

The coupling effect of polymeric vanadyl species greatly promotes the NH3-SCR de-NOx process over vanadia-based catalysts.
Abstract
The structure of dispersed vanadyl species plays a crucial role in the selective catalytic reduction (SCR) of NO with NH3 over vanadia-based catalysts. Here, we demonstrate that the polymeric vanadyl species have a markedly higher NH3-SCR activity than the monomeric vanadyl species. The coupling effect of the polymeric structure not only shortens the reaction pathway for the regeneration of redox sites but also substantially reduces the overall reaction barrier of the catalytic cycle. Therefore, it is the polymeric vanadyl species, rather than the monomeric vanadyl species, that determine the NH3-SCR activity of vanadia-based catalysts, especially under low-temperature conditions. The polymeric vanadia-based SCR mechanism reported here advances the understanding of the working principle of vanadia-based catalysts and paves the way toward the development of low vanadium–loading SCR catalysts with excellent low-temperature activity.
INTRODUCTION
Nitrogen oxides (NOx), as key precursor pollutants inducing the formation of acid rain, photochemical smog, and haze, are mainly emitted from power plants and mobile diesel vehicles. Selective catalytic reduction of NOx with NH3 (i.e., NH3-SCR) over vanadia-based catalysts is one of the most widely adopted techniques for the removal of NOx from stationary and mobile sources (1–4). The lower exhaust temperature of advanced combustion engines requires that catalysts be active at lower temperatures (5, 6). Therefore, it is highly desirable to develop vanadia-based SCR catalysts with excellent low-temperature activity to meet future emission regulations, which requires an in-depth understanding of the active center and the reaction mechanism (2, 7, 8).
For the commercial vanadia-based catalysts, previous studies revealed that dispersed vanadyl species, serving as the active moieties for NOx reduction, are mainly present as isolated monomeric and polymeric vanadyl species (9–12), the distributions of which are closely related to the content of V2O5 (13, 14). As for V2O5/TiO2 catalysts, in situ laser Raman analysis performed by Went et al. (12) confirmed that monomeric species predominated at low vanadia loading of 1.3 weight % (wt %) V2O5, at 80% of vanadyl species. With increasing vanadia loading from 1.3 to 3.0 wt %, the fraction of monomeric species decreased monotonically, while the percentage of polymeric species increased from 20 to 33%. Vanadia-based catalysts with 2 to 3 wt % V2O5 are commonly used in industrial applications for mobile source emission control (15). Moreover, for commercial V2O5/TiO2 catalysts, sulfate species are commonly present in the anatase TiO2 used. As revealed by Nam’s group (16), sulfate can enhance the transformation of isolated vanadyl species to polymeric vanadyl species. Therefore, the polymeric vanadyl species may play an important role in the removal of NOx from mobile sources. However, currently proposed NH3-SCR schemes over vanadia-based catalysts mainly focus on the reaction process occurring on isolated monomeric vanadyl species (1, 7, 8, 17), without considering the polymerization of reactive sites and their coupling effects. The working principle of polymeric vanadyl species remains unclear.
Here, using a TiO2 support pretreated with sulfate species, we successfully obtained V2O5/TiO2 catalysts with varying proportions of polymeric and monomeric vanadyl species, which exhibited marked differences in NH3-SCR activity. By combining the results of experimental measurements with density functional theory (DFT) calculations, we found a polymeric vanadyl-based NH3-SCR mechanism, which is energetically more favorable than the reaction schemes based on monomeric vanadyl species. Therefore, it is the polymeric vanadyl species, rather than monomeric vanadyl species, that determine the activity of vanadia-based catalysts, especially under low-temperature conditions. This finding shows that the surface structure of dispersed vanadia plays a critical role in the NH3-SCR reaction. In addition, the experimental approach used here provides an efficient way to develop high-performance low vanadium–loading SCR catalysts.
RESULTS
Catalytic activity
It is well known that vanadia-based catalysts are widely applied because of their excellent sulfur resistance. We pretreated TiO2 with (NH4)2SO4 to regulate the amount of sulfur on the surface, and we obtained a series of V2O5/TiO2 catalysts with different amounts of sulfur, denoted as 1V/xSTi (x wt % represents the sulfate content). For comparison with these sulfur-containing samples, we also prepared samples without sulfur by treating the TiO2 support with NH3 [i.e., Ti-NH3 and 1V/(Ti-NH3) samples]. Because of the reduction of sulfate by NH3, x-ray photoelectron spectroscopy (XPS) results showed that there was no sulfur in the Ti-NH3 sample (table S1), while a small quantity of sulfur was detected in the 1V/(Ti-NH3) sample owing to the existence of traces of sulfur in the vanadia precursor (NH4VO3). With increasing sulfur content, the specific surface area decreased slightly (table S2). There was no change in the crystalline structure, and all these samples crystallized in the anatase structure (fig. S1).
As shown in Fig. 1A, the catalytic activity of the V2O5/TiO2 catalysts in the SCR of NO with NH3 increased with increasing sulfur content. The highest NH3-SCR activity was obtained over 1V/9STi and 1V/12STi samples, which yielded 90% NOx conversion at 300°C under a high gas hourly space velocity (GHSV) of 200,000 hour–1. The purpose of this study is to elucidate the fundamental NH3-SCR principles of vanadia-based catalysts; thus, we used a simplified V2O5/TiO2 catalytic system without introducing any promoter components. A comparison of the activity with that of previously reported catalysts (18, 19) shows that the activity of our 1V/9STi catalyst, expressed in terms of reaction rate and turnover frequency (TOF), is comparable to that of many commercial V/WTi catalysts (table S3). The selectivity toward N2 was maintained at about 98% over the whole temperature range (fig. S2). Moreover, the 1V/9STi sample exhibited excellent resistance to SO2 and H2O (fig. S3). To confirm the effect of sulfur content on SCR activity, we treated the support with NH3 to remove the sulfur, and the obtained 1V/(Ti-NH3) sample showed the lowest catalytic activity.
Fig. 1. Catalytic activity and catalyst characterization.
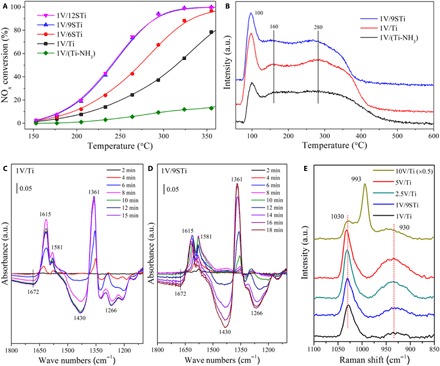
(A) NOx conversion as a function of temperature in the feed gas of 500 parts per million (ppm) NO/500 ppm NH3/5.0 vol % O2/N2 (200,000 hour−1). (B) NH3-TPD of vanadia-based catalysts after 1% NH3/N2 adsorption at 50°C for 1 hour, followed by Ar purging. a.u., arbitrary units. (C and D) In situ DRIFTS of NO + O2 adsorption on 1V/Ti and 1V/9STi catalysts pretreated with NH3 at 150°C with the NH3-adsorbed samples as background. (E) Raman results of vanadia-based catalysts under an oxygen flow at 400°C.
Reactivity of surface acid sites
It has been well established that the occurrence of NH3-SCR requires both acid and redox sites on the vanadia-based catalysts working together (1, 20, 21). Therefore, there are basically two possibilities, i.e., a change in the acid sites or the redox sites, to explain the enhancement of catalytic performance induced by sulfur introduction. To elucidate whether such enhancement derives from a change in the acid sites on the catalyst, we conducted temperature-programmed desorption of NH3 (NH3-TPD) (Fig. 1B) and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) (Fig. 1, C and D) studies. The adsorption of NH3 on Brønsted acid sites would result in the formation of NH4+, which is generally less thermally stable than NH3 bound to Lewis acid sites (22–24). Therefore, the low-, middle-, and high-temperature peaks of the NH3-TPD curves are attributed to the desorption of physisorbed NH3, NH3 bound to weak Brønsted acid sites, and NH3 bound to strong Brønsted and Lewis acid sites, respectively. The NH3-TPD result (Fig. 1B) shows that a high sulfur content on the catalysts increases the amount of Brønsted acid sites, whereas it decreases the amount of Lewis acid sites, in accordance with our DRIFTS data on NH3 adsorption (fig. S4A) and previous literature (25).
We investigated the reactivity of adsorbed NH3 toward NO + O2 using in situ DRIFTS (Fig. 1, C and D). The samples were exposed to NH3 until adsorption saturation and then flushed with N2 for collection of the background spectrum. After NO + O2 was introduced to the samples, the DRIFTS data were collected. The occurrence of negative infrared (IR) peaks indicates the consumption of adsorbed NH3 species. The NH3 adsorbed on Lewis acid sites and ad-NOx exhibit characteristic IR peaks in the same regions (1200 to 1300 cm–1 and 1500 to 1650 cm–1); hence, it is difficult to differentiate these species. For the 1V/TiO2 sample (Fig. 1C), the negative peaks at 1430 and 1672 cm–1 due to NH4+ on Brønsted acid sites appeared after the flow of NO + O2 for 5 min at 150°C and became more negative over time. This result indicates that during the first 5 min, the NH3 coordinated to Lewis acid sites participated in the NH3-SCR reaction, and then the NH4+ on Brønsted acid sites were consumed. The low reactivity of NH4+ bound on Brønsted acid sites was also observed for the 1V/9STi sample (Fig. 1D). The occurrence of the asymmetric O=S=O stretching vibration peaks of sulfate species at 1361 cm–1 (26, 27) denotes the consumption of NH3 species adsorbed on sulfate sites. Similar results were also obtained at 250°C (fig. S4, B and C). Ferri and co-workers (7) have also reported that NO reacts predominantly with NH3 coordinated to Lewis acid sites during NH3-SCR on the V2O5-WO3-TiO2 catalyst, while Brønsted acid sites are not involved in the catalytic cycle and mainly serve as an NH3 pool to replenish the Lewis acid sites, which is consistent with our experimental results.
As revealed above, sulfuration increases the amount of Brønsted acid sites, whereas it decreases the amount of Lewis acid sites. Moreover, the NH3 coordinated to Lewis acid sites preferentially participates in the NH3-SCR reaction (7). Therefore, this result suggests that the introduction of sulfate changes not only the acid sites but also other active centers (i.e., the redox sites), and the enhancement of NH3-SCR reactivity may result mainly from changes to the redox sites.
Structure of surface redox sites
We used Raman spectroscopy to determine the structure of dispersed surface vanadyl species. According to the literature (16, 28, 29), the experimentally observed bands at 1030, 993, and 930 cm–1 are characteristic of monomeric vanadyl species, crystalline V2O5, and polymeric vanadyl species, respectively. Therefore, the evident enhancement of the band at 930 cm–1 from the 1V/Ti sample to the 1V/9STi sample indicates that the introduction of sulfate causes the polymerization of monomeric vanadyl species under low vanadium loading (Fig. 1E). To confirm the Raman spectroscopy result, we conducted nuclear magnetic resonance (NMR) measurements. In general, the effect of 51V electronic shielding increases with the decrease of its isotropic chemical shift. The 51V NMR spectra (fig. S5) show that, with increasing sulfur content, the intensity of the central-band peak at lower chemical shifts (i.e., −655 ppm) was significantly increased, suggesting that the polymerization of vanadyl species was enhanced with sulfate introduction (30). The transformation of monomeric vanadyl to polymeric vanadyl species caused by sulfate introduction may be due to the surface sites of TiO2 being partially occupied by sulfate, which enables the vanadyl species to be close to each other (16). The XPS results (table S4 and fig. S6) show that the surface sulfate species do not have a significant effect on the valence state of the V ions; rather, they simply change the structure of dispersed vanadyl species.
NH3-SCR reaction pathways
To elucidate the mechanism underlying the activity difference between monomeric and polymeric vanadyl species at the atomic scale, we carried out DFT calculations (see Figs. 2 to 4). According to the computational results, the NH3-SCR de-NOx reaction over vanadia/TiO2 catalysts proceeds via the Eley-Rideal mechanism, and both the redox sites and the acid sites are involved in the SCR process. The results of DRIFT spectra (fig. S4A) and DFT calculations (fig. S7, A and B) show that NH3 is preferentially adsorbed on surface Ti sites (31). The adsorbed NH3 is activated by the transfer of an H atom to the vanadyl species and subsequently reacts with NO in the gas phase, resulting in the formation of an intermediate nitrosamide (NH2NO, denoted by the IR signals at 1505 cm−1 in fig. S4D) (7) and a V-OH or V-OH2 group (A→C, F→H, K→M, and P→R). Then, the NH2NO intermediate is decomposed into N2 and H2O (C→E, H→J, M→O, and R→T). Gas-phase O2 replenishes the consumed surface oxygen on the vanadyl species (J→K) (8). When the V=O groups are regenerated, a catalytic cycle is completed. The overall reaction barrier on the dimeric vanadyl species is predicted to be 1.34 eV, 0.21 eV lower than that on the monomeric vanadyl species (Fig. 2). According to transition state theory (32), at 493.15 K (220°C), this reduction of barriers would induce an increase in reaction rate of up to two orders of magnitude (table S5). This result indicates that the dimeric vanadyl species, rather than the monomeric vanadyl species, determine the NH3-SCR activity of vanadia-based catalysts, which is consistent with our experimental results (Fig. 1, A and E).
Fig. 2. Energy profile of the entire NH3-SCR process.
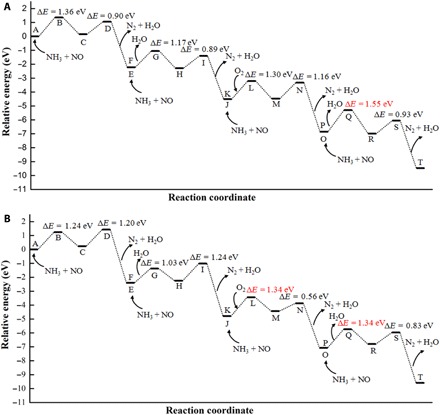
(A) Pathways over monomeric vanadia/TiO2 surfaces. (B) Pathways over dimeric vanadia/TiO2 surfaces. The letters at each state correspond to the structures in Figs. 3 and 4. The barrier in the rate-determining step is marked in red font.
Fig. 4. Optimized geometries of the reactant, transition states, intermediate, and product for all elementary steps in the NH3-SCR mechanism over the dimeric vanadia/TiO2 surfaces.
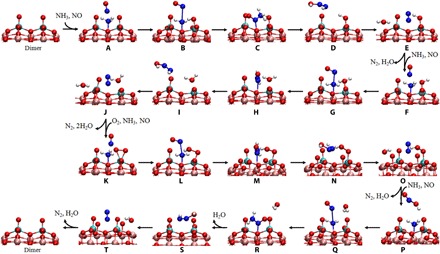
All legends are the same as those in Fig. 3.
It is noteworthy that a VOOH intermediate would be formed when an H atom transfers from the adsorbed NH3 to the adsorbed O2 on the vanadyl species (K→O). The VOOH intermediate is converted into the O=V-OH structure instantaneously for the monomeric vanadyl species (O→P in Fig. 3). However, the existence of an adjacent vanadyl enhances the thermal stability and lifetime of the VOOH intermediate due to the formation of a hydrogen bond between the VOOH group and the adjacent V=O group (fig. S7C), which allows a barrierless reaction between the VOOH intermediate and NO to occur on the polymeric vanadyl species. The regeneration of redox sites, as well as the formation of a nitrous acid molecule (HNO2), is achieved at this step (O→P in Fig. 4). Yet on the monomeric vanadyl species, the regeneration of redox sites does not occur until the P→R process (Fig. 3). In the following process, the reaction of adsorbed NH3 with HNO2 is energetically more favorable than the reaction of adsorbed NH3 with NO (P→T in Fig. 2). Therefore, the coupling effect of the polymeric vanadyl species not only shortens the reaction pathway for the regeneration of redox sites but also substantially reduces the overall reaction barrier of the catalytic cycle, which therefore greatly accelerates the NH3-SCR reaction. On the basis of the DFT calculations and experimental evidence, the entire NH3-SCR mechanism over the monomeric and dimeric vanadia/TiO2 surfaces was deduced and is summarized in Fig. 5.
Fig. 3. Optimized geometries of the reactant, transition states, intermediate, and product for all elementary steps in the NH3-SCR mechanism over the monomeric vanadia/TiO2 surfaces.
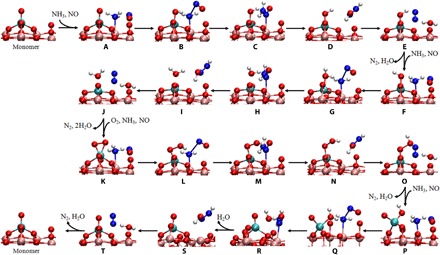
Red, pink, cyan, blue, and white circles denote O, Ti, V, N, and H atoms, respectively.
Fig. 5. Mechanism of the standard NH3-SCR reaction.
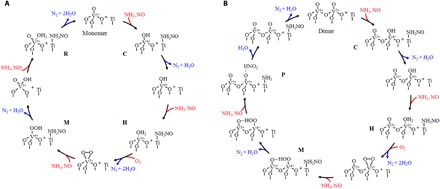
(A) Reactions over monomeric vanadia/TiO2 surfaces. (B) Reactions over dimeric vanadia/TiO2 surfaces. Reactants are marked in red, and products are marked in blue. The letters at each state correspond to the structures in Figs. 3 and 4.
DISCUSSION
Our study indicates that the surface structure of dispersed vanadia significantly affects the NH3-SCR activity of vanadia-based catalysts. Owing to the difference in elementary reaction steps, the polymeric vanadyl species exhibit markedly higher activity than the monomeric vanadyl species. The polymeric vanadia-based SCR mechanism reported here advances the understanding of the working principle of vanadia-based catalysts. In the removal of NOx from mobile sources, catalysts with high vanadium loading have been prepared to enhance the low-temperature activity. However, the high vanadium loading would result in a decrease of the thermal stability and an enhancement of SO2 oxidation. Here, we successfully obtained low vanadium–loading catalysts with excellent low-temperature SCR activity, which paves the way toward solving this problem.
MATERIALS AND METHODS
Catalyst synthesis and activity test
The SO42−-pretreated TiO2 supports were prepared by the wet impregnation method. The (NH4)2SO4 was dissolved in distilled water before TiO2 was added to the solution. The mixture was agitated for 1 hour, and then the moisture was evaporated at 60°C using a rotary vacuum evaporator before drying overnight at 100°C. The mixture was calcined in air for 3 hours at 300°C. The resulting samples were labeled as xSTi, where x is the loading amount of SO42−. 1V/xSTi catalysts were prepared by the wet impregnation method using ammonium metavanadate (1.0 wt % vanadium pentoxide) and calcined xSTi and were finally calcined in air for 3 hours at 500°C.
Before the NH3-SCR activity tests, the catalysts were pressed, crushed, and sieved to 40 to 60 mesh. The activity tests were carried out in a fixed-bed quartz flow reactor at atmospheric pressure. The reaction conditions were controlled as follows: 500 ppm NO, 500 ppm NH3, 5 volume percent (vol %) O2, 100 ppm SO2 (when used), 10 vol % H2O (when used), and N2 balance. Under ambient conditions, the total flow rate was 500 ml/min, and the GHSV was 200,000 hour−1. The effluent gas, including NO, NH3, NO2, and N2O, was continuously analyzed by a Thermo Antaris IGS FTIR gas analyzer equipped with a heated, low-volume, multiple-path gas cell (2 m).
Catalyst characterization
The surface area and pore characteristics of the catalysts were obtained from N2 adsorption/desorption analysis at −196°C using a Quantachrome Quadrasorb SI-MP. Before the N2 physisorption, the catalysts were degassed at 300°C for 5 hours. The surface area was determined by the Brunauer-Emmett-Teller equation in the 0.05 to 0.35 partial pressure range. The pore volume and average pore diameter were determined by the Barrett-Joyner-Halenda method from the desorption branches of the isotherms.
XPS spectra of the catalysts were recorded on a scanning X-ray microprobe (Axis Ultra, Kratos Analytical Ltd.) using Al Kα radiation (1486.7 eV). All the binding energies were calibrated using the C 1s peak (binding energy = 284.8 eV) as standard.
NH3-TPD experiments were performed using a quadrupole mass spectrometer (HPR-20, Hiden Analytical Ltd.) to record the signal of NH3 [mass/charge (m/z) ratio = 15 for NH]. Before TPD experiments, the samples (150 mg) were pretreated at 400°C in a flow of 20 vol % O2/N2 (50 ml min−1) for 0.5 hours and cooled down to room temperature. The samples were then exposed to a flow of 1% NH3/N2 (50 ml min−1) at 50°C for 1 hour, followed by Ar purging for 1 hour. Last, the temperature was raised to 600°C in Ar flow at a rate of 10°C min−1.
Raman spectra were measured on a LabRAM HR 800 Raman spectrometer using a 532-nm laser as the excitation source. The spectrometer was equipped with an in situ reaction cell (PIKE Technologies), in which samples can be heated to 500°C in a gas flow. In our experiments, all catalysts were heated from room temperature to 400°C under an oxygen flow and then the Raman spectra were acquired.
The 51V solid-state NMR experiments were performed at 11.7 T on a Bruker Avance III 500 spectrometer with a resonance frequency of 131.6 MHz, using a 1.9-mm HX double-resonance probe at a spinning rate of 40 kHz. The NMR spectra of 51V were acquired using a Hahn-echo pulse sequence with a π/2 pulse width of 1.5 μs. For our samples with 1 wt % V2O5 loading, 60,000 scans with a 0.3-s recycle delay were used. The 51V chemical shift was referenced to V2O5 at −610 ppm.
In situ DRIFTS measurements
In situ DRIFTS experiments were performed on a Fourier transform infrared (FTIR) spectrometer (Nicolet Nexus 670) equipped with a Smart Collector and an MCT/A detector cooled by liquid nitrogen. The reaction temperature was controlled precisely by an Omega programmable temperature controller. Before each experiment, the sample was pretreated at 300°C for 0.5 hours in a flow of 20 vol % O2/N2 and then cooled down to 150 or 250°C. The background spectra were collected in flowing N2 and automatically subtracted from the sample spectrum. The reaction conditions were controlled as follows: total flow rate (300 ml min−1), 500 ppm NH3 and/or 500 ppm NO, 5 vol % O2, and N2 balance. All spectra were recorded by accumulating 100 scans with a resolution of 4 cm−1.
Computational details
Geometries and energies were calculated using the Perdew-Burke-Ernzerhof functional (33) with van der Waals correction proposed by Grimme (i.e., DFT-D2 method) (34), as implemented in the Vienna ab initio simulation package (VASP 5.4.1) (35). The projector augmented wave method was used to describe the interaction between the ions and the electrons (36). The energy cutoff of the plane wave was set to 400 eV. A (2 × 4) supercell of the anatase (101) surface with two stoichiometric TiO2 layers (about 11 Å × 15 Å × 6 Å; see fig. S7, D and E) was used as the substrate. A vacuum gap of 12 Å was used to avoid the periodic image interaction normal to the surface. During the geometrical optimization, the bottom TiO2 layer was fixed at its bulk position, while all other atoms were allowed to relax, until the forces on each atom were smaller than 0.02 eV Å−1. Only the Γ point of the Brillouin zone was sampled. The Gaussian smearing method with a smearing width of 0.2 eV was used to accelerate the convergence of integration at the Brillouin zone. The reaction pathways and transition states were traced by the climbing image nudged elastic band method with a spring constant of 5.0 eV Å−2 (37, 38). No obvious differences in the structural properties (<0.002 Å) and energy barriers (<0.002 eV) were observed when k points were increased from the Γ point only to a 2 × 1 × 1 k-point mesh and the energy cutoff from 400 to 600 eV (table S6), indicating that the calculations had converged, and the computational settings used here were reliable for describing the studied reactions. The coupling effect between two adjacent vanadyl species was generally applicable in dimeric and higher-order polymeric vanadia structures. During the NH3-SCR of NO over the polymeric vanadyl species, the coupling effect between the two adjacent vanadyl species (i.e., within a dimer unit of vanadia) at the reaction site accelerated the whole catalytic cycle, and hence, it could be expected that dimeric and higher-order polymeric vanadia would have similar effects on the SCR reaction. The dimeric vanadyl species is the basic structural unit of various polymeric vanadia structures and can reasonably represent the coupling effect in polymeric vanadia structures; hence, it was used as the model in our DFT calculations.
Supplementary Material
Acknowledgments
We thank F. Deng, J. Xu, Q. Wang, and X. Zhao for help in NMR measurement and data analysis. The NMR experiment was performed at the Wuhan Institute of Physics and Mathematics, CAS. Funding: This work was supported by the National Natural Science Foundation of China (21637005 and 21607149), the National Key R&D Program of China (2016YFC0205301), and the K. C. Wong Education Foundation. Author contributions: H.H. conceived and supervised the project. G.H. conducted DFT calculations. Z.L., Y. Yang, K.L., X.S., Z.Y., and W.S. performed the experiments. G.H., Z.L., and Y. Yu wrote the manuscript. All the authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/11/eaau4637/DC1
Fig. S1. XRD patterns of vanadia/TiO2 samples.
Fig. S2. N2 selectivity as a function of temperature in the feed gas of 500 ppm NO/500 ppm NH3/5.0 vol % O2/N2 (200,000 hour−1).
Fig. S3. Effect of H2O and SO2 on NOx conversion over the 1V/9STi catalyst at 350°C in the feed gas of 500 ppm NO/500 ppm NH3/5.0 vol % O2/N2 (200,000 hour−1).
Fig. S4. DRIFT spectra of NH3 and NO + O2 adsorption on the vanadia/TiO2 catalysts.
Fig. S5. NMR spectra of vanadia/TiO2 samples.
Fig. S6. V 2p XPS spectra of vanadia/TiO2 samples.
Fig. S7. Models used in DFT calculations.
Table S1. Relative surface atomic concentrations of vanadia/TiO2 samples obtained from XPS.
Table S2. N2 physisorption results of vanadia/TiO2 samples.
Table S3. A comparison of reaction rates and TOFs between our catalyst and reported commercial catalysts for the SCR of NO with NH3 at 200°C.
Table S4. Vanadium valence distribution in vanadia/TiO2 samples obtained from XPS.
Table S5. NH3-SCR de-NOx reaction rate constants (k) at 493.15 K (220°C) over the monomeric and dimeric vanadia/TiO2 surfaces.
Table S6. DFT-calculated structural parameters and energy barriers (ΔE) for the formation of the first NH2NO intermediate over the monomeric vanadia/TiO2 surfaces (i.e., the A→C process in Figs. 2A and 3) with different computational settings.
REFERENCES AND NOTES
- 1.Topsøe N. Y., Mechanism of the selective catalytic reduction of nitric oxide by ammonia elucidated by in situ online Fourier transform infrared spectroscopy. Science 265, 1217–1219 (1994). [DOI] [PubMed] [Google Scholar]
- 2.Zhu M., Lai J. K., Tumuluri U., Wu Z., Wachs I. E., Nature of active sites and surface intermediates during SCR of NO with NH3 by supported V2O5–WO3/TiO2 catalysts. J. Am. Chem. Soc. 139, 15624–15627 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Busca G., Lietti L., Ramis G., Berti F., Chemical and mechanistic aspects of the selective catalytic reduction of NOx by ammonia over oxide catalysts: A review. Appl. Catal. B 18, 1–36 (1998). [Google Scholar]
- 4.Li J., Chang H., Ma L., Hao J., Yang R. T., Low-temperature selective catalytic reduction of NOx with NH3 over metal oxide and zeolite catalysts—A review. Catal. Today 175, 147–156 (2011). [Google Scholar]
- 5.Paolucci C., Khurana I., Parekh A. A., Li S., Shih A. J., Li H., Di Iorio J. R., Albarracin-Caballero J. D., Yezerets A., Miller J. T., Delgass W. N., Ribeiro F. H., Schneider W. F., Gounder R., Dynamic multinuclear sites formed by mobilized copper ions in NOx selective catalytic reduction. Science 357, 898–903 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Nie L., Mei D., Xiong H., Peng B., Ken Z., Hernandez X. I. P., DeLariva A., Wang M., Engelhard M. H., Kovarik L., Datye A. K., Wang Y., Activation of surface lattice oxygen in single-atom Pt/CeO2 for low-temperature CO oxidation. Science 358, 1419–1423 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Marberger A., Ferri D., Elsener M., Kröcher O., The significance of Lewis acid sites for the selective catalytic reduction of nitric oxide on vanadium-based catalysts. Angew. Chem. Int. Ed. 55, 11989–11994 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Zhu M., Lai J.-K., Tumuluri U., Ford M. E., Wu Z., Wachs I. E., Reaction pathways and kinetics for selective catalytic reduction (SCR) of acidic NOx emissions from power plants with NH3. ACS Catal. 7, 8358–8361 (2017). [Google Scholar]
- 9.Kwon D. W., Park K. H., Hong S. C., Influence of VOx, surface density and vanadyl species on the selective catalytic reduction of NO by NH3 over VOx/TiO2 for superior catalytic activity. Appl. Catal. A 499, 1–12 (2015). [Google Scholar]
- 10.Youn S., Jeong S., Kim D. H., Effect of oxidation states of vanadium precursor solution in V2O5/TiO2 catalysts for low temperature NH3 selective catalytic reduction. Catal. Today 232, 185–191 (2014). [Google Scholar]
- 11.Yun D., Herrera J. E., A novel methodology for in situ redox active site titration of TiO2-supported vanadia during ethanol partial oxidation catalysis. J. Catal. 350, 72–85 (2017). [Google Scholar]
- 12.Went G. T., Leu L.-j., Bell A. T., Quantitative structural analysis of dispersed vanadia species in TiO2 (anatase)-supported V2O5. J. Catal. 134, 479–491 (1992). [Google Scholar]
- 13.Li C., Identifying the isolated transition metal ions/oxides in molecular sieves and on oxide supports by UV resonance Raman spectroscopy. J. Catal. 216, 203–212 (2003). [Google Scholar]
- 14.Went G. T., Oyama S. T., Bell A. T., Laser Raman spectroscopy of supported vanadium oxide catalysts. J. Phys. Chem. 94, 4240–4246 (1990). [Google Scholar]
- 15.Marberger A., Elsener M., Ferri D., Kröcher O., VOx surface coverage optimization of V2O5/WO3-TiO2 SCR catalysts by variation of the V loading and by aging. Catalysts 5, 1704–1720 (2015). [Google Scholar]
- 16.Choo S. T., Lee Y. G., Nam I.-S., Ham S.-W., Lee J.-B., Characteristics of V2O5 supported on sulfated TiO2 for selective catalytic reduction of NO by NH3. Appl. Catal. A 200, 177–188 (2000). [Google Scholar]
- 17.Arnarson L., Falsig H., Rasmussen S. B., Lauritsen J. V., Moses P. G., A complete reaction mechanism for standard and fast selective catalytic reduction of nitrogen oxides on low coverage VOx/TiO2(001) catalysts. J. Catal. 346, 188–197 (2017). [Google Scholar]
- 18.Hu W. S., Gao X., Deng Y. W., Qu R., Zheng C. H., Zhu X. B., Cen K. F., Deactivation mechanism of arsenic and resistance effect of SO42− on commercial catalysts for selective catalytic reduction of NOx with NH3. Chem. Eng. J. 293, 118–128 (2016). [Google Scholar]
- 19.Kim M. H., Ham S.-W., Determination of N2O emissions levels in the selective reduction of NOx by NH3 over an on-site-used commercial V2O5–WO3/TiO2 catalyst using a modified gas cell. Top. Catal. 53, 597–607 (2010). [Google Scholar]
- 20.Topsoe N. Y., Dumesic J. A., Topsoe H., Vanadia/titania catalysts for selective catalytic reduction of nitric oxide by ammonia: II studies of active sites and formulation of catalytic cycles. J. Catal. 151, 241–252 (1995). [Google Scholar]
- 21.Wang C., Yang S., Chang H., Peng Y., Li J., Dispersion of tungsten oxide on SCR performance of V2O5—WO3/TiO2: Acidity, surface species and catalytic activity. Chem. Eng. J. 225, 520–527 (2013). [Google Scholar]
- 22.Lian Z., Liu F., He H., Liu K., Nb-doped VOx/CeO2 catalyst for NH3-SCR of NOx at low temperatures. RSC Adv. 5, 37675–37681 (2015). [Google Scholar]
- 23.Roy S., Viswanath B., Hegde M. S., Madras G., Low-temperature selective catalytic reduction of NO with NH3 over Ti0.9M0.1O2-δ (M = Cr, Mn, Fe, Co, Cu). J. Phys. Chem. C 112, 6002–6012 (2008). [Google Scholar]
- 24.Jin R., Liu Y., Wu Z., Wang H., Gu T., Low-temperature selective catalytic reduction of NO with NH3 over MnCe oxides supported on TiO2 and Al2O3: A comparative study. Chemosphere 78, 1160–1166 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Zhao H., Bennici S., Shen J., Auroux A., Nature of surface sites of V2O5–TiO2/SO42− catalysts and reactivity in selective oxidation of methanol to dimethoxymethane. J. Catal. 272, 176–189 (2010). [Google Scholar]
- 26.Long R. Q., Yang R. T., Selective catalytic reduction of nitrogen oxides by ammonia over Fe3+-exchanged TiO2-pillared clay catalysts. J. Catal. 186, 254–268 (1999). [Google Scholar]
- 27.Liu F., Asakura K., He H., Shan W., Shi X., Zhang C., Influence of sulfation on iron titanate catalyst for the selective catalytic reduction of NOx with NH3. Appl. Catal. B 103, 369–377 (2011). [Google Scholar]
- 28.Besselmann S., Löffler E., Muhler M., On the role of monomeric vanadyl species in toluene adsorption and oxidation on V2O5/TiO2 catalysts: A Raman and in situ DRIFTS study. J. Mol. Catal. A Chem. 162, 401–411 (2000). [Google Scholar]
- 29.Christodoulakis A., Machli M., Lemonidou A. A., Boghosian S., Molecular structure and reactivity of vanadia-based catalysts for propane oxidative dehydrogenation studied by in situ Raman spectroscopy and catalytic activity measurements. J. Catal. 222, 293–306 (2004). [Google Scholar]
- 30.Hu J. Z., Xu S., Li W.-Z., Hu M. Y., Deng X., Dixon D. A., Vasiliu M., Craciun R., Wang Y., Bao X., Peden C. H. F., Investigation of the structure and active sites of TiO2 nanorod supported VOx catalysts by high-field and fast-spinning 51V MAS NMR. ACS Catal. 5, 3945–3952 (2015). [Google Scholar]
- 31.Giraud F., Geantet C., Guilhaume N., Gros S., Porcheron L., Kanniche M., Bianchi D., Experimental microkinetic approach of de-NOx by NH3 on V2O5/WO3/TiO2 catalysts. 1. Individual heats of adsorption of adsorbed NH3 species on a sulfate-free TiO2 support using adsorption isobars. J. Phys. Chem. C 118, 15664–15676 (2014). [Google Scholar]
- 32.Canneaux S., Bohr F., Henon E., KiSThelP: A program to predict thermodynamic properties and rate constants from quantum chemistry results. J. Comput. Chem. 35, 82–93 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Perdew J. P., Burke K., Ernzerhof M., Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996). [DOI] [PubMed] [Google Scholar]
- 34.Grimme S., Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Kresse G., Furthmüller J., Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996). [DOI] [PubMed] [Google Scholar]
- 36.Kresse G., Joubert D., From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999). [Google Scholar]
- 37.Henkelman G., Uberuaga B. P., Jónsson H., A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000). [Google Scholar]
- 38.He G., Ma J., He H., Role of carbonaceous aerosols in catalyzing sulfate formation. ACS Catal. 8, 3825–3832 (2018). [Google Scholar]
- 39.Johnson E. R., Keinan S., Mori-Sánchez P., Contreras-García J., Cohen A. J., Yang W., Revealing noncovalent interactions. J. Am. Chem. Soc. 132, 6498–6506 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu T., Chen F., Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012). [DOI] [PubMed] [Google Scholar]
- 41.M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09, Revision D.01 (Gaussian Inc., 2013).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/11/eaau4637/DC1
Fig. S1. XRD patterns of vanadia/TiO2 samples.
Fig. S2. N2 selectivity as a function of temperature in the feed gas of 500 ppm NO/500 ppm NH3/5.0 vol % O2/N2 (200,000 hour−1).
Fig. S3. Effect of H2O and SO2 on NOx conversion over the 1V/9STi catalyst at 350°C in the feed gas of 500 ppm NO/500 ppm NH3/5.0 vol % O2/N2 (200,000 hour−1).
Fig. S4. DRIFT spectra of NH3 and NO + O2 adsorption on the vanadia/TiO2 catalysts.
Fig. S5. NMR spectra of vanadia/TiO2 samples.
Fig. S6. V 2p XPS spectra of vanadia/TiO2 samples.
Fig. S7. Models used in DFT calculations.
Table S1. Relative surface atomic concentrations of vanadia/TiO2 samples obtained from XPS.
Table S2. N2 physisorption results of vanadia/TiO2 samples.
Table S3. A comparison of reaction rates and TOFs between our catalyst and reported commercial catalysts for the SCR of NO with NH3 at 200°C.
Table S4. Vanadium valence distribution in vanadia/TiO2 samples obtained from XPS.
Table S5. NH3-SCR de-NOx reaction rate constants (k) at 493.15 K (220°C) over the monomeric and dimeric vanadia/TiO2 surfaces.
Table S6. DFT-calculated structural parameters and energy barriers (ΔE) for the formation of the first NH2NO intermediate over the monomeric vanadia/TiO2 surfaces (i.e., the A→C process in Figs. 2A and 3) with different computational settings.