

Abstract
Pirfenidone, a pyridone compound, is an effective and novel antifibrotic agent. In this article, we describe the design, synthesis and activity evaluation of novel antifibrotic agents, 1-(substituted aryl)-5-trifluoromethyl-2(1H) pyridones modified with carbohydrate. Most of the title compounds exhibited comparable or better inhibitory activity than fluorofenidone. Notably, compound 19a demonstrated the highest cell-based inhibitory activity against NIH 3T3 (IC50 = 0.17 mM).
Keywords: antifibrotic, pirfenidone, fluorofenidone, carbohydrate modified, glucose
1. Introduction
Fibrosis of organs and tissues (e.g., heart, lungs, liver, kidneys, blood vessel and skin) is an important pathological change associated with many diseases that are major causes of human morbidity and mortality [1,2]. Generally, fibrosis can occur as a consequence of different pathological conditions, such as tissue damage, inflammatory diseases, foreign implants, and tumors. It is clear that fibrosis of different organs and tissues possesses the same feature of fibroblast accumulation and excess deposition of extracellular matrix (ECM), which leads to distorted organ architecture and function [3,4]. There are various cytokines, chemokines, and growth factors involved in the development of fibrosis [5]. In spite of the increased knowledge about the pathogenesis of fibrosis and the different mediators involved, there is still no effective treatment for fibrosis.
Pirfenidone (5-methyl-1-phenyl-2(1H)-pyridone, Figure 1, compound 1a), is an effective and novel antifibrotic agent that is potentially effective for the treatment of idiopathic pulmonary fibrosis (IPF) [6] that was launched in Japan in 2008. Pirfenidone can inhibit fibroblast proliferation and collagen synthesis, and it ameliorates bleomycin-induced and cyclophosphamide-induced lung fibrosis [7,8]. In order to improve the antifibrotic activity of pirfenidone, a series of pirfenidone derivatives (Figure 1) were synthesized [9,10,11,12]. Fluorofenidone (Figure 1, compound 1b), an analogue of pirfenidone, shows equivalent antifibrotic activity, lower toxicity and longer half-life than the parent compound pirfenidone. However, the activities of pirfenidone and fluorofenidone are relatively weak; their effective doses are 500 mg/kg for the treatment of renal fibrosis in rats and the regular dose of pirfenidone for Japanese patients with IPF is 400–600 mg three-times daily [6,13]. The metabolized products have no in vivo activity, which could be the cause of high effective doses of pirfenidone and fluorofenidone. Replacement of the methyl group with trifluoromethyl (Figure 1) could protect the derivative from metabolism, but trifluoromethyl substitution increases lipid solubility and toxicity. Therefore, it is necessary to prepare more effective antifibrotic agents with novel chemical structures and improved water solubility.
Figure 1.

Structures of Pirfenidone and analogues.
The aim of this study was to prepare antifibrotic compounds with higher water solubility. Because carbohydrates are a kind of polyhydroxy compounds with no toxicity and high water solubility, attachment of a carbohydrate moiety should increase hydrophilicity [14]. The modification with carbohydrate also influences the pharmacokinetic properties of the modified compounds. Recent advancements in molecular glycobiology have facilitated the development of effective glycodrugs [15]. A series of N-substituted 1-(4-amino-2-chlorophenyl)-5-trifluoromethyl-2(1H) pyridones were previously synthesized and tested for NIH 3T3 inhibitory activity [10]. Therefore, we selected amines 15–19 (Table 1, R = H) as parent compounds for modification with monosaccharides. Herein, we report the design, synthesis and biological evaluation of carbohydrate-modified 1-(substituted aryl)-5-trifluoromethyl-2(1H) pyridones. The structures of the final target compounds are shown in Table 1. We explored the SAR using methyl 6-deoxy-α-D-glucopyranoside, methyl 6-deoxy-α-D-mannopyranoside and methyl 6-deoxy-β-D-galactofuranoside.
Table 1.
NIH 3T3 inhibitory activity.
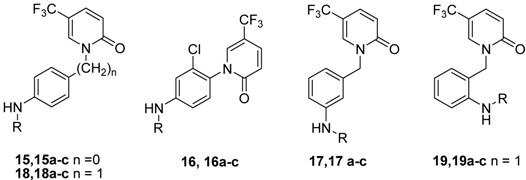 |
| Compound | R | IC50 (mM) * | Compound | R | IC50 (mM) * |
|---|---|---|---|---|---|
| 1b | -- | 4.18 | 18 | H | 1.31 |
| 15a | 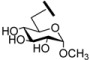 |
3.93 | 18a | 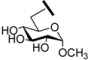 |
2.47 |
| 15b | 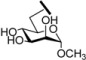 |
5.68 | 18b | 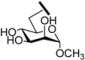 |
13.17 |
| 15c | 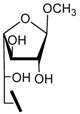 |
8.40 | 18c | 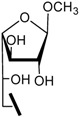 |
3.30 |
| 16 | H | 0.46 | 19 | H | 1.71 |
| 16a | 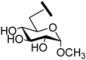 |
N.D. a | 19a | 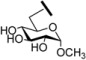 |
0.17 |
| 16b | 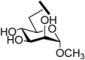 |
1.13 | 19b | 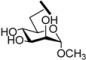 |
0.79 |
| 16c | 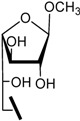 |
N.D. a | 19c | 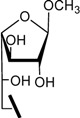 |
0.84 |
| 17a | 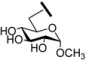 |
2.63 | |||
| 17b | 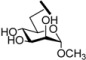 |
5.81 | |||
| 17c | 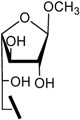 |
0.42 |
* p < 0.05; a N.D.: not determined.
2. Results and Discussion
The preparation of methyl 2,3,4-tri-O-acetyl-6-aldehydo-α-D-gluco-hexodialdo-1,5-pyranoside (8) and methyl 2,3,4-tri-O-acetyl-6-aldehydo-α-D-manno-hexodialdo-1,5-pyranoside (12) is described in Scheme 1. Methyl 2,3,4-tri-O-acetyl-6-O-trityl-α-D-glucopyranoside (6) was prepared directly from methyl α-D-glucopyranoside in two steps. Removal of trityl protection was accomplished by treatment with boron trifluoride etherate in methanol and methylene chloride to produce compound 7[16]. Subsequently selective oxidation of a primary hydroxyl group to an aldehydo group was performed at room temperature with a mild oxidation reagent, 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) and [bis(acetoxy)iodo]benzene (BAIB) [17]. Using the same method, compound 12 was obtained from methyl α-D-mannopyranoside 9. Galactose aldehyde can be prepared with a isopropylidene moiety as a protective group (Scheme 1). Isopropylidenation of D-galactose with sulfuric acid-copper sulfate as catalyst in acetone produced 1,2;3,4-di-O-isopropylidene-D-galactose (13) in 58.3% yield [18]. The conversion of compound 13 to 14 was performed with the same method used in the conversion of compound 7 to 8. The structure of compounds 7–8 and 11–14 were in accordance with their reported spectroscopic data [19,20,21].
Scheme 1.

Synthesis of compounds 8, 12 and 14.
The synthesis of 15a-19a and 15b-19b is shown in Scheme 2. Nucleophilic addition of the aldehyde 8 or 12 with amine 15–19 [10] in methanol and acetic acid followed by dehydration produced the corresponding imines, which were further reduced with sodium cyanoborohydride to give the corresponding secondary amines [22]. Amines were converted to the desired compounds 15a-19a and 15b–19b by treatment with sodium methoxide in methanol. The synthesis of 15c–19c is also described in Scheme 2. Using the same method [22], secondary amines 14a–e were prepared. Treatment of 14a–e with a solution of 0.5 M HCl/CH3OH under room temperature, caused removal of the isopropylidene groups and the formation of methyl β-D-galactofuranosides 15c–19c as the main product. Under these conditions, deprotection of 14a also gave a minor amount of the corresponding galactopyranoside (5.7%) except for 15c, but deprotection of 14b–e gave little of the corresponding galactopyranosides (<1%).
Scheme 2.

Synthesis of compounds 15a-19a, 15b-19b and 15c-19c.
Biological Assay
A cell-based fibrosis inhibition assay was performed to evaluate the inhibitory effects of the prepared compounds on NIH 3T3 cells. NIH 3T3 (mouse embryonic lung fibroblast, State Key Laboratory of Genetics) cells were seeded on 96-well plates (Costar) in DMEM containing 10% NBS (3 × 104 cell per well in 100 μL medium) in humidified air with 5% CO2 at 37 °C. Different concentrations of test compounds and fluorofenidone were added to the medium, and DMSO was used as a solvent for the test compounds and fluorofenidone, which were applied at a final concentration of 0.1% (v/v) in cell culture medium. Cells were then incubated for 48 hours, followed by detecting with MTT spectrophotometry. Briefly, 5 mg/mL MTT (100 μL) was added to each well, and incubated for 4 hours at 37 °C. All liquid in each well was then discarded and DMSO (150 μL) was added. The OD at 570 nm was detected by spectrometry. IC50 was determined by nonlinear regression analysis using GraphPad PRISM.
Of the aryl amines connected with a carbohydrate that were tested, compound 19a exhibited the highest inhibitory activity against NIH 3T3 (IC50 = 0.17 mM) among the five aryl amines modified with glucose. When the carbohydrate moiety was mannose, 19b appeared to have the highest inhibitory activity (IC50 = 0.79 mM). When the carbohydrate moiety was galactose, 17c demonstrated the highest inhibitory activity (IC50 = 0.42 mM), while 19c showed two-fold lower inhibitory activity (IC50 = 0.84 mM) than 17c. Thus, among the five aryl amines that were modified with carbohydrate, 19 yielded more active compounds than the other aryl amines.
The same aryl amine-type compounds were also explored. Compound 15a demonstrated better inhibitory activity than 15b and 15c (IC50 = 3.93 mM for 15a, IC50 = 5.68 mM for 15b, IC50 = 8.40 mM for 15c); compound 17c showed the highest inhibitory activity among compounds 17a–c (IC50 = 2.63 mM for 17a, IC50 = 5.81 mM for 17b, IC50 = 0.42 mM for 17c); compound 18a appeared to have significantly higher activity than 18b and slightly better activity than 18c in NIH 3T3 inhibition (IC50 = 2.47 mM for 18a, IC50 = 13.17 mM for 18b, IC50 = 3.30 mM for 18c); compound 19a exhibited better activity than either 19b or 19c (IC50 = 0.17 mM for 19a, IC50 = 0.79 mM for 19b, IC50 = 0.84 mM for 19c). These results indicate that among the three carbohydrates used for the modification of pirfenidone analogues, glucose appeared to be a better candidate (e.g., 19a, IC50 = 0.17 mM), although some compounds that were modified with galactose also had high inhibitory activity (e.g., 17c, IC50 = 0.42 mM).
Compounds 17, 18, and 19 differ in the NHR (R= various carbohydrate moieties) positions. When NHR was modified at the ortho position, the antifibrotic activities of all three compounds were increased, especially compound 19a which showed ten-fold higher activity compared with compound 19 (R = H); when NHR was modified at the para position, the antifibrotic activities were decreased; when NHR was modified at the meta position, compound 17c showed better antifibrotic activities compared with 17a and 17b. Among these compounds, compound 19a, that is NHR modified with glucose at the ortho position, exhibited the highest antifibrotic activity.
When a CH2 group was used to link the substituted phenyl ring with 5-trifluoromethyl-2(1H)-pyridone (Table 1, compounds 18a–c and 19a–c), some of the resulting products showed obviously higher NIH 3T3 inhibitory activities (Table 1, compound 19a–c), perhaps due to the increased flexibility between two substituted aryl rings.
Some modifications of pirfenidone analogues with different monosaccharides appeared to increase the inhibitory activity. For example, compounds 19a–c, which were prepared by modification of pirfenidone analogue 19 with a carbohydrate ring, showed a significant increase of inhibitory activity in NIH 3T3 cells. Compound 19a demonstrated ten-fold higher activity compared with pirfenidone analogue 19, but modifications of 16 and 18 with carbohydrates led to decreases in inhibitory activities against NIH 3T3 cells.
3. Experimental
3.1. General
Unless specified, all reagents and starting materials were purchased from commercial sources and used as received. Solvents were purified following standard literature procedures. Analytical TLC was performed on silica gel60 F254 precoated on glass plates, with detection by fluorescence and/or by staining with 5% concentrated sulphuric acid in EtOH. 1H- (400 MHz) and 13C-NMR spectra (100 MHz) were recorded on a Bruker DRX-400 spectrometer at 25 °C. Chemical shifts (δ) for 1H and 13C spectra are expressed in ppm relative to internal Me4Si as standard. Signals were abbreviated as s, singlet; bs, broad singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Sugar signals were numbered as customary. ESI mass spectra and high resolution mass spectrometry (HRMS) were recorded using an Agilent Technologies 1100 Series instrument (ESI ionization). Optical rotations were measured in a 1.00 dm tube with an Optical Activity AA-10R polarimeter in methanol or chloroform.
3.2. General Procedure for the Synthesis of Compounds 14a–e
Amine (compounds 15–19, 0.5 mmol) was added to a solution of compounds 14 (155 mg, 0.6 mmol) in dry methanol (2 mL) and acetic acid (1.5 mL) under stirring and under a nitrogen atmosphere at room temperature. After 10 min NaCNBH3 (40 mg, 0.6 mmol) was added. The solution was stirred at room temperature for 30 min. After completion of the reaction (TLC 3:1 petroleum ether b.p. 60–90 °C-EtOAc), the methanol was evaporated under reduced pressure, the reaction mixture was diluted with CH2Cl2 (5 mL) and washed with saturated NaHCO3 (3 × 5 mL) and with water (5 mL). The organic phase was dried over sodium sulphate and the solvent was evaporated under reduced pressure to give a crude mass which was purified by flash chromatography (4:1 petroleum ether b.p. 60–90 °C-EtOAc).
1,2;3,4-di-O-Isopropylidene-6-deoxy-6-(4-(5-trifluoromethyl-2(1H)-pyridone-1-yl)-anilino)-α-D-galactopyranose (14a). Compound 15 (127 mg, 0.5 mmol) was used to prepare compound 14a (205 mg, 82.3%) as a colorless syrup; [α]25D −75 (c 0.10, CHCl3); 1H-NMR (CDCl3): δ 7.72 (s, 1H), 7.46 (dd, J1 = 2.8 Hz, J2 = 9.6 Hz, 1H), 7.15–7.13 (m, 2H), 6.73–6.68 (m, 3H), 5.56 (d, J = 5.2 Hz, 1H, H-1), 4.64 (dd, J1 = 2.4 Hz, J2 = 8.0 Hz, 1H, H-3), 4.34 (dd, J1 = 2.4 Hz, J2 = 5.2 Hz, 1H, H-2), 4.26 (dd, J1 = 2.0 Hz, J2 = 8.0 Hz, 1H, H-4), 4.04–4.00 (m, 1H, H-5), 3.40–3.37 (m, 2H, H-6), 1.48 (s, 3H, CH3), 1.44 (s, 3H, CH3), 1.37 (s, 3H, CH3), 1.33 (s, 3H, CH3); 13C-NMR(CDCl3): δ 162.1 (C=O), 148.7, 138.3, 138.2, 135.0, 129.7, 122.0, 113.2, 109.5, 109.1, 108.7, 96.4 (C-1), 71.6, 70.8, 70.6 and 65.7 (C-2, C-3, C-4, C-5), 43.9 (C-6), 26.0 (CH3), 26.0 (CH3), 24.9 (CH3), 24.4 (CH3); ESI-MS: Calcd for C24H27N2O6F3: 497.2[M+H]+, found: 497.2[M+H]+.
1,2;3,4-di-O-Isopropylidene-6-deoxy-6-(3-chloro-4-(5-trifluoromethyl-2(1H)-pyridone-1-yl)-anilino)-α-D-galactopyranose (14b). Compound 16 (145 mg, 0.5 mmol) was used to prepare compound 14b (195 mg, 73.6%) as a colorless syrup; [α]25D −59 (c 0.10, CHCl3); 1H-NMR (CDCl3): δ 7.58 (s, 1H), 7.51 (dd, J1 = 2.8 Hz, J2 = 9.6 Hz, 1H), 7.10 (dd, J1 = 1.6 Hz, J2 = 8.8 Hz, 1H), 6.78 (d, J = 2.4 Hz, 1H), 6.71 (d, J = 2.8 Hz, 1H), 6.63–6.60 (m, 1H), 5.56 (d, J = 5.2 Hz, 1H, H-1), 4.64 (dd, J1 = 2.4 Hz, J2 = 8.0 Hz, 1H, H-3), 4.35 (dd, J1 = 2.4 Hz, J2 = 5.2 Hz, 1H, H-2), 4.26 (dd, J1 = 2.0 Hz, J2 = 8.0 Hz, 1H, H-4), 4.02–3.99 (m, 1H, H-5), 3.38–3.35 (m, 2H, H-6), 1.48 (s, 3H, CH3), 1.47 (s, 3H, CH3), 1.37 (s, 3H, CH3), 1.34 (s, 3H, CH3); 13C-NMR (CDCl3): δ 161.7 (C=O), 150.0, 138.7, 135.5, 132.0, 129.2, 126.5, 122.3, 113.3, 112.2, 112.1, 109.6, 108.8, 96.4 (C-1), 71.6, 70.8, 70.6 and 65.6 (C-2, C-3, C-4, C-5), 43.8 (C-6), 26.0 (CH3), 24.9 (CH3), 24.4 (CH3); ESI-MS: Calcd for C24H26N2O6F3Cl: 531.2 [M+H]+, found: 531.1 [M+H]+.
1,2;3,4-di-O-Isopropylidene-6-deoxy-6-(3-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-α-D-galactopyranose (14c). Compound 17 (135 mg, 0.5 mmol) was used to prepare compound 14c(192 mg, 75.3%) as a colorless syrup; [α]25D −47 (c 0.10, CHCl3); 1H-NMR (CDCl3): δ 7.63 (s, 1H), 7.41 (d, J = 1.6 Hz, 1H), 7.18–7.14 (m, 1H), 6.67–6.59 (m, 4H), 5.54 (d, J = 4.8 Hz, 1H, H-1), 5.04 (m, 2H, CH2), 4.62 (dd, J1 = 1.6 Hz, J2 = 8.0 Hz, 1H, H-3), 4.33 (t, J = 2.4 Hz, 1H, H-2), 4.25 (d, J = 7.6 Hz, 1H, H-4), 3.98 (d, J = 4.8 Hz, 1H, H-5), 3.40–3.29 (m, 2H, H-6), 1.47 (s, 3H, CH3), 1.37 (s, 3H, CH3), 1.36 (s, 3H, CH3), 1.31 (s, 3H, CH3); 13C-NMR (CDCl3): δ 161.9 (C=O), 148.8, 136.7, 136.1, 134.8, 130.0, 121.5, 117.3, 113.1, 109.5, 108.7, 96.4 (C-1), 71.7, 70.8, 70.6 and 65.7 (C-2, C-3, C-4, C-5), 52.3 (CH2), 43.9 (C-6), 26.0 (CH3), 25.8 (CH3), 24.9 (CH3), 24.4 (CH3); ESI-MS: Calcd for C25H29N2O6F3: 511.2 [M+H]+, found: 511.2 [M+H]+.
1,2;3,4-di-O-Isopropylidene-6-deoxy-6-(4-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-α-D-galactopyranose (14d). Compound 18 (135 mg, 0.5 mmol) was used to prepare compound 14d (207 mg, 81.2%) as a colorless syrup; [α]25D −68 (c 0.10, CHCl3); 1H-NMR (CDCl3): δ 7.60 (s, 1H), 7.39 (dd, J1 = 2.0 Hz, J2 = 9.6 Hz, 1H), 7.15 (d, J = 8.4 Hz, 2H), 6.65–6.62 (m, 3H), 5.54 (d, J= 4.8 Hz, 1H, H-1), 5.00 (s, 2H, CH2), 4.62 (dd, J1 = 2.0 Hz, J2 = 8.0 Hz, 1H, H-3), 4.33–4.31 (m, 1H, H-2), 4.25–4.23 (m, 1H, H-4), 3.99 (t, J = 6.0 Hz, 1H, H-5), 3.41–3.30 (m, 2H, H-6), 1.47 (s, 3H, CH3), 1.36 (s, 3H, CH3), 1.33 (s, 3H, CH3), 1.30 (s, 3H, CH3); 13C-NMR (CDCl3): δ 162.0 (C=O), 148.4, 136.4, 136.3, 134.7, 134.6, 130.1, 123.5, 121.4, 113.6, 109.5, 108.7, 96.4 (C-1), 71.7, 70.8, 70.6 and 65.6 (C-2, C-3, C-4, C-5), 52.1 (CH2), 43.9 (C-6), 26.0 (CH3), 25.7 (CH3), 24.9 (CH3), 24.4 (CH3); ESI-MS: Calcd for C25H29N2O6F3: 511.2 [M+H]+, found: 511.2 [M+H]+.
1,2;3,4-di-O-Isopropylidene-6-deoxy-6-(2-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-α-D-galactopyranose (14e). Compound 19 (135 mg, 0.5 mmol) was used to prepare compound 14e (210 mg, 82.4%) as a colorless syrup; [α]25D −62 (c 0.10, CHCl3); 1H-NMR (CDCl3): δ 7.68 (s, 1H), 7.42 (dd, J1 = 2.0 Hz, J2 = 9.6 Hz, 1H), 7.25 (m, 1H), 7.14 (d, J = 7.2 Hz, 1H), 6.72–6.64 (m, 3H), 5.44 (d, J = 5.2 Hz, 1H, H-1), 5.20–4.95 (m, 2H, CH2), 4.57 (dd, J1 = 2.0 Hz, J2 = 8.0 Hz, 1H H-3), 4.27 (dd, J1 = 2.0 Hz, J2 = 5.2 Hz, 1H H-2), 4.21 (d, J = 8.8 Hz, 1H, H-4), 3.94 (t, J = 6.4 Hz, 1H, H-5), 3.50–3.43 (m, 1H, H-6), 3.36–3.31 (m, 1H, H-6), 1.47 (s, 3H, CH3), 1.34 (s, 3H, CH3), 1.26 (d, 6H, 2xCH3); 13C-NMR (CDCl3): δ 162.4 (C=O), 146.6, 136.1, 136.1, 135.0, 131.3, 130.5, 121.3, 119.3, 116.9, 111.6, 110.3, 109.3, 108.5, 96.4 (C-1), 71.4, 70.8, 70.6 and 65.1 (C-2, C-3, C-4, C-5), 49.1 (CH2), 43.6 (C-6), 26.0 (CH3), 25.6 (CH3), 24.9 (CH3), 24.4 (CH3); ESI-MS: Calcd for C25H29N2O6F3: 511.2 [M+H]+, found: 511.2 [M+H]+.
3.3. General Procedure for the Synthesis of Compounds 15a–19a and 15b–19b
Amine (compound 15–19, 0.5 mmol) was added to a solution of compounds 8 or 12 (0.6 mmol) in dry methanol (2 ml) and acetic acid (1.5 mL) under stirring and under nitrogen atmosphere at room temperature. After 10 min NaCNBH3 (40 mg, 0.6 mmol) was added. The solution was stirred at room temperature for 30 min. After completion of the reaction (TLC 3:1 petroleum ether b.p. 60–90 °C-EtOAc), the methanol was evaporated under reduced pressure, the reaction mixture was diluted with CH2Cl2 (5 mL) and washed with saturated NaHCO3 (3 × 5 mL) and with water (5 mL). The organic phase was dried over sodium sulphate and the solvent was evaporated under reduced pressure to give a crude mass which was dissolved in 0.05 M NaOMe/MeOH (5 mL) and stirred at ambient temperature for 30 min. The stirring reaction mixture was neutralized with prewashed Amberlite IR 120-H+. The resin was filtered off and the solvent was evaporated under reduced pressure. The crude product was purified by flash chromatography (7:1 CHCl3-MeOH).
Methyl 6-deoxy-6-(4-(5-trifluoromethyl-2(1H)-pyridone-1-yl)-anilino)-α-D-glucopyranoside (15a). Compound 15 (127 mg, 0.5 mmol) and 8 (190 mg, 0.6 mmol) were used to prepare compound 15a (143 mg, 66.5%) as a colorless syrup; [α]25D −63 (c 0.10, MeOH); 1H-NMR (MeOH-d4) δ 7.97 (s, 1H), 7.66 (dd, J1 = 2.4 Hz, J2 = 9.6 Hz, 1H), 7.01 (d, J = 8.8 Hz, 2H), 6.76 (d, J = 8.4 Hz, 2H), 6.64 (d, J = 9.2 Hz, 1H), 4.62 (d, J = 3.6 Hz, 1H, H-1), 3.71–3.63 (m, 1H), 3.59–3.55 (m, 2H), 3.38–3.35 (m, 1H), 3.28 (s, 3H), 3.22–3.17 (m, 2H); 13C-NMR (MeOH-d4) δ 164.5 (C=O), 151.0, 140.6, 140.5, 137.3, 130.3, 128.1, 122.2, 113.6, 101.2 (C-1), 75.1, 73.6, 73.5 and 71.4 (C-2, C-3, C-4, C-5), 55.5 (OCH3), 45.7 (C-6); HRMS: Calcd for C19H21N2O6F3 [M+H]+ 431.1431, found 431.1425.
Methyl 6-deoxy-6-(3-chloro-4-(5-trifluoromethyl-2(1H)-pyridone-1-yl)-anilino)-α-D-glucopyranoside (16a). Compound 16 (145 mg, 0.5 mmol) and 8 (190 mg, 0.6 mmol) were used to prepare compound 16a (152 mg, 65.5%) as a colorless syrup; [α]25D −34 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 7.92 (s, 1H), 7.70 (dd, J1 = 2.8 Hz, J2 = 9.6 Hz, 1H), 7.08 (d, J = 8.8 Hz, 2H), 6.84 (m, 1H), 6.68 (m, 2H), 4.62 (d, J = 3.6 Hz, 1H, H-1), 3.63–3.60 (m, 1H), 3.59–3.51 (m, 2H), 3.38–3.34 (m, 1H), 3.29 (s, 3H, OCH3), 3.26–3.17 (m, 3H); 13C-NMR (MeOH-d4): δ 164.1 (C=O), 152.5, 141.2, 137.8, 132.7, 130.2, 126.7, 122.5, 113.8, 112.8, 101.2 (C-1), 75.1, 73.6, 73.3 and 71.6 (C-2, C-3, C-4, C-5), 55.6 (OCH3), 45.5 (C-6); HRMS: Calcd for C19H21N2O6F3Cl: 465.1041 [M+H]+, found: 465.1038 [M+H]+.
Methyl 6-deoxy-6-(3-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-α-D-glucopyranoside (17a). Compound 17 (135 mg, 0.5 mmol) and 8 (190 mg, 0.6 mmol) were used to prepare compound 17a (150 mg, 67.6%) as a colorless syrup; [α]25D −45 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 8.06 (s, 1H), 7.55 (dd, J1 = 2.4 Hz, J2 = 9.6 Hz, 1H), 7.02 (t, J = 8.0 Hz, 1H), 6.61–6.58 (m, 3H), 6.49 (d, J = 7.6 Hz, 1H), 5.03 (s, 2H, CH2), 4.57 (d, J = 2.8 Hz, 1H, H-1), 3.62–3.48 (m, 3H), 3.35–3.12 (m, 1H), 3.16 (s, 3H, OCH3), 3.19–3.06 (m, 2H); 13C-NMR (MeOH-d4): δ 161.2 (C=O), 147.8, 136.6, 136.6, 135.0, 134.1, 127.8, 123.3, 120.6, 118.9, 114.6, 111.3, 111.0, 108.7, 108.4, 98.2 (C-1), 72.2, 70.7, 70.7 and 68.19 (C-2, C-3, C-4, C-5), 52.5 (OCH3), 50.9 (CH2), 43.0 (C-6); HRMS: Calcd for C20H23N2O6F3: 445.1587 [M+H]+, found: 445.1583 [M+H]+.
Methyl 6-deoxy-6-(4-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-α-D-glucopyranoside (18a). Compound 18 (135 mg, 0.5 mmol) and 8 (190 mg, 0.6 mmol) were used to prepare compound 18a (155 mg, 69.8%) as a colorless syrup; [α]25D −54 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 8.03 (s, 1H), 7.54 (dd, J1 = 2.8 Hz, J2 =9.6 Hz, 1H), 7.07 (d, J = 8.4 Hz, 2H), 6.64 (d, J = 4.4 Hz, 2H), 6.56 (d, J = 9.6 Hz, 1H), 4.98 (s, 2H, CH2), 4.57 (d, J = 3.6 Hz, 1H, H-1), 3.63–3.58 (m, 1H), 3.55–3.49 (m, 2H), 3.34–3.31 (m, 1H), 3.24–3.23 (m, 1H), 3.18 (s, 3H, OCH3), 3.15–3.06 (m, 2H); 13C-NMR (MeOH-d4): δ 162.8 (C=O), 148.9, 137.7, 137.7, 135.5, 129.3, 123.5, 120.3, 113.1, 99.8 (C-1), 73.7, 72.2, 72.2 and 69.6 (C-2, C-3, C-4, C-5), 54.0 (OCH3), 52.1 (CH2), 44.5 (C-6); HRMS: Calcd for C20H23N2O6F3: 467.1400 [M+Na]+, found: 465.1391 [M+Na]+.
Methyl 6-deoxy-6-(2-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-α-D-glucopyranoside (19a). Compound 19 (135 mg, 0.5 mmol) and 8 (190 mg, 0.6 mmol) were used to prepare compound 19a (147 mg, 66.2%) as a colorless syrup; [α]25D −57 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 7.90 (s, 1H), 7.55 (dd, J1 = 1.6 Hz, J2 =7.6 Hz, 1H), 7.15 (m, 2H), 6.70 (d, J = 8.0 Hz, 1H), 6.61 (m, 2H), 5.06 (m, 2H, CH2), 4.51 (d, J = 3.6 Hz, 1H, H-1), 3.67–3.59 (m, 1H), 3.52–3.47 (m, 2H), 3.37–3.31 (m, 1H), 3.22 (s, 1H), 3.16–3.10 (m, 2H), 3.02 (s, 3H, OCH3); 13C-NMR (MeOH-d4): δ 162.9 (C=O), 146.6, 137.0, 137.0, 135.6, 131.0, 130.0, 127.3, 120.3, 119.5, 116.6, 111.1, 110.6, 99.8 (C-1), 73.7, 72.2, 72.1 and 69.0 (C-2, C-3, C-4, C-5), 54.0 (OCH3), 48.2 (CH2), 44.2 (C-6); HRMS: Calcd for C20H23N2O6F3: 445.1587 [M+H]+, found: 445.1586 [M+H]+.
Methyl 6-deoxy-6-(4-(5-trifluoromethyl-2(1H)-pyridone-1-yl)-anilino)-α-D-mannopyranoside (15b). Compound 15 (127 mg, 0.5 mmol) and 12 (190 mg, 0.6 mmol) were used to prepare compound 15b (140 mg, 65.1%) as a colorless syrup; [α]25D +82 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 7.98 (s, 1H), 7.67 (dd, J1 = 2.4 Hz, J2 = 9.6 Hz, 1H), 7.08 (d, J = 8.8 Hz, 2H), 6.76 (d, J = 8.4 Hz, 2H), 6.65 (d, J = 9.2 Hz, 1H), 4.61 (d, J= 1.2 Hz, 1H, H-1), 3.76 (s, 1H), 3.64–3.59 (m, 3H), 3.54–3.51 (m, 1H), 3.32–3.28 (m, 1H), 3.26 (s, 3H, OCH3), 13C-NMR (MeOH-d4): δ 164.5 (C=O), 151.0, 140.6, 140.5, 140.5, 137.3, 130.2, 128.1, 126.2, 123.6, 122.2, 113.0, 111.5, 111.1, 102.8(C-1), 72.6, 72.1, 72.1 and 69.9 (C-2, C-3, C-4, C-5), 55.2 (OCH3), 45.5 (C-6); HRMS: Calcd for C19H21N2O6F3: 431.1431 [M+H]+, found: 431.1425 [M+H]+.
Methyl 6-deoxy-6-(3-chloro-4-(5-trifluoromethyl-2(1H)-pyridone-1-yl)-anilino)-α-D-mannopyranoside (16b). Compound 16 (145 mg, 0.5 mmol) and 12 (190 mg, 0.6 mmol) were used to prepare compound 16b (145 mg, 62.5%) as a colorless syrup; [α]25D −59 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 7.93 (s, 1H), 7.71 (dd, J1 = 2.4 Hz, J2 = 9.6 Hz, 1H), 7.09 (d, J = 2.2 Hz, 2H), 6.84 (dd, J1 = 0.4 Hz, J2 = 1.6 Hz, 1H), 6.71–6.66 (m, 2H), 4.60 (d, J = 1.6 Hz, 1H, H-1), 3.76 (s, 1H), 3.64–3.57 (m, 3H), 3.52–3.48 (m, 1H), 3.30–3.26 (m, 2H), 3.28 (s, 3H, OCH3); 13C-NMR (MeOH-d4): δ 162.7 (C=O), 151.1, 139.8, 139.7, 136.4, 131.3, 128.8, 125.3, 124.7, 122.1, 121.1, 112.3, 111.4, 111.4, 110.2, 109.9, 101.4 (C-1), 71.2, 70.9, 70.7 and 68.4 (C-2, C-3, C-4, C-5), 53.9 (OCH3), 43.8 (C-6); HRMS: Calcd for C19H21N2O6F3Cl: 465.1041 [M+H]+, found: 465.1039 [M+H]+.
Methyl 6-deoxy-6-(3-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-α-D-mannopyranoside (17b). Compound 17 (135 mg, 0.5 mmol) and 12 (190 mg, 0.6 mmol) were used to prepare compound 17b (154 mg, 69.4%) as a colorless syrup; [α]25D −83 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 8.08 (s, 1H), 7.60 (dd, J1 = 2.8 Hz, J2 = 9.6 Hz, 1H), 7.06 (dd, J1 = 7.6 Hz, J2 = 8.0 Hz, 1H), 6.63–6.60 (m, 3H), 6.52 (d, J = 7.2 Hz, 1H), 5.07 (s, 2H, CH2), 4.57 (d, J = 1.6 Hz, 1H, H-1), 3.74 (t, 1H), 3.62–3.56 (m, 3H), 3.49–3.46 (m, 1H), 3.27–3.25 (m, 1H), 3.17 (s, 3H, OCH3); 13C-NMR (MeOH-d4): δ 162.7 (C=O), 149.3, 138.1, 138.1, 136.5,135.7, 129.3, 124.8, 120.4, 116.0, 112.7, 112.3, 110.2, 109.9, 101.3 (C-1), 71.2, 70.7, 70.3 and 68.6 (C-2, C-3, C-4, C-5), 53.7 (OCH3), 52.4 (CH2), 44.2 (C-6); HRMS: Calcd for C20H23N2O6F3: 445.1587 [M+H]+, found: 445.1585 [M+H]+.
Methyl 6-deoxy-6-(4-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-α-D-mannopyranoside (18b). Compound 18 (135 mg, 0.5 mmol) and 12 (190 mg, 0.6 mmol) were used to prepare compound 18b (150 mg, 67.6%) as a colorless syrup; [α]25D −67 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 8.06 (s, 1H), 7.58 (dd, J1 = 2.8 Hz, J2 = 2.4 Hz, 1H), 7.11 (d, J = 8.4 Hz, 2H), 6.65 (d, J = 8.4 Hz, 2H), 6.58 (d, J = 9.6 Hz, 1H), 5.01 (s, 2H, CH2), 4.57 (d, J= 1.6 Hz, 1H, H-1), 3.73 (t, 1H), 3.61–3.55 (m, 3H), 3.51–3.46 (m, 1H), 3.27–3.25 (m, 3H), 3.19 (s, 3H, OCH3); 13C-NMR (MeOH-d4): δ 164.2 (C=O), 150.4, 139.1, 139.1, 136.9,130.7, 126.2, 124.9, 123.5, 121.7, 114.4, 111.6, 111.2, 102.7 (C-1), 72.6, 72.1, 71.8 and 70.1 (C-2, C-3, C-4, C-5), 55.1 (OCH3), 53.5 (CH2), 45.7 (C-6); HRMS: Calcd for C20H23N2O6F3: 445.1587 [M+H]+, found: 445.1589 [M+H]+.
Methyl 6-deoxy-6-(2-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-α-D-mannopyranoside (19b). Compound 19 (135 mg, 0.5 mmol) and 12 (190 mg, 0.6 mmol) were used to prepare compound 19b (151 mg, 68.0%) as a colorless syrup; [α]25D −72 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 7.95 (s, 1H), 7.58 (d, J = 9.6 Hz 1H), 7.23–7.17 (m, 2H), 6.72–6.70 (m, 2H), 6.65 (t, J = 7.6 Hz, 1H), 5.19–5.09 (m, 2H, CH2), 4.62 (s, 1H, H-1), 3.75 (s, 1H), 3.69–3.60 (m, 3H), 3.46–3.42 (m, 1H), 3.34–3.27 (m, 2H), 3.14 (s, 3H, OCH3); 13C-NMR (MeOH-d4): δ 163.0 (C=O), 146.7, 137.1, 137.0, 136.9, 135.7, 130.9, 130.1, 124.6, 121.9, 120.2, 119.5, 116.5, 110.9, 110.9, 110.6, 101.5(C-1), 71.3, 71.0, 70.0 and 68.5 (C-2, C-3, C-4, C-5), 53.8 (OCH3), 43.8 (C-6); HRMS: Calcd for C20H23N2O6F3: 445.1587 [M+H]+, found: 445.1581 [M+H]+.
3.4. General Procedure for the Synthesis of Compounds 15c–19c
Compound 14a–e (0.2 mmol) was added to a solution of 0.5 M HCl/MeOH (5 mL) under stirring and under nitrogen atmosphere at room temperature. The progress of reaction was monitored by TLC (6:1CHCl3-MeOH). After completion of the reaction the solvent was evaporated under reduced pressure, the residue was purified by flash chromatography (7:1CHCl3-MeOH).
Methyl 6-deoxy-6-(4-(5-trifluoromethyl-2(1H)-pyridone-1-yl)-anilino)-β-D-galactofuranoside (15c). Compound 14a (100 mg, 0.2 mmol) was used to prepare compound 15c (45 mg, 52.3%) as a colorless syrup; [α]25D −48 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 7.93 (s, 1H), 7.62 (dd, J1 = 2.8 Hz, J2 = 9.6 Hz, 1H), 7.02 (d, J = 8.4 Hz, 2H), 6.66 (d, J = 8.8 Hz, 2H), 6.58 (d, J = 9.6 Hz, 1H), 4.69 (s, 1H, H-1), 3.85–3.77 (m, 4H), 3.34–3.13 (m, 5H); 13C-NMR (MeOH-d4): δ 163.3 (C=O), 149.7, 139.4, 136.1, 129.0, 127.0, 121.0, 114.8, 112.4, 110.3, 109.9, 109.3 (C-1), 83.9, 82.0, 77.6 and 68.8 (C-2, C-3, C-4, C-5), 54.3 (OCH3), 46.3 (C-6); HRMS: Calcd for C19H21N2O6F3: 431.1424 [M+H]+, found: 431.1420 [M+H]+.
Methyl 6-deoxy-6-(3-chloro-4-(5-trifluoromethyl-2(1H)-pyridone-1-yl)-anilino)-β-D-galactofuranoside (16c). Compound 14b (106 mg, 0.2 mmol) was used to prepare compound 16c (42 mg, 45.2%) as a yellow syrup; [α]25D −64 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 7.95 (s, 1H), 7.69 (dd, J1 = 2.8 Hz, J2 = 9.6 Hz, 1H), 7.07 (d, J = 8.8 Hz, 1H), 6.79 (t, J = 2.4 Hz, 1H), 6.66–6.63 (m, 2H), 4.73 (d, J = 1.6 Hz, 1H, H-1), 3.96–3.95 (m, 1H), 3.87–3.85 (m, 2H), 3.83–3.78 (m, 1H), 3.38–3.35 (m, 1H), 3.32 (s, 3H, OCH3),3.25–3.23 (m, 1H), 3.22–3.16 (m, 1H); 13C-NMR (MeOH-d4): δ 162.7 (C=O), 150.9, 139.7, 136.4, 131.3, 128.9, 125.2, 112.0, 111.2, 109.1, 83.6, 81.7, 77.3 and 68.5 (C-2, C-3, C-4, C-5), 54.0 (OCH3), 45.8 (C-6); HRMS: Calcd for C19H20N2O6F3Cl: 465.1035 [M+H] +, found: 465.1038 [M+H]+.
Methyl 6-deoxy-6-(3-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-β-D-galactofuranoside (17c). Compound 14c (100 mg, 0.2 mmol) was used to prepare compound 17c (52 mg, 58.4%) as a yellow syrup; [α]25D −34 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 8.07 (s, 1H), 7.58 (dd, J1 = 2.8 Hz, J2 = 9.6 Hz, 1H), 7.05–7.01 (m, 1H), 6.60–6.55 (m, 3H), 6.48 (d, J = 7.6 Hz, 1H), 5.04 (s, 2H, CH2), 4.71 (d, J= 1.2 Hz, 1H, H-1), 3.95–3.93 (m, 1H), 3.87–3.83 (m, 2H), 3.80–3.79 (m, 1H), 3.30 (s, 3H, OCH3), 3.24 (m, 1H), 3.15–3.10 (m, 1H); 13C-NMR (MeOH-d4): δ 164.1(C=O), 150.7, 139.5, 138.0, 137.1, 130.7, 121.8, 117.2, 113.6, 113.4, 111.7, 110.5, 85.3, 83.2, 78.9 and 70.0 (C-2, C-3, C-4, C-5), 55.3 (OCH3), 53.8 (CH2), 47.7 (C-6); HRMS: Calcd for C20H23N2O6F3: 445.1581 [M+H]+, found: 445.1587 [M+H]+.
Methyl 6-deoxy-6-(4-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-β-D-galactofuranoside (18c). Compound 14d (100 mg, 0.2 mmol) was used to prepare compound 18c (47 mg, 52.8%) as a yellow syrup; [α]25D −75 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 8.06 (s, 1H), 7.56 (dd, J1 = 2.8 Hz, J2 = 9.6 Hz, 1H), 7.09 (d, J = 8.4 Hz, 2H), 6.61–6.54 (m, 3H), 4.98 (s, 2H, CH2), 4.70 (d, J = 2.0 Hz, 1H, H-1), 3.94–3.91 (m, 1H), 3.85–3.82 (m, 2H), 3.80–3.76 (m, 1H), 3.29 (s, 3H, OCH3),3.25–3.23 (m, 3H), 3.16–3.11 (m, 1H); 13C-NMR (MeOH-d4): δ 162.8 (C=O), 148.9, 137.8, 135.5, 129.4, 123.4, 120.3, 112.6, 109.1, 83.7, 81.9, 77.4 and 68.6 (C-2, C-3, C-4, C-5), 54.0 (OCH3), 52.1 (CH2), 46.2 (C-6); HRMS: Calcd for C20H23N2O6F3: 467.1400 [M+Na]+, found: 465.1380 [M+Na]+.
Methyl 6-deoxy-6-(2-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-β-D-galactofuranoside (19c). Compound 14e (100 mg, 0.2 mmol) was used to prepare compound 19c (49 mg, 55.1%) as a yellow syrup; [α]25D −72 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 7.97 (s, 1H), 7.57 (dd, J1 = 2.8 Hz, J2 = 7.2 Hz, 1H), 7.16–7.10 (m, 2H), 6.68 (d, J = 8.0 Hz, 1H), 6.63–6.59 (m, 2H), 5.09 (s, 2H, CH2), 4.73 (s, 1H, H-1), 3.97–3.96 (m, 1H), 3.88–3.84 (m, 3H), 3.36–3.17 (m, 5H); 13C-NMR (MeOH-d4): δ 163.0 (C=O), 146.5, 137.3, 137.2, 135.7, 130.4, 129.9, 120.3, 119.5, 116.5, 110.8, 109.1, 84.0, 81.8, 77.5 and 68.5 (C-2, C-3, C-4, C-5), 54.0 (OCH3), 48.5 (CH2), 46.2 (C-6); HRMS: Calcd for C20H23N2O6F3: 445.1581 [M+H]+, found: 445.1583 [M+H]+.
4. Conclusions
In conclusion, we report the design, synthesis and biological evaluation of some carbohydrate-modified 1-(substituted aryl)-5-trifluoromethyl-2(1H) pyridones. Our studies suggest that some modifications of pirfenidone analogues with carbohydrates appear to increase the inhibitory activity against NIH 3T3 cell proliferation. Among the compounds tested, compound 19a, which was synthesized by modification of pirfenidone analogue 19 with glucose, demonstrated the highest cell-based inhibitory activity (IC50 = 0.17 mM).
Footnotes
Sample Availability: Samples of the compounds are available from the authors.
References and Notes
- 1.du Bois R.M. Strategies for treating idiopathic pulmonary fibrosis. Nat. Rev. Drug Dis. 2010;9:129–140. doi: 10.1038/nrd2958. [DOI] [PubMed] [Google Scholar]
- 2.Balsano1 C., Alisi A., Nobili V. Liver fibrosis and therapeutic strategies: The goal for improving metabolism. Curr. Drug Targets. 2009;10:505–512. doi: 10.2174/138945009788488459. [DOI] [PubMed] [Google Scholar]
- 3.Krenning G., Zeisberg E.M., Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J. Cell Physiol. 2010;225:631–637. doi: 10.1002/jcp.22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weber K.T. Fibrosis and hypertensive heart disease. Curr. Opin. Cardiol. 2000;5:264–272. doi: 10.1097/00001573-200007000-00010. [DOI] [PubMed] [Google Scholar]
- 5.Wick G., Backovic A., Rabensteiner E., Plank N., Schwentner C., Sgonc R. The immunology of fibrosis: Innate and adaptive responses. Trends Immunol. 2010;31:110–119. doi: 10.1016/j.it.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Azuma A. Pirfenidone: Antifibrotic agent for idiopathic pulmonary fibrosis. Expert Rev. Resp. Med. 2010;4:301–310. doi: 10.1586/ers.10.32. [DOI] [PubMed] [Google Scholar]
- 7.Iyer S.N., Hyde D.M., Giri S.N. Anti-inflammatory effect of pirfenidone in the bleomycin-hamster model of lung inflammation. Inflammation. 2000;24:477–491. doi: 10.1023/A:1007068313370. [DOI] [PubMed] [Google Scholar]
- 8.Spond J., Case N., Chapman R.W., Crawley Y., Egan R.W., Fine J., Hey J.A., Kreutner W., Kung T., Wang P., Minnicozzi M. Inhibition of experimental acute pulmonary inflammation by pirfenidone. Pulm. Pham. Ther. 2003;16:207–214. doi: 10.1016/S1094-5539(03)00026-9. [DOI] [PubMed] [Google Scholar]
- 9.Tao L., Hu G. 1-(substituted phenyl)-5-methyl-2-(1H) pyridone in the manufacture of medicaments for treating fibrosis in organs or tissues. WO 2006/108354 A1. PCT Int. Appl. 2000 Oct 19;
- 10.Hu G., Tao L., Chen J. Preparation methods and uses of 1-(substituted aryl)-5-trifluoromethyl-2-(1H) pyridone compounds and their salts. WO2010/135972 A1. PCT Int. Appl. 2010 Dec 2;
- 11.Tao L., Zhang J., Hu G., Chen Z., Gong J. Effects of 1-(3-fluorophenyl)-5-methyl-2-(1H)-pyridone on renal fibroblast in rats. J. Cent. South Univ. (Med. Sci.) 2004;29:139–141. [PubMed] [Google Scholar]
- 12.Peng Z., Hu G., Shen H., Wang L., Ning W., Xie Y., Wang N., Li B., Tang Y., Tao L. Fluorofenidone attenuates collagen I and transforming growth factor-β1 expression through a nicotinamide adenine dinucleotide phosphate oxidase-dependent way in NRK-52E cells. Nephrology. 2009;14:565–572. doi: 10.1111/j.1440-1797.2009.01129.x. [DOI] [PubMed] [Google Scholar]
- 13.Shimizu T., Fukagawa M., Kuroda T., Hata S., Iwasaki Y., Nemoto M., Shirai K., Yamauchi S., Margolin S.B., Shimizu F., Kurokawa K. Pirfenidone prevents collagen accumulation in the remnant kidney in rats with partial nephrectomy. Kidney Int. Suppl. 1996;63:S239–S243. [PubMed] [Google Scholar]
- 14.Stallforth P., Lepenies B., Adibekian A., Seeberger P.H. Carbohydrates: A frontier in medicinal chemistry. J. Med. Chem. 2009;52:5561–5577. doi: 10.1021/jm900819p. [DOI] [PubMed] [Google Scholar]
- 15.Kren V., Martínková L. Glycosides in medicine: “The role of glycosidic residue in biological activity”. Curr. Med. Chem. 2001;8:303–1328. doi: 10.2174/0929867013372193. [DOI] [PubMed] [Google Scholar]
- 16.Reynolds D.D., Evans W.L. The synthesis of certain oligosaccharide acetates in the mannose series. J. Am. Chem. Soc. 1940;62:66–69. doi: 10.1021/ja01858a014. [DOI] [Google Scholar]
- 17.Sixta G., Hofinger A., Kosma P. Synthesis of spacer-containing chlamydial disaccharides as analogues of the α-Kdop-(2→8)-α-Kdop-(2→4)-α-Kdop trisaccharide epitope. Carbohydr. Res. 2007;342:576–585. doi: 10.1016/j.carres.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 18.Klier A.H., Alves R.J., Auxiliadora M., Prado F., de Souza Filho J.D., D’Accorso N.B. Synthesis of new five membered nitrogen containing heterocycles bearing D-galactose side chains. Syn. Comm. 2000;30:4361–4374. doi: 10.1080/00397910008087059. [DOI] [Google Scholar]
- 19.Lee H.H., Hodgson Philip G., Bernacki R.J., Korytnyk W., Sharma M. Analogs of cell surface carbohydrates. Synthesis of D-galactose derivatives having an ethynyl, vinyl or epoxy residue at C-5. Carbohydr. Res. 1998;176:59–72. doi: 10.1016/0008-6215(88)84057-6. [DOI] [PubMed] [Google Scholar]
- 20.Terreni M., Salvetti R., Linati L., Fernandez-Lafuente R., Fernandez-Lorento G., Bastida A., Guisan J.M. Regioselective enzymatic hydrolysis of acetylated pyranoses and pyranosides using immobilised lipases. An easy chemoenzymatic synthesis of α- and β-D-glucopyranose acetates bearing a free secondary c-4 hydroxyl group. Carbohydr. Res. 2002;337:1615–1622. doi: 10.1016/S0008-6215(02)00113-1. [DOI] [PubMed] [Google Scholar]
- 21.Horrobin T., Tran C.H., Crout D. Esterase-catalysed regioselective 6-deacylation of hexopyranose per-acetates, acid-catalysed rearrangement to the 4-deprotected products and conversions of these into hexose 4- and 6-sulfates. J. Chem. Soc. Perkin Trans. 1. 1998:1069–1080. [Google Scholar]
- 22.Romero D.L., Morge R.A., Biles C., Berrios-Pena N., May P.D., Palmer J.R., Johnson P.D., Smith H.W., Busso M., Tan C.K., et al. Discovery, synthesis, and bioactivity of bis(heteroaryl)piperazines. 1. A novel class of non-nucleoside HIV-1 reverse transcriptase inhibitors. J. Med. Chem. 1994;37:999–1014. doi: 10.1021/jm00033a018. [DOI] [PubMed] [Google Scholar]