

Abstract
Deciphering how an odorant activates an odorant receptor (OR) and how changes in specific OR residues affect its responsiveness are central to understanding how we smell. A joint approach combining site-directed mutagenesis and functional assays with computational modeling has been used to explore the signaling mechanics of OR7D4. In this OR, a genetic polymorphism affects our perception of androstenone. A total of 0.12 ms molecular simulations predicted that, similarly to observations from other G protein-coupled receptors with known experimental structures, an activation pathway connects the ligand and G protein binding site. The 3D model activation mechanism correlates with in vitro data and notably predicts that the OR7D4 WM variant does not activate. Upon activation, an OR-specific sequence motif is the convergence point of the mechanism. Our study suggests that robust homology modeling can serve as a powerful tool to capture OR dynamics related to smell perception.
Keywords: olfactory receptor, functional assays, molecular dynamics, GPCR, activation
Graphical Abstract
The selective activation dynamics of the odorant receptor 7D4 is captured by computational modeling. The role of a specific sequence motif in helix six is described.
Our sense of smell is triggered by the activation of odorant receptors (ORs) expressed on the surface of olfactory sensory neurons. Smelling relies on a so-called combinatorial code of OR activations.[1] Our repertoire of ~400 ORs endow us with spectacular discriminatory power. ORs represent more than 3% of our protein coding genes and belong to the class A G protein-coupled receptor (GPCR) family. Structurally, class A GPCRs are made up of seven transmembrane helices (TMs), named TM1 to TM7, which form a bundle within the cell membrane. Upon activation, an allosteric signal is transmitted from the ligand to the G protein binding site over a distance of more than 20 Å (~5 helix turns).[2] The intracellular part of TM6 then tilts outward with respect to its position in the inactive state.[3]
Mammalian OR sequences show a highly conserved and OR-specific RxKAFSTCxSH motif in the intracellular part of TM6, suggesting a crucial role in the control of OR activation. In non-olfactory class A GPCRs, only the second lysine is conserved.[4] Odorant receptors activation dynamics still remains to be uncovered but a significant body of work has focused on the molecular details of OR activation, especially concerning the interaction between OR and ligands.[5] The strength of molecular modeling approaches on GPCRs was demonstrated by a groundbreaking study based on homology modeling and virtual screening, which deorphanized GPR68 and GPR65.[6] By applying this to ORs, we recently demonstrated how certain mutations alter receptor function or dynamics.[4, 7]
The human OR7D4, which is narrowly tuned to detect the testosterone metabolites androstadienone and androstenone, is a spectacular example of how natural sequence variations can affect activation and subsequent odor perception. People with two copies of the most common genetic variant (OR7D4-RT or wt) tend to describe the androstenone smell as sickening, putrid, foul or urine-like. However, people who possess the unresponsive R88W/T133M variant (OR7D4-WM) tend to report the smell to be more pleasant.[8] This polymorphism correlates with a differential preference for pork containing varying amounts of androstenone,[9] which has a consequent effect on the percentage of castrated pigs in European countries.[10]
To evaluate how an OR sequence affects its activation dynamics we have used a joint approach combining functional assays with molecular dynamics (MD) simulations. Mouse and human genomes account for ~1500 ORs, making the OR family suited to build robust homology models. In this study, a 3D model of OR7D4 was built following a previously published protocol.[4]
The sequence alignment was performed by comparing residue conservation between the ORs and crystallized class A GPCRs. Manual adjustments were made to recover constraints imposed by the 141 in vitro data points that were tested by site-directed mutagenesis in various ORs, including OR7D4.[4, 7, 11] In summary, they represent 96 distinct positions within the sequence of OR7D4 (31% of the 312 residues, see Supporting Information). They cover the seven TMs as well as the orthosteric binding site and unambiguously confirmed our sequence alignment.
More specifically, in this study we performed 27 site-directed mutations on 16 sites to assess the final 3D model of OR7D4. Figure 1 highlights the positions of conserved motifs and alanine-scanning functional assays. Nine mutations interrogated the binding cavity, motifs putatively involved in the transmission of the signal, and segments in contact with the G protein. In mammalian ORs, higher sequence-specific conservation suggests the existence of points of contact on the faces of the TM3, TM6 and TM7 helices. Our model demonstrated that most of these conserved residues are facing each other. Consistently, the responses of the corresponding mutant ORs are strongly affected (Figure 1b). The two control A116T3.44 and G253A6.49 mutant ORs (see [12] for numbering), whose side chains are not involved in any TM contact, show an unaffected response. Neither the wt nor the mutant ORs showed a luciferase response upon decenal stimulation (See Supporting Information).
Figure 1.
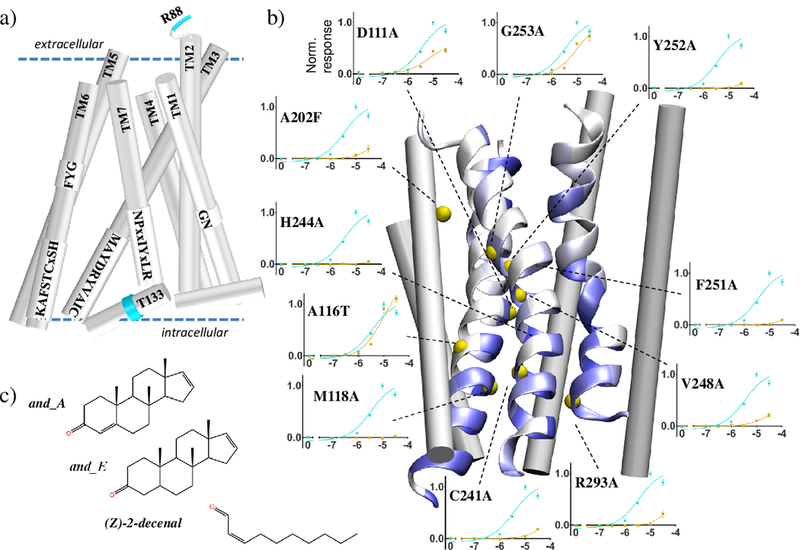
OR7D4 3D model with the loops omitted for clarity a) Location of the conserved sequence motifs as predicted by the model. Residues R88 and T133 are highlighted in cyan. b) 3D model of OR7D4 with TM3, TM6 and TM7 shown as helices with residues colored by conservation (from white (low) to blue (high)). Yellow spheres indicate the position of residues for which androstadienone dose-response curves are obtained. For each position the mutant OR (yellow curve) is compared to the wt (blue curve). Normalized responses are shown as means and s.e.m. (n= 3). Stimulations with androstenone induce receptor responses similar to that of androstadienone (SI). c) Chemical structures of androstadienone (and_A), androstenone (and_E) and Z-(2)-decenal (decenal).
And_A, And_E and decenal were docked to the apo receptor prior to microsecond timescale MD simulations. As expected, the model predicts a much larger binding energy of the receptor for the two agonists than for the control (see Supporting Information), consistent with the notion that agonists must reach a certain affinity to trigger OR activation.[13]
From a structural point of view, the binding cavity is made up of residues belonging to TM3, TM5, TM6 and TM7, which is consistent with those identified in GPCRs with a known experimental structure.[14] Considering the ORs specifically, the key residues forming the binding cavity are consistent with those identified by previous modeling approaches.[3b, 7, 15] In the OR7D4 model, the A2025.42 methyl side-chain points towards the ligand and allows for the binding of the bulky agonists. Accordingly, the A2025.42F mutant OR is unresponsive to agonists in vitro.
The model also predicts that A2797.42 points towards the binding cavity. Its mutation to a bulkier and hydrophilic residue (A→D) was reported to abolish the in vitro response of the OR upon agonist stimulation.[16]
Starting from the same initial model structure of OR7D4, three unconstrained MD simulations of ~5 µs were performed for each of these eight systems. At total, OR7D4 dynamics were investigated for ~0.12 ms and compared to MD simulations of the β2-adrenergic receptor (β2-AR) bound to an agonist (PDB id: 3SN6) or an antagonist (PDB id: 2RH1). The models provided mechanistic hypotheses that were further tested in vitro.
Interestingly, for experimentally-activated systems, back and forth movements involving TM6 and the intracellular loop 3 (ICL3) open a cleft in the intracellular domain of the receptor. When these movements are observed, the open and closed structures of OR7D4 are highly similar to those observed in the β2-AR, as shown in Figure 2. This intracellular cleft is typically used as a benchmark to discriminate between active and inactive conformations of class A GPCRs.[17] The root mean square deviation (rmsd), with respect to the crystal structure of the β2-AR in its active and inactive state, correlates with this structural feature.
Figure 2.
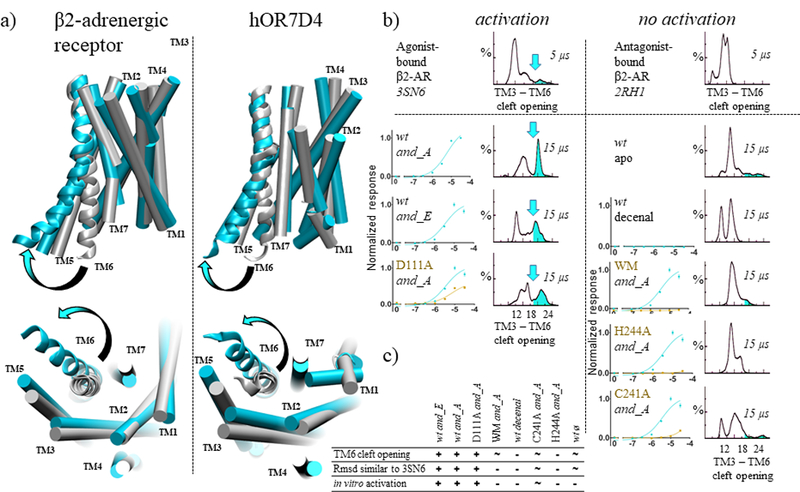
OR7D4 intracellular cleft opening is similar to that of β2-adrenergic receptor (β2-AR). (a) Comparisons between the β2-AR X-ray structure in its inactive (grey, PDB id: 2RH1) and active state (cyan, PDB id: 3SN6) and typical structures of OR7D4 sampled during the simulations. The TM domains of the receptor are shown as cylinders, except TM6 which is shown as a ribbon. Loops are omitted for clarity. The cleft opening involving the intracellular part of TM6 is indicated with an arrow. (b) in vitro activations are correlated with a cleft opening during MD simulations. The TM3-TM6 cleft opening is measured by the distance between the Cα atoms of the ionic-lock residues, (R3.50 and E6.30 in β2-AR and D3.49 and K6.30 in OR7D4). A 5 µs MD simulation of the β2-AR in its active state bound to an agonist (3SN6) samples cleft opening events, while the inactive state (2RH1, antagonist-bound) does not. Similarly, in MD simulations of three OR7D4 systems where in vitro data confirms activation, structures with an open intracellular cleft are observed. The opening is highlighted by a cyan arrow on the distance distribution. Alternatively, such a cleft is rarely, if ever, observed in the five systems where in vitro data does not report any activation. Normalized responses are shown as means and s.e.m. (n= 3). (c) Cleft opening and rmsd similarity with β2-AR active state (PDB id 3SN6) correlate with the in vitro response, represented as “+” when the feature is clearly observed, “−” when it is not. A “~” represents an intermediate observation of the considered feature.
Structures with conformations closer to the active state of the β2-AR were specifically monitored for systems where the receptor is shown to respond in vitro (Figure 2b and rmsd analysis in the Supporting Information). We have shown that this also applies to constitutively active mutants of a mouse OR, where the K2346.30 residue was crucial for the activation likelihood.[7]
The MD of agonist-bound OR7D4 compares with those performed on the β2-AR bound to an agonist. The latter captures the typical cleft opening involving TM6, while this was never observed during the simulation with an antagonist (Figure 2b, top). Agonist binding shifts the equilibrium between different receptor states from an inactive to an active-dominant one. This feature is almost absent for systems where in vitro data do not report any receptor response or where the response is considered negligible (C241A6.37, H244A6.40 and WM bound to and_A, the wt receptor in its apo form or bound to decenal, figure 2c).
In the OR7D4 agonist-bound systems, fluctuations between these β2-AR active-like and inactive-like models (Supporting Information) suggest that the activation dynamics involving TM6 are captured by the model. In all cases, a correlation between in vitro response and TM6 dynamics is demonstrated (Figure 2c). Even though the homology model is not as accurate as an X-ray structure, it nonetheless selectively captures the nature of the bound ligand, and the subtle variations within the receptor sequence that affect the equilibrium between the inactive and active state. The RT to WM changes, although far removed from the cavity or the G protein binding site, affect the dynamics of the receptor and hamper the activation signal from being translated from the binding cavity to the intracellular part of TM6.
From a mechanistic point of view, the model recovers the typical features of class A GPCRs activation (Figure 3a). Upon activation, residue Y6.48 shifts towards P5.50, consistent with data reported on the muscarinic 2 or the cannabinoid 1 receptors.[18] Also, the TM6 cleft opening is accompanied by an inward shift of Y5.58 as typically reported on the β2-AR.[19] More generally, series of complex allosteric movements propagate the activation signal through a dynamic network involving the whole protein structure, comprising the loops (see Supporting Information).
Figure 3.
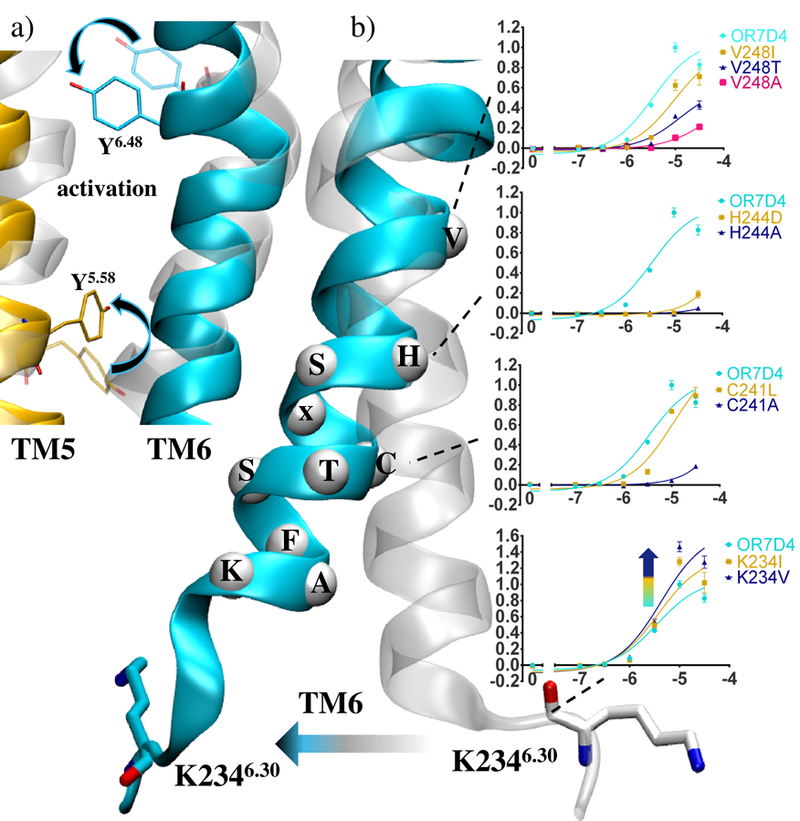
a) The mechanism of activation involves reorganisation of TM5 and TM6 residues Y6.48 and Y5.58. b) and_A dose-response curves involving the RxKAFSTCxSHTM6 motif residues. Normalized responses are shown as means with s.e.m. (n= 3). Comparison between the open (cyan) and closed (transparent) conformations of OR7D4.
A comparison between inactive and active structures revealed differential receptor dynamics involving the OR-specific RxKAFSTCxSHTM6 motif. As emphasized in Figure 3, this motif forms a rigid helical segment whose conformational change is associated with activation. Similar structural behavior is observed in the β2-AR and more generally in class A GPCRs, which show a conserved lysine (K6.30) that is involved in the canonical ionic-lock at the junction between TM6 and ICL3.[18a, 20] In luciferase assays probing OR7D4 mutants with a modified intracellular TM6, mutations introducing a hydrophobic residue at position K2346.30 are likely to prevent the formation of the ionic-lock between TM6 and TM3.[7] Consistently, the K234I or V mutant ORs are more responsive than the wt (Figure 3b).
Moving towards the extracellular end of this conserved motif, H244A and H244D or C241A mutations abolish the response to agonists, which is consistent with a conservation of 98% in human OR sequences. Away from the RxKAFSTCxSH motif, mutations are less deleterious to the receptor function. Mutations at position 2486.44 differentially modulate the responsiveness of the receptor suggesting a role in communicating the signal from the binding pocket to the convergence point represented by this TM6 motif.
In this study, a robust homology model of OR7D4 was analyzed using MD simulations of its apo form or bound form with androstenone and androstadienone, two known agonists, and also to (Z)-2-decenal as a negative control. The intriguing WM variant associated with a differential perception of boar taint was also studied together with two additional control mutants.
The model of OR7D4 is consistent with conservation analyses and in vitro functional assays on 27 mutant ORs obtained by site-directed mutagenesis at 16 different sites. In total, the dynamics of OR7D4 variants were explored for ~0.12 ms. The model more frequently explored structures showing the typical features of an active (open) state, while in vitro data assessed receptor activation.
Conversely, when considered in its apo form, bound to a non-agonist or mutated at deleterious sites (WM, C241A, H244A), the receptor was mostly modeled in a closed (inactive) state. Thus, starting from this single model of the receptor we were able to discriminate agonists of the human OR7D4 from non-agonists. This was achieved by monitoring the allosteric movement of the TM3-TM6 inter-helical cleft, the metric most representative of a GPCR preorganized for recruiting the G protein. A total of 24 independent MD simulations were run and 18 of them were consistent with in vitro data, leading to a predictive power of 75%. For each system studied, at worse, only one MD out of three led to either a false positive (apo-wt, C241A, and WM) or a false negative (And_A, And_E, and D111A). This highlights the importance of enhanced sampling through multiple MDs to prevent erroneous conclusions on the nature of the bound ligand.
Shifts between open and closed conformations involve rearrangements of the mammalian OR-specific motif within TM6, with respect to TM3. This RxKAFTCxSH motif thus plays a pivotal role in activation, consistent with its high conservation across mammalian ORs.[4] The position of this highly conserved motif emphasizes its importance in the control of both OR activation and specificity for the olfactory G protein (Golf).
In the WM variant bound to androstadienone, while mutations are far removed from the orthosteric binding site or the G protein coupling area, the receptor does not show any intracellular cleft opening. This highlights the crucial role of residues that do not belong to the binding cavity in the dynamics of GPCRs in general. Similarly, genetic analysis of “super smellers” of androstenone suggested a link with a single nucleotide polymorphism in TM2 of OR7D4 (S84N). This higher detection threshold is related to the mutant OR, which is more responsive to the agonists.[8] This emphasizes the extreme complexity of GPCR signaling mechanisms. Subtle sequence modifications affect OR activation and, in this case, can spectacularly impact smell perception and food preferences.
Supplementary Material
Acknowledgements
We thank Dr. T. Abaffy and Dr. X. Cong for critical reading of the manuscript. This work is supported by grants from the National Institute on Deafness and Other Communication Disorders, National Institute of Health Grants DC005782 and DC012095, National Science Foundation (NSF) Grants 1515801, and 1515930 (to H.M.), from Agence Nationale de la Recherche as part of NSF/NIH/ANR Collaborative Research in Computational Neuroscience and from UCA IDEX (to J.G).
Footnotes
Experimental Section
Experimental details on conservation analysis, molecular modeling and functional assays are given in Supporting information.
Contributor Information
Claire A. de March, Institute of Chemistry - Nice, UMR 7272 CNRS – Université Côte d’Azur, 06108 Nice cedex, France, Dept. of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, NC 27710, USA.
Jérémie Topin, Institute of Chemistry - Nice, UMR 7272 CNRS – Université Côte d’Azur, 06108 Nice cedex, France.
Elise Bruguera, Dept. of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, NC 27710, USA
Gleb Novikov, Institute of Chemistry - Nice, UMR 7272 CNRS – Université Côte d’Azur, 06108 Nice cedex, France
Kentaro Ikegami, Dept. of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, NC 27710, USA, Dept. of Biotechnology and Life Science, Tokyo University of Agriculture and Technology, Tokyo, Japan.
Hiroaki Matsunami, Dept. of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, NC 27710, USA.
Jérôme Golebiowski, Institute of Chemistry - Nice, UMR 7272 CNRS – Université Côte d’Azur, 06108 Nice cedex, France, Dept. of Brain & Cognitive Sciences, DGIST, 333, Techno JungAng Daero, HyeongPoong Myeon, Daegu, 711-873, Rep. of Korea.
References
- [1].a) Buck L, Axel R, Cell 1991, 65, 175–187; [DOI] [PubMed] [Google Scholar]; b) Malnic B, Godfrey PA, Buck LB, Proc Natl Acad Sci U S A 2004, 101, 2584–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rosenbaum DM, et al. , Science 2007, 318, 1266–1273. [DOI] [PubMed] [Google Scholar]
- [3].a) Gether U, Kobilka BK, J. Biol. Chem 1998, 273, 17979–17982; [DOI] [PubMed] [Google Scholar]; b) Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP, Hubbell WL, Proc Natl Acad Sci U S A 2008, 105, 7439–7444; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Manglik A, et al. , Cell 2015, 161, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].de March CA, Kim S-K, Antonczak S, Goddard WA, Golebiowski J, Protein Sci 2015, 24, 1543–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Geithe C, Protze J, Kreuchwig F, Krause G, Krautwurst D, Cell. Mol. Life Sci 2017, 74, 4209–4229; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li S, Ahmed L, Zhang R, Pan Y,Matsunami, Burger JL, Block E, Batista VS, Zhuang H, J. Am. Chem. Soc 2016; [DOI] [PubMed]; c) Wolf S, Jovancevic N, Gelis L, Pietsch S, Hatt H, Gerwert K, Sci Rep 2017, 7, 16007; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Baud O, Yuan S, Veya L, Filipek S, Vogel H, Pick H, Sci Rep 2015, 5, 14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Huang XP, et al. , Nature 2015, 527, 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].de March CA, Yu Y, Ni MJ, Adipietro KA, Matsunami H, Ma M, Golebiowski J, J. Am. Chem. Soc 2015, 137, 8611–8616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Keller A, Zhuang H, Chi Q, Vosshall LB, Matsunami H, Nature 2007, 449, 468–472. [DOI] [PubMed] [Google Scholar]
- [9].Lunde K, Egelandsdal B, Skuterud E, Mainland JD, Lea T, Hersleth M, Matsunami H, PLoS ONE 2012, 7, e35259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Blanch M, Panella-Riera N, Chevillon P, Furnols M, Gil M, Gil J, Kallas Z, Oliver M, Meat science 2012, 90, 572–578. [DOI] [PubMed] [Google Scholar]
- [11].Yu Y, de March CA, Ni MJ, Adipietro KA, Golebiowski J, Matsunami H, Ma M, Proc. Natl. Acad. Sci. USA 2015, 112, 14966–14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ballesteros JA, Weinstein H, in Methods in Neurosciences, Vol. 25 (Ed.: Stuart CS), Academic Press, 1995, pp. 366–428. [Google Scholar]
- [13].Topin J, de March CA, Charlier L, Ronin C, Antonczak S, Golebiowski J, Chem. Eur. J 2014, 20, 10227–10230. [DOI] [PubMed] [Google Scholar]
- [14].a) Venkatakrishnan AJ, et al. , Nature 2016, 536, 484–487; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Latorraca NR, Venkatakrishnan AJ, Dror RO, Chem. Rev 2016. [DOI] [PubMed]
- [15].de March CA, Kim S-K, Antonczak S, Goddard WA, Golebiowski J, Protein Science 2015, 24, 1543–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Keller A, Zhuang HY, Chi QY, Vosshall LB, Matsunami H, Nature 2007, 449, 468–U466. [DOI] [PubMed] [Google Scholar]
- [17].Deupi X, Standfuss J, Curr. Opin. Struct. Biol 2011, 21, 541–551. [DOI] [PubMed] [Google Scholar]
- [18].a) Miao Y, Nichols SE, Gasper PM, Metzger VT, McCammon JA, Proc. Natl. Acad. Sci. USA 2013, 110, 10982–10987; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hua T, et al. , Cell 2016, 167, 750–762 e714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, Xu H, Borhani DW, Shaw DE, Proc. Natl. Acad. Sci. USA 2011, 108, 18684–18689; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nygaard R, et al. , Cell 2013, 152, 532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shan J, Khelashvili G, Mondal S, Mehler EL, Weinstein H, PLoS Comput Biol 2012, 8, e1002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.