

Abstract
LUBAC modulates signalling by various immune receptors. In TNF signalling, linear (also known as M1) ubiquitin enables full gene-activation and prevents cell death. However, the mechanisms underlying cell-death prevention remain ill-defined. We show that LUBAC activity enables TBK1 and IKKε recruitment to and activation at the TNFR1-signalling complex (TNFR1-SC). Whilst exerting only limited effects on TNF-induced gene-activation, TBK1/IKKε are essential to prevent TNF-induced cell death. Mechanistically, TBK1/IKKε phosphorylate RIPK1 in the TNFR1-SC, thereby preventing RIPK1-kinase-activity-dependent cell death. This activity is essential in vivo, as it prevents TNF-induced lethal shock. Strikingly, NEMO/IKKγ, which mostly, but not exclusively, binds to the TNFR1-SC via M1-ubiquitin, mediates recruitment of the adaptors TANK and NAP1/AZI2 which are constitutively associated with TBK1/IKKε and TBK1, respectively. We here discover a previously unrecognised TBK1/IKKε-mediated cell-death checkpoint and uncover an essential survival function for NEMO by enabling recruitment and activation of these noncanonical IKKs to prevent TNF-induced cell death.
Introduction
TNF-receptor 1 (TNFR1) signalling involves the formation of two distinct complexes1. The plasma-membrane-associated TNFR1-signalling complex (TNFR1-SC), also termed complex-I, is responsible for gene-activation whereas the subsequently formed cytosolic complex-II induces cell death. Under physiological conditions, TNFR1 stimulation induces gene-activation rather than cell death. However, aberrant TNF-induced signalling causes several auto-immune pathologies and cancer-related inflammation2, 3. Therefore, defining the molecular checkpoints which determine the different TNFR1 signalling outputs is critical to understand the biology of inflammation.
Linear ubiquitination is crucial for multiple immune-receptor signalling pathways4, 5. The linear-ubiquitin chain assembly complex (LUBAC) is the only known E3 ligase capable of forming linear-ubiquitin, also termed M1-ubiquitin, linkages. LUBAC is composed of three core proteins, heme-oxidized IRP2 ubiquitin ligase 1 (HOIL-1), SHANK-associated RH-domain-interacting protein (SHARPIN) and the catalytically active HOIL-1-interacting protein (HOIP)6–9. The essence of LUBAC and M1-ubiquitination has been demonstrated by the embryonic lethality of mice lacking HOIL-1, HOIP or LUBAC activity10–12 which is caused by aberrant TNFR1-mediated signalling10, 12.
LUBAC recruitment to complex-I enables full gene-activation whilst preventing cell death8, 10, 13, 14. TNFR1-recruited LUBAC M1-ubiquitinates several TNFR1-SC components, including RIPK1, NEMO, TNFR1 and TRADD8, 14, 15. In complex-I, M1-ubiquitin acts as a recruitment platform for adaptors such as NEMO, the regulatory subunit of the inhibitor of κB (IκB) kinase (IKK) complex. Although NEMO can be recruited to the TNFR1-SC in the absence of M1-ubiquitin, this is much weaker than in its presence13. Accordingly, NEMO binding to TNFR1-SC-contained M1-ubiquitin enables full activation of the canonical IKKs, IKKα and IKKβ16. When activated, they phosphorylate IκB, causing nuclear translocation of NF-κB, thereby initiating gene transcription. Independently thereof, IKKα/β prevent TNF-induced cell death by phosphorylating RIPK1. This limits RIPK1’s ability to promote formation of complex-II17. Phosphorylation events on RIPK1, mediated by the p38 target MK2, also restrict RIPK1’s capacity to trigger complex-II formation and, consequently, cell death18–20. Notably, IKKα/β and p38/MK2 both also mediate gene-expression downstream of TAK1 which is required for TNF-induced gene-activation16, 21–26.
TBK1 and IKKε are two closely related kinases which are homologous to the canonical kinases IKKα and IKKβ27, 28. Various adaptors recruit them to distinct immune signalling complexes29. Concomitant TBK1 and IKKε ablation abolishes the activation of interferon signalling by various immune-receptors, including TLR3/4, or after viral infection30, 31. Here we show that by phosphorylating RIPK1 in complex-I, TBK1 and IKKε serve an essential function in TNF signalling, providing a physiologically relevant cell-death-restricting checkpoint which depends on M1-ubiquitination and NEMO.
Results
Effective TBK1 and IKKε recruitment to and activation in complex-I require LUBAC activity
To elucidate how LUBAC modulates TNF signalling, we compared the composition of purified TNFR1-SCs from wild-type and HOIP-deficient A549 cells by mass spectrometry (MS)14. This confirmed that in HOIP-KO cells, HOIL-1, SHARPIN, CYLD and SPATA214, 32 as well as the A20/Abin1/Abin2 complex are missing from complex-I, whereas NEMO is still recruited to it, albeit poorly (Figure 1a and Supplementary table 2)14, 32 16, 25, 33–36. Importantly however, TBK1, IKKε and one of their known adaptors, TANK37, were detected in complex-I in control but not HOIP-KO cells (Figure 1a).
Figure 1. LUBAC mediates recruitment and activation of the non-canonical kinases TBK1 and IKKε in the TNFR1-SC.
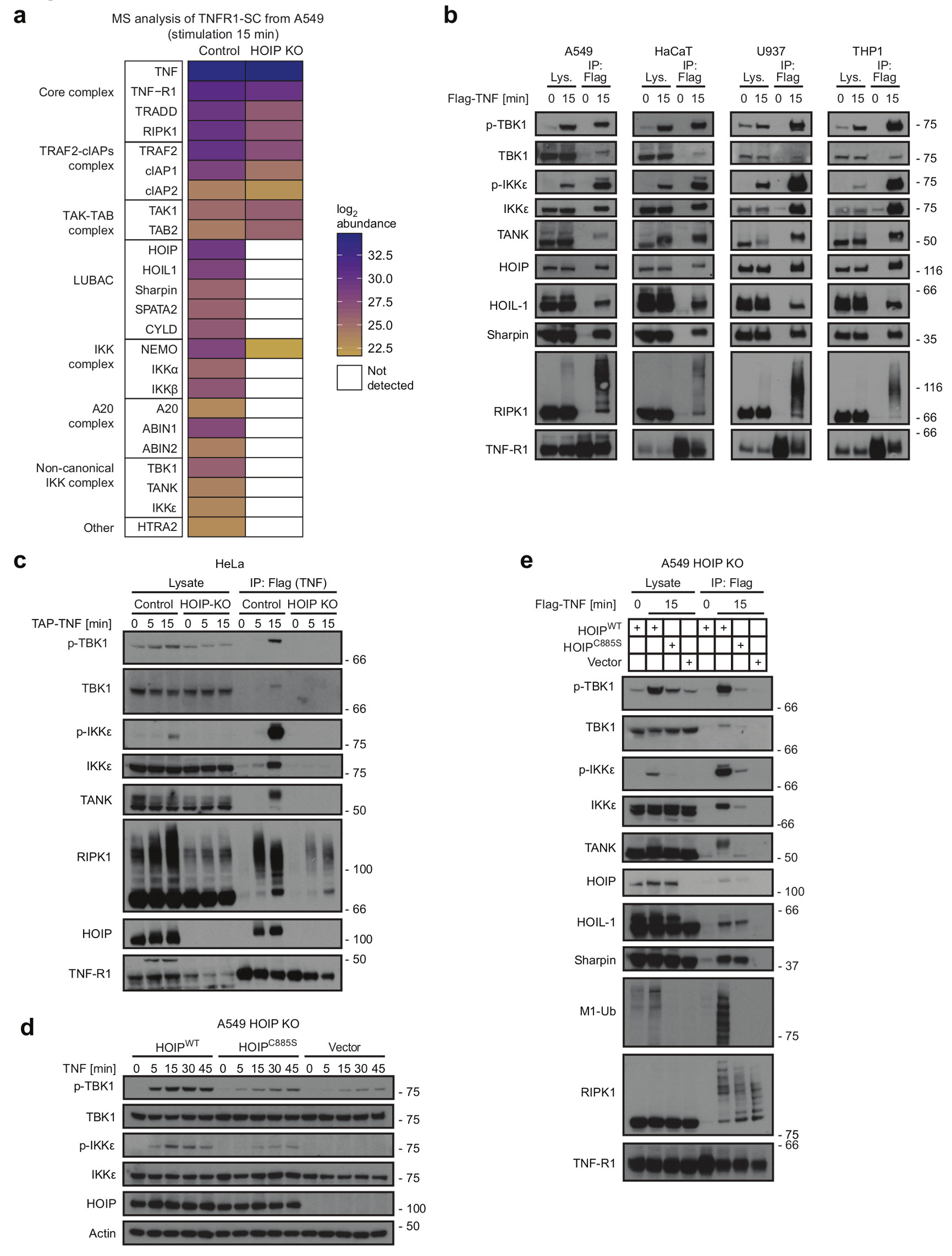
(a) A549 HOIP Control or HOIP KO cells were stimulated with TAP-TNF (500 ng/mL). TNFR1-SC was purified in a two-step immunoprecipitation via TAP-Tag and analysed by LC-MS/MS. Mean protein abundance for two independent experiments and two LC-MS analyses is plotted as indicated. Raw data can be accessed on the ProteomeXchange Consortium via the PRIDE70 partner repository with the dataset identifier PXD008497, analysed data is available in Supplementary table 2 (b) A549, HaCaT, U937 and THP-1 cells were stimulated with FLAG-TNF (1 µg/mL) for the indicated times. TNFR1-SC was purified and analysed by western blotting. (c) HeLa HOIP control or HOIP KO cells were stimulated with TAP-TNF (500 ng/mL) and subjected to TNFR1-SC isolation and western blot analysis. (d) HOIP-deficient A549 -KO cells were reconstituted with empty vector, HOIPWT or enzymatically inactive HOIPC885S and were stimulated with TNF (200 ng/mL) for the indicated times and lysates were analysed by western blotting. (e) HOIP-deficient A549 reconstituted with empty vector, HOIPWT or enzymatically inactive HOIPC885S were stimulated with FLAG-TNF (1 µg/mL) for the indicated time. Lysates and TNFR1-SC were analysed by western blot. (b-e) One representative experiment out of two is shown. Unprocessed original scans of blots are shown in Supplementary Figure 7.
Western blot analysis revealed that TBK1 and IKKε are recruited to and strongly phosphorylated within the native TNFR1-SC on the activatory Ser17238 in various HOIP-proficient cell lines, whereas their activation in corresponding HOIP-deficient cells is very weak (Figure 1b, c and Supplementary Figure 1a,b). Thus, TBK1 and IKKε are bona fide components of complex-I and LUBAC enables their recruitment.
Using HOIP-deficient HeLa and A549 cells reconstituted with wild-type (HOIPWT) or catalytically-inactive HOIP (HOIPC885S)39, we determined that effective TBK1/IKKε recruitment to complex-I requires the M1-ubiquitin-forming activity of LUBAC as TBK1/IKKε recruitment was strongly diminished in HOIP-deficient HeLa and A549 cells whether or not HOIPC885S was re-expressed in them (Figure 1d,e, Supplementary Figure 1c,d).
The role of TBK1 and IKKε in TNF-induced gene-activation is limited
As TBK1 and IKKε are crucial for gene-expression by various immune-receptor complexes30, 40–42, we evaluated whether these kinases influenced TNF-induced gene-activation by generating TBK1/IKKε/TNF-triple-knockout (TKO) L929 cells. However, absence of TBK1 and IKKε did not significantly affect TNF-induced gene-activatory signalling and, if anything, slightly increased IkB-α phosphorylation (Supplementary Figure 2a), in line with the previously proposed role of TBK1/IKKε as negative regulators of IKKα/β activation43. Similarly, neither in MEFs nor A549 cells treatment with the TBK1/IKKε-specific inhibitor MRT6730743 (MRT) exerted any significant effects on TNF-induced activation of MAPKs or NF-κB (Figure 2a and Supplementary Figure 2b). To evaluate whether TBK1/IKKε affect gene-induction upon TNFR1 stimulation, we performed an unbiased RNA-Seq analysis upon TNF- versus TNF/MRT-stimulation, also including TNF/TPCA-1 which, as an IKKα/β-inhibiting control, is known to profoundly affect TNF-induced gene-expression44.
Figure 2. Inhibition of TBK1/IKKε exerts only minor effects on TNF-induced gene-activatory signalling.
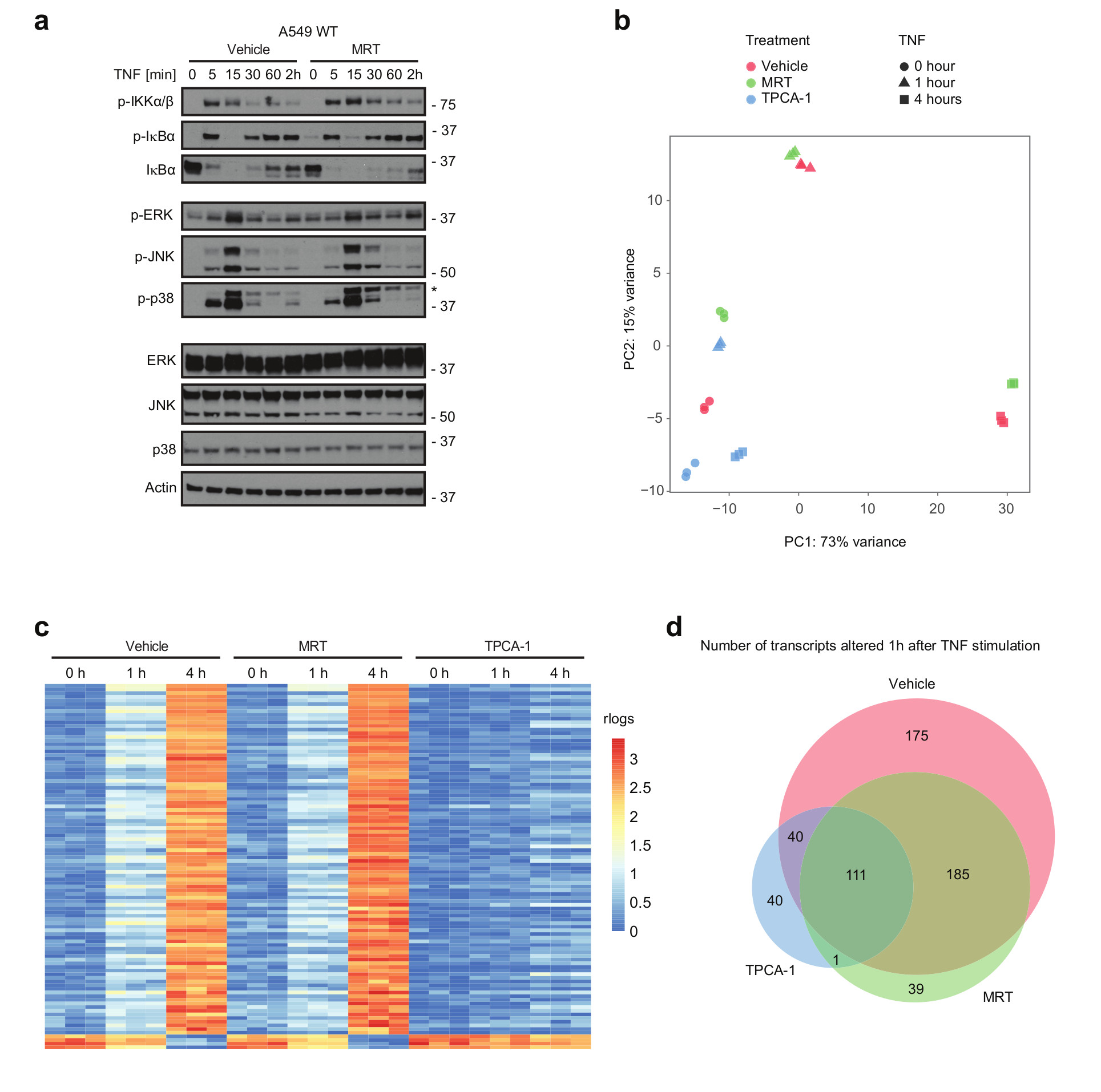
(a-d) A549 WT cells were pre-incubated with either vehicle (DMSO) or MRT for 30 min, followed by stimulation with TNF (200 ng/mL) for the indicated times. (a) Lysates were analysed by western blotting. One representative experiment out of two is shown. * staining from previous p-JNK. Unprocessed original scans of blots are shown in Supplementary Figure 7 (b-d) Cells were then lysed, their total RNA extracted and RNA-Seq analysis performed. Samples from three independent experiments were obtaineded and analysed. (b) Principal-component analysis (PCA) of A549 samples based on transcriptome-wide expression level data is shown. (c) The heatmap illustrates the major change of expression across the dataset. The genes selected to be shown were the 100 most highly correlated with PC1 (see Fig 2b). For clarity of comparison the 'rlog' expression data of each row was zeroed at time-point 0 hr and then scaled by the standard deviation. The RNA-Seq raw dataset for b and c are available in the SRA repository and can be accessed by using the following BioProject accession: PRJNA422567 or SRA accession: SRP126844 (https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP126844). (d) The Venn diagram represents the number of all transcripts significantly regulated upon 1 hr of TNF-stimulation in vehicle, MRT- or TPCA-1 -treated samples and the transcript overlap between those three groups. Corresponding transcripts can be found in supplementary table 3. Differential RNA-seq expression statistics (p-values) on contrasting biological triplicates, corresponding to samples obtained from three independent experiments (groups as in b-d) were estimated using DESeq2. Adjusted p-value statistics were calculated with the Benjamoni-Hochberg and IHW adjustment.
Principal-component analysis revealed that TNF drastically modulated gene-expression, with TNF-treated clearly segregating from untreated samples. Whilst the effect of IKKα/β-inhibition on TNF-induced gene-expression was substantial, that of TBK1/IKKε-inhibition was surprisingly limited (Figure 2b) with the top 100 most altered transcripts being highly similar between TNF-stimulated control and TNF/MRT-treated cells but significantly different in TNF/TPCA-1-treated cells (Figure 2c and Supplementary Figure 2c). Notably, the majority of transcripts with significantly altered expression at the 1-hour time-point in control cells were similarly modulated in TNF/MRT-, yet not in TNF/TPCA-1-treated cells (Figure 2d and Supplementary Table 3). Thus, in contrast to the role of TBK1 and IKKε in other immune signalling pathways as drivers of gene-induction40, 41, their role in TNF-induced gene-expression is rather limited.
TBK1/IKKε prevent TNF-induced, RIPK1-kinase-activity-dependent cell death
We next assessed the role of TBK1 and IKKε in TNF-induced cell death. Strikingly, treatment with MRT or another TBK1/IKKε-specific inhibitor, BX-79545, drastically sensitised MEFs and L929 cells to TNF-induced death (Figure 3 a,b and Supplementary Figure 3a). Intriguingly, in both cases cell death was prevented by the RIPK1 kinase inhibitor Nec-1s (Figure 3a, b and Supplementary Figure 3a). Interestingly, MRT-induced spontaneous death of L929 cells was inhibited by the TNF-blocker ENBREL®/etanercept (Supplementary Figure 3b). Creating TNF-KO MEFs and L929 cells in which we further deleted TBK1, IKKε or both kinases enabled us to genetically assess their cell-death-preventative function. In line with the results on their inhibition, concomitant deletion of TBK1 and IKKε also strongly sensitised these cells to TNF-induced death (Figure 3c and Supplementary Figure 3c, d). Thus, pharmacologic inhibition or genetic ablation of TBK1 and IKKε sensitises cells to TNF-induced death.
Figure 3. Inhibition of TBK1/IKKε sensitises cells to TNF-induced RIPK1 activity-dependent cell death downstream of LUBAC.

(a) MEF TNF-/- and (b) L929 cells were treated with TNF (500 ng/mL and 50 ng/mL, respectively) in the presence or absence of MRT and Nec-1s. (c) MEF TNF-/- cells of the indicated genotype were stimulated with TNF (500 ng/mL) for 6h. Loss of cell viability was determined using the Cell Titer Glo (CTG) assay. Mean +/- SEM of n=3 independent experiments. Lysates of untreated cells were analysed by Western blot. Unprocessed original scans of blots are shown in Supplementary Figure 7. (d-h) MEFs of the indicated genotype were treated with TNF (500 ng/mL) in the presence or absence of the indicated compounds. (a, b and d-h) Cell death was measured in function of time by SytoxGreen positivity. The RFU mean of 4 technical replicates of one representative experiment out of three independent experiments is represented. Representative images of indicated measurements are depicted with corresponding percentage of dead cells. Cell counting was performed manually using ImageJ. White bar in microscopy images equals 200 µm. Raw data are provided in Supplementary table 1.
We next investigated the cell death modality induced by TBK1/IKKε inhibition. Whilst pre-treatment with the pan-caspase inhibitor zVAD substantially sensitised MEFs to TNF alone, sensitisation to TNF/MRT was significantly higher (Figure 3d and Supplementary Figure 3e). Importantly, RIPK1-kinase-dead MEFs were resistant to MRT-induced death sensitisation by TNF or TNF/zVAD (Figure 3e,f). In line with the finding that LUBAC is required for effective TNF-induced TBK1/IKKε activation, TBK1/IKKε inhibition did not sensitise HOIP-deficient MEFs to TNF- or TNF/zVAD-induced cell death (Figure 3g, h). Moreover, MRT-mediated sensitisation to TNF-induced necroptosis also occurred in RIPK3-expressing human A549 cells (Supplementary Figure 3f-h). Notably, MEFs were also sensitised to TNF-induced death in the presence of cycloheximide, implying that protein synthesis is not necessary for TBK1/IKKε-inhibition-mediated sensitisation (Supplementary Figure 3i,j). MRT also sensitised primary Bone-Marrow-Derived Macrophages (BMDMs) to TNF-induced RIPK1-kinase-activity-dependent death (Supplementary Figure 4a). Again, a substantial percentage of MRT-only-treated BMDMs were killed by endogenous TNF (Supplementary Figure 4b). Interestingly, BMDMs were exclusively sensitised to TNF-induced apoptosis (Supplementary Figure 4c). Accordingly, absence of the necroptosis-mediating pseudokinase MLKL did not diminish TNF/MRT-induced BMDM death whilst caspase-8/MLKL co-ablation abrogated it (Supplementary Figure 4d, e).
Together, these results demonstrate that inhibition or absence of TBK1 and IKKε promotes TNF-induced apoptosis and/or necroptosis, depending on the ability of a given cell type to undergo death by the respective modality. Importantly, in either case cell death induction requires RIPK1 activity.
TBK1/IKKε prevent TNF-induced RIPK1 activation and ensuing complex-II formation
TNFR1-SC assembly precedes formation of complex-II which contains RIPK1, FADD, FLIP, Caspase-8, RIPK3, and MLKL. Depending on cellular context and relative expression of pro- and anti-apoptotic and -necroptotic proteins15, 46–48, complex-II can trigger apoptosis or necroptosis. RIPK1 kinase activity mediates complex-II formation and is, therefore, generally required for TNF-induced cell death17.
Since RIPK1’s kinase activity is required for TNF-induced cell death when TBK1/IKKε are absent or inhibited, we assessed whether TBK1/IKKε prevent cytotoxicity by keeping RIPK1 from mediating complex-II formation. We treated A549 cells expressing TAP-tagged RIPK3 with TNF/zVAD or TNF/zVAD/BX-795 before immunoprecipitating RIPK3. TBK1/IKKε inhibition markedly enhanced RIPK3 association with Caspase-8/FADD, phosphorylated MLKL (p-MLKL) and phosphorylated RIPK1 (p-RIPK1) (Figure 4a). Enhanced RIPK3 binding to caspase-8 and p-MLKL was also observed in RIPK3-expressing HeLa cells and, decisively, concomitant RIPK1 inhibition prevented complex-II formation (Figure 4b).
Figure 4. Inhibition of TBK1/IKKε leads to TNF-induced RIPK1 activation and increased complex II formation.
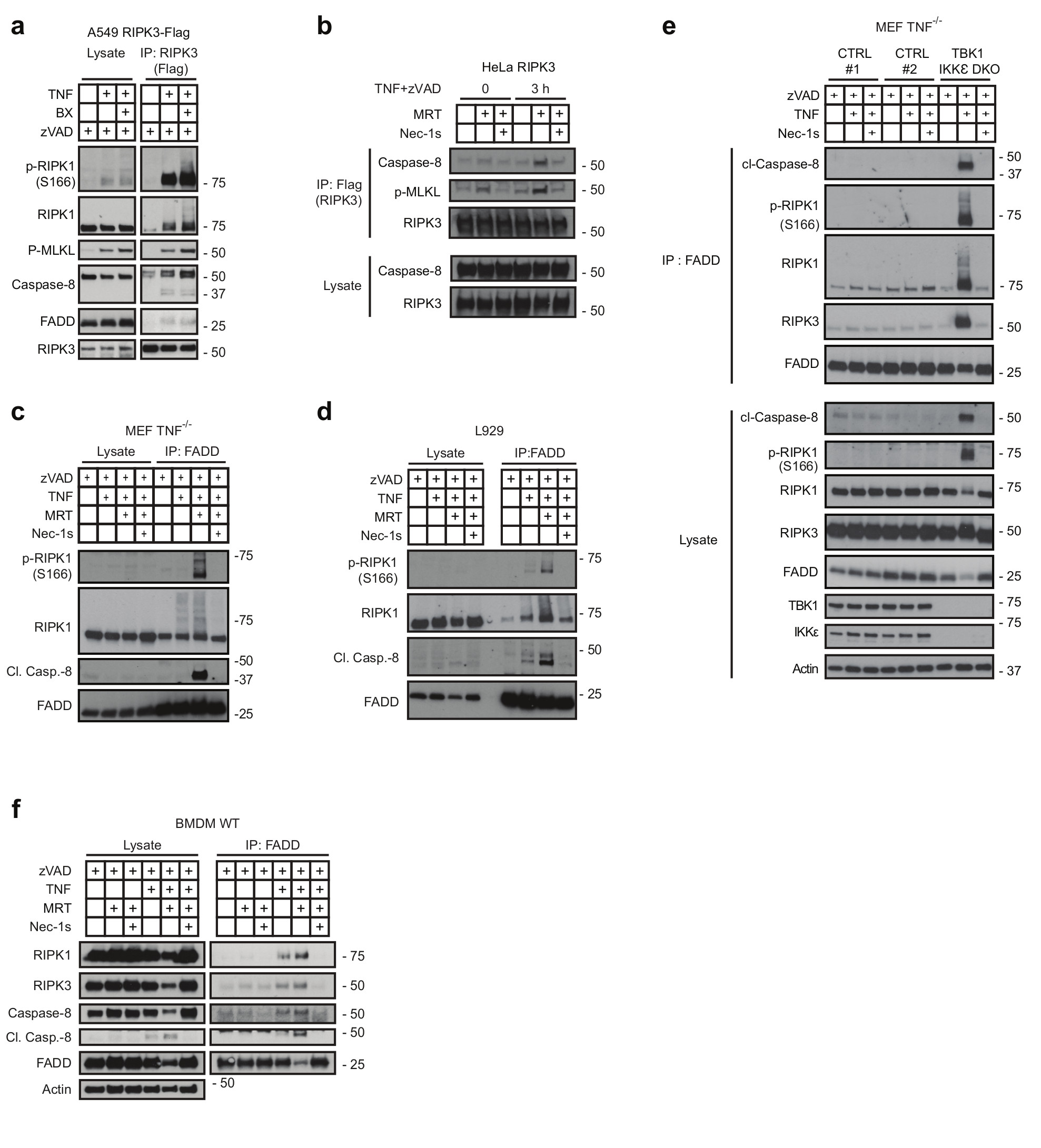
(a) A549 and (b) HeLa cells both overexpressing FLAG-tagged RIPK3 were treated with or without BX-795 and zVAD (a) or MRT, zVAD and Nec-1s (b) and were stimulated with TNF (500 ng/mL) for 3 hours. Complex II was then FLAG-immunoprecipitated and analysed by western blot. (c-e) MEFs (c), L929 (d), MEF TNF-/- ;TBK1/IKKε DKO cells and corresponding MEF TNF-/- control cells (e) and primary BMDMs (f) were pre-treated with MRT in combination with zVAD and Nec-1s as indicated and stimulated with TNF (500 ng/mL), for either 6 hours (c, e, f) or 4 hours (d), respectively. Complex II was then immunoprecipitated with a FADD antibody and analysed by western blot. (a-f) One representative experiment is shown out of two independent experiments. Unprocessed original scans of blots are shown in Supplementary Figure 7.
In accordance with the increased cell death observed upon TBK1/IKKε inhibition, L929 cells and MEFs also displayed increased RIPK1 activation and complex-II formation upon TNF/zVAD/MRT co-treatment (Figure 4c,d). Importantly, genetic TBK1/IKKε co-ablation in MEFs (Figure 4e) and TNF/MRT-treatment of primary BMDMs (Figure 4f) also substantially enhanced complex-II formation. In all cases, complex-II formation was RIPK1-kinase-activity-dependent (Figure 4b-f). Hence, TBK1 and IKKε prevent TNF-induced cell death by restricting RIPK1 auto-activation and consequent complex-II formation.
TBK1/IKKε mediate phosphorylation of RIPK1 in the TNFR1-SC
We hypothesised that the negative effect of TBK1/IKKε on RIPK1 auto-phosphorylation might be achieved via TNF-induced RIPK1 phosphorylation mediated by these kinases. Thus, we determined whether we could detect any TBK1/IKKε-dependent phosphorylation of RIPK1. After immunoprecipitating RIPK1 from L929 cells treated with TNF or TNF/BX-795 in the presence of zVAD, we incubated the resulting immunoprecipitates with or, as a control, without the pan-deubiquitinase USP2 to completely deubiquitinate RIPK1. This was done with or without λ-phosphatase to uncover any TBK1/IKKε-activity-requiring RIPK1 phosphorylation (Figure 5a). USP2 treatment resulted in the collapse of most modified high-molecular-weight onto low-molecular-weight forms of RIPK1. The intermediate-molecular-weight forms remaining after USP2 treatment completely collapsed onto the band of unmodified RIPK1 upon treatment with λ-phosphatase, demonstrating that phosphorylation accounts for the TBK1/IKKε-dependent mobility shift of deubiquitinated RIPK1. Interestingly, TNF/BX-795 co-treatment reduced the RIPK1 mobility shift as compared to TNF treatment. Most importantly, this shift was not seen upon λ-phosphatase treatment, demonstrating that TBK1/IKKε inhibition specifically prevents RIPK1phosphorylation (Figure 5a).
Figure 5. TBK1 and IKKε phosphorylate RIPK1 both in vitro and at the TNFR1-SC, providing a physiologically relevant cell death checkpoint.

(a) L929 pre-treated +/- BX-795 were stimulated with TNF (1 µg/mL)/zVAD for 15 minutes before RIPK1-pulldown. RIPK1-IP treated with USP2 and Lambda-phosphatase as indicated was analysed by western blot. (b) L929 cells were stimulated with TAP-TNF for 15 minutes (1 µg/mL) +/- MRT. TNFR1-SC was FLAG-immunoprecipitated. Samples were first separated by pI on IPG-strips pH 3-10 NL, followed by SDS-PAGE and western blot analysis. (c) Indicated MEFs were treated with FLAG-TNF (1 µg/mL)/Nec-1s, followed by FLAG-IP. TNFR1-SC treated with USP2 and Lambda-phosphatase as indicated was analysed by western blot. (d,e) GST-tagged TBK1 (d) and IKKε (e) were incubated with GST-tagged RIPK1 in a kinase assay +/- indicated inhibitors before western blot analysis. (f) Phosphosites identified by LC-MS/MS from one kinase assay using GST-tagged RIPK1,Nec-1s and IKKε. Raw data is accessible on ProteomeXchange Consortium via PRIDE70 partner repository with dataset identifier PXD008518, analysed data in Supplementary table 4.(g) MEFs TNF-/- pre-treated with indicated inhibitors were stimulated with FLAG-TNF (1 µg/mL)/Nec-1s for 15 minutes, followed by FLAG-IP. TNFR1-SCs treated with USP2 and Lambda-phosphatase as indicated were analysed via western blotting. * unspecific bands (h) MEFs TNF-/- pre-treated with indicated inhibitors were stimulated with FLAG-TNF (1 µg/mL)/Nec1-s for 15 minutes, followed by FLAG-IP. TNFR1-SCs treated with USP2 were first separated by pI on IPG-strips pH 4-7 followed by SDS-PAGE and western blot analysis. (i) MEFs TNF-/- stimulated with indicated inhibitors for 30 minutes were treated with TNF (500 ng/mL) for 7 hours. (j) L929 pre-incubated with Nec-1s for 15 minutes and further incubated with indicated inhibitors for 30 minutes were treated with TNF (50 ng/mL) for 4 hours. (i,j) Loss of cell viability was determined by CTG assay. Mean +/-SEM of n=4 (i) or mean +/-SEM of n=3 (j) independent experiments. (k) Cumulative survival rates of mice following TNFα-induced shock in presence of indicated inhibitors were compared using log-rank Mantel-Cox test. MRT vs TNF+MRT: p<0.001; TNF vs TNF+MRT: p<0.0001; TNF+MRT vs TNF+MRT+Nec-1s: p=0.0061. Unprocessed blots are shown in Supplementary Figure 7, raw data in Supplementary table 1. One experiment representative of two (a-c, g, h) or three (d, e) independent experiments is shown.
To assess whether TBK1/IKKε-dependent RIPK1 phosphorylation occurs in the TNFR1-SC, we immunoprecipitated complex-I following TNF versus TNF/MRT treatment, before evaluating the status of RIPK1 phosphorylation by 2D-gel electrophoresis. This revealed that TBK1/IKKε inhibition reduced the low-pI forms of RIPK1 upon TNF-stimulation in complex-I (Figure 5b). Importantly, RIPK1 phosphorylation in complex-I was also reduced in TNF/TBK1/IKKε TKO MEFs, TNF/TBK1 DKO and TNF/TBK1/IKKε TKO L929 cells (Figure 5c and Supplementary Figure 5a). We therefore conclude that TBK1 and IKKε promote RIPK1 phosphorylation within the native TNFR1-SC.
Performing in-vitro kinase assays to test whether TBK1/IKKε can directly phosphorylate RIPK1 revealed that both kinases were able to do so (Figure 5d, e). MS analysis of the in-vitro kinase assay employing recombinant IKKε and RIPK1 in the presence of Nec-1s showed that RIPK1 was phosphorylated on numerous residues, as also confirmed by 2D-gel electrophoresis (Figure 5f, Supplementary Figure 5b and Supplementary Table 4). Of note, certain of these residues were reported to be phosphorylated by other kinases17–20.
Together, these results show that TBK1/IKKε mediate RIPK1 phosphorylation on multiple residues in complex-I, thereby preventing RIPK1 auto-phosphorylation and consequent formation of complex-II.
TBK1/IKKε phosphorylate RIPK1 independently from IKKα/β and MK2
TNF-induced RIPK1 activation is controlled by IKKα/β- and MK2-mediated phosphorylation, both acting downstream of TAK117–20. These phosphorylation events function as checkpoints to inhibit RIPK1-dependent complex-II formation. We next determined whether TBK1/IKKε-mediated RIPK1 phosphorylation represents a distinct cell-death-preventing checkpoint. We purified the TNFR1-SC from cells treated with TNF together with different inhibitors or combinations thereof targeting TBK1/IKKε (MRT), IKKα/β (TPCA-1), MK2 (PF-3644022) and TAK1 (7-oxozeanol) before treating the immunoprecipitated complexes with USP2 to visualise phosphorylation events. This analysis confirmed that inhibition of TAK1, MK2 and/or IKKα/β reduces RIPK1 phosphorylation in complex-I. Importantly, however, TBK1/IKKε phosphorylated RIPK1 independently from MK2 and IKKα/β as only combined inhibition of TBK1/IKKε, either with that of TAK1 or with those of MK2 and IKKα/β, abrogated RIPK1 phosphorylation (Figure 5g).
2D-gel analysis of complex-I further revealed that several low-pI forms of RIPK1 disappeared upon addition of MRT in conditions under which all other known phosphorylation events are inhibited (Figure 5h). Thus, TBK1/IKKε-activity in complex-I promotes phosphorylation of RIPK1 at multiple sites which are not phosphorylated by other RIPK1-inhibiting kinases. In line with a distinct checkpoint mediated by TBK1/IKKε, their inhibition sensitised MEFs and L929 cells to RIPK1-dependent TNF-induced death even in the presence of MK2 and/or IKKα/β inhibitors (Figure 5i, j). Thus, TBK1/IKKε-mediated RIPK1 phosphorylation constitutes a cell-death checkpoint different from the previously described ones. Furthermore, these results strongly suggest that TBK1/IKKε directly phosphorylate RIPK1 in the TNFR1-SC.
TBK1/IKKε protect from TNF-induced, RIPK1-kinase-activity-dependent lethal shock in vivo
To reveal whether the cell-death checkpoint identified herein is functionally relevant in vivo, we employed TNF at an established sublethal dose17, 20 in a murine model of TNF-induced shock that requires RIPK1-kinase-activity-dependent cell death49, 50. As expected, none of the mice succumbed following treatment with TNF alone. Strikingly, however, TNF/MRT co-treatment resulted in highly significant reduction in survival, with 50% lethality only 8.5 hours after injection and an overall survival of less than 20%. Importantly, concomitant RIPK1 inhibition completely prevented TNF/MRT-induced lethality (Figure 5k). Hence, inhibition of RIPK1 by TBK1/IKKε is essential for protection from TNF-induced lethal shock.
NEMO-recruited TANK and NAP1 bring TBK1 and IKKε to the TNFR1-SC
Having identified that the control of TNF-induced cell death by TBK1 and IKKε is essential, we determined the biochemical mechanism of their recruitment to complex-I. TBK1 and IKKε are known to associate with various adaptors which recruit them to different signalling platforms29. One such adaptor is TANK51 which is recruited to complex-I in a HOIP-activity-dependent manner (Figure 1). Using TANK-KO cells, we observed that IKKε recruitment was abrogated in these cells and that TBK1 recruitment was reduced but still occurred (Figures 6a, b). Thus, IKKε recruitment entirely relies on TANK, whereas TBK1 is recruited by TANK and at least one additional adaptor.
Figure 6. NEMO acts upstream of the adaptors TANK and NAP1 which recruit TBK1 and IKKε, or TBK1-only, respectively, to the TNFR1-SC.

(a) TANK-deficient A549 cell clones and control cells were treated with TNF (200 ng/mL) for the indicated times. Lysates were analysed by western blot. (b) TANK KO or corresponding control A549 cells were stimulated with FLAG-TNF (500 ng/mL) for the indicated times. The purified TNFR1-SC and lysates were analysed by western blot. (c) A549 TBK1 KO cells were reconstituted with TBK1WT-TAP. The TBK1 interactome was purified in a two-step immunoprecipitation via TAP-Tag and analysed by LC-MS/MS. One experiment was analysed. Raw data can be accessed on the ProteomeXchange Consortium via the PRIDE70 partner repository with the dataset identifier PXD010777 and analysed data is available in Supplementary table 5. (d) A549 TBK1 KO cells were reconstituted with TBK1WT-TAP or the catalytically inactive mutant TBK1D135N-TAP and treated with 500 ng/mL TNF as indicated. The purified TBK1-associated complex and lysates were analysed by western blot. (e) HOIP-deficient A549 cells reconstituted with HOIP WT, enzymatically inactive HOIP C885S or vector control were stimulated with FLAG-TNF (500 ng/mL) for the indicated times. The purified TNFR1-SC and lysates were analysed by western blot. (f) HeLa cells deficient in TANK, concomitantly deficient in TANK and NAP1 (TANK/NAP1 DKO), or corresponding control cells were stimulated with TAP-TNF (500 ng/mL) and subjected to immunoprecipitation via Flag. The purified TNFR1-SC and lysates were analysed by western blot. (g) NEMO KO and corresponding WT MEFs were stimulated with TNF (200 ng/mL) for the indicated times. Lysates were analysed by western blot. (h) A549 control and NEMO KO cells were stimulated with HA-TNF (1 µg/mL) and subjected to immunoprecipitation via HA. The purified TNFR1-SC and lysates were analysed by western blot. (i) A549 NEMO KO cells reconstituted with NEMOWT, NEMOΔTBD or vector control were stimulated with HA-TNF (1 µg /mL) for the indicated times. The purified TNFR1-SC and lysates were analysed by western blot. One experiment representative of two (a,b, d-g) or three independent experiments (h,i). Unprocessed original scans of blots are shown in Supplementary Figure 7.
After ruling out Optineurin, a suggested TNFR1-SC component52, as responsible for TANK-independent TBK1 recruitment to complex-I (Supplementary Figure 6a), we sought to identify the additional adaptor(s) for TBK1 in an unbiased manner by studying the TBK1 interactome using TBK1-KO cells re-expressing TAP-tagged TBK1 and performing MS analysis on affinity-purified TBK1 obtained therefrom. With NAP1, SINTBAD, TANK and TRAF2 this analysis only identified known TBK1 interactors37 (Figure 6c, d and Supplementary table 5). Whilst TRAF2 was recruited via TRADD independently of HOIP, and SINTBAD was not identified in the complex (Figure 6e), NAP1 was recruited to complex-I in a LUBAC-activity-dependent manner (Figure 6e). Using TANK/NAP1-DKO HeLa and A549 cells revealed that TBK1 recruitment was completely and nearly completely absent from these cells, respectively (Figure 6f and Supplementary Figure 6b). In line with these results, absence of TANK and NAP1 increased the auto-phosphorylation of RIPK1 in complex-I (Supplementary Figure 6c). Thus, TANK and NAP1 are together responsible for the vast majority of TBK1 recruitment to complex-I.
As TANK was shown to interact with NEMO53, which is recruited to complex-I via M1-ubiquitin36, we next evaluated the role of NEMO in TANK recruitment. This revealed that NEMO not only recruits TANK but also NAP1 and, in consequence, brings IKKε and TBK1 to complex-I in both, A549 cells and MEFs (Figure 6g, h and Supplementary Figure 6d). Moreover, re-expression of wild-type NEMO, but not of a deletion mutant unable to bind TANK53, NEMOΔTBD, restored recruitment of TANK, NAP1, TBK1 and IKKε to complex-I (Figure 6i). Interestingly, IKKα/β–deficiency or inhibition as well as TAK1 inhibition reduced TNF-induced TBK1 activation, and the additional treatment with MRT further dampened it (Supplementary Figure 6e-g). Thus, NEMO recruitment to complex-I which is mostly, but not exclusively, M1-ubiquitin-dependent34, 36, 54 recruits TANK and NAP1 which in turn bring TBK1/IKKε and TBK1, respectively, to the TNFR1-SC. Their activation is partly IKKα/β- and partly auto-phosphorylation-dependent.
Together, these results show that NEMO mediates recruitment of both, canonical and non-canonical IKKs to complex-I which all provide essential checkpoints on RIPK1, preventing untoward TNF-induced death.
Discussion
A major focus of current research on TNF-induced cell death is placed on the regulation of RIPK1, the central kinase in this process. Various post-translational modifications (PTMs) of RIPK1 are at the core of this regulation50, 55–58.
Currently, RIPK1 is thought to be kept in check in the cytosol under unstimulated conditions59. Upon TNF-stimulation, RIPK1 is recruited to complex-I13 where it is rapidly post-translationally modified, including by cIAP1/2-mediated K63/K11/K48-linked ubiquitination and LUBAC-catalysed M1-ubiquitination. M1-ubiquitin promotes complex-I recruitment of NEMO together with its associated kinases, IKKα/β. Apart from activating NF-kB, IKKα/β inactivate RIPK1 by phosphorylation, preventing its translocation to complex-II. Recently, the existence of an additional cell death checkpoint involving the p38 MAPK target MK2 was described18–20.
Whilst TBK1 and IKKε were shown to be activated upon TNF-stimulation45, 52, their function in TNF signalling remained enigmatic. Here we identify a NEMO-dependent checkpoint that controls TNF-induced cell death by TBK1/IKKε-mediated phosphorylation of RIPK1 which is largely dependent on M1-ubiquitination and functionally independent from known cell death checkpoints (Supplementary figure 6h). It was recently proposed that TBK1-mediated phosphorylation of RIPK1 on T189 impairs its substrate-binding capacity60. Our results, however, show that TBK1 and IKKε phosphorylate RIPK1 on multiple residues. It therefore appears that the TBK1/IKKε-mediated regulation of RIPK1 is more complex. Further investigation will be required to define the contribution of the different TBK1/IKKε-mediated phosphorylation events to keeping RIPK1 in check.
Interestingly, TBK1-deficient mice are embryonically lethal at E14.5 due to aberrant TNFR1-induced cell death30, 31, a phenotype similar, but not identical, to that of NEMO-deficient animals44, 61. Hence, it was initially assumed that TBK1 activates TNF-induced NF-κB-dependent gene-expression. However, preventing TBK1/IKKε activity exerts only limited effects on TNF-induced gene-expression. Instead, we discovered that, by phosphorylating RIPK1 on multiple sites within complex-I, TBK1/IKKε control a physiologically essential cell death checkpoint. Accordingly, their pharmacologic inhibition or genetic ablation results in TNF-induced cell death, both in cell lines and in vivo, as a consequence of unleashed RIPK1 activity and aberrant complex-II formation.
Intending to elucidate the mechanism how TBK1/IKKε exert their function, we made the surprising discovery that NEMO mediates TBK1/IKKε activation in complex-I. Mechanistically, NEMO enables recruitment of TANK, which brings both TBK1 and IKKε to the complex, and of NAP1, which brings in additional TBK1. These results are striking, as they demonstrate that NEMO serves an essential survival function beyond regulating IKKα/β. Intriguingly, NEMO was previously shown to exert cell-death-preventing functions independently of NF-kB17, 62. Our results provide an explanation for this observation as the combined activity of IKKα/β, TAK1/MK2 and TBK1/IKKε is required to keep RIPK1 in check. Of note, NEMO mutations are causative for a spectrum of diseases, including incontinentia pigmenti, ectodermal dysplasia and immunodeficiency, which are thought to be caused by deficiency in activating IKKα/β63. On the basis of our results, one may consider that aberrant TNF-induced cell death caused by lack of TBK1/IKKε activity may participate in disease progression in patients with certain NEMO mutations.
Our results prompt the question as to what could be the evolutionary advantage of having three distinct checkpoints, all focussed on RIPK1, a single component in the TNFR1 pathway. TNF is induced as one of the first cytokines in response to various cellular stressors64. It is tempting to speculate that this triple safe-guard mechanism might serve to ensure that, should anything go wrong – perhaps as the result of a pathogen-mediated targeted intervention65 – with any of three of the arguably most crucial signalling pathways for innate and adaptive immunity, this is sensed as early as possible during innate immune signalling. The inability to properly phosphorylate RIPK1 in response to TNF would serve this purpose, with the resulting outcome of TNF signalling, the untoward death of the cell, triggering an alternative route to inflammation.
We here identify a crucial role for TBK1/IKKε in preventing TNF-induced cell death. So far, these kinases have mainly been considered as modulators of gene-expression, mostly in IFN responses40, 41 and autophagy66, 67. However, certain pathologies have been associated with mutations or altered expression of TBK1/IKKε, including neuro-inflammatory diseases, e.g. ALS or FTD68, and various types of cancer69. Based on our results, diseases caused by NEMO deficiency should be added to this list. It shall be interesting to evaluate to which extent aberrant TNF-induced, RIPK1-mediated cell death caused by deregulated TKB1/IKKε activity participates in initiation or progression of these diseases.
Online methods
Recombinant Proteins, Cells and Cell Lines
Human full-length GST-RIPK1 was from Abnova, GST-IKKε was from Thermo Fisher and GST-TBK1 was from Sigma-Aldrich. Untagged TNF, HA-TNF and TAP-TNF were produced and purified as described in 14. Etanercept (ENBREL®) was purchased from Pfizer.
WT cancer cell lines used herein were purchased from the ATCC. Generation and reconstitution of HOIP, TANK, NAP1, OPTINEURIN, NEMO, TBK1 and IKKε, TNF knock-out cancer cell lines and generation of the different knock-out MEFs were performed as described earlier10, 14, using sgRNA sequences presented in supplemental table 6. Newly generated KO cells were validated by sequencing. A549 and HeLa cells were transduced to express a TAP-tagged form of RIPK3. Briefly, we used a C-terminal TAP-tag consisting of 2x Strep-tag II sequence followed by a PreScission cleavage site and 1x Flag-tag. Coding sequence of human RIPK3 was inserted into the retroviral MSCV vector, followed by an internal ribosome entry site (IRES) and the open reading frame of EGFP. This vector was transfected using Lipofectamine 2000 in Phoenix-AMPHO cells. One day after transfection, the medium was replaced, and viral supernatants were collected at day two and three. Viral supernatants were passed through 0.45 μm filter, added to HeLa or A549 cells at 60% confluence in the presence of polybrene at 6 μg/ml before cells were subjected to spinfection (2500 rpm, 45 min, 30°C). EGFP-positive cells were isolated using MoFlo FACS (Beckman Coulter) to more than 95% purity two days after infection. HOIP KO HeLa and A549 cells re-expressing HOIPWT, HOIPC885S, Empty pBabe or MSCV vector, as well as TBK1 KO A549 cells re-expressing TBK1 WT or TBK1 D135N were generated as described previously39. A549 NEMO KO cells were transduced with pBabe puro to reexpress NEMOWT or NEMO ΔTBD (200-250 aa deletion). Corresponding primers can be found in supplementary table 6. All cell lines were regularly tested for mycoplasma using the MycoAlert™ Mycoplasma Detection Kit (LONZA).
Inhibitors and Antibodies
The following inhibitors were used at the indicated final concentration in vitro, unless otherwise specified in the figure or figure legends: MRT-67307 (Sigma-Aldrich, 2 µM), BX-795 (Invivogen, 1 µM), TPCA-1 (Tocris Bioscience, 5 µM), PF-3644022 (Tocris Bioscience, 1 µM), 7-oxozeanol (Tocris Bioscience, 1 µM), Nec-1s (Biovision, 10 µM) zVAD-fmk (Abcam, 20 µM), cycloheximide (Sigma-Aldrich).
All antibodies used in this study are listed in supplementary table 7 including information regarding dilutions and validation.
Retroviral Transduction of Cells
Coding sequences of HOIP-WT, HOIP C885S, TBK1 WT or TBK1 D135N were inserted into the retroviral MSCV vector containing GFP as selection marker. Upon infection, cells were sorted using MoFlo FACS (Beckman Coulter).
Tandem Affinity Purification
Samples were either directly lysed in the case of TBK1-TAP-expressing or corresponding control cells or, in the case of A549 cells expressing HOIP WT or HOIP KO (7.5x108 cells each), cells were first stimulated with TAP-TNF for 15 min. Cells were subsequently solubilised in IP-lysis buffer, cleared by centrifugation (13,000 rpm, 30 min, 4°C), incubated overnight with 100 μl of anti-Flag M2-Agarose beads (Sigma), washed three times with IP-lysis buffer and proteins were eluted overnight in IP lysis buffer containing 150 μg/ml 3x Flag peptide (Sigma) and 20 U/ml PreScission Protease (GE Healthcare). Samples were subsequently subjected to a second affinity precipitation using Strep-Tactin resin (QIAGEN) overnight at 4°C and eluted with 5 mM biotin. Proteins were precipitated using 2-D Clean-Up Kit (GE Healthcare), resuspended in ice cold denaturation buffer (100 mM ammonium bicarbonate, 8M urea), reduced with 4mM dithiothreitol at 56ºC for 30 min, alkylated with 8mM iodoacetamide at 22ºC in the dark, and 4mM dithiothreitol was used to neutralize the excess of iodoacetamide. Proteins were digested first with endoproteinase Lys-C (Wako Chemicals) for 4 hours at 37ºC. Subsequently samples were diluted to 2M urea, and further digested with sequencing grade trypsin (Promega) for 15 hours at 37ºC. Samples were desalted with microspin columns filled with SEM SS18V silica (The Nest Group), eluted with 50% acetonitrile 0.1% TFA, evaporated to dryness at 30ºC, and resolubilised in 20 µL water containing 10% formic acid. 1 µL of the resulting peptidic solution was used for LC-MS analysis.
In Vitro Kinase Assays
For IKKε in vitro kinase assays 250 ng recombinant RIPK1 and 125 ng of recombinant IKKε were mixed in 30 µl kinase buffer (20 mM HEPES (pH 7.5), 10 mM MgCl2, 2 mM DTT, phosphatase inhibitor (PhosSTOP™), EDTA-free protease inhibitor cocktail, 20 µM ATP) and incubated for 30 min at 30°C. For TBK1 in vitro kinase assay equal amounts of recombinant proteins were used, i.e.250 ng of recombinant RIPK1 and TBK1. Incubation time was 2 hrs at 30°C. Kinase buffer was the same as for IKKε kinase assay but included in addition 100 mM NaCl and the ATP concentration was 100 µM. Kinase reactions were denatured by addition of 4X reducing sample buffer (200 µM DTT).
For the MS experiment, 2 µg of recombinant RIPK1 and 1 µg of recombinant IKKƐ were used, and the assay was performed as described above. Proteins were separated and purified on an SDS-PAGE gel. Respective protein bands were excised, and proteins were reduced with 5mM TCEP in 50 mM triethylammonium bicarbonate (TEAB) at 37°C for 20 min, alkylated with 10 mM chloroacetamide in 50 mM TEAB at ambient temperature for 20 min in the dark, and digested with 1:10 trypsin to protein ratio in-gel at 37°C for 4 h. Protein gel bands were redigested to extract any remaining peptides. Samples were evaporated to dryness at 30°C and resolubilised in 0.1% formic acid.
Mass spectrometry
nLC-MS/MS was performed on a Q Exactive Orbitrap Plus interfaced to a NANOSPRAY FLEX ion source and coupled to an Easy-nLC 1000 (Thermo Scientific). Peptides were separated on a 24 cm fused silica emitter, 75 µm diameter, packed in-house with Reprosil-Pur 200 C18-AQ, 2.4 µm resin (Dr. Maisch) using a linear gradient from 5% to 30% Acetonitrile/ 0.1% Formic acid over 10min (for TBK1 AP-MS) or 30 min (for TNFR-SC AP-MS and kinase assay), at a flow rate of 250 nL/min. Precursor ions were measured in a data-dependent mode in the orbitrap analyser at a resolution of 70,000 and a target value of 3e6 ions. The ten most intense ions from each MS1 scan were isolated, fragmented in the HCD cell, and measured in the orbitrap at a resolution of 17,500. The proteomic raw data has been deposited on the ProteomeXchange Consortium via the PRIDE70 partner repository with the dataset identifier PXD008497 (TNFR1-SC analysis), PXD010777 (TBK1 analysis), and PXD008518 (RIPK1 kinase assay). Analysis of these data is provided in Supplementary tables 2, 4 and 5.
Protein and phosphosite identification
Raw data were analysed with MaxQuant version 1.5.2.8 where they were searched against the human UniProt database (http://www.uniprot.org/, downloaded 22/10/2015 (for TNFR-SC AP-MS) and 05/10/2017 (for TBK1 AP-MS and kinase assay) using default settings. Carbamidomethylation of cysteines was set as fixed modification, and oxidation of methionines and acetylation at N-termini were set as variable modifications. Phosphorylation (STY) was set as a variable modification for the kinase assay. Enzyme specificity was set to trypsin with maximally 2 missed cleavages allowed. To ensure high confidence identifications, PSMs, peptides, and proteins were filtered at a less than 1% false discovery rate (FDR). Proteins identified with a single peptide were filtered out. Label-free quantification in MaxQuant was used to quantify the AP-MS data.
Processing of TNFR1-SC AP-MS data
Proteins quantified in unstimulated controls were regarded as contaminants and filtered out. Proteins detected in more than 15 experiments in the Contaminant Repository for Affinity Purification Mass Spectrometry Data (CRAPome) were filtered out. Proteins not present in the TNF-stimulated parental cells were filtered out. A protein-protein interaction network was generated in the STRING protein-protein interaction database using known interactions from curated databases and experimentally determined at default settings. Proteins not connected in the interaction network in were filtered out.
Processing of TBK1 AP-MS data
Proteins quantified in TBK1 knock out cells were filtered out. Proteins detected in more than 15 experiments in the Contaminant Repository for Affinity Purification Mass Spectrometry Data (CRAPome) were filtered out. A protein-protein interaction network was generated in the STRING protein-protein interaction database using known interactions from curated databases and experimentally determined at default settings. Proteins not connected in the interaction network in were filtered out.
Processing of RIPK1 kinase assay data
Three raw files for each sample type (50% of 1st digestion, 10% of second digestion, 90% of second digestion) were searched and grouped in MaxQuant to produce one output file. Non-RIPK1 phosphosites were filtered out from the ‘phosphoSTY.txt ’ results file. Phosphorylation profiles of RIPK1 with and without IKKƐ were compared. Phosphosites detected in RIPK1+IKKƐ that were absent from RIPK1 alone were taken forward. Phosphosite localisation was carried out using the ‘Phospho (STY) Probabilities’ column. Serines, threonines or tyrosines with probabilities: <0.1 were considered not likely to be phosphorylated, ≥0.1 and <0.75 were considered to be possible phosphosites, and ≥0.75 were considered to be likely phosphosites.
Cell Activation and Immunoprecipitation
For TNFR1-SC preparation, cells were washed with PBS, resuspended in serum-free medium and stimulated with FLAG- or TAP-TNF for the indicated times. Cells were lysed in IP-lysis buffer (30 mM Tris-HCl, pH 7.4, 120 mM NaCl, 2 mM EDTA, 2 mM KCl, 10% Glycerol, 1% Triton X-100, 50 mM NaF, 5 mM Na3VO4, 1x COMPLETE protease-inhibitor cocktail (Roche)) at 4°C for 1 hour. FLAG-TNF or TAP-TNF (500ng) was added to the lysates of non-stimulated control samples. Subsequently, the lysates were centrifuged at 13,300 rpm for 20 min and the TNFR1-SC was immunoprecipitated using M2-Agarose beads (Sigma) overnight at 4°C. The following day, the beads were washed three times with 1 ml IP-lysis buffer and proteins were eluted by boiling in reducing sample buffer. Samples were analyzed by western blotting. For isolation of retrovirally expressed TAP-tagged proteins, cells were stimulated with untagged TNF as indicated and subjected to immunoprecipitation with M2-Agarose beads. Immunoprecipitation with specific antibodies for RIPK1 (BD, 610459) or FADD (Santa Cruz, H-181) was performed by antibody coupling to protein A/G-Agarose beads (Santa Cruz) for 3 hours at room temperature. De-ubiquitination and/or de-phosphorylation was performed on some immunoprecipitates as indicated in the figure legends. For de-ubiquitination beads were resuspended in DUB-buffer (50 mM HEPES, pH7.6, 150 mM NaCl, 5 mM DTT) and 1 µM of the recombinant deubiquitinase USP-2 (Boston Biochem) was added. The assay was performed at 30°C for 1 hour. For samples in which additional removal of phosphorylation was carried out, 400 U Lambda-phosphatase was added where indicated and reactions were further incubated at 37°C for 30 min.
SDS-PAGE, Western blot
Proteins were separated using 4 – 15% Mini- or Midi-PROTEAN®-TGX™-gels (BioRad) with Tris/glycine/SDS running buffer. Proteins were transferred on Mini- or Midi- 0.2 µm nitrocellulose membranes (BioRad transfer packs) using the Trans-Blot® Turbo™ Transfer System from BioRad. Proteins were detected with antibodies as indicated.
Isolation of Bone Marrow Derived Macrophages (BMDMs)
For preparation of BMDMs, 6-12 weeks old mice were sacrificed. Hind legs were removed and bones were separated from muscle tissue. Femur and tibia were opened on each site and bone marrow was flushed out using a 25-gauge needle and syringe. Cells were then resuspended in RPMI medium containing 10%FCS, 1% penicillin/streptomycin (Invitrogen) and 10% conditioned medium from L929 cells and passed through a cell strainer. Subsequently, cells were plated in a 12-or 24 well plate. The conditioned medium was replaced every two days and cells were incubated for seven days before the experiment.
RNA Sequencing Analysis
A549 WT cells were pre-treated with either vehicle (DMSO), MRT or TPCA-1 followed by TNF-stimulation (200 ng/mL) for either 0h, 1h or 4hrs. Triplicates for each stimulation time and type of treatment were performed. Cells were then lysed and their total RNA extracted using the RNeasy minikit (Qiagen, 74104) according to the manufacturer’s instruction. To generate the library, samples were processed using the KAPA mRNA HyperPrep Kit (p/n KK8580) according to the manufacturer’s instructions. Briefly, mRNA was isolated from total RNA using Oligo dT beads to pull down poly-adenylated transcripts. The purified mRNA was fragmented using chemical hydrolysis (heat and divalent metal cation) and primed with random hexamers. Strand-specific first strand cDNA was generated using Reverse Transcriptase in the presence of Actinomycin D. The second cDNA strand was synthesised using dUTP in place of dTTP, to mark the second strand. The resultant cDNA is then “A-tailed” at the 3’ end to prevent self-ligation and adapter dimerisation. Truncated adaptors, containing a T overhang are ligated to the A-Tailed cDNA. Successfully ligated cDNA molecules were then enriched with limited cycle PCR. All pipetting steps and incubations are performed by automation on the Hamilton StarLet liquid handler, with the exception of the high temperature fragmentation and the limited cycle PCR. Libraries to be multiplexed in the same run are pooled in equimolar quantities, calculated from Qubit and Bioanalyser fragment analysis. Samples were sequenced on the NextSeq 500 instrument (Illumina, San Diego, US) using a either a 43bp or 81bp paired end run. Run data were demultiplexed and converted to fastq files using Illumina’s bcl2fastq Conversion Software v2.19. Next, the expression of Illumina paired RNA-Seq transcript counts was quantified using kallisto software and a GRCh38 transcript model. The data was imported to the R statistical environment and summarised at the gene level (i.e transcript counts summed) using tximport. Statisical transformations (e.g the rlog used in Fig 2c and supplementary figure 2c) and analysis of differential expression performed with DESeq2. Multiple testing adjustments of differential expression utilised the Benjamini Hochberg and IHW adjustments. Transcripts which were significantly changed between 0 and 1 hr of TNF-stimulation in each group (ctr, MRT-treated and TPCA-1-treated) were illustrated using BioVenn software.
The RNA-Seq raw dataset generated during the current study is available in the SRA repository and can be accessed by using the following BioProject accession: PRJNA422567 or SRA accession: SRP126844 (https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP126844). Specific analysis of transcripts significantly changed at 1 hour, corresponding to Figure 2d is provided in Supplementary Table 3.
Cell Death Analysis
Cells were seeded the day before the experiment at 90 000 cells per well of a 24 well plate or 150 000 cells per well of a 12 well plate. The following day cells were pretreated with the indicated inhibitors and treated with recombinant human TNFα (50 ng/ml-500 ng/ml) in the presence of 5 µM SYTOX® Green (Thermofisher). Dead cells were imaged in real-time for the indicated duration of time in either 1 hr or 2 hrs intervals via fluorescence signals using the IncuCyte FLR. Percentage of cell death was calculated counting the number of dead cells manually from a representative recorded image recorded at the last time-point for each sample using ImageJ.
CellTiter-Glo® Luminescent Cell Viability Assay
Cells were seeded the day before the experiment with either 10 000 cells/well of a 96 well plate in triplicates or 90 000 cells/well of a 24 well plate. The next day cells were pre-treated with the indicated inhibitors, followed by treatment with recombinant human TNF (50 ng/ml for L929 and 500 ng/ml for MEFS, or the indicated dose). Cells were subsequently lysed and treated according to the manufacturer’s manual. Luminescence was measured using Mithras LB 940 Multimode Microplate Reader and viability was calculated via normalisation to untreated samples.
2D Gel Electrophoresis
TNFR1-SC was eluted from M2-Agarose beads in 300µl lysis buffer containing 5U/ml PreScission (GE Healthcare Life Sciences) and 250mg/ml 3XFLAG-peptide (SIGMA) for 12 hrs at 4 °C. A second elution step was carried out for 6 hrs and both elution volumes were pooled. Eluted TNFR1-SC was then prepared for 2D electrophoresis by using 2-D Clean-Up Kit (GE Healthcare Life Sciences). The pellet from clean-up step was resuspended in 125 µl 2D rehydration solution (7 M urea, 2 M thiourea, 4% CHAPS, 0.5% Carrier ampholyte (IPG buffer of respective pH), 20 mM DTT, 0.002% Bromophenol Blue), before being loaded by cup loading onto either pH 3–10NL or pH 4–7 rehydrated 7cm Immobiline Drystrip (GE Healthcare) and run on Ettan IPGphor 3 system using manufacturer’s recommended settings. The Immobiline strip was then washed with equilibration buffer (GE Healthcare) containing 65 mM DTT and 135 mM Iodoacetamide for 15 min each. Equilibrated gel strips were then loaded and run on NuPAGE™ 4-12% Bis-Tris ZOOM™ Protein Gels, followed by western blotting.
Mice injections and monitoring for TNFα-induced shock
Six- to eight-week old female C57BL/6N mice received each two intraperitoneal (i.p.) injections (total volume per mouse was 300 µL) at 15 minute intervals of either vehicle, 120 µg Nec-1s, 300 µg MRT or a combination thereof, followed by one intravenous (i.v.) injection (total volume per mouse was 150 µl) of either vehicle or 5 µg murine TNF. Inhibitors and TNF were reconstituted and diluted in endotoxin-free PBS. Animals were under permanent observation. Six to ten mice were used per group, as specifically indicated for each experiment. Approval of the study protocol used for animal experiments by the local authorities for the use of the TNF-shock model was obtained by the German Approval of Animal Act committee, application No V244-7224.121-4. All in vivo experiments were performed according to institutional, national, and European ethical animal regulations (Protection of Animals Act).
Code availability
For RNAseq analysis, the data was imported to the R statistical environment and summarised at the gene level (i.e transcript counts summed) using tximport. Statisical transformations (e.g the rlog used in Fig 2c and supplementary figure 2c) and analysis of differential expression performed with DESeq2. Multiple testing adjustments of differential expression utilised the Benjamini Hochberg and IHW adjustments. The corresponding computational code for analysis is available upon request directly with the corresponding author.
Statistics and reproducibility
For the vast majority of the western blot analyses, results shown are representative of at least two independent experiments, as indicated in each legend. All statistical analyses were performed with GraphPad Prism V6 software (Graphpad). Cell death data are presented as RFU mean of technical replicates of Sytox® Green positive cells for one representative experiment out of three independent experiments, as specified in the legends. For loss of viability data, values are expressed as mean percentage of loss of viability +/- SD for 3 to 4 independent experiments as specified for each panel in the legends. Survival curves of the in vivo experiment were compared using log-rank Mantel-Cox test. Significance between the samples is indicated in the figure. For the RNA seq experiments, statistical analyses are specified in the “RNA sequencing analysis” section.
Supplementary Material
Acknowledgements
We thank all members of the Walczak group for useful technical advice and fruitful scientific discussions and Alexis Betrancourt and Justyna Kadluczka for excellent technical support. We thank Mathieu Bertrand for IKKα/β KO MEFs, Manolis Pasparakis for NEMO KO MEFs and John Bertin for RIPK1KD MEFs. We thank Tony Brooks from the ICH Genetics & Genomic Medicine Programme (London, UK) for RNA-Seq services. This work was supported by a Wellcome Trust Investigator Award (096831/Z/11/Z), an ERC Advanced Grant (294880) and a Cancer Research UK programme grant (A17341) awarded to H.W., a Czech Science Foundation grant (17-27355Y) awarded to P.D. and a BBSRC CASE studentship (BB/J013129/1) awarded to M.R. This work also received support from a CRUK–UCL Centre grant (C416/A25145), the CRUK Cancer Immunotherapy Network Accelerator (CITA) Award (C33499/A20265) and the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
Footnotes
Author contributions
HW conceived the project. E.L., P.D., E.R., M.R., S.K, A.v.M and A.L. designed and performed experiments and analysed the data. S.S., A.B., and K.W. performed the mass spectrometry experiments and analysed the obtained data. S.S. supervised the mass spectrometric experiments. H.D. and A.C. performed experiments. D.M. generated essential tools for the study. S.H. analysed the RNA-Seq data. E.L., E.R., P.D. and H.W. wrote the manuscript.
Competing interests
The authors declare that they have no financial and non-financial competing interests.
Conflict of interest
H.W. is co-founder and shareholder of Apogenix AG. Otherwise the authors declare that they have no conflict of interest.
Data availability
The raw data for RNA-Seq analysis (Figure 2, supplementary Figure 2c, and supplementary table 3) and the proteomic raw data (Figure 1a, Figure 5f and supplementary tables 2, 4 and 5) are available as described below:
The RNA-Seq raw dataset generated during the current study are available in the SRA repository and can be accessed by using the following BioProject accession: PRJNA422567 or SRA accession: SRP126844 (https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP126844).
The proteomic raw data has been deposited on the ProteomeXchange Consortium via the PRIDE70 partner repository with the dataset identifier PXD008497 (TNFR1-SC analysis), PXD010777 (TBK1 analysis), and PXD008518 (RIPK1 kinase assay).
Source data for the graphs of all other experiments in this study are available in Supplementary table1 and unprocessed scans for Western blot are displayed in Supplementary figure 7. Publicly available tools have been used for RNAseq analysis as specified in the online methods and corresponding computational code is available upon request directly with the authors.
References
- 1.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 2.Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nature reviews Rheumatology. 2016;12:49–62. doi: 10.1038/nrrheum.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nature reviews Immunology. 2015;15:362–374. doi: 10.1038/nri3834. [DOI] [PubMed] [Google Scholar]
- 4.Hrdinka M, Gyrd-Hansen M. The Met1-Linked Ubiquitin Machinery: Emerging Themes of (De)regulation. Mol Cell. 2017;68:265–280. doi: 10.1016/j.molcel.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 5.Shimizu Y, Taraborrelli L, Walczak H. Linear ubiquitination in immunity. Immunological reviews. 2015;266:190–207. doi: 10.1111/imr.12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirisako T, et al. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006;25:4877–4887. doi: 10.1038/sj.emboj.7601360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ikeda F, et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-kappaB activity and apoptosis. Nature. 2011;471:637–641. doi: 10.1038/nature09814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerlach B, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471:591–596. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- 9.Tokunaga F, et al. SHARPIN is a component of the NF-kappaB-activating linear ubiquitin chain assembly complex. Nature. 2011;471:633–636. doi: 10.1038/nature09815. [DOI] [PubMed] [Google Scholar]
- 10.Peltzer N, et al. HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell reports. 2014;9:153–165. doi: 10.1016/j.celrep.2014.08.066. [DOI] [PubMed] [Google Scholar]
- 11.Emmerich CH, et al. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc Natl Acad Sci U S A. 2013;110:15247–15252. doi: 10.1073/pnas.1314715110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peltzer N, et al. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature. 2018;557:112–117. doi: 10.1038/s41586-018-0064-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haas TL, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. 2009;36:831–844. doi: 10.1016/j.molcel.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 14.Draber P, et al. LUBAC-Recruited CYLD and A20 Regulate Gene Activation and Cell Death by Exerting Opposing Effects on Linear Ubiquitin in Signaling Complexes. Cell reports. 2015;13:2258–2272. doi: 10.1016/j.celrep.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kupka S, Reichert M, Draber P, Walczak H. Formation and removal of poly-ubiquitin chains in the regulation of tumor necrosis factor-induced gene activation and cell death. FEBS J. 2016;283:2626–2639. doi: 10.1111/febs.13644. [DOI] [PubMed] [Google Scholar]
- 16.Rahighi S, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell. 2009;136:1098–1109. doi: 10.1016/j.cell.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Dondelinger Y, et al. NF-kappaB-Independent Role of IKKalpha/IKKbeta in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol Cell. 2015;60:63–76. doi: 10.1016/j.molcel.2015.07.032. [DOI] [PubMed] [Google Scholar]
- 18.Menon MB, et al. p38MAPK/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nature cell biology. 2017;19:1248–1259. doi: 10.1038/ncb3614. [DOI] [PubMed] [Google Scholar]
- 19.Jaco I, et al. MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Mol Cell. 2017 doi: 10.1016/j.molcel.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dondelinger Y, et al. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nature cell biology. 2017;19:1237–1247. doi: 10.1038/ncb3608. [DOI] [PubMed] [Google Scholar]
- 21.Kotlyarov A, et al. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nature cell biology. 1999;1:94–97. doi: 10.1038/10061. [DOI] [PubMed] [Google Scholar]
- 22.Kanayama A, et al. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell. 2004;15:535–548. doi: 10.1016/j.molcel.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 23.Wang C, et al. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 24.Adhikari A, Xu M, Chen ZJ. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene. 2007;26:3214–3226. doi: 10.1038/sj.onc.1210413. [DOI] [PubMed] [Google Scholar]
- 25.Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected] Nature cell biology. 2006;8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- 26.Salmeron A, et al. Direct phosphorylation of NF-kappaB1 p105 by the IkappaB kinase complex on serine 927 is essential for signal-induced p105 proteolysis. The Journal of biological chemistry. 2001;276:22215–22222. doi: 10.1074/jbc.M101754200. [DOI] [PubMed] [Google Scholar]
- 27.Tojima Y, et al. NAK is an IkappaB kinase-activating kinase. Nature. 2000;404:778–782. doi: 10.1038/35008109. [DOI] [PubMed] [Google Scholar]
- 28.Peters RT, Liao SM, Maniatis T. IKKepsilon is part of a novel PMA-inducible IkappaB kinase complex. Mol Cell. 2000;5:513–522. doi: 10.1016/s1097-2765(00)80445-1. [DOI] [PubMed] [Google Scholar]
- 29.Helgason E, Phung QT, Dueber EC. Recent insights into the complexity of Tank-binding kinase 1 signaling networks: the emerging role of cellular localization in the activation and substrate specificity of TBK1. FEBS Lett. 2013;587:1230–1237. doi: 10.1016/j.febslet.2013.01.059. [DOI] [PubMed] [Google Scholar]
- 30.Hemmi H, et al. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. The Journal of experimental medicine. 2004;199:1641–1650. doi: 10.1084/jem.20040520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. The Journal of experimental medicine. 2004;199:1651–1658. doi: 10.1084/jem.20040528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kupka S, et al. SPATA2-Mediated Binding of CYLD to HOIP Enables CYLD Recruitment to Signaling Complexes. Cell reports. 2016;16:2271–2280. doi: 10.1016/j.celrep.2016.07.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Komander D, et al. Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO reports. 2009;10:466–473. doi: 10.1038/embor.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laplantine E, et al. NEMO specifically recognizes K63-linked poly-ubiquitin chains through a new bipartite ubiquitin-binding domain. EMBO J. 2009;28:2885–2895. doi: 10.1038/emboj.2009.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 36.Hadian K, et al. NF-kappaB essential modulator (NEMO) interaction with linear and lys-63 ubiquitin chains contributes to NF-kappaB activation. The Journal of biological chemistry. 2011;286:26107–26117. doi: 10.1074/jbc.M111.233163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chau TL, et al. Are the IKKs and IKK-related kinases TBK1 and IKK-epsilon similarly activated? Trends in biochemical sciences. 2008;33:171–180. doi: 10.1016/j.tibs.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 38.Kishore N, et al. IKK-i and TBK-1 are enzymatically distinct from the homologous enzyme IKK-2: comparative analysis of recombinant human IKK-i, TBK-1, and IKK-2. The Journal of biological chemistry. 2002;277:13840–13847. doi: 10.1074/jbc.M110474200. [DOI] [PubMed] [Google Scholar]
- 39.Lafont E, et al. The linear ubiquitin chain assembly complex regulates TRAIL-induced gene activation and cell death. Embo j. 2017 doi: 10.15252/embj.201695699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fitzgerald KA, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nature immunology. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 41.Sharma S, et al. Triggering the interferon antiviral response through an IKK-related pathway. Science (New York, N.Y.) 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 42.Clement JF, Meloche S, Servant MJ. The IKK-related kinases: from innate immunity to oncogenesis. Cell research. 2008;18:889–899. doi: 10.1038/cr.2008.273. [DOI] [PubMed] [Google Scholar]
- 43.Clark K, et al. Novel cross-talk within the IKK family controls innate immunity. The Biochemical journal. 2011;434:93–104. doi: 10.1042/BJ20101701. [DOI] [PubMed] [Google Scholar]
- 44.Schmidt-Supprian M, et al. NEMO/IKK gamma-deficient mice model incontinentia pigmenti. Mol Cell. 2000;5:981–992. doi: 10.1016/s1097-2765(00)80263-4. [DOI] [PubMed] [Google Scholar]
- 45.Clark K, Plater L, Peggie M, Cohen P. Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IkappaB kinase epsilon: a distinct upstream kinase mediates Ser-172 phosphorylation and activation. The Journal of biological chemistry. 2009;284:14136–14146. doi: 10.1074/jbc.M109.000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feng S, et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cellular signalling. 2007;19:2056–2067. doi: 10.1016/j.cellsig.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 47.Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes & development. 1999;13:2514–2526. doi: 10.1101/gad.13.19.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O'Donnell MA, et al. Caspase 8 inhibits programmed necrosis by processing CYLD. Nature cell biology. 2011;13:1437–1442. doi: 10.1038/ncb2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duprez L, et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35:908–918. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 50.Polykratis A, et al. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. Journal of immunology (Baltimore, Md. : 1950) 2014;193:1539–1543. doi: 10.4049/jimmunol.1400590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pomerantz JL, Baltimore D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. Embo j. 1999;18:6694–6704. doi: 10.1093/emboj/18.23.6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wagner SA, Satpathy S, Beli P, Choudhary C. SPATA2 links CYLD to the TNF-α receptor signaling complex and modulates the receptor signaling outcomes. EMBO J. 2016;35:1868–1884. doi: 10.15252/embj.201694300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chariot A, et al. Association of the adaptor TANK with the I kappa B kinase (IKK) regulator NEMO connects IKK complexes with IKK epsilon and TBK1 kinases. The Journal of biological chemistry. 2002;277:37029–37036. doi: 10.1074/jbc.M205069200. [DOI] [PubMed] [Google Scholar]
- 54.Dynek JN, et al. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. Embo j. 2010;29:4198–4209. doi: 10.1038/emboj.2010.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 56.Peltzer N, Darding M, Walczak H. Holding RIPK1 on the Ubiquitin Leash in TNFR1 Signaling. Trends Cell Biol. 2016;26:445–461. doi: 10.1016/j.tcb.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 57.Annibaldi A, Meier P. Checkpoints in TNF-Induced Cell Death: Implications in Inflammation and Cancer. Trends in molecular medicine. 2017 doi: 10.1016/j.molmed.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 58.Ting AT, Bertrand MJ. More to Life than NF-kappaB in TNFR1 Signaling. Trends in immunology. 2016;37:535–545. doi: 10.1016/j.it.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vanden Berghe T, Kalai M, van Loo G, Declercq W, Vandenabeele P. Disruption of HSP90 function reverts tumor necrosis factor-induced necrosis to apoptosis. The Journal of biological chemistry. 2003;278:5622–5629. doi: 10.1074/jbc.M208925200. [DOI] [PubMed] [Google Scholar]
- 60.Xu D, et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell. 2018;174:1477–1491.e1419. doi: 10.1016/j.cell.2018.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rudolph D, et al. Severe liver degeneration and lack of NF-kappaB activation in NEMO/IKKgamma-deficient mice. Genes & development. 2000;14:854–862. [PMC free article] [PubMed] [Google Scholar]
- 62.Legarda-Addison D, Hase H, O'Donnell MA, Ting AT. NEMO/IKKgamma regulates an early NF-kappaB-independent cell-death checkpoint during TNF signaling. Cell death and differentiation. 2009;16:1279–1288. doi: 10.1038/cdd.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maubach G, Schmadicke AC, Naumann M. NEMO Links Nuclear Factor-kappaB to Human Diseases. Trends in molecular medicine. 2017;23:1138–1155. doi: 10.1016/j.molmed.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 64.Udalova I, Monaco C, Nanchahal J, Feldmann M. Anti-TNF Therapy. Microbiology spectrum. 2016;4 doi: 10.1128/microbiolspec.MCHD-0022-2015. [DOI] [PubMed] [Google Scholar]
- 65.Zhao W. Negative regulation of TBK1-mediated antiviral immunity. FEBS Lett. 2013;587:542–548. doi: 10.1016/j.febslet.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nature immunology. 2009;10:1215–1221. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 67.Pilli M, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012;37:223–234. doi: 10.1016/j.immuni.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ahmad L, Zhang SY, Casanova JL, Sancho-Shimizu V. Human TBK1: A Gatekeeper of Neuroinflammation. Trends in molecular medicine. 2016;22:511–527. doi: 10.1016/j.molmed.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Verhelst K, Verstrepen L, Carpentier I, Beyaert R. IkappaB kinase epsilon (IKKepsilon): a therapeutic target in inflammation and cancer. Biochemical pharmacology. 2013;85:873–880. doi: 10.1016/j.bcp.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vizcaino JA, et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44:11033. doi: 10.1093/nar/gkw880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.