Sugimoto T, et al. 2015. Forcible destruction of severely misfolded mammalian glycoproteins by the non-glycoprotein ERAD pathway. J. Cell Biol 211(4):775–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ninagawa S, Okada T, Sumitomo Y, Kamiya Y, Kato K, et al. 2014. EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J. Cell Biol 206(3):347–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ohsaki Y, Cheng J, Fujita A, Tokumoto T, Fujimoto T. 2006. Cytoplasmic lipid droplets are sites of convergence of proteasomal and autophagic degradation of apolipoprotein B. Mol. Biol. Cell 17(6):2674–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ohsaki Y, Cheng J, Suzuki M, Fujita A, Fujimoto T. 2008. Lipid droplets are arrested in the ER membrane by tight binding of lipidated apolipoprotein B-100. J. Cell Sci 121:2415–22 [DOI] [PubMed] [Google Scholar]
- 130.Okuda-Shimizu Y, Hendershot LM. 2007. Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require Herp. Mol. Cell 28(4):544–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Olzmann JA, Kopito RR. 2011. Lipid droplet formation is dispensable for endoplasmic reticulum-associated degradation. J. Biol. Chem 286(32):27872–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Olzmann JA, Kopito RR, Christianson JC. 2013. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb. Perspect. Biol 5(9):a013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Olzmann JA, Richter CM, Kopito RR. 2013. Spatial regulation of UBXD8 and p97/VCP controls ATGL-mediated lipid droplet turnover. PNAS 110(4):1345–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ou J, Tu H, Shan B, Luk A, DeBose-Boyd RA, et al. 2001. Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. PNAS 98(11):6027–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pearce MM, Wormer DB, Wilkens S, Wojcikiewicz RJ. 2009. An endoplasmic reticulum (ER) membrane complex composed of SPFH1 and SPFH2 mediates the ER-associated degradation of inositol 1,4,5-trisphosphate receptors. J. Biol. Chem 284(16):10433–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Petris G, Casini A, Sasset L, Cesaratto F, Bestagno M, et al. 2014. CD4 and BST-2/tetherin proteins retro-translocate from endoplasmic reticulum to cytosol as partially folded and multimeric molecules. J. Biol. Chem 289(1):1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Plemper K, Bohmler S, Bordallo J, Sommer T. 1997. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature 388(6645):891–95 [DOI] [PubMed] [Google Scholar]
- 138.Ploegh HL. 2007. A lipid-based model for the creation of an escape hatch from the endoplasmic reticulum. Nature 448(7152):435–38 [DOI] [PubMed] [Google Scholar]
- 139.Pol A, Gross SP, Parton RG. 2014. Biogenesis of the multifunctional lipid droplet: lipids, proteins, and sites. J. Cell Biol 204(5):635–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Prinz WA. 2014. Bridging the gap: membrane contact sites in signaling, metabolism, and organelle dynamics. J. Cell Biol 205(6):759–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. 2008. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab 8(6):512–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS, Goldstein JL. 2007. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Oxysterols block transport by binding to Insig. PNAS 104(16):6511–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rapoport TA. 2007. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 450(7170):663–69 [DOI] [PubMed] [Google Scholar]
- 144.Ruggiano A, Foresti O, Carvalho P. 2014. ER-associated degradation: protein quality control and beyond. J. Cell Biol 204(6):869–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rutledge AC, Qiu W, Zhang R, Kohen-Avramoglu R, Nemat-Gorgani N, Adeli K. 2009. Mechanisms targeting apolipoprotein B100 to proteasomal degradation: evidence that degradation is initiated by BiP binding at the N terminus and the formation of a p97 complex at the C terminus. Arterioscler. Thromb. Vasc. Biol 29(4):579–85 [DOI] [PubMed] [Google Scholar]
- 146.Sato BK, Schulz D, Do PH, Hampton RY. 2009. Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Mol. Cell 34(2):212–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sato R, Goldstein JL, Brown MS. 1993. Replacement of serine-871 of hamster 3-hydroxy-3-methylglutaryl-CoA reductase prevents phosphorylation by AMP-activated kinase and blocks inhibition of sterol synthesis induced by ATP depletion. PNAS 90(20):9261–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sato T, Sako Y, Sho M, Momohara M, Suico MA, et al. 2012. STT3B-dependent posttranslational N-glycosylation as a surveillance system for secretory protein. Mol. Cell 47(1):99–110 [DOI] [PubMed] [Google Scholar]
- 149.Sato T, Susuki S, Suico MA, Miyata M, Ando Y, et al. 2007. Endoplasmic reticulum quality control regulates the fate of transthyretin variants in the cell. EMBO J 26(10):2501–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Satoh T, Chen Y, Hu D, Hanashima S, Yamamoto K, Yamaguchi Y. 2010. Structural basis for oligosaccharide recognition of misfolded glycoproteins by OS-9 in ER-associated degradation. Mol. Cell 40(6):905–16 [DOI] [PubMed] [Google Scholar]
- 151.Schliebs W, Girzalsky W, Erdmann R. 2010. Peroxisomal protein import and ERAD: variations on a common theme. Nat. Rev. Mol. Cell Biol 11(12):885–90 [DOI] [PubMed] [Google Scholar]
- 152.Schumacher MM, Elsabrouty R, Seemann J, Jo Y, DeBose-Boyd RA. 2015. The prenyltransferase UBIAD1 is the target of geranylgeraniol in degradation of HMG CoA reductase. eLife 4:e05560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sekijima Y, Wiseman RL, Matteson J, Hammarstrom P, Miller SR, et al. 2005. The biological and chemical basis for tissue-selective amyloid disease. Cell 121(1):73–85 [DOI] [PubMed] [Google Scholar]
- 154.Sever N, Song BL, Yabe D, Goldstein JL, Brown MS, DeBose-Boyd RA. 2003Insig-dependentubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J. Biol. Chem 278(52):52479–90 [DOI] [PubMed] [Google Scholar]
- 155.Sever N, Yang T, Brown MS, Goldstein JL, DeBose-Boyd RA. 2003Accelerated degradation of HMG CoA reductase mediated by binding of Insig-1 to its sterol-sensing domain. Mol. Cell 11(1):25–33 [DOI] [PubMed] [Google Scholar]
- 156.Shao S, Hegde RS. 2011. Membrane protein insertion at the endoplasmic reticulum. Annu. Rev. Cell Dev. Biol 27:25–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Shearer AG, Hampton RY. 2004. Structural control of endoplasmic reticulum-associated degradation: effect of chemical chaperones on 3-hydroxy-3-methylglutaryl-CoA reductase. J. Biol. Chem 279(1):188–96 [DOI] [PubMed] [Google Scholar]
- 158.Shearer AG, Hampton RY. 2005. Lipid-mediated, reversible misfolding of a sterol-sensing domain protein. EMBO J 24(1):149–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Shelness GS, Ledford AS. 2005. Evolution and mechanism of apolipoprotein B-containing lipoprotein assembly. Curr. Opin. Lipidol 16(3):325–32 [DOI] [PubMed] [Google Scholar]
- 160.Shrimal S, Cherepanova NA, Gilmore R. 2015. Cotranslational and posttranslocational N-glycosylation of proteins in the endoplasmic reticulum. Semin. Cell Dev. Biol 41:71–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Simons K, Ikonen E. 2000. How cells handle cholesterol. Science 290(5497):1721–26 [DOI] [PubMed] [Google Scholar]
- 162.Simons K, Toomre D. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol 1(1):31–39 [DOI] [PubMed] [Google Scholar]
- 163.Simons K, Vaz WL. 2004. Model systems, lipid rafts, and cell membranes. Annu. Rev. Biophys. Biomol. Struct 33:269–95 [DOI] [PubMed] [Google Scholar]
- 164.Skalnik DG, Narita H, Kent C, Simoni RD. 1988. The membrane domain of 3-hydroxy-3-methylglutaryl-coenzyme A reductase confers endoplasmic reticulum localization and sterol-regulated degradation onto β-galactosidase. J. Biol. Chem 263(14):6836–41 [PubMed] [Google Scholar]
- 165.Song BL, DeBose-Boyd RA. 2004. Ubiquitination of 3-hydroxy-3-methylglutaryl-CoA reductase in permeabilized cells mediated by cytosolic E1 and a putative membrane-bound ubiquitin ligase. J. Biol. Chem 279(27):28798–806 [DOI] [PubMed] [Google Scholar]
- 166.Song BL, Sever N, DeBose-Boyd RA. 2005. Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol. Cell 19(6):829–40 [DOI] [PubMed] [Google Scholar]
- 167.Stagg HR, Thomas M, van den Boomen D, Wiertz EJ, Drabkin HA, et al. 2009. The Trc8 E3 ligase ubiquitinates MHC class I molecules before dislocation from the ER. J. Cell Biol 186(5):685–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Steck TL, Lange Y. 2010. Cell cholesterol homeostasis: mediation by active cholesterol. Trends Cell Biol 20(11):680–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Stein A, Ruggiano A, Carvalho P, Rapoport TA. 2014. Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell 158(6):1375–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Stevenson J, Luu W, Kristiana I, Brown AJ. 2014. Squalene mono-oxygenase, a key enzyme in cholesterol synthesis, is stabilized by unsaturated fatty acids. Biochem J 461(3):435–42 [DOI] [PubMed] [Google Scholar]
- 171.Stone SJ, Levin MC, Farese RV. 2006. Membrane topology and identification of key functional amino acid residues of murine acyl-CoA:diacylglycerol acyltransferase-2. J. Biol. Chem 281(52):40273–82 [DOI] [PubMed] [Google Scholar]
- 172.Sun LP, Seemann J, Goldstein JL, Brown MS. 2007. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in SCAP inaccessible to COPII proteins. PNAS 104(16):6519–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sun S, Shi G, Sha H, Ji Y, Han X, et al. 2015. IRE1α is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat. Cell Biol 17:1546–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Suzuki M, Otsuka T, Ohsaki Y, Cheng J, Taniguchi T, et al. 2012. Derlin-1 and UBXD8 are engaged in dislocation and degradation of lipidated ApoB-100 at lipid droplets. Mol. Biol. Cell 23(5):800–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Tannous A, Patel N, Tamura T, Hebert DN. 2015. Reglucosylation by UDP-glucose:glycoprotein glucosyltransferase 1 delays glycoprotein secretion but not degradation. Mol. Biol. Cell 26(3):390–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tannous A, Pisoni GB, Hebert DN, Molinari M. 2015. N-linked sugar-regulated protein folding and quality control in the ER. Semin. Cell Dev. Biol 41:79–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Thibault G, Ng DT. 2012. The endoplasmic reticulum-associated degradation pathways of budding yeast. Cold Spring Harb. Perspect. Biol 4(12):a013193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Tirosh B, Furman MH, Tortorella D, Ploegh HL. 2003. Protein unfolding is not a prerequisite for endoplasmic reticulum-to-cytosol dislocation. J. Biol. Chem 278(9):6664–72 [DOI] [PubMed] [Google Scholar]
- 179.Toyama T, Kudo N, Mitsumoto A, Hibino Y, Tsuda T, Kawashima Y. 2007. Stearoyl-CoA desaturase activity is elevated by the suppression of its degradation by clofibric acid in the liver of rats. J. Pharmacol. Sci 103(4):383–90 [DOI] [PubMed] [Google Scholar]
- 180.Tsai YC, Leichner GS, Pearce MM, Wilson GL, Wojcikiewicz RJ, et al. 2012. Differential regulation of HMG-CoA reductase and Insig-1 by enzymes of the ubiquitin-proteasome system. Mol. Biol. Cell 23(23):4484–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Tyler RE, Pearce MM, Shaler TA, Olzmann JA, Greenblatt EJ, Kopito RR. 2012. Unassembled CD147 is an endogenous endoplasmic reticulum-associated degradation substrate. Mol. Biol. Cell 23(24):4668–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ushioda R, Hoseki J, Araki K, Jansen G, Thomas DY, Nagata K. 2008. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 321(5888):569–72 [DOI] [PubMed] [Google Scholar]
- 183.Ushioda R, Hoseki J, Nagata K. 2013. Glycosylation-independent ERAD pathway serves as a backup system under ER stress. Mol. Biol. Cell 24(20):3155–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Van de Weijer ML, Bassik MC, Luteijn RD, Voorburg CM, Lohuis MA, et al. 2014. A high-coverage shRNA screen identifies TMEM129 as an E3 ligase involved in ER-associated protein degradation. Nat. Commun 5:3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Van den Berg B, Clemons WM, Collinson I, Modis Y, Hartmann E, et al. 2004. X-ray structure of a protein-conducting channel. Nature 427(6969):36–44 [DOI] [PubMed] [Google Scholar]
- 186.Van den Boomen DJ, Lehner PJ. 2015. Identifying the ERAD ubiquitin E3 ligases for viral and cellular targeting ofMHC class I. Mol. Immunol 68(2):106–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Van den Boomen DJ, Timms RT, Grice GL, Stagg HR, Skødt K, et al. 2014. TMEM129 is a Derlin-1 associated ERAD E3 ligase essential for virus-induced degradation of MHC-I. PNAS 111(31):11425–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Voorhees RM, Fernandez IS, Scheres SH, Hegde RS. 2014. Structure of the mammalian ribosome–Sec61 complex to 3.4 Å resolution. Cell 157(7):1632–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Walther TC, Farese RV. 2012. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem 81:687–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wang CW, Lee SC. 2012. Theubiquitin-like (UBX)-domain-containing protein Ubx2/Ubxd8 regulates lipid droplet homeostasis. J. Cell Sci 125:2930–39 [DOI] [PubMed] [Google Scholar]
- 191.Wang Q, Li L, Ye Y. 2008. Inhibition of p97-dependent protein degradation by eeyarestatin I. J. Biol. Chem 283(12):7445–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Wang Q, Liu Y, Soetandyo N, Baek K, Hegde R, Ye Y. 2011. A ubiquitin ligase-associated chaperone holdase maintains polypeptides in soluble states for proteasome degradation. Mol. Cell 42(6):758–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ward CL, Omura S, Kopito RR. 1995. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83(1):121–27 [DOI] [PubMed] [Google Scholar]
- 194.Wilfling F, Wang H, Haas JT, Krahmer N, Gould TJ, et al. 2013. Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev. Cell 24(4):384–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wojcikiewicz RJ, Pearce MM, Sliter DA, Wang Y. 2009. When worlds collide: IP3 receptors and the ERAD pathway. Cell Calcium 46(3):147–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wong J, Quinn CM, Gelissen IC, Brown AJ. 2008. Endogenous 24(S),25-epoxycholesterol fine-tunes acute control of cellular cholesterol homeostasis. J. Biol. Chem 283(2):700–7 [DOI] [PubMed] [Google Scholar]
- 197.Xu C, Ng DT. 2015. Glycosylation-directed quality control of protein folding. Nat. Rev. Mol. Cell Biol 16(12):742–52 [DOI] [PubMed] [Google Scholar]
- 198.Xu Y, Cai M, Yang Y, Huang L, Ye Y. 2012. SGTA recognizes a noncanonical ubiquitin-like domain in the Bag6–Ubl4A–Trc35 complex to promote endoplasmic reticulum-associated degradation. Cell Rep 2(6):1633–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Xu Y, Liu Y, Lee JG, Ye Y. 2013. A ubiquitin-like domain recruits an oligomeric chaperone to a retrotranslocation complexin endoplasmic reticulum-associated degradation. J. Biol. Chem 288(25):18068–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Yabe D, Brown MS, Goldstein JL. 2002. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. PNAS 99(20):12753–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Yabe D, Xia ZP, Adams CM, Rawson RB. 2002. Three mutations in sterol-sensing domain of SCAP block interaction with Insig and render SREBP cleavage insensitive to sterols. PNAS 99(26):16672–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. 2004. A membrane protein complex mediates retrotranslocation from the ER lumen into the cytosol. Nature 429(6994):841–47 [DOI] [PubMed] [Google Scholar]
- 203.Yen CL, Stone SJ, Koliwad S, Harris C, Farese RV. 2008. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res 49(11):2283–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Yu H, Kaung G, Kobayashi S, Kopito RR. 1997. Cytosolic degradation of T-cell receptor α chains by the proteasome. J. Biol. Chem 272(33):20800–4 [DOI] [PubMed] [Google Scholar]
- 205.Yu H, Kopito RR. 1999. The role of multiubiquitination in dislocation and degradation of the α subunit of the T cell antigen receptor. J. Biol. Chem 274(52):36852–58 [DOI] [PubMed] [Google Scholar]
- 206.Zanetti G, Pahuja KB, Studer S, Shim S, Schekman R.2012. COPII and the regulation of protein sorting in mammals. Nat. Cell Biol 14(1):20–28 [DOI] [PubMed] [Google Scholar]
- 207.Zehmer JK, Bartz R, Bisel B, Liu P, Seemann J, Anderson RG. 2009. Targeting sequences of UBXD8 and AAM-B reveal that the ER has a direct role in the emergence and regression of lipid droplets. J. Cell Sci 122:3694–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Zelcer N, Sharpe LJ, Loregger A, Kristiana I, Cook EC, et al. 2014. The E3 ubiquitin ligase MARCH6 degrades squalene monooxygenase and affects 3-hydroxy-3-methyl-glutaryl coenzyme A reductase and the cholesterol synthesis pathway. Mol. Cell Biol 34(7):1262–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Zettl M, Adrain C, Strisovsky K, Lastun V, Freeman M. 2011. Rhomboid family pseudoproteases use the ER quality control machinery to regulate intercellular signaling. Cell 145(1):79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Zhang T, Xu Y, Liu Y, Ye Y. 2015. Gp78 functions downstream of Hrd1 to promote degradation of misfolded proteins of the endoplasmic reticulum. Mol. Biol. Cell 26(24):4438–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Zhang ZR, Bonifacino JS, Hegde RS. 2013. Deubiquitinases sharpen substrate discrimination during membrane protein degradation from the ER. Cell 154(3):609–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zhou M, Wu X, Huang LS, Ginsberg HN. 1995. Apoprotein B100, an inefficiently translocated secretory protein, is bound to the cytosolic chaperone, heat shock protein 70. J. Biol. Chem 270(42):25220–24 [DOI] [PubMed] [Google Scholar]