

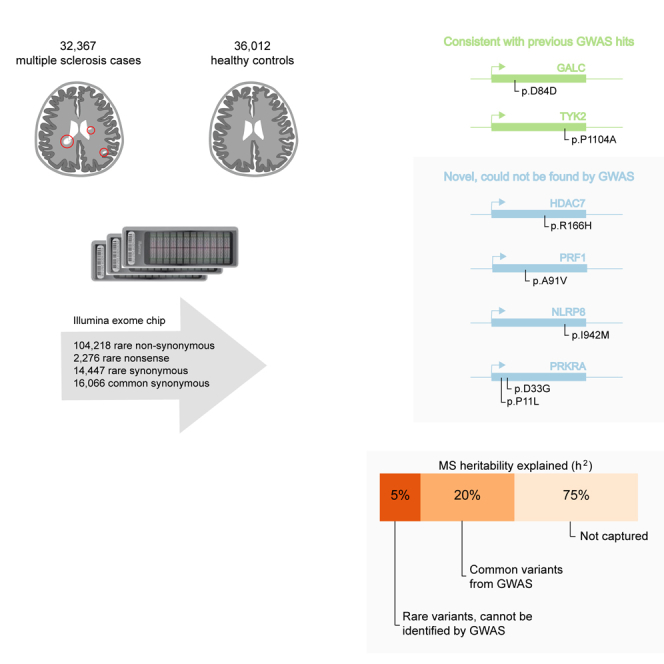
Summary
Multiple sclerosis is a complex neurological disease, with ∼20% of risk heritability attributable to common genetic variants, including >230 identified by genome-wide association studies. Multiple strands of evidence suggest that much of the remaining heritability is also due to additive effects of common variants rather than epistasis between these variants or mutations exclusive to individual families. Here, we show in 68,379 cases and controls that up to 5% of this heritability is explained by low-frequency variation in gene coding sequence. We identify four novel genes driving MS risk independently of common-variant signals, highlighting key pathogenic roles for regulatory T cell homeostasis and regulation, IFNγ biology, and NFκB signaling. As low-frequency variants do not show substantial linkage disequilibrium with other variants, and as coding variants are more interpretable and experimentally tractable than non-coding variation, our discoveries constitute a rich resource for dissecting the pathobiology of MS.
Graphical Abstract
Highlights
-
•
Almost 20% of MS risk heritability can be attributed to common genetic variants
-
•
We show that nearly 5% of heritability is explained by coding low-frequency variants
-
•
We identify four novel genes driving risk independently of common-variant signals
-
•
These genes would not have been found by common-variant studies
In a large multi-cohort study, unexplained heritability for multiple sclerosis is detected in low-frequency coding variants that are missed by GWAS analyses, further underscoring the role of immune genes in MS pathology.
Introduction
Multiple sclerosis (MS; MIM 126200) is an autoimmune disease of the central nervous system and a common cause of neurologic disability in young adults (Compston and Coles, 2008). It is most prevalent in individuals of northern European ancestry and—in line with other complex, common disorders—shows substantial heritability (Binder et al., 2016), with a sibling standardized incidence ratio of 7:1 (Westerlind et al., 2014). Over the last 15 years, we have identified 233 independent, common-variant associations mediating disease risk by genome-wide association studies (GWASs) of increasing sample size (Andlauer et al., 2016, Australia and New Zealand Multiple Sclerosis Genetics Consortium (ANZgene), 2009, Baranzini et al., 2009, Beecham et al., 2013, De Jager et al., 2009, International Multiple Sclerosis Genetics Consortium et al., 2011, International Multiple Sclerosis Genetics Consortium et al., 2017, Jakkula et al., 2010, Martinelli-Boneschi et al., 2012, Nischwitz et al., 2010, Patsopoulos et al., 2011, Sanna et al., 2010, Burton et al., 2007). In our most recent meta-analysis of 14,802 MS cases and 26,703 controls, these effects—including 32 mapping to classical human leukocyte antigen (HLA) alleles and other variation in the major histocompatibility (MHC) locus (International Multiple Sclerosis Genetics Consortium et al., 2017, Moutsianas et al., 2015, Patsopoulos et al., 2013)—account for 7.5% of h2g, the heritability attributable to additive genetic effects captured by genotyping arrays, with a total of 19.2% of h2g attributable to all common variants in the autosomal genome (International Multiple Sclerosis Genetics Consortium et al., 2017). MS is thus a prototypical complex disease with a substantial portion of heritability determined by hundreds of common genetic variants, each of which explain only a small fraction of risk (Sawcer et al., 2014).
As with other common, complex diseases where large GWASs have been conducted, we find that although common variants (minor allele frequency [MAF] > 5%) account for the bulk of trait heritability, they cannot account for its entirety. Identifying the source of this unexplained heritability has thus become a major challenge (Manolio et al., 2009). Two hypotheses are frequently advanced: some common variants show epistatic (i.e., non-additive) interactions so that they contribute more risk in combination than each does alone, and a portion of risk is due to rare variants that cannot be imputed via linkage disequilibrium to common variants present on genotyping arrays and are therefore invisible to heritability calculations based on such arrays. The only evidence we have found for epistatic interactions between common MS risk variants is between two HLA haplotype families in the MHC locus (Moutsianas et al., 2015). This lack of epistatic interactions is consistent with other common, complex diseases, both of the immune system and beyond (Altshuler et al., 2008). We have also found no evidence that mutations in individual families drive disease risk in genome-wide linkage analyses of 730 MS families with multiple affected members (Sawcer et al., 2005). These results indicate that neither epistasis between known risk variants nor mutations in a limited number of loci are major sources of MS risk. They do not, however, preclude a role for variants present in the population at low frequencies, which cannot be imputed but are likely to individually contribute moderate risk.
Here, we report our assessment of the contribution of low-frequency variation in gene coding regions to MS risk. We conducted a meta-analysis of 120,991 low-frequency coding variants across all autosomal exons, including 104,218 non-synonymous and 2,276 nonsense variants, which are more likely to have a phenotypic effect. We analyzed a total of 32,367 MS cases and 36,012 controls drawn from centers across Australia, 10 European countries, and multiple US states, which we genotyped either on the Illumina HumanExome Beadchip (exome chip) or on a custom array (the MS chip), incorporating the exome chip content (International Multiple Sclerosis Genetics Consortium et al., 2017), and which satisfied our stringent quality control filters (Figure S1 and Table S1). The exome array is a cost-efficient alternative to exome sequencing, capturing approximately 88% of low-frequency and rare-coding variants present in 33,370 non-Finnish Europeans included in the Exome Aggregation Consortium (MAFs between 0.0001 and 0.05; Figure S1), and <5% of the extremely rare alleles present at even lower frequencies. Our study was well powered, with 80% power to detect modest effects at low frequency (odds ratio [OR] = 1.15 at MAF = 5%) and rare variants (OR = 1.5 at MAF = 0.5%) at a significance threshold of p < 3.5 × 10−7 (Bonferroni correction for the total number of variants genotyped).
Figure S1.

Data Quality Overview, Related to STAR Methods
(A) QC process. We assembled 42 cohorts of data (either entire country-level collections or groups of samples processed as a batch; Table S1). We called common variant genotypes with the standard algorithm provided by Illumina (GenCall), and low-frequency variants with zCall, an algorithm specifically developed to call these variants on the exome chip (Goldstein et al., 2012). We performed initial quality control on each cohort separately to account for variation between batches and cohorts (upper gray region), then merged cohorts into 13 country-level strata. To ensure that these strata were uniform we then performed stringent quality control on each stratum (lower gray region) to produce our final dataset.
(B) the exome chip captures a large fraction of ExAC (release version 1) low-frequency miss-sense variants. The exome chip captures the majority of variants present in ExAC (Lek et al., 2016) down to a minor allele frequency ∼0.0005, below which a large number of variants is observed (left). Thus, the overall coverage at very rare alleles (5 × 10−4 > MAF > 1.5 × 10−5, corresponding to a single allele seen in 33,370 non-Finnish European individuals in ExAC) is low (right).
Results
We first assessed the contribution of individual variants to MS risk by conducting a meta-analysis of association statistics across 14 country-level strata (Figure 1 and Table S1). We used linear mixed models to correct for population structure in 13 of these strata, estimated from the 16,066 common, synonymous coding variants present on the exome chip (i.e., variants with MAF > 5% in our samples). We included population structure-corrected summary statistics for the remaining cohort (from Germany), which has been previously described (Dankowski et al., 2015). As expected, we saw a strong correlation between effect size and variant frequency, with rarer alleles exerting larger effects (Figure S2). We found significant association between MS risk and seven low-frequency coding variants in six genes outside the extended MHC locus on chromosome 6 (Table 1 and Figure S3). Two of these variants (TYK2 p.Pro1104Ala, overall MAF 4.1% in our samples; GALC p.Asp84Asp, overall MAF 3.9%) are in regions identified by our latest MS GWAS and show linkage disequilibrium with the common-variant associations we have previously reported (International Multiple Sclerosis Genetics Consortium et al., 2011). The remaining associations are novel, with the variants neither in linkage disequilibrium nor physical proximity to common variant association signals and thus not imputable in our GWASs (Table S2).
Figure 1.

Rare-Coding Variants Are Associated to Multiple Sclerosis Risk in a Multi-cohort Study
(A–C) We analyzed 120,991 low-frequency non-synonymous coding variants across all autosomal exons in 32,367 MS cases and 36,012 controls drawn across the International Multiple Sclerosis Genetics Consortium centers. We find evidence for association with both common variants with combined MAF > 5% (A) and with rare variants across the autosomes (B). We sourced samples from Australia, 10 European countries, and the United States (C).
See also Figures S2 and S3.
Figure S2.

Low-Frequency Variant Association Statistic Characteristics, Related to Figure 1
(A) effect sizes increase at low minor allele frequency. We conducted a meta-analysis of 120,991 low-frequency coding variants across all autosomal exons, concentrating on non-synonymous variants which are more likely to have a phenotypic effect. We analyzed a total of 32,367 MS cases and 36,012 controls in thirteen strata. Here, we show that estimates of effect size (β or log odds ratio, y axis) increase at low allele frequency (number of minor alleles present in control samples, x axis). Because many low-frequency variants are not present in all cohorts, we stratify these data by number of cohorts in which a variant is polymorphic (subplots). Rarer variants have larger estimated effect sizes and are present in fewer cohorts.
(B) forest plots for genome-wide significant low-frequency variants. Seven variants in six genes are significant in our analysis (p < 3.5 × 10−7, Bonferroni correction for the total number of variants genotyped). Two of these (TYK2 p.Pro1104Ala and GALC p.Asp84Asp), are in linkage disequilibrium with known GWAS hits. Studies are ordered by increasing sample size.
Table 1.
Coding Variants Associated to Multiple Sclerosis Risk
| Chr | Position | rsID | Minor Allele | MAF | Studies Observed | P Value | OR | LCI | UCI | Gene | AA Change |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 14 | 88452945 | rs11552556 | A | 3.9% | 14 | 5.759E−14 | 0.95 | 0.93 | 0.97 | GALC | Synonymous D84D |
| 19 | 10463118 | rs34536443 | G | 4.1% | 13 | 6.282E−13 | 0.95 | 0.93 | 0.97 | TYK2 | Missense P1104A |
| 10 | 72360387 | rs35947132 | A | 5.0% | 14 | 1.043E−10 | 1.04 | 1.02 | 1.06 | PRF1 | Missense A91V |
| 2 | 179315031 | rs61999302 | T | 5.6% | 12 | 6.467E−10 | 0.95 | 0.93 | 0.97 | PRKRA | Missense D33G |
| 2 | 179315726 | rs62176112 | A | 5.6% | 12 | 6.633E−10 | 0.95 | 0.93 | 0.97 | PRKRA | Missense P11L |
| 19 | 56487619 | rs61734100 | C | 0.2% | 9 | 1.925E−07 | 0.78 | 0.67 | 0.91 | NLRP8 | Missense I942M |
| 12 | 48191247 | rs148755202 | T | 1.4% | 14 | 2.597E−07 | 0.94 | 0.91 | 0.98 | HDAC7 | Missense R166H |
We analyzed 120,991 low-frequency non-synonymous coding variants across all autosomal exons in 32,367 MS cases and 36,012 controls drawn from centers across Australia, 10 European countries, and multiple US states. Genome positions are relative to hg19. The two variants in PRKRA are in linkage disequilibrium (R2 = 1, D` = 1 in the 1000 Genomes European samples). These variants lie in common variant risk loci found in our previous GWAS (International Multiple Sclerosis Genetics Consortium et al., 2017).
Figure S3.

Patterns of Association for Common and Rare Variants in Seven Genome-wide Significant Loci, Related to Figure 1
Plots are centered on the seven variants reported in Figure 1 and Table 1. Each show LD and association signal of low frequency variants (circles, this study), and common variants from our most recent GWAS (squares, 14,802 MS cases and 26,703 controls; International Multiple Sclerosis Genetics Consortium et al., 2017) and the ImmunoChip meta-analysis (diamonds; Beecham et al., 2013). For GALC and TYK2, our most associated variants, rs11552556 and rs34536443 respectively, capture the common variant signals we have previously reported (panels A and G). For the remaining loci, our most associated variants show no LD to other variants, with no evidence of association in our common variant studies (panels B-F).
We were struck by the observation that the minor allele is protective in six of the seven cases in Table 1, a trend we also observe at less stringent significant thresholds (Figure S2). This pattern is unusual in common-variant studies: for example, in our most recent GWAS, 101/200 non-MHC effects showed that the minor allele increases risk. To test if this phenomenon is due to our strata containing more cases than controls, we randomly resampled 4,000 affected and 4,000 unaffected samples in our three largest strata and calculated association statistics as for our main analysis. In this symmetric design, we found no bias toward protective minor alleles at even modest levels of significance (Table S3). Thus, low-frequency variants do not preferentially decrease MS risk rather than increase it.
Though we are able to identify individual low-frequency variants associated with MS risk, we recognize that we cannot detect all such variants at genome-wide significance, even in a study of this magnitude. We thus sought to quantify the overall contribution of low-frequency coding variation to MS risk. We used a restricted maximum-likelihood approach to model heritability attributable to genotypic variation across the genome that was initially developed for common-variant analyses (Yang et al., 2011) and later shown to also perform well for rare variants, as in the present case (Mancuso et al., 2016). In each of the 13 strata that comprise our data, we estimated the proportion of heritability explained by common (MAF > 5%) and low-frequency (MAF ≤ 5%; Table S4) variants on the exome arrays (Yang et al., 2011). We included genotype-derived principal components to further control for population stratification. By meta-analyzing these estimates across the twelve strata where the restricted maximum likelihood model converged, we found that low-frequency variants explain 11.34% (95% confidence interval 11.33%–11.35%) of the observed difference between cases and controls (mean estimate 4.1% on the liability scale; Figure 2). We further partitioned the low-frequency variants into intermediate (5% > MAF ≥ 1%) and rare (MAF < 1%) and found that the latter alone explain 9.0% (95% confidence interval 8.9%–9.1%) on the observed scale (mean estimate 3.2% on the liability scale; Figure 2). We note that six of the eight genome-wide significant variants presented in Table 1 are of intermediate frequency and thus are not included in the rare category. We capture the majority, though not all, of known common risk variants to some extent with the common variants on the exome chip (Table S5); our analysis therefore adequately, though imperfectly, models this portion of the frequency spectrum. Our results thus indicate that many more non-synonymous rare variants contribute to MS risk but are not individually detectable at genome-wide thresholds, even in large studies like ours.
Figure 2.

Rare Variants Explain a Substantial Portion of Multiple Sclerosis Heritability
We estimated the MS risk heritability explained by common variants (MAF > 5%) and low-frequency non-synonymous coding variation (MAF < 5%) in each of 13 cohorts genotyped on the exome chip using genome-wide complex trait analysis (GCTA; top). By meta-analyzing these estimates across cohorts, we found that low-frequency variants explain 11.34% of heritability on the observed scale, which corresponds to 4.1% on the liability scale (right top). After dividing the low-frequency variants into intermediate (5% > MAF > 1%) and rare (MAF < 1%; bottom), we found that the latter alone explains 9.0% heritability on the observed scale (3.2% on the liability scale; bottom right). Meta-analysis confidence intervals are small and visually occluded by the mean estimate plot characters. Cohorts (abbreviations as in Table S1) are ordered by sample size, with the percentage of the overall sample size shown in each subplot title. We could not obtain estimates for either model for our Finnish cohort (see STAR Methods; not shown), or for the three-component model for our Belgian cohort (bottom, top row, fourth from left). Both cohorts are small, which may explain the failure to converge.
In this study, we show that low-frequency coding variation explains a fraction of MS risk that cannot be attributed to common variants across the genome. We capture most, but not all, low-frequency missense variants (Figure S1), suggesting our heritability estimates for low-frequency and rare variation are conservative. This broadly agrees with previous reports that such variants contribute to complex traits, including Alzheimer’s disease (Sims et al., 2017) and schizophrenia (Purcell et al., 2014), where heritability modeling similar to ours supports a role for rare variants. Studies of quantitative phenotypes shared by the entire population, such as height (Marouli et al., 2017), serum lipid levels (Liu et al., 2017), and blood cell traits (Chami et al., 2016, CHARGE Consortium Hematology Working Group, 2016) have also reported novel associations to low-frequency coding variants outside the large number of known GWAS loci in each trait. However, a meta-analysis of different type 2 diabetes study designs found no associations outside common-variant GWAS regions (Fuchsberger et al., 2016), though this may be due to the heterogeneity of sample ascertainment and study design. In aggregate, therefore, our results and these past studies demonstrate that rare coding variants contribute a fraction of common, complex trait heritability. These results also agree with both theoretical expectation and empirical observations that low-frequency coding variants are under natural selection and are unlikely to increase in frequency in the population (Nelson et al., 2012, Schoech et al., 2017, Zeng et al., 2018). Thus, some portion of disease-associated variants, and hence the genes they influence, may not be detectable with conventional GWAS designs.
The newly discovered genes have clear immunological functions, confirming that MS pathogenesis is primarily driven by immune dysfunction. The associated polymorphisms show negligible linkage disequilibrium with other variants (Table S2), so the genes harboring them are likely to be relevant to disease. PRF1 encodes perforin, a key component of the granzyme-mediated cytotoxicity pathways used by several lymphocyte populations. In addition to cytotoxic lymphocytes and natural killer (NK) cells (House et al., 2015), perforin-dependent cytotoxicity is also seen in CD4+FOXP3+ regulatory T cells (Tregs), which show aberrant, T helper-like IFNγ secretion in MS patients (Dominguez-Villar et al., 2011). The MS risk variant rs35947132 (p.Ala91Val) is associated with a decrease in target cell-killing efficiency and increases in IFNγ secretion by NK cells (House et al., 2015), which aligns with the aberrant Treg phenotype observed in MS. This decreased cytotoxicity efficiency will prolong average cell-cell interactions with target cells, and such extended interactions are known to increase T cell-receptor-mediated signaling and induce changes to T cell phenotypes, especially secretion of IFNγ and other cytokines (Constant et al., 1995). Similarly, HDAC7 encodes the class II histone deacetylase 7, which potentiates the repressive effects of FOXP3, the master regulator governing naive CD4+ T cell development into Tregs (Bettini et al., 2012, Li et al., 2007). It also regulates T cell survival during their development in the thymus (Kasler et al., 2011). PRKRA encodes protein kinase interferon-inducible double-stranded RNA-dependent activator; in response to double-stranded RNA due to virus infection, it heterodimerizes with protein kinase R to inhibit EIF2a-dependent translation, resulting in upregulation of nuclear factor κB (NFκB) signaling, interferon production, and eventually, apoptosis (Sadler and Williams, 2008). NFκB-mediated signaling is a core feature of MS pathogenesis, which we have shown to be altered by at least one MS-associated variant (Housley et al., 2015) and may be the relevant mechanism for this gene. Finally, NLRP8 is an intracellular cytosolic receptor active in innate immune responses; the Ile942Met MS risk variant rs61734100 is detected only in individuals with European ancestry in ExAC, consistent with the higher prevalence of MS in European ancestry populations.
Discussion
Broadly, therefore, our results show that low-frequency genetic variation explains a portion of MS risk and that this variation impacts genes not detectable by common-variant association studies. Our heritability modeling demonstrates that more low-frequency and rare-variant associations remain to be discovered, though larger sample sizes will be required to increase statistical power. Recent attention has focused on changes to the adaptive immune system as pathogenic for MS, particularly to functional changes in helper T cell subsets and B cells after they have been released from the thymus and bone marrow, respectively, into the peripheral blood stream. These processes remain important to pathogenesis and are supported by a wealth of data, including our own GWAS (International Multiple Sclerosis Genetics Consortium et al., 2017). However, two of the four new genes we report (PRKRA and NLRP8) have clear functions in innate immunity, and HDAC7 plays a central role in the development of T cells in the thymus. Roles for both innate immune function and thymic development in MS pathogenesis are also supported by pathway analyses of our most recent GWAS data (International Multiple Sclerosis Genetics Consortium et al., 2017), an independent observation due to the lack of linkage disequilibrium (LD) between the variants in this study and those in our GWAS and the non-overlapping sample collections. Our data thus expand the scope of immune function relevant to MS pathogenesis.
The mechanisms whereby our newly discovered variants alter MS risk will require detailed experimental dissection: even when we can directly implicate specific genes and variants, these can have diverse consequences across multiple cell types. For example, perforin 1 has key—and potentially distinct—roles in cytotoxic T cells, regulatory helper T cells, NK cells, and other cell types. Both the effects of the variant on each of these functions and their relevance to MS pathogenesis will thus require demonstration, as is the case for the genes central to IFNγ biology, Treg function, and the NFκB signaling pathway.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited Data | ||
| Genotype data | This paper (Not all data are available at EGA—please contact dac@imsgc.net for access to the entire dataset) | EGAS00001003195 |
| Software and Algorithms | ||
| Plink v1.9 | Purcell et al., 2007 | https://www.cog-genomics.org/plink/1.9/ |
| GCTA | Yang et al., 2011 | http://cnsgenomics.com/software/gcta |
| EIGENSOFT | Price et al., 2006 | https://github.com/DReichLab/EIG |
| The R Project for Statistical Computing | R Development Core Team, 2017 | https://www.R-project.org |
| QC and analysis pipeline | This paper | https://github.com/cotsapaslab/IMSGCexomechip |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Chris Cotsapas (chris.cotsapas@yale.edu).
Experimental Model and Subject Details
We assembled a total of 76,140 samples (36,219 cases, 38,629 controls and 1,292 samples with missing phenotype information) from across the International MS Genetics Consortium (IMSGC; Table S1). All individuals gave informed consent at enrolment, and recruitment was monitored by research ethics boards in Australia: University of Tasmania; Bond University; University of Sydney. Belgium: Katholieke Universiteit, Leuven. Canada: McGill University, Montreal. Denmark: University of Copenhagen. Finland: University of Helsinki. France: Hôpital Pitié-Salpêtrière, Paris; Hôpital Neurologique Pierre Wertheimer, Bron; Université de Nantes. Germany: University of Lübeck; Max Planck Institute of Psychiatry, Munich; Technische Universität München; Johannes Gutenberg University-Medical Center, Mainz; Klinikums at Augsburg, Hanover and Großhadern Munich; Universitätsklinikums of Hamburg, Erlangen, Gießen/Marburg, Leipzig, Köln, Münster, Heidelberg, Rostock, and Tübingen, the Universität Ulm. Greece: University of Larissa. Italy: University of Eastern Piedmont, Novara; Ospedale Maggiore, Novara; San Raffaele Scientific Institute, Milan; University of Milan. Netherlands: Erasmus MC, Rotterdam; VU University Medical Center, Amsterdam. Norway: University of Bergen; University of Oslo. Spain: Universitat Autònoma de Barcelona. Sweden: Karolinska Institutet, Stockholm. Switzerland: University Hospital Zurich. United States of America: Yale University, New Haven CT; Brigham & Women’s Hospital, Boston MA; the University of Miami, Miami FL; UCSF and USB San Francisco, CA; Kaiser Permanente Divison of Research, Oakland, CA; Johns Hopkins University Baltimore MD; Washington University St Louis, St Louis MO; Vanderbilt University Medical Center, Nashville TN; Brigham Young University, Provo, UT; Case Western Reserve University, Cleveland, OH; The University of Pennsylvania and the Children’s Hospital of Philadelphia, PA; Columbia University Medical Center, New York, NY. United Kingdom: MRC Biostatistics Unit, Cambridge; University of Cambridge; Keele University; King’s College London; University of Oxford; and University College London.
Method Details
We genotyped these either on the Illumina HumanExome Beadchip (exome chip) or on a previously described custom array (International Multiple Sclerosis Genetics Consortium et al., 2017) including the exome chip content, both manufactured by Illumina Inc. We called genotypes both with Illumina’s default algorithm, gencall, and zCall, specifically developed to call low-frequency variants where all three groups of genotypes may not be observed (Goldstein et al., 2012).
An overview of our quality control process is shown in Figure S1; we used PLINK (Purcell et al., 2007) for all analyses unless otherwise noted. Briefly, we first excluded samples with low genotyping rate, extreme heterozygosity rate, inconsistent genotypic and recorded sex; we also removed closely related samples, keeping the relative with least missing data. Next, we removed population outliers by calculating genotype principal components using 16,066 common variants in linkage disequilibrium (r2 < 0.1) across the exome. We used EIGENSOFT 6 (Price et al., 2006) and FlashPCA (Abraham and Inouye, 2014) for cohorts with more than 10.000 individuals. We next removed variants with > 3% gencall missing data rate for variants with minor allele frequency MAF > 5%, or > 1% zCall missing data rate for variants with MAF < 5%. We also removed variants out of Hardy-Weinberg equilibrium (p < 10−5). Next, we removed samples with high similarity in missing genotypes (“identity by missingness”) indicative of production artifact, and samples with missing phenotype information. Finally, we again removed any remaining population outliers using projection principal component analysis. We calculated 30 principal components for 1,092 individuals in 1000 Genomes reference populations, again using the 16,066 common variants in linkage disequilibrium (r2 < 0.1) across the exome. We then projected the IMSGC samples into this space and excluded individuals more than six standard deviations from loading means as previously described (Price et al., 2006). We performed the projection and outlier detection and removal steps a total ten times to gradually remove more subtle population outliers.
We compiled cases and controls into strata for analysis as shown in Table S1. In total, we removed 17,938/76,140 (24%) samples either due to low data quality or as population outliers, leaving a final dataset of 27,891 cases and 30,298 controls in 13 strata (Figure S1 and Table S1). Separately, we included summary statistics from 4,476 MS cases and 5,714 controls from Germany, genotyped on the exome chip as previously described (Dankowski et al., 2015), giving us a total of 32,367 MS cases and 36,012 controls for analysis.
Quantification and Statistical Analysis
Exome chip coverage of ExAC variants
To assess how thoroughly the exome chip assesses low-frequency coding variation genome-wide, we compared it to the list of variants reported by the Exome Aggregation Consortium, ExAC (Lek et al., 2016), in their data release version 1. We filtered their summary table of all ExAC variants (available at ftp://ftp.broadinstitute.org/pub/ExAC_release/release1/manuscript_data/ExAC.r1.sites.vep.table.gz and last accessed 15 November 2017) for nonsynonymous coding variants passing their quality control, with at least one minor allele observed in non-Finnish European samples. We identified which of these variants are represented on the exome chip by comparing genomic coordinates.
Univariate association analysis
We used mixed linear models for association analysis, as implemented in GCTA (Yang et al., 2011). In each of our 13 genotype-level strata, we calculated genetic relatedness matrices from 16,066 common, noncoding variants (overall MAF > 0.05) in linkage equilibrium (all pairwise r2 < 0.1) present on the exome chip, and with these calculated univariate association statistics for each autosomal variant present on the exome chip. To further control for population stratification, we also calculated genotypic principal components with the 16,066 common variants, and included these as covariates to the association analysis. We also included genotypic sex and chip type as covariates. We combined statistics across strata using inverse-variance-weighted meta-analysis, also as implemented in GCTA (Yang et al., 2011). As the bulk of exome chip variants are not common and do not show appreciable linkage disequilibrium, we controlled for multiple tests with a Bonferroni correction for the number of low-frequency variants, to give a genome-wide significance threshold of p < 3.58 × 10e-7 (0.05/139,764 variants with a combined MAF < 0.05 in controls and a heterogeneity index I2 < 50 in our meta-analysis).
Heritability estimation
We used GCTA to calculate the heritability attributable to groups of variants in each of our 13 genotype-level strata (Yang et al., 2011). In each stratum, we ran two sets of models: a two-component model, estimating the heritability attributable to common and low-frequency (MAF ≤ 0.05) variants; and a three component model with rare (MAF ≤ 0.01), intermediate (0.01 < MAF ≤ 0.05), and common variants. In all strata, common variants are the set of 16,066 independent variants (overall MAF > 0.05) used for population stratification calculations in the univariate analysis above. We computed genetic relatedness matrices for each component of each model, then calculated narrow-sense heritability (h2) with 100 iterations of constrained restricted maximum likelihood (REML) fitting, assuming a disease prevalence of 0.001. We also included the principal components of population structure computed for the univariate analysis as covariates. As anticipated, several of the smaller cohorts presented fitting issues: no models converged for FIN; both three-component and two-component fits for UCSF2, and the three-component model for GRE would not converge under constraint and so were run without constraints; and the three-component model for BEL converged on two exactly equally likely solutions after 10,000 iterations. For the latter, we chose the most conservative estimates of variance explained. We combined these estimates with inverse variance-weighted meta-analysis.
Data and Software Availability
Meta-analysis summary statistics are available at http://imsgc.net/. Due to varying privacy laws across countries, some of our genotype data are available from the European Genome-phenome Archive (deposited under accession EGAS00001003195), with the remainder available directly from participating centers. A single request for all data access may be submitted to the IMSGC Data Access Committee (dac@imsgc.net). Our QC and analysis pipeline is available at https://github.com/cotsapaslab/IMSGCexomechip.
Acknowledgments
Australia
Sample collection and genotyping was supported by NHMRC project grant APP605511.
Belgium
B.D., R.L., and P.V.D. are Clinical Investigators of the Research Foundation Flanders (FWO-Vlaanderen). A.G. and B.D. are supported by the Research Fund KU Leuven (C24/16/045) and the Research Foundation Flanders (FWO-Vlaanderen) (G.0734.15). The SiGN study was funded by a cooperative agreement grant from the US National Institute of Neurological Disorders and Stroke, National Institutes of Health (U01-NS069208).
Germany
We would like to thank Verena Grummel and Nadine Miksch for technical support. We thank Sabine Fleischer for continuous and extensive support with biobanking logistics. This work was supported by the German Ministry for Education and Research (BMBF, “German Competence Network Multiple Sclerosis” [KKNMS]) [01GI0916, 01GI0917] and the Munich Cluster for Systems Neurology (SyNergy). The KORA study was initiated and financed by the Helmholtz Zentrum München—German Research Center for Environmental Health, which is funded by the BMBF and by the State of Bavaria. Furthermore, KORA research was supported within the Munich Center of Health Sciences (MC-Health), Ludwig-Maximilians-Universität, as part of LMUinnovativ. The popgen 2.0 network is supported by a grant from the German Ministry for Education and Research (01EY1103). The Heinz Nixdorf Recall Study was supported by the Heinz Nixdorf Foundation Germany, the BMBF, and the Deutsche Forschungsgemeinschaft (DFG; ER 155/6-1, ER 155/6-2).
Italy
Sample collection and genotyping was supported by the Italian Foundation for Multiple Sclerosis (FISM grants, special project “Immunochip” 2011/R/1, 2015/R/10) and Fondazione Cariplo (grant 2010-0728).
Norway
The Norwegian MS and control samples were collected and funded in collaboration between the multiple sclerosis research group in at Oslo University Hospital/University in Oslo, the Norwegian MS Registry and Biobank in Bergen, and the Norwegian Bone Marrow Registry, supported by the Oslo MS Association, Bergen MS Society, Odda MS Society, and the Research Council of Norway (grant 240102_Harbo).
Sweden
Sample collection and genotyping was supported by the Swedish Medical Research Council; Swedish Research Council for Health, Working Life, and Welfare; Knut and Alice Wallenberg Foundation; AFA insurance; Swedish Brain Foundation; the Swedish Association for Persons with Neurological Disabilities; and AstraZeneca Science for Life grant.
UK
This study makes use of data generated as part of the Wellcome Trust Case Control Consortium 2 project (085475/B/08/Z and 085475/Z/08/Z), including data from the British 1958 Birth Cohort DNA collection (funded by the Medical Research Council grant G0000934 and the Wellcome Trust grant 068545/Z/02) and the UK National Blood Service controls (funded by the Wellcome Trust). The study was supported by the Cambridge National Institute for Health Research (NIHR) Biomedical Research Centre, UK Medical Research Council (G1100125) and the UK MS Society (861/07). We thank the NIHR and NHS Blood and Transplant. TwinsUK is funded by the Wellcome Trust, Medical Research Council, European Union, and the NIHR-funded BioResource, Clinical Research Facility, and Biomedical Research Centre based at Guy’s and St. Thomas’s NHS Foundation Trust in partnership with King’s College London. We thank the volunteers from the Oxford Biobank (https://www.oxfordbiobank.org.uk/) for their participation. The recall process was supported by the NIHR Oxford Biomedical Research Centre Programme. The recall process was supported by the NIHR Oxford Biomedical Research Centre Programme. We thank Neil Robertson, Sam Loveless, Richard Reynolds, and John Zajicek for contributing case samples from the UK. The study makes use of material from the UK MS Society Tissue Bank (grant no 910/09) provided by Richard Reynolds.
USA
We thank the Biorepository Facility and the Center for Genome Technology laboratory personnel (specifically Sandra West, Simone Clarke, Daniela Martinez, and Patrice Whitehead) within the John P. Hussman Institute for Human Genomics at the University of Miami for centralized DNA handling and genotyping for this project. Related funding support is from the US National Multiple Sclerosis Society (grant RG-4680-A-1) and the NIH (R01-NS096212, R01-NS049477, and R01-NS026799). The IMSGC wishes to acknowledge William and Lois Edgerly, John and Elaine Carlos, Martha Crowninshield, and William and Cindy Fowler, whose enduring committments were critical in creation of the consortium.
Author Contributions
B.D., M.B.D., R. Lemmens, P.V.D., F.S., P.S.S., H.U., L.W.T., J.S., I.C.-R., B.F., L.G.-N., S.V., V.P., D.R.B., C.G., A.B., C. Heesen, T.K., R. Linker, F. Paul, M. Stange, B. Tackenberg, F.T.B., C.W., H. Wiendl, B.W., U. Zettl, U. Ziemann, H.T., R.G., B.H., B.K., C.M.L., F.L., E.D., C.A., L.F.B., G.C., F.E., L.F., C. Comi, D.G., M.A.L., J.M., R.H., C.E.T., K.-M.M., E.G.C., B.A.L., A. Spurkland, M.C., X.M., L.A., J.H., M.J., F. Piehl, I.J., R.M., M. Sospedra, C. Hawkins, S.K., P.A.C., B.A.C.C., A. Cross, M.F.D., J.L.H., S. Delgado, M. Dembele, K.E., K.C.F., E.L., C.P.M., M.A.P.-V., L.P., C.S., H. Weiner, T.O., G.H., B. Taylor, L.T., J.C., D.B., H.F.H., A.J.I., S.L.H., A. Compston, G.S., F.Z., L.F.B., F.M.-B., S. D’Alfonso, A.O., J.L.M., S.J.S., J.R.O., P.L.D.J., I. Kockum, and D.A.H. acquired DNA samples. A.G., H.B.S., H.U., L.W.T., T.W., I.C.-R., V.D., B.F., D.R.B., V.G., C.M.L., N.B., E.M., M. Sorosina, S.D.B., B.A.L., P.S., I.J., M.B., S.J.C., I. Konidari, C.M., B. Taylor, D.B., H.F.H., L.F.B., F.M.-B., S. D’Alfonso, A.O., J.L.M., J.R.O., P.L.D.J., and I. Kockum processed DNA samples. H.B.S., F.S., T.W., I.C.-R., V.D., B.F., M.L., D.C., J.M., R.H., C.v.D., M.B., P.H., F.K., J.K., G.L., M.N., H.H., I. Konidari, B. Taylor, J.C., H.F.H., L.F.B., A.O., J.L.M., S.J.S., and P.L.D.J. genotyped DNA samples. M.M., N.A.P., A.H.B., T.D., T.F.M.A., C.v.D., M.B., M.F.D., A. Santaniello, P.I.W.d.B., M.A.P., J.G., B. Taylor, S.E.B., A.Z., J.L.M., S.J.S., P.L.D.J., and C. Cotsapas analyzed data for this study. M.M., N.A.P., A.H.B., T.D., A.G., B.D., H.B.S., T.W., J.S., I.C.-R., B.F., M.L., P.-A.G., V.P., D.R.B., A.B., C. Heesen, T.K., R. Linker, F. Paul, M. Stange, B. Tackenberg, F.T.B., C.W., H. Wiendl, B.W., U. Zettl, U. Ziemann, H.T., R.G., V.G., B.H., C.M.L., E.D., C.A., N.B., E.M., L.B., G.C., D.C., F.E., C. Comi, D.G., M.A.L., M. Sorosina, J.M., R.H., S.D.B., E.G.C., P.S., I.J., R.M., M. Sospedra, M.B., C. Hawkins, P.H., S.K., F.K., J.K., G.L., M.N., S.J.C., B.A.C.C., M.F.D., J.L.H., S. Delgado, H.H., I. Konidari, C.P.M., C.S., C.M., G.H., D.B., H.F.H., A.J.I., F.Z., L.F.B., F.M.-B., S. D’Alfonso, A.Z., A.O., J.L.M., S.J.S., P.L.D.J., I. Kockum, D.A.H., and C. Cotsapas acquired or analyzed clinical, demographic, or genotypic data. M.M., N.A.P., J.L.M., P.L.D.J., and C. Cotsapas designed the project. M.M. and C. Cotsapas wrote the manuscript with input and approval from all authors.
Declaration of Interests
Kjell-Morten Myhr has received unrestricted research grants to his institution and scientific advisory board or speakers honoraria from Amirall, Biogen Idec, Novartis, Merck, Roche and Teva; and has participated in clinical trials organized by Biogen Idec, Merck, Novartis and Roche.
Annette Bang Oturai has served on scientific advisory boards for Biogen Idec; has received research support from Novartis and Biogen Idec; has received speaker honoraria from Biogen Idec, Novartis and TEVA; and has received support for congress participation from, Merck Serono, TEVA, Biogen, Novartis and Genzyme.
Finn Sellebjerg has served on scientific advisory boards for Biogen Idec, Genzyme, Merck Serono, Novartis, Sanofi-Aventis and Teva, has been on the steering committee of a clinical trial sponsored by Merck Serono, and served as consultant for Biogen Idec and Novo Nordisk; has received support for congress participation from Biogen Idec, Novartis, Genzyme (Sanofi-aventis) and Teva; has received speaker honoraria from Bayer Schering, Biogen Idec, Genzyme, Merck Serono, Novartis, Sanofi-Aventis and Schering-Plough. His laboratory has received research support from Biogen Idec, Bayer Schering, Merck Serono, Sanofi-Aventis and Novartis.
Per Soelberg Sorensen has served on scientific advisory boards Biogen Idec, Merck Serono, Novartis, Genmab, TEVA, Elan, GSK, has been on steering committees or independent data monitoring boards in clinical trials sponsored by Merck Serono, Genmab, TEVA, GSK, Bayer Schering, and he has received funding of travel for these activities; has served as Editor-in-Chief of the European Journal of Neurology, and is currently editorial board member for Multiple Sclerosis Journal, European Journal of Neurology, Therapeutic Advances in Neurological Disorders and; has received speaker honoraria from Biogen Idec, Merck Serono, TEVA, Bayer Schering, Sanofi-aventis, Genzyme, and Novartis; and has received payment for writing and reviewing manuscript from IBI Consulting, a division of Informa plc. His department has received research support from Biogen Idec, Bayer Schering, Merck Serono, TEVA, Baxter, Sanofi-Aventis, BioMS, Novartis, Bayer, RoFAR, Roche, Genzyme, from the Danish Multiple Sclerosis Society, the Danish Medical Research Council, and the European Union Sixth Framework Programme: Life sciences, Genomics and Biotechnology for health.
Helle Bach Søndergaard has received support for congress participation from TEVA and Genzyme.
Henrik Ullum has received honoraria for lecturing from Roche.
Peter Calabresi has received research support from Annexon, Medimmune, Genzyme and Biogen Idec.
Hanne F Harbo has received travel support and honoraria from Biogen Idec, Sanofi-Genzyme, Merck, Novartis, Roche, and Teva and an unrestricted research grant from Novartis.
Benjamin Knier has received research support from Novartis.
Frederik Piehl has received research support from Biogen Idec, Novartis, and Genzyme.
Tania Kümpfel has received travel expenses and speaker honoraria from Bayer Healthcare, Teva Pharma, Merck, Novartis Pharma, Sanofi-Aventis/Genzyme, CLB Behring, Roche Pharma and Biogen as well as grant support from Bayer-Schering AG, Novartis and Chugai Pharma.
Ralf Gold has received speaker and board honoraria from Baxter, Bayer Schering, Biogen Idec, CLB
Behring, Genzyme, Merck Serono, Novartis, Roche, Stendhal, Talecris, and TEVA. His department received grant support from Bayer Schering, BiogenIdec, Genzyme, Merck Serono, Novartis, TEVA. He possesses stock options from Merck Serono and Roche.
Thomas Werge has acted as lecturer and scientific advisory to H. Lundbeck A/S
Stephen L Hauser serves on the board of trustees for Neurona and on scientific advisory boards for Annexon, Bionure, Symbiotix, and Alector, and has received travel reimbursement and writing assistance from F. Hoffmann-La Roche Ltd for CD20-related meetings and presentations.
Roland Martin received unrestricted grant support from Biogen Idec and Novartis, and compensation for advice or lecturing by Biogen Idec, Novartis, Sanofi Genzyme, Hoffmann La Roche, Neuway and CellProtect. He is a cofounder and co-owner of Cellerys.
Felix Luessi has served on advisory boards for Roche Pharma and received travel support from Teva Pharma.
Sandra Vukusic has received consulting and lecturing fees, travel grants and unconditional research support from Biogen Idec, Geneuro, Genzyme, MedDay, Merck Serono, Novartis, Roche, Sanofi Aventis and Teva Pharma.
Frauke Zipp has recently received research grants and/or consultation funds from Genzyme, Merck Serono, Roche, Novartis, Sanofi-Aventis, Celgene, ONO and Octapharma.
Bénédicte Dubois and An Goris have received travel/consulting fees and/or research funding from Novartis, Merck-Serono and Roche. B.D. has received consulting fees and/or funding from Biogen Idec, Sanofi-Aventis and Teva
Elizabeth G Celius has received personal compensation for serving on scientific advisory boards for Almirall, Biogen Idec, Merck, Novartis, Genzyme and Teva, has received speaker honoraria from Biogen Idec, Genzyme, Novartis, Merck and Teva, and her department has received unrestricted research grants from Novartis and Genzyme.
Heinz Wiendl receives honoraria for acting as a member of Scientific Advisory Boards and as consultant for Biogen, Evgen, MedDay Pharmaceuticals, Merck Serono, Novartis, Roche Pharma AG, Sanofi-Genzyme, as well as speaker honoraria and travel support from Alexion, Biogen, Cognomed, F. Hoffmann-La Roche, Gemeinnützige Hertie-Stiftung, Merck Serono, Novartis, Roche Pharma AG, Sanofi-Genzyme, TEVA, and WebMD Global. Prof. Wiendl is acting as a paid consultant for Abbvie, Actelion, Biogen, IGES, Novartis, Roche, Sanofi-Genzyme, and the Swiss Multiple Sclerosis Society. His research is funded by the German Ministry for Education and Research (BMBF), Deutsche Forschungsgesellschaft (DFG), Else Kröner Fresenius Foundation, Fresenius Foundation, Hertie Foundation, NRW Ministry of Education and Research, Interdisciplinary Center for Clinical Studies (IZKF) Muenster and RE Children’s Foundation, Biogen GmbH, GlaxoSmithKline GmbH, Roche Pharma AG, and Sanofi-Genzyme.
Friedemann Paul has received honoraria and research support from Alexion, Bayer, Biogen Idec, Chugai, MerckSerono, Novartis, Genyzme, MedImmune, Shire, Teva, and serves on scientific advisory boards for Alexion, MedImmune and Novartis. He has received funding from Deutsche Forschungsgemeinschaft (DFG Exc 257), Bundesministerium für Bildung und Forschung (Competence Network Multiple Sclerosis), Guthy Jackson Charitable Foundation, EU Framework Program 7, National Multiple Sclerosis Society of the USA, and serves on advisory boards and steering committees for Novartis and MedImmune.
Antonios Bayas received personal compensation from Merck Serono, Biogen, Bayer Vital, Novartis, TEVA, Roche and Sanofi/Genzyme and grants for congress trips and participation from Biogen, TEVA, Novartis, Sanofi/Genzyme, and Merck Serono; none related to this work
Giancarlo Comi has received personal compensation, not related to the submitted work, for consulting services and/or speaking activities from Novartis, Teva, Sanofi Genzyme, Merck, Biogen Idec, Roche, Almirall, Celgene, Forward Pharma, Medday and Excemed.
Brigitte Wildemann has received grants from the German Ministry of Education and Research, grants from Dietmar Hopp Founfation, grants from Klaus Tschira Foundation, personal fees from Bayer Healthcare, personal fees from Biogen Idec, grants and personal fees from Merck Serono, grants and personal fees from Novartis, grants and personal fees from Sanofi Genzyme, grants and personal fees from TEVA, outside the submitted work.
Martin Stangel has received honoraria for scientific lectures or consultancy from Bayer Healthcare, Biogen, CSL Behring, Grifols, MedDay, Merck-Serono, Novartis, Roche, Sanofi-Genzyme, Shire, and Teva. His institution received research support from Bayer Healthcare, Biogen, Genzyme, Merck-Serono, Novartis, and Teva. He is on the editorial board of PLoS ONE and Multiple Sclerosis International.
None of this is related to this manuscript.
Clemens Warnke has received consulting fees from Novartis and Biogen Idec unrelated to this study.
Paul IW de Bakker is an employee of Vertex Pharmaceuticals and holds stock therein.
Jan Hillert has received research support from Biogen Idec, Merck and Roche.
Xavier Montalban has received speaking honoraria and travel expenses for participation in scientific meetings, has been a steering committee member of clinical trials or participated in advisory boards of clinical trials in the past years with Actelion, Bayer, Biogen, Celgene, Hoffmann-La Roche, Merck, Novartis, Sanofi-Genzyme and Teva Pharmaceutical.
Björn Tackenberg has received personal speaker honoraria and consultancy fees as a speaker and advisor from Alexion, Bayer Healthcare, Biogen, Celegene, CSL Behring, GRIFOLS, Merck Serono, Novartis, Octapharma, Roche, Sanofi Genzyme, TEVA and UCB Pharma. His University received unrestricted research grants from Biogen, Novartis, TEVA, Bayer Healthcare, CSL-Behring, GRIFOLS, Octapharma, Sanofi Genzyme and UCB Pharma.
Federica Esposito has received consulting fees from Novartis, Almirall and Genzyme
Tomas Olsson has received advisory board/lecture compensations and/ or unrestricted MS research grants from: Biogen Idec, Novartis, Genzyme, Roche and Merck. None of which have any relevance to the current study.
Alastair Compston has received honoraria and travel support from Sanofi/Genzyme.
Howard Weiner reports grants from National Institutes of Health, the National Multiple Sclerosis Society, Verily Life Sciences, EMD Serono, Biogen Idec, Teva Pharmaceuticals, Sanofi/Genzyme, Novartis, Genentech, and Tilos Therapeutics; and personal fees from Tiziana Life Sciences, Genentech, IM Therapeutics, MedDay Pharmaceuticals, Tilos Therapeutics, and vTv Therapeutics. All these support efforts were unrelated to the present work.
Philip L De Jager has received honoraria for acting as a member of Scientific Advisory Boards for Sanofi/Genzyme, Celgene, Roche, and Biogen Idec; and has received research support from Biogen Idec, Roche and Genentech.
David A Hafler has received consulting fees from Compass Therapeutics, EMD Serono, Genentech, Medimmune, Merck Sharp & Dohme, Mylan Pharmaceuticals, Novartis Pharmaceuticals, Proclara Bioscience, Sanofi Genzyme. He has received research support from Genentech and Bristol-Myers Squibb.
Chris Cotsapas has received research support from Biogen Idec.
The remaining authors declare no competing interests.
Published: October 18, 2018
Footnotes
Supplemental Information includes three figures and five tables and can be found with this article online at https://doi.org/10.1016/j.cell.2018.09.049.
Contributor Information
International Multiple Sclerosis Genetics Consortiumchris.cotsapas@yale.edu:
Mitja Mitrovič, Nikolaos A. Patsopoulos, Ashley H. Beecham, Theresa Dankowski, An Goris, Bénédicte Dubois, Marie B. D’hooghe, Robin Lemmens, Philip Van Damme, Helle Bach Søndergaard, Finn Sellebjerg, Per Soelberg Sorensen, Henrik Ullum, Lise W. Thørner, Thomas Werge, Janna Saarela, Isabelle Cournu-Rebeix, Vincent Damotte, Bertrand Fontaine, Lena Guillot-Noel, Mark Lathrop, Sandra Vukusik, Pierre-Antoine Gourraud, Till F.M. Andlauer, Viola Pongratz, Dorothea Buck, Christiane Gasperi, Antonios Bayas, Christoph Heesen, Tania Kümpfel, Ralf Linker, Friedemann Paul, Martin Stangel, Björn Tackenberg, Florian Then Bergh, Clemens Warnke, Heinz Wiendl, Brigitte Wildemann, Uwe Zettl, Ulf Ziemann, Hayrettin Tumani, Ralf Gold, Verena Grummel, Bernhard Hemmer, Benjamin Knier, Christina M. Lill, Felix Luessi, Efthimios Dardiotis, Cristina Agliardi, Nadia Barizzone, Elisabetta Mascia, Luisa Bernardinelli, Giancarlo Comi, Daniele Cusi, Federica Esposito, Laura Ferrè, Cristoforo Comi, Daniela Galimberti, Maurizio A. Leone, Melissa Sorosina, Julia Mescheriakova, Rogier Hintzen, Cornelia van Duijn, Charlotte E. Theunissen, Steffan D. Bos, Kjell-Morten Myhr, Elisabeth G. Celius, Benedicte A. Lie, Anne Spurkland, Manuel Comabella, Xavier Montalban, Lars Alfredsson, Pernilla Stridh, Jan Hillert, Maja Jagodic, Fredrik Piehl, Ilijas Jelčić, Roland Martin, Mireia Sospedra, Maria Ban, Clive Hawkins, Pirro Hysi, Seema Kalra, Fredrik Karpe, Jyoti Khadake, Genevieve Lachance, Matthew Neville, Adam Santaniello, Stacy J. Caillier, Peter A. Calabresi, Bruce A.C. Cree, Anne Cross, Mary F. Davis, Jonathan L. Haines, Paul I.W. de Bakker, Silvia Delgado, Marieme Dembele, Keith Edwards, Kathryn C. Fitzgerald, Hakon Hakonarson, Ioanna Konidari, Ellen Lathi, Clara P. Manrique, Margaret A. Pericak-Vance, Laura Piccio, Cathy Schaefer, Cristin McCabe, Howard Weiner, Jacqueline Goldstein, Tomas Olsson, Georgios Hadjigeorgiou, Bruce Taylor, Lotti Tajouri, Jac Charlesworth, David R. Booth, Hanne F. Harbo, Adrian J. Ivinson, Stephen L. Hauser, Alastair Compston, Graeme Stewart, Frauke Zipp, Lisa F. Barcellos, Sergio E. Baranzini, Filippo Martinelli-Boneschi, Sandra D’Alfonso, Andreas Ziegler, Annette Oturai, Jacob L. McCauley, Stephen J. Sawcer, Jorge R. Oksenberg, Philip L. De Jager, Ingrid Kockum, David A. Hafler, and Chris Cotsapas
Supplemental Information
We used our scaffold of 16,066 common, independent variants to impute each of the 200 common MS risk variants reported in our latest GWAS (International Multiple Sclerosis Genetics Consortium et al., 2017), in each of our three largest cohorts. We can impute 162, 156, and 162 variants in Sweden, UK/Australia and USA-Boston with an INFO score > = 0.25; these numbers drop to 72, 67, and 75 of 200 at INFO > = 0.5 and 22, 20, and 20 at INFO > = 0.75, respectively.
References
- Abraham G., Inouye M. Fast principal component analysis of large-scale genome-wide data. PLoS ONE. 2014;9:e93766. doi: 10.1371/journal.pone.0093766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler D., Daly M.J., Lander E.S. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andlauer T.F.M., Buck D., Antony G., Bayas A., Bechmann L., Berthele A., Chan A., Gasperi C., Gold R., Graetz C. d. Sci. Adv. 2016;2 doi: 10.1126/sciadv.1501678. e1501678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Australia and New Zealand Multiple Sclerosis Genetics Consortium (ANZgene) Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat. Genet. 2009;41:824–828. doi: 10.1038/ng.396. [DOI] [PubMed] [Google Scholar]
- Baranzini S.E., Galwey N.W., Wang J., Khankhanian P., Lindberg R., Pelletier D., Wu W., Uitdehaag B.M., Kappos L., Polman C.H., GeneMSA Consortium Pathway and network-based analysis of genome-wide association studies in multiple sclerosis. Hum. Mol. Genet. 2009;18:2078–2090. doi: 10.1093/hmg/ddp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beecham A.H., Patsopoulos N.A., Xifara D.K., Davis M.F., Kemppinen A., Cotsapas C., Shah T.S., Spencer C., Booth D., Goris A., International Multiple Sclerosis Genetics Consortium (IMSGC) Wellcome Trust Case Control Consortium 2 (WTCCC2) International IBD Genetics Consortium (IIBDGC) Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013;45:1353–1360. doi: 10.1038/ng.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettini M.L., Pan F., Bettini M., Finkelstein D., Rehg J.E., Floess S., Bell B.D., Ziegler S.F., Huehn J., Pardoll D.M., Vignali D.A. Loss of epigenetic modification driven by the Foxp3 transcription factor leads to regulatory T cell insufficiency. Immunity. 2012;36:717–730. doi: 10.1016/j.immuni.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder M.D., Fox A.D., Merlo D., Johnson L.J., Giuffrida L., Calvert S.E., Akkermann R., Ma G.Z.M., Perera A.A., Gresle M.M., ANZgene Common and Low Frequency Variants in MERTK Are Independently Associated with Multiple Sclerosis Susceptibility with Discordant Association Dependent upon HLA-DRB1∗15:01 Status. PLoS Genet. 2016;12:e1005853. doi: 10.1371/journal.pgen.1005853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton P.R., Clayton D.G., Cardon L.R., Craddock N., Deloukas P., Duncanson A., Kwiatkowski D.P., McCarthy M.I., Ouwehand W.H., Samani N.J., Wellcome Trust Case Control Consortium. Australo-Anglo-American Spondylitis Consortium (TASC) Biologics in RA Genetics and Genomics Study Syndicate (BRAGGS) Steering Committee. Breast Cancer Susceptibility Collaboration (UK) Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 2007;39:1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chami N., Chen M.-H., Slater A.J., Eicher J.D., Evangelou E., Tajuddin S.M., Love-Gregory L., Kacprowski T., Schick U.M., Nomura A. Exome Genotyping Identifies Pleiotropic Variants Associated with Red Blood Cell Traits. Am. J. Hum. Genet. 2016;99:8–21. doi: 10.1016/j.ajhg.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHARGE Consortium Hematology Working Group Meta-analysis of rare and common exome chip variants identifies S1PR4 and other loci influencing blood cell traits. Nat. Genet. 2016;48:867–876. doi: 10.1038/ng.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compston A., Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- Constant S., Pfeiffer C., Woodard A., Pasqualini T., Bottomly K. Extent of T cell receptor ligation can determine the functional differentiation of naive CD4+ T cells. J. Exp. Med. 1995;182:1591–1596. doi: 10.1084/jem.182.5.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankowski T., Buck D., Andlauer T.F.M., Antony G., Bayas A., Bechmann L., Berthele A., Bettecken T., Chan A., Franke A., German Competence Network for Multiple Sclerosis (KKNMS) Successful Replication of GWAS Hits for Multiple Sclerosis in 10,000 Germans Using the Exome Array. Genet. Epidemiol. 2015;39:601–608. doi: 10.1002/gepi.21933. [DOI] [PubMed] [Google Scholar]
- De Jager P.L., Jia X., Wang J., de Bakker P.I.W., Ottoboni L., Aggarwal N.T., Piccio L., Raychaudhuri S., Tran D., Aubin C., International MS Genetics Consortium Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat. Genet. 2009;41:776–782. doi: 10.1038/ng.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Villar M., Baecher-Allan C.M., Hafler D.A. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat. Med. 2011;17:673–675. doi: 10.1038/nm.2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchsberger C., Flannick J., Teslovich T.M., Mahajan A., Agarwala V., Gaulton K.J., Ma C., Fontanillas P., Moutsianas L., McCarthy D.J. The genetic architecture of type 2 diabetes. Nature. 2016;536:41–47. doi: 10.1038/nature18642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein J.I., Crenshaw A., Carey J., Grant G.B., Maguire J., Fromer M., O’Dushlaine C., Moran J.L., Chambert K., Stevens C., Swedish Schizophrenia Consortium. ARRA Autism Sequencing Consortium zCall: a rare variant caller for array-based genotyping: genetics and population analysis. Bioinformatics. 2012;28:2543–2545. doi: 10.1093/bioinformatics/bts479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- House I.G., Thia K., Brennan A.J., Tothill R., Dobrovic A., Yeh W.Z., Saffery R., Chatterton Z., Trapani J.A., Voskoboinik I. Heterozygosity for the common perforin mutation, p.A91V, impairs the cytotoxicity of primary natural killer cells from healthy individuals. Immunol. Cell Biol. 2015;93:575–580. doi: 10.1038/icb.2015.1. [DOI] [PubMed] [Google Scholar]
- Housley W.J., Fernandez S.D., Vera K., Murikinati S.R., Grutzendler J., Cuerdon N., Glick L., De Jager P.L., Mitrovic M., Cotsapas C., Hafler D.A. Genetic variants associated with autoimmunity drive NFκB signaling and responses to inflammatory stimuli. Sci. Transl. Med. 2015;7:291ra93. doi: 10.1126/scitranslmed.aaa9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Multiple Sclerosis Genetics Consortium. Patsopoulos N., Baranzini S.E., Santaniello A., Shoostari P., Cotsapas C., Wong G., Beecham A.H., James T., Replogle J. The Multiple Sclerosis Genomic Map: Role of peripheral immune cells and resident microglia in susceptibility. bioRxiv. 2017 [Google Scholar]
- International Multiple Sclerosis Genetics Consortium. Wellcome Trust Case Control Consortium 2. Sawcer S., Hellenthal G., Pirinen M., Spencer C.C., Patsopoulos N.A., Moutsianas L., Dilthey A., Su Z. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakkula E., Leppä V., Sulonen A.-M., Varilo T., Kallio S., Kemppinen A., Purcell S., Koivisto K., Tienari P., Sumelahti M.-L. Genome-wide association study in a high-risk isolate for multiple sclerosis reveals associated variants in STAT3 gene. Am. J. Hum. Genet. 2010;86:285–291. doi: 10.1016/j.ajhg.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasler H.G., Young B.D., Mottet D., Lim H.W., Collins A.M., Olson E.N., Verdin E. Histone deacetylase 7 regulates cell survival and TCR signaling in CD4/CD8 double-positive thymocytes. J. Immunol. 2011;186:4782–4793. doi: 10.4049/jimmunol.1001179. [DOI] [PubMed] [Google Scholar]
- Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., Exome Aggregation Consortium Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Samanta A., Song X., Iacono K.T., Bembas K., Tao R., Basu S., Riley J.L., Hancock W.W., Shen Y. FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc. Natl. Acad. Sci. USA. 2007;104:4571–4576. doi: 10.1073/pnas.0700298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.J., Peloso G.M., Yu H., Butterworth A.S., Wang X., Mahajan A., Saleheen D., Emdin C., Alam D., Alves A.C., Charge Diabetes Working Group. EPIC-InterAct Consortium. EPIC-CVD Consortium. GOLD Consortium. VA Million Veteran Program Exome-wide association study of plasma lipids in >300,000 individuals. Nat. Genet. 2017;49:1758–1766. doi: 10.1038/ng.3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso N., Rohland N., Rand K.A., Tandon A., Allen A., Quinque D., Mallick S., Li H., Stram A., Sheng X., PRACTICAL consortium The contribution of rare variation to prostate cancer heritability. Nat. Genet. 2016;48:30–35. doi: 10.1038/ng.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A., Hunter D.J., McCarthy M.I., Ramos E.M., Cardon L.R., Chakravarti A. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marouli E., Graff M., Medina-Gomez C., Lo K.S., Wood A.R., Kjaer T.R., Fine R.S., Lu Y., Schurmann C., Highland H.M., EPIC-InterAct Consortium. CHD Exome+ Consortium. ExomeBP Consortium. T2D-Genes Consortium. GoT2D Genes Consortium. Global Lipids Genetics Consortium. ReproGen Consortium. MAGIC Investigators Rare and low-frequency coding variants alter human adult height. Nature. 2017;542:186–190. doi: 10.1038/nature21039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli-Boneschi F., Esposito F., Brambilla P., Lindström E., Lavorgna G., Stankovich J., Rodegher M., Capra R., Ghezzi A., Coniglio G. A genome-wide association study in progressive multiple sclerosis. Mult. Scler. 2012;18:1384–1394. doi: 10.1177/1352458512439118. [DOI] [PubMed] [Google Scholar]
- Moutsianas L., Jostins L., Beecham A.H., Dilthey A.T., Xifara D.K., Ban M., Shah T.S., Patsopoulos N.A., Alfredsson L., Anderson C.A., International IBD Genetics Consortium (IIBDGC) Class II HLA interactions modulate genetic risk for multiple sclerosis. Nat. Genet. 2015;47:1107–1113. doi: 10.1038/ng.3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M.R., Wegmann D., Ehm M.G., Kessner D., St Jean P., Verzilli C., Shen J., Tang Z., Bacanu S.-A., Fraser D. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012;337:100–104. doi: 10.1126/science.1217876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nischwitz S., Cepok S., Kroner A., Wolf C., Knop M., Müller-Sarnowski F., Pfister H., Roeske D., Rieckmann P., Hemmer B. Evidence for VAV2 and ZNF433 as susceptibility genes for multiple sclerosis. J. Neuroimmunol. 2010;227:162–166. doi: 10.1016/j.jneuroim.2010.06.003. [DOI] [PubMed] [Google Scholar]
- Patsopoulos N.A., Esposito F., Reischl J., Lehr S., Bauer D., Heubach J., Sandbrink R., Pohl C., Edan G., Kappos L., Bayer Pharma MS Genetics Working Group. Steering Committees of Studies Evaluating IFNβ-1b and a CCR1-Antagonist. ANZgene Consortium. GeneMSA. International Multiple Sclerosis Genetics Consortium Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann. Neurol. 2011;70:897–912. doi: 10.1002/ana.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsopoulos N.A., Barcellos L.F., Hintzen R.Q., Schaefer C., van Duijn C.M., Noble J.A., Raj T., Gourraud P.A., Stranger B.E., Oksenberg J., IMSGC. ANZgene Fine-mapping the genetic association of the major histocompatibility complex in multiple sclerosis: HLA and non-HLA effects. PLoS Genet. 2013;9:e1003926. doi: 10.1371/journal.pgen.1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S.M., Moran J.L., Fromer M., Ruderfer D., Solovieff N., Roussos P., O’Dushlaine C., Chambert K., Bergen S.E., Kähler A. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team . R Foundation for Statistical Computing; 2017. R: A language and environment for statistical computing. [Google Scholar]
- Sadler A.J., Williams B.R.G. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna S., Pitzalis M., Zoledziewska M., Zara I., Sidore C., Murru R., Whalen M.B., Busonero F., Maschio A., Costa G. Variants within the immunoregulatory CBLB gene are associated with multiple sclerosis. Nat. Genet. 2010;42:495–497. doi: 10.1038/ng.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawcer S., Ban M., Maranian M., Yeo T.W., Compston A., Kirby A., Daly M.J., De Jager P.L., Walsh E., Lander E.S., International Multiple Sclerosis Genetics Consortium A high-density screen for linkage in multiple sclerosis. Am. J. Hum. Genet. 2005;77:454–467. doi: 10.1086/444547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawcer S., Franklin R.J.M., Ban M. Multiple sclerosis genetics. Lancet Neurol. 2014;13:700–709. doi: 10.1016/S1474-4422(14)70041-9. [DOI] [PubMed] [Google Scholar]
- Schoech A., Jordan D., Loh P.-R., Gazal S., O’Connor L., Balick D.J., Palamara P.F., Finucane H., Sunyaev S.R., Price A.L. Quantification of frequency-dependent genetic architectures and action of negative selection in 25 UK Biobank traits. bioRxiv. 2017 doi: 10.1038/s41467-019-08424-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims R., van der Lee S.J., Naj A.C., Bellenguez C., Badarinarayan N., Jakobsdottir J., Kunkle B.W., Boland A., Raybould R., Bis J.C., ARUK Consortium. GERAD/PERADES, CHARGE, ADGC, EADI Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 2017;49:1373–1384. doi: 10.1038/ng.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerlind H., Ramanujam R., Uvehag D., Kuja-Halkola R., Boman M., Bottai M., Lichtenstein P., Hillert J. Modest familial risks for multiple sclerosis: a registry-based study of the population of Sweden. Brain. 2014;137:770–778. doi: 10.1093/brain/awt356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Lee S.H., Goddard M.E., Visscher P.M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J., de Vlaming R., Wu Y., Robinson M.R., Lloyd-Jones L.R., Yengo L., Yap C.X., Xue A., Sidorenko J., McRae A.F. Signatures of negative selection in the genetic architecture of human complex traits. Nat. Genet. 2018;50:746–753. doi: 10.1038/s41588-018-0101-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
We used our scaffold of 16,066 common, independent variants to impute each of the 200 common MS risk variants reported in our latest GWAS (International Multiple Sclerosis Genetics Consortium et al., 2017), in each of our three largest cohorts. We can impute 162, 156, and 162 variants in Sweden, UK/Australia and USA-Boston with an INFO score > = 0.25; these numbers drop to 72, 67, and 75 of 200 at INFO > = 0.5 and 22, 20, and 20 at INFO > = 0.75, respectively.