

Abstract
Neurofibromatosis type 1 is a familial genetic syndrome associated with a predisposition to develop peripheral and central nervous system neoplasms. We have previously reported on a subset of gliomas developing in these patients with morphologic features resembling subependymal giant cell astrocytoma, but the molecular features of these tumors remain undefined. A total of 14 tumors were studied and all available slides were reviewed. Immunohistochemical stains and telomere-specific FISH were performed on all cases. In addition, next generation sequencing was performed on 11 cases using a platform targeting 644 cancer-related genes. The average age at diagnosis was 28 years (range: 4–60, 9F/5M). All tumors involved the supratentorial compartment. Tumors were predominantly low grade (n=12), with 2 high grade tumors, and displayed consistent expression of glial markers. Next generation sequencing demonstrated inactivating NF1 mutations in 10 (of 11) cases. Concurrent TSC2 and RPTOR mutations were present in two cases (1 sporadic and 1 neurofibromatosis type 1-associated). Interestingly, alternative lengthening of telomeres was present in 4 (of 14) (29%) cases. However, an ATRX mutation associated with aberrant nuclear ATRX expression was identified in only 1 (of 4) cases with alternative lenghtening of telomeres. Mutations in the DNA helicase RECQL4 (n=2) and components of the Fanconi anemia complementation group (FANCD2, FANCF, FANCG) (n=1) were identified in two alternative lenghtening of telomeres -positive/ATRX intact cases. Other variants involved genes related to NOTCH signaling, DNA maintenance/repair pathways, and epigenetic modulators. There were no mutations identified in DAXX, PTEN, PIK3C genes, TP53, H3F3A, HIST1H3B or in canonical hotspots of IDH1, IDH2, or BRAF. A subset of subependymal giant cell astrocytoma-like astrocytomas are alternative lenghtening of telomeres -positive and occur in the absence of ATRX alterations, thereby suggesting mutations in other DNA repair/maintenance genes may also facilitate alternative lenghtening of telomeres. These findings suggest that subependymal giant cell astrocytoma -like astrocytoma represents a biologically distinct group that merits further investigation.
Keywords: Neurofibromatosis, Neurofibromatosis type 1, subependymal giant cell astrocytoma, TSC, alternative lengthening of telomeres, ATRX, RECQL4
Introduction
Neurofibromatosis type 1 is a familial genetic syndrome associated with a predisposition to develop neoplasms in the peripheral and central nervous system. In the central nervous system, both high grade and low grade gliomas, predominantly astrocytomas, may be encountered. The biological basis of these tumors, other than the consistent demonstration of NF1 gene inactivation, remains to be defined (1). In a large series of neurofibromatosis type 1 -associated gliomas, we identified 6 tumors with unusually broad processes and prominent nucleoli, resembling subependymal giant cell astrocytoma (2). Subsequent immunophenotypic analysis demonstrated that these tumors not only consistently expressed markers of glial differentiation, but also neuronal markers, another feature typical of subependymal giant cell astrocytoma (3). In addition to the variable positivity with neuronal markers, activation of the mTOR pathway was present in these tumors to a greater extent than in other neurofibromatosis type 1 -associated gliomas (4). This represents another feature in common with subependymal giant cell astrocytoma which usually develops in the setting of tuberous sclerosis complex, a genetic syndrome caused by alterations in TSC1 or TSC2 resulting in constitutive mTOR pathway activation (5).
Our knowledge of the biologic basis of low and high grade gliomas in children and adults has increased in recent years due to large comprehensive sequencing studies (6–9). All cancers require a mechanism for telomere maintenance, which occurs predominantly through expression of telomerase. A subset of cancers rely instead on the telomerase independent mechanism, termed alternative lengthening of telomeres, to maintain their telomeres. Prior studies have linked alterations in the alpha thalassemia/mental retardation syndrome X-linked (ATRX) or death domain-associated protein (DAXX) genes with the alternative lenghtening of telomeres phenotype in cancer (10). More recently, concurrent alternative lenghtening of telomeres and ATRX loss have been demonstrated in diffuse and high grade neurofibromatosis type 1 -associated gliomas (11), and NF1 and ATRX mutations coexist in subsets of pediatric high grade gliomas (12). In the current study, we revisit the morphologic features of this distinctive subset of gliomas resembling subependymal giant cell astrocytoma, mostly associated with neurofibromatosis type 1, and analyze the phenotypic and molecular genetic features.
Materials and Methods
Patients and Samples
A total of 12 tumors developing in patients with a clinical diagnosis of neurofibromatosis type 1 (per NIH guidelines) were initially indentified. Two additional tumors were identified among 342 low grade astrocytomas/gliomas (0.6%) evaluated by one of the authors (FJR), which shared the same morphologic features. One tumor developed in a patient lacking any clinical evidence of a genetic syndrome, and the second developed in a patient not known to have a syndrome but with limited clinical history. Tumors were graded using principles outlined in the 2016 WHO Classification when feasible (13) as well as prior criteria developed for anaplasia in pilocytic astrocytomas (14), a tumor type that is overrepresented in patients with neurofibromatosis type 1. In brief, we described tumors as high grade based on the presence of brisk mitotic activity, defined as having 5 or more mitoses per 10 high power fields, with or without necrosis. The study was performed under institutional review board approval and adhered to all its guidelines. Clinical information was abstracted from retrospective chart review. All histologic and immunohistochemical slides performed during routine workup of the pathology were evaluated.
Telomere-specific Fluorescence in situ hybridization (FISH) and chromogenic in situ hybridization (CISH)
Alternative lenghtening of telomeres determination was made using previously published criteria for telomere-specific FISH (10, 15) using whole formalin-fixed paraffin embedded processed unstained slides. Deparaffinized slides were subjected to hydration and steamed (25 min) in citrate buffer (Vector Laboratories), dehydrated, and then hybridized with a Cy3-labeled peptide nucleic acid (PNA) probe complementary to the mammalian telomere repeat sequence [(N-terminus to C-terminus) CCCTAACCCTAACCCTAA]. For FISH experiments, an Alexa Fluor-488–labeled PNA probe specific to human centromeric DNA repeats (ATTCGTTGGAAACGGGA; CENP-B–binding sequence) was included in the hybridization solution to serve as a positive control for efficiency of the hybridization experiment. Slides were counterstained with DAPI following post-hybridization washes.
In three alternative lenghtening of telomeres -positive cases, co-immunolabeling with the promyelocytic leukemia (PML) gene product was performed. Slides were incubated for 2 hours at room temperature with the primary antibody (Rabbit polyclonal, 1:250, Bethyl, Cat # A301-167A) followed by a 30 min room temperature incubation with the secondary antibody (Alexa Fluor 647, 1:100).
Telomere-specific CISH was performed in two cases using previously described methods (11), with hydration and hybridization steps as described above. Endogenous peroxidase activity was blocked with Dual Endogenous Enzyme-Blocking Agent (Dako) for 10 minutes, followed by incubation with an anti-Cy3/Cy5 antibody (monoclonal ab52060, Abcam, 1:2500) for 1 hour at room temperature. This was followed by incubation with an anti-mouse secondary antibody (Leica Microsystems) for 30 minutes, and detected with 3,3′-diaminobenzidine (DAB, Sigma-Aldrich) after 10 minutes. As a final step, sections were counterstained with hematoxylin, rehydrated, and mounted. Positive calls were made by the presence of distinct ultrabright signals using FISH or CISH. Alternative lenghtening of telomeres was tested by FISH in 12 cases and by CISH in 2 cases.
Immunohistochemistry
All immunohistochemical staining obtained as part of the clinical diagnostic workup were re-reviewed as part of this study. Additional immunohistochemical studies were systematically performed as needed (e.g. missing or equivocal results) using the following antibodies: ATRX (Rabbit polyclonal, 1:200 dilution, catalog# HPA001906 Sigma-Aldrich), H3-K27M (Rabbit polyclonal, 1:400 dilution, catalog# ABE419, Millipore Sigma), p16 (CINtec®, prediluted, catalog# 705-4713, Roche), IDH1-R132H (Clone H09, 1:100 dilution, catalog# DIA H09 LM, Dianova), p53 (clone BP53-11, prediluted, catalog# 760-2542, Ventana) and PTEN (clone 6H2.1, 1:100 dilution, catalog# PM278AA, BioCare). These antibodies were tested on automated instruments (BenchMark, Ventana Medical Systems, Tucson, AZ, USA) with a protocol that included deparaffinization, hydration, antigen retrieval, incubation with primary antibody, and detection per manufacturer’s instructions. Immunohistochemistry for DAXX was performed manually using a rabbit polyclonal antibody (1:100 dilution, catalog# HPA008736, Atlas Antibodies). Primary antibody incubation was performed for 2 hours at room temperature followed by secondary antibody incubation (Leica Microsystems) for 30 min and detection with 3,3′-diaminobenzidine (DAB, Sigma-Aldrich) after 10 min. To confirm ATRX and DAXX immunoreactivity, positivity in internal non-neoplastic cell components was required in all cases scored.
Targeted Next Generation Sequencing
Areas enriched with tumor were scraped from adjacent 5-micron thick formalin-fixed paraffin-embedded sections (3 to 7 unstained slides per sample) and transferred to 2 ml tubes for DNA extraction. Extraction and purification was performed with the automated Siemens Tissue Preparation System (Siemens Healthcare Diagnostics, Inc., Tarrytown, NY). Genomic DNA was quantified using the Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA). DNA libraries were prepared using Agilent SureSelect-XT reagents (Agilent Technologies, Inc., Santa Clara, CA) with genomic regions of interest captured by means of an Agilent custom-designed bait set covering the full coding regions of 644 cancer associated genes (exon 1 poorly covered for some genes). The libraries were sequenced to an average unique read depth of greater than 500X using Sequencing by Synthesis (SBS) 2 x 100 base pairs (bp) paired-end cluster generation on the Illumina HiSeq 2500 plataform (Illumina, Inc., San Diego, CA). FASTQ files were generated from Binary Cluster Files (.bcl) using the Illumina bcl2fastq v1.8.4 software with parameters set as per vendor’s specifications. FASTQ files were aligned to the human genome reference hg19 (GRCh37) using the Burrows-Wheeler Aligner v0.7.10 algorithm with default settings. BAM files (.bam) were generated using Picard Tools v1.119 and variant calling was performed using in-house variant caller algorithm (MDLVC v5.0) cross referenced with HaplotypeCaller (Genome Analysis Tool Kit 3.3) under discovery mode in the coding regions of target genes. All variant calls were inspected using Integrated Genomics Viewer v2.3.4 (IGV; Broad Institute, MIT Harvard, Cambridge, MA) and annotated with dbSNP v150 and COSMIC v82 databases. Common single nucleotide polymorphisms (SNPs; population allele frequency >50%) were excluded from analysis. Matched normal (tissue or peripheral blood) was not available for comparison.
Mean target coverage of the full coding regions (including exon 1) of ATRX, DAXX, and NF1 was 688 reads (330 to 930 average base read depth), 811 reads (406 to 1129 average base read depth), and 823 reads (423 to 1089 average base read depth), respectively, across all cases.
Statistical analyses
Variables were described using proportions, ranges, means, and medians.
Results
Subependymal giant cell astrocytoma -like astrocytomas have distinct clinicopathologic features
Patients included 9 females and 5 males, with a mean age at diagnosis 28 years (range 4–60). All tumors occurred in the supratentorial compartment, predominantly the hemispheres, with the frontal lobe being the most common site (n=8). Histologically, the tumors were moderately cellular and characterized by cells with plump eosinophilic/glassy cytoplasm and large nuclei with macronucleoli, arranged in nests or short fascicles (Figure 1). The architecture was mostly compact (n=9) but partially infiltrative in five cases, based on hematoxylin and eosin stained sections or neuronal immunostains (i.e.neurofilament protein, synaptophysin) identifying underlying axons and/or entrapped neurons. No cases were diffusely infiltrating. Pilocytic features (e.g. Rosenthal fibers, eosinophilic granular bodies, hypereosinophilic cell processes) were focally present in 7 cases, but were not a dominant feature (Figure 1). Ependymoma or astroblastoma-like perivascular pseudorosettes (n=6), chronic inflammation (n=10), microcysts (n=8), and microcalfications (n=2) were variably present. Tumors were predominantly low grade (n=12) (Figure 1) with inconspicuous mitotic activity (<1 mitosis per 10 high power fields) in most cases (n=9), although up to 4 mitoses per 10 high power fields were found in a single case. Microvascular proliferation (n=3) and non-pseudopalisading necrosis (n=2) were present in a minority of cases. In contrast, two tumors were high grade (Figure 2) based on the presence of brisk mitotic activity (6 and 7 mitoses per 10 high power fields, respectively), microvascular proliferation, and necrosis. One of these two (case 7) had partial piloid features, including bipolar processes and microcysts, but these features were not dominant. Reticulin staining performed in five cases was limited to vessels and demonstrated no significant pericellular deposition.
Figure 1. Morphologic features of low grade neurofibromatosis type 1 -associated subependymal giant cell astrocytoma -like astrocytoma.

Large cells with voluminous eosinophilic cytoplasm and macronucleoli were distinctive features of all tumors (a). A vague fascicular architecture was seen in a subset of cases (b). Pilocytic features were not conspicuous, but included rare eosinophilic granular bodies (arrows) in a subset (c). Perivascular pseudorosettes were occasionally encountered (d). Pleomorphism and more overt ganglioid morphology was rare (e). Reticulin staining was limited to the microvasculature (f).
Figure 2. Morphologic features of high grade subependymal giant cell astrocytoma -like astrocytoma.

Two tumors were high grade, but preserved a solid pattern of growth (arrows) (a). These tumors displayed identical morphologic features as the other cases (b), but in addition brisk mitotic activity (arrow) (c) and necrosis (d).
All tumors expressed at least 1 glial marker (GFAP 10/10, OLIG2 8/8, S100 4/4), while neuronal marker expression was less consistent in frequency and extent (synaptophysin 6/10, chromogranin 3/6, NeuN 0/3) (Figure 3). P53 immunolabeling was either focal (6 of 9) or strong and diffuse (3 of 9). CD34 was negative in all 5 cases assessed. Additional immunohistochemical results included absence of IDH1 (R132H) (6 of 6), BRAF (V600E) (4 of 4), and H3K27M mutant proteins (2 of 2). Additionally, there was loss of p16 expression in 4 cases (partial n=3, complete n=1), while PTEN was retained in 3 (of 3) cases. Ki67 labeling index was predominantly low (n=9) with only a few moderate (n=2) to high (n=1) cases, ranging from <1 to 20%. For each case, the clinicopathologic features are outlined in Table 1.
Figure 3. Immunophenotypic features of low grade subependymal giant cell astrocytoma -like astrocytoma.

The immunohistochemical profile of subependymal giant cell astrocytoma -like astrocytoma is that of a predominantly glial tumor, based on GFAP (a), OLIG2 (b) and S100 (c) expression. Immunopositivity with neuronal markers like synaptophsin (d) and chromogranin (e) was more limited. Partial p16 loss was also frequent (f).
Table 1.
Clinicopathologic Features of SEGA-like astrocytoma
| CASE | age | sex | location | radiology | NF1 status | Clinical | Other NF1 features | Grade | surgery | Treatment | overall survival (days) | Follow-up |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 20 | F | Frontal lobe | heterogeneously enhancing | NF1 | New onset seizure | NA | Low grade | Resection | NA | NA | NA |
| 2 | 15 | M | R frontal lobe | 1.9 cm mass heterogeneous T2 signal | NF1 | Bilateral optic gliomas treated with chemo and/or rad; slow growing r frontal lobe lesion | Bilateral OPG | Low grade | Resection | NA | NA | NA |
| 3 | 17 | M | L frontal lobe | 2 cm enhacing mass slowly growing | NF1 | Seizures | NA | Low grade | GTR | Observation | 277 | NED |
| 4 | 24 | F | R frontal lobe | Solid an cystic mass with heterogeneous enhancement and mass effect | NF1 | NA | NA | Low grade | NA | NA | NA | |
| 5 | 53 | F | Lateral ventricle | focal enhancement, involvement of corpus callosum | NF1 | Headaches and blurry vision | OPG, neurofibromas | Low grade | STR | none | NA | Poor clinical course after surgery, died. |
| 6 | 20 | F | L frontal lobe | Heterogeneously enhancing mural noducle, cystic 5.8x5.6x4.3 cm mass | NF1 | worsening headaches, bilateral papilledema, binasal visual field deficits | Café au lait spots, plexiform neurofibroma, cutaneous neurofibromas | Low grade | GTR | Observation | 281 | NED |
| 7 | 25 | M | L frontal lobe | Solitary, partially cystic mass with heterogeneous enhancement and vasogenic edema | NF1 | NA | NA | High grade | Resection | NA | NA | NA |
| 8 | 9 | M | Hypothalamus | 4.2x3.2x3.6 cm mass with peripheral enhancement | NF1 | Morning headaches and vomiting | NA | Low grade | endoscopic biopsy | Carboplatin-Vincristine | 31 | undergoing chemo |
| 9 | 35 | M | R temporo-occipital lobe | 5.2x3.8x3.8 cm enhancing mass | Sporadic | GTC seizure | None | Low grade | GTR | observation | 97 | seizure activity but no tumor on f/u imaging |
| 10 | 46 | F | R temporal lobe | Large avascular mass involving the entire temporal lobe with associated uncal herniation | NF1 | gait unsteadiness, fainting x 8mo | Multiple neurofibromas and café au lait spots family history, café au lait spots, genu valgus deformity, OPG Pheochromocytoma, multiple neurofibromas, café au lait spot, spine deformities Multiple cutaneous neurofibromas, Lisch nodules, café au lait spots, freckling, plexiform neurofibroma |
Low grade | STR | irradiation | 81 | died of complications of surgery |
| 11 | 4 | F | L lateral ventricle | 4 cm peripherally enhancing mass with edema | NF1 | Headaches | Low grade | STR | irradiation, BCNU subsequently | 769 | Progression, DOD | |
| 12 | 49 | F | R frontal lobe | 1-CT scan: large cystic mass with adjacent edema 2-Angiogram: Highly vascular cystic mass involving much of the R frontal lobe | NF1 | generalized severe headaches, nausea, lightheadedness | High grade | STR | irradiation | 5417 | progressive decline probably related to radiation, died. | |
| 13 | 25 | F | L lateral ventricle | Nodular, partially enhancing mass; no parenchymal T2 signal | NF1 | Episodic vertigo, headache and unsteadiness | Low grade | GTR | Observation | NA | postop recovery | |
| 14 | 60 | F | Frontal lobe | 4x3.5x3.5 cm enhancing mass with associated edema in caudate head | NA | NA | NA | Low grade | STR | NA | 650 | DEAD |
NA=not available, GTR=gross total resection, STR=subtotal resection, NED=no evidence of disease, OPG=optic pathway glioma
Molecular genetic features of subependymal giant cell astrocytoma -like astrocytoma
Next generation sequencing was successful in 11 cases and technically failed in 3 due to poor DNA quality. All gene variants are listed by case in Supplementary Table 1 and by gene in Supplementary Table 2, including mutation type and variant allele frequencies. Relevant gene alterations are outlined in Table 2. A total of 205 gene variants were detected (average 19 gene variants per case, range 12–27). The majority of tumors had inactivating mutations in the NF1 gene, including one of the 2 patients lacking a clinical diagnosis of neurofibromatosis type 1 (per NIH guidelines). The other case is described below. When looking at Log2R ratios of normalized read depths, changes consistent with copy number loss in the NF1 gene were identifiedin 3 cases, with one additional case having two NF1 mutations (Table 2).
Table 2.
Molecular and Key Immunophenotypic Features of SEGA-like astrocytomas
| CASE | age | sex | NF1 status | ALT | ATRX IHC | DAXX IHC | NF1 | ATRX | DAXX | Gene variants of interest |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 20 | F | NF1 | neg | retained | retained | NF1 p.L180Yfs | WT | WT | BRCA1, FANCC, KMT2D, MAGEA1, RPN1, SMO, TAF1, XPC |
| 2 | 15 | M | NF1 | Positive | retained | retained | NF1 p.T2423Nfs | WT | WT | EPHB6, KMT2D, NTRK3, PDE4DIP, PDGFRB, PTPN6, RECQL4, RPN1, RPTOR, TSC2 |
| 3 | 17 | M | NF1 | neg | retained | retained | NF1 p.Y2171X | WT | WT | ALK, DCC, FN1, RNF213, SMARCA4 |
| 4 | 24 | F | NF1 | Positive | retained | retained | NF1 p.G1190Afs | WT | WT | CCT6B, FOXO3, MKI67, MSH3, NSD1, PASK, RB1, SMARCA1 |
| 5 | 53 | F | NF1 | neg | retained | NP | NF1 p.Y2264Tfs and NF1 p.G1219R | ATRX p.N1214I | WT | ARID1B, BCOR, CCT6B, DDX10, FN1, FOXO3, KMT2C, MAGEA1, NSD1, PARP1, PASK, PDE4DIP, SAMD9, STK36, XPC |
| 6 | 20 | F | NF1 | Positive | retained | retained | NF1 splice mutation (c.3198-2A>G) | WT | WT | ASMTL, DTX1, FANCD2, FANCF, FANCG, FN1, FOXO3, GNAS, KMT2D, NOTCH1, PASK, PDE4DIP, RECQL4, SAMD9, SPEN |
| 7 | 25 | M | NF1 | Positive | retained (but aberrant pattern) | retained | NF1 p.L2323X | ATRX p.N1860S (100% VAF) | WT | ARID1B, BCORL1, DTX1, KMT2C, KMT2D, NOTCH4, PDE4DIP, SMO |
| 8 | 9 | M | NF1 | neg | retained | NP | NF1 p.R720Gfs | WT | WT | AKAP9, ASXL1, BLM, CASP8, DDX10, NOTCH1, PASK, PDE4DIP |
| 9 | 35 | M | Sporadic | neg | retained | retained | WT1 | WT | WT | AKAP9, ASMTL, FGFR4, IGF2R, KMT2C, NOTCH1, NOTCH2, PER1, RPTOR, TAF1L, TSC2, WRN |
| 10 | 46 | F | NF1 | neg | Failed | NP | Failed | Failed | Failed | Failed |
| 11 | 4 | F | NF1 | neg | NP | NP | Failed | Failed | Failed | Failed |
| 12 | 49 | F | NF1 | Neg | equivocal | NP | Failed | Failed | Failed | Failed |
| 13 | 25 | F | NF1 | neg | retained | NP | NF1 p.S285Qfs1 | WT | WT | ARHGAP26, BRCA2, EPHB6, KMT2D, NCOR1, SPEN, STK36 |
| 14 | 60 | F | Sporadic | neg | retained | retained | NF1 p.R815Gfs1 | WT | WT | BCORL1, BRCA2, CASP8, IDH1, MKI67, NCOR1, PER1, TAF1 |
NP, not performed; WT, wild type;
These cases had additional changes consistent with NF1 copy number loss
Additional gene variants of interest present in two or more cases involved pathways related to apoptosis (CASP8, FOXO3A), cell adhesion (FN1), cell proliferation (MKI67), circadian rhythms (PER1), receptor tyrosine kinases and oncogenic signaling (IGF2R, AKAP9, EPHB4/EPHB6, GNAS, PASK, PDE4DIP, SMO, STK36), NOTCH signaling (DTX1, NOTCH1/2/4), DNA/RNA maintenance/repair (BRCA1/BRCA2, DDX10, Fanconi Anemia Genes (FANCs), MSH3, RECQL4, XPC), protein chaperones (CCT6B), epigenetic regulators/methyltransferases (ASMTL, BCOR, BCORL1, KAT6A, KMT2C/KMT2D, MLLT4/6/10), transcription (MAGEA1, NSD1, SPEN, TAF1, TAF1L), chromatin components or remodeling (ARID1B, NCOR1, SMARCA1/SMARCA4), and protein degradation (RPN1).
One case, without a clinical history of neurofibromatosis type 1 or tuberous sclerosis, had a largely extraventricular temporo-occipital tumor with similar morphologic features to the other tumors, reminiscent of subependymal giant cell astrocytoma (Figure 4). Next generation sequencing did not reveal NF1 mutations, but rather a truncating mutation in TSC2 (p.G305*, variant allele frequency 48%) and a rare RPTOR p.R789H variant (variant allele frequency 58%, rs374232748, allele frequency 0.005% ExAC). Interestingly, TSC2 (p.P1786S, rs371543182, allele frequency 0.001% ExAC) and RPTOR (p.V290I, rs374127851, AF 0.002% ExAC) variants were also present in a single neurofibromatosis type 1 -associated case with an NF1 mutation.
Figure 4. Imaging and morphologic features of sporadic subependymal giant cell astrocytoma -like astrocytoma.

One case developed arose in the absence of clinical features of neurofibromatosis type 1. MRI shows a well circumscribed intraparenchymal mass, heterogeneously T2 hypointense (a,c) with avid contrast enhancement (b,d) in the right parieto-occipital region, with a rim of vasogenic edema. The mass abuts the right occipital horn and exerts mass effect, displacing it anteriorly and partially effacing it. Note that there has been previous surgical resection. Morphologic features were similar to neurofibromatosis type 1 -associated subependymal giant cell astrocytoma -like astrocytoma (e), including GFAP expression (f). A truncating mutation in TSC2 was identified in the tumor tissue.
Glioma associated genes
Variants in genes associated with diffuse gliomas were rare in this cohort. Specifically, there were no mutations in canonical IDH1, IDH2, or BRAF hotspots, nor were there alterations in EGFR, PTEN, PIK3C genes, H3F3A, HIST1H3B, or TP53 (Supplementary Table 2). A variant in IDH1 (p.V178I, variant allele frequency 46%, rs34218846, 5% ExAC) was detected in a single case, but this most likely represents an uncommon germline variant. A single variant was also observed in RB1 (p.Q631K, variant allele frequency 44%) in a region that is typically highly conserved among vertebrates and primates (Genomic Evolutionary Rate Profiling score 5.97; UCSC Genome Browser, https://genome.ucsc.edu).
Alternative Lengthening of Telomeres in subependymal giant cell astrocytoma -like astrocytoma may develop in the absence of ATRX and DAXX alterations
Four cases (of 14; 29%) were alternative lenghtening of telomeres -positive, as evaluated by telomere-specific FISH (Figure 5 and Table 2). One of these cases (case 7) had an ATRX mutation (p.N1860S) and an aberrant pattern of ATRX immunoreactivity, with scattered cell nuclei showing decreased positivity but containing large positive granules (Figure 5). The other 3 cases lacked ATRX and DAXX mutations and demonstrated preserved ATRX and DAXX immunoreactivity. PML was found to be colocalized with large telomeric foci, as determined by telomere FISH and immunofluorescence (Figure 5h, inset). These observations support the possibility that alterations in other genes may also facilitate alternative lenghtening of telomeres. Within these 3 alternative lenghtening of telomeres -positive/ATRX-intact cases, gene variants of possible interest include the DNA helicase RECQL4 (2 of 3 cases). The two variants identified in our study are located in the c-terminal PARP-1 interaction site (Figure 6a). When looking at the Cancer Genome Atlas (TCGA) data via the cBioPortal for Cancer Genomics website (www.cbioportal.org) (16, 17), RECQL4 mutations were identified in 514 (1.2%) samples across cancer subtypes (Figure 6b) but were absent in diffuse gliomas. Other variant of possible relevance in this group of alternative lenghtening of telomeres +/ATRX intact subependymal giant cell astrocytoma -like astrocytomas involve components of the Fanconi anemia complementation group (FANCD2, FANCF, and FANCG) (n=1) and SMARCA1 (n=1).
Figure 5. Morphologic and immunophenotypic features of subependymal giant cell astrocytoma -like astrocytoma with alternative lengthening of telomeres.
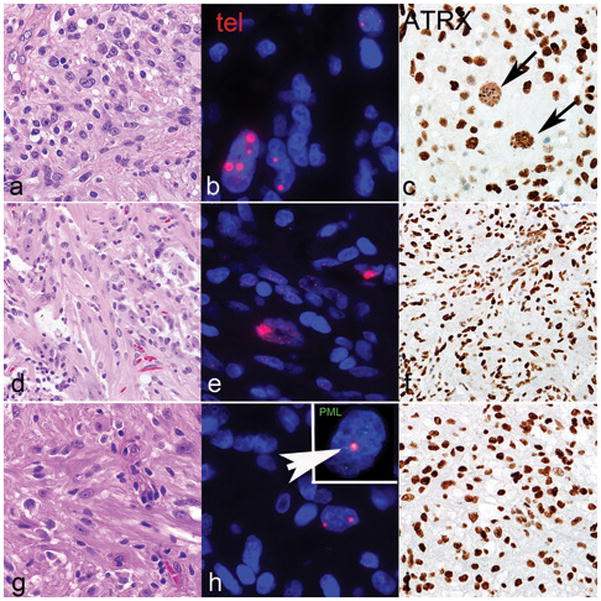
A subset of cases with subependymal giant cell astrocytoma -like morphology (a, d, g) were alternative lenghtening of telomeres -positive, as assessed by ultrabright telomere-specific FISH signals (b, e, h). ATRX demonstrated an aberrant pattern of staining in a subset of cells (arrows, c) and a concurrent ATRX mutation in case 7. The remaining cases had intact ATRX immunoreactivity and were wild type for the ATRX gene (f, i). Telomeric foci colocalized with PML bodies (arrow) (h, inset). Case 7=a,b,c; Case 2=d,e,f; Case 6=g,h,i.
Figure 6. RECQL4 mutations.
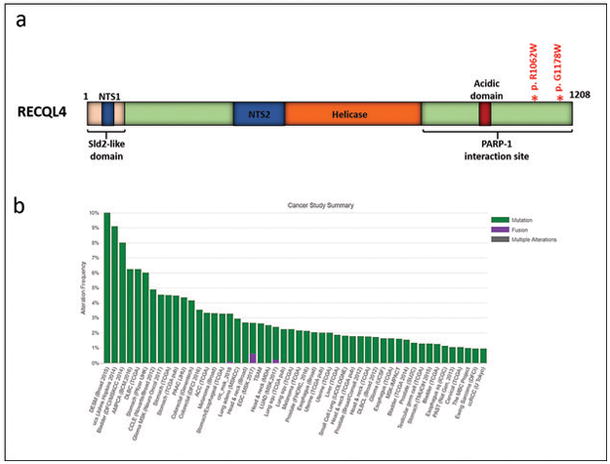
Mutations in RECQL4 involved the PARP-1 interacting domain in two subependymal giant cell astrocytoma -like astrocytoma (a) (modified from (32–34)). RECQL4 mutations occur at low frequencies across cancer types represented in the Cancer Genome Atlas (http://www.cbioportal.org/) (16, 17).
Outcomes
Clinical follow-up was available in 8 cases. Two patients died secondary to complications around the time of surgery, while one patient with a high grade tumor died 15 years after a subtotal tumor resection, most likely from complications of radiation therapy. Two additional patients died with progressive disease approximately two years after subtotal resections. Three patients are alive without evidence of disease following 9 months, 9 months, and 3 months of observation after gross total resections, and one patient is stable, undergoing chemotherapy 1 month after surgery.
Discussion
In the current study, we describe distinctive phenotypic features of an astrocytoma, frequently neurofibromatosis type 1 -associated, that overlap with those of the better-known subependymal giant cell astrocytoma which develops in patients with tuberous sclerosis complex. These features include the presence of cells with voluminous, often glassy cytoplasm, and large nuclei with macronucleoli (18). Of interest, one of the tumors was predominantly extraventricular and developed in a patient lacking clinical evidence of neurofibromatosis type 1 or tuberous sclerosis complex, but had a truncating TSC2 mutation in the sequenced tumor tissue. Subependymal giant cell astrocytoma generally develops within the lateral ventricles near the foramen of Monro in patients with tuberous sclerosis complex, and demonstrates an excellent response to mTOR inhibitors (19). Extraventricular locations in patients with tuberous sclerosis have been reported, although they are distinctly uncommon and essentially limited to the pediatric literature (20). Therefore, when considering a diagnosis of extraventricular subependymal giant cell astrocytoma, alternative tumor types must be carefully considered first. In our experience, separating subependymal giant cell astrocytoma -like astrocytoma from extraventricular subependymal giant cell astrocytoma is not possible on a morphologic basis; therefore a careful clinical history, looking for specific criteria of neurofibromatosis type 1 or tuberous sclerosis complex may be advisable in these patients.
The consistent molecular genetic finding in this set of tumors is the presence of NF1 gene alterations in all cases tested. Copy number loss was found in 3 cases, with an additional case having two inactivating NF1 mutations consistent with homozygous inactivation. Interestingly, the cases with changes consistent with copy number loss included the tumor developing in the absence of a documented clinical history of neurofibromatosis type 1 syndrome, which suggests that NF1 loss may play a basic role in this unique phenotype. Although, a second NF1 hit was not identified in the remaining cases, the sensitivity of many next generation sequencing platforms for the identification of copy number alterations is still limited.
Prior published studies have linked alterations in ATRX or DAXX genes with alternative lenghtening of telomeres in a subset of cancers (10). Loss of ATRX function leads to abnormal methylation and gene expression patterns, histone composition, as well as chromosome missegregation (21, 22). In the nucleus, ATRX cooperates with the molecular chaperone DAXX to incorporate the H3.3 histone variant in heterochromatic regions, including telomeres (23). ATRX mutations and concurrent alternative lenghtening of telomeres are associated with specific molecular subgroups of brain tumors (24). We have previously demonstrated that ATRX loss and alternative lenghtening of telomeres are frequent features of diffuse and high grade astrocytomas developing in patients with neurofibromatosis type 1 (11). In our previous study, alternative lenghtening of telomeres was present in 8 (of 12) diffuse/high grade gliomas compared to only 2 (of 14) low grade circumscribed gliomas, while ATRX loss was identified in 7 (of 12) diffuse/high grade gliomas compared to 2 (of 14) low grade circumscribed gliomas (11). We have also previously developed diagnostic criteria for the rare PAs with anaplasia (14), approximately 25% of which develop in neurofibromatosis type 1 patients (14). In addition, we demonstrated an association with CDKN2A deletions and increased PI3K/AKT signaling in these tumors (25). Recent studies have reported a high frequency of CDKN2A, MAPK pathway, and ATRX alterations, as well as alternative lenghtening of telomeres, in pilocytic astrocytomas with anaplasia (26, 27).
In the present study, we found alternative lenghtening of telomeres in 4 cases, but only 1 had a pathogenic ATRX mutation and an aberrant pattern of ATRX immunopositivity. Evolving evidence suggests that alterations in genes other than ATRX and DAXX may lead to or facilitate the development of alternative lenghtening of telomeres. For example, alternative lenghtening of telomeres -associated features have been induced through knockdown of ASF1 (28), and proteomic analyses have identified important roles for DNA polymerase eta (Pol η) in alternative lenghtening of telomeres (29). Similarly, FANCD2 and BLM, two enzymes involved in DNA repair and replication, seem to limit the telomere instability resulting from alternative lenghtening of telomeres (30). Of interest, one of the alternative lenghtening of telomeres -positive cases with wildtype ATRX had variants in multiple components of the Fanconi anemia complementation group (FANCD2, FANCF, and FANCG), while another case that lacked alternative lenghtening of telomeres demonstrated a BLM gene variant. More recently, Mukherjee et al. demonstrated that IDH1 mutations and ATRX loss cooperated to induce alternative lenghtening of telomeres in the context of RAP1 (member of the telomere-protective shelterin complex) and XRCC1 (component of the non-homologous end joining repair pathway) downregulation (31).
Interestingly, two alternative lenghtening of telomeres -positive/ATRX intact cases had mutations in RECQL4. RECQL4 encodes an ATP-dependent DNA helicase that is part of a group of the RecQ helicase protein family, which also includes WRN and BLM, and plays critical roles in genome maintenance and stability (32–34). Germline mutations in RECQL4 are associated with Rothmund-Thomson syndrome and related disorders. These patients develop poikiloderma, juvenile cataracts, and a predisposition to develop osteosarcoma (35). It must be noted that osteosarcoma is one of the human cancers with the highest frequency of alternative lenghtening of telomeres (36, 37), and indeed cancer is the leading cause of death in patients with germline RECQL4 mutations (32). Many of the pathogenic mutations that have been described involve the helicase and the PARP1 interaction domains, and usually spare the N-terminus (33). The interaction with PARP1 appears to play a role in RECQL4 localization to the nucleolus in response to oxidative stress (38). Interestingly, experiments have shown that RECQL4 associates with activity on telomeric D-loops and telomeric substrates containing thymine glycol, suggesting a role in telomere maintenance (39). We also found a gene variant in SMARCA1 in an alternative lenghtening of telomeres -positive/ATRX-intact case. SMARCA1 is involved in chromatin dynamics (40). While potentially interesting, determining any functional role(s) of this gene, as well as RECQL4, in alternative lenghtening of telomeres will require future formal testing in functional experiments.
Our data supports the notion that next generation sequencing efforts are helpful in the genetic characterization of subependymal giant cell astrocytoma -like astrocytomas. Except for one neurofibromatosis type 1 -associated case, which had TSC2 and RPTOR gene variants accounting for this peculiar subependymal giant cell astrocytoma -like morphology, genetic hits in mTOR pathway components were not identified. These results suggest that increased mTOR pathway activity in these tumors may be a result of NF1 loss, as has been described in neurofibromatosis type 1 -associated tumors (41). Additionally, some of the morphologic features overlap with pleomorphic xanthoastrocytoma, which has also been reported in neurofibromatosis type 1 patients (42). However, subependymal giant cell astrocytoma -like astrocytoma lacks significant pleomorphism, only rarely has eosinophilic granular bodies and does not demonstrate CD34 expression or increased reticulin deposition, in contrast to pleomorphic xanthoastrocytoma.
The clinical outcome of the patients in our series appears to be heterogeneous. Approximately half of patients with follow-up did well even after observation, but a subset died from complications of treatment and two others with progression after subtotal resections. Therefore, the outcome in some of these patients could justify a WHO grade II designation, as in pleomorphic xanthoasrocytoma but precise grade assignment will require further studies and increased awareness of the entity. Observation may be advisable when a gross total resection is feasible in these subependymal giant cell astrocytoma-like asrocytomas, given that neurofibromatosis type 1 patients are particularly sensitive to the negative effects of irradiation. It is noteworthy that a minority (two patients in our cohort) demonstrated high grade features. Interestingly, one of these two patients survived for 15 years after diagnosis, and appeared to develop progressive decline related to treatment (irradiation) rather than tumor recurrence (case 12). Although it is tempting to interpret these tumors as glioblastomas, IDH wildtype, we do not agree with this approach. Given the presence of bland áreas, occasional piloid features (case 7), and relatively compact architecture, a more relevant comparison is to pilocytic astrocytoma with anaplasia which even in the presence of necrosis had a clinical outcome better than glioblastoma and closer to historic cohorts of anaplastic astrocytoma (WHO grade III) (14), and in a more recent study similar to patients with IDH mutant glioblastomas, but better than IDH wildtype glioblastomas (26). Strict, reproducible grading criteria is still pending for these rare tumors.
In summary, we expand our experience with subependymal giant cell astrocytoma -like astrocytoma, revisit its clinicopathologic features, and provide novel genetic data in this unique primary central nervous system tumor. Future studies will further clarify the underlying biology of these tumors in a more precise manner, and hopefully develop novel therapeutic approaches for these patients.
Supplementary Material
Acknowledgments
This work was supported in part by NIH grant 5T32CA193145-02 (D.N.P.), the Pilocytic/Pilomyxoid Fund, including Lauren’s First and Goal, and the Stick it to Brain Tumors Annual Women’s Ice Hockey Tournament (F.J.R.); the A Kids’ Brain Tumor Cure Foundation/Pediatric low grade astrocytoma foundation (F.J.R), NIH grant P30 CA006973 to the Sidney Kimmel Comprehensive Cancer Center (PI: W. Nelson).
References
- 1.Gutmann DH, McLellan MD, Hussain I, et al. Somatic neurofibromatosis type 1 (NF1) inactivation characterizes NF1-associated pilocytic astrocytoma. Genome Res. 2013;23:431–9. doi: 10.1101/gr.142604.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodriguez FJ, Perry A, Gutmann DH, et al. Gliomas in neurofibromatosis type 1: a clinicopathologic study of 100 patients. J Neuropathol Exp Neurol. 2008;67:240–9. doi: 10.1097/NEN.0b013e318165eb75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lopes MB, Altermatt HJ, Scheithauer BW, Shepherd CW, VandenBerg SR. Immunohistochemical characterization of subependymal giant cell astrocytomas. Acta Neuropathol. 1996;91:368–75. doi: 10.1007/s004010050438. [DOI] [PubMed] [Google Scholar]
- 4.Jentoft M, Giannini C, Cen L, et al. Phenotypic variations in NF1-associated low grade astrocytomas: possible role for increased mTOR activation in a subset. Int J Clin Exp Pathol. 2010;4:43–57. [PMC free article] [PubMed] [Google Scholar]
- 5.Chan JA, Zhang H, Roberts PS, et al. Pathogenesis of tuberous sclerosis subependymal giant cell astrocytomas: biallelic inactivation of TSC1 or TSC2 leads to mTOR activation. J Neuropathol Exp Neurol. 2004;63:1236–42. doi: 10.1093/jnen/63.12.1236. [DOI] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Research N. Brat DJ, Verhaak RG, et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med. 2015;372:2481–98. doi: 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang J, Wu G, Miller CP, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. 2013;45:602–12. doi: 10.1038/ng.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones DT, Hutter B, Jager N, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45:927–32. doi: 10.1038/ng.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sturm D, Witt H, Hovestadt V, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–37. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 10.Heaphy CM, de Wilde RF, Jiao Y, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425. doi: 10.1126/science.1207313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodriguez FJ, Vizcaino MA, Blakeley J, Heaphy CM. Frequent alternative lengthening of telomeres and ATRX loss in adult NF1-associated diffuse and high-grade astrocytomas. Acta Neuropathol. 2016;132:761–3. doi: 10.1007/s00401-016-1619-0. [DOI] [PubMed] [Google Scholar]
- 12.Salloum R, McConechy MK, Mikael LG, et al. Characterizing temporal genomic heterogeneity in pediatric high-grade gliomas. Acta Neuropathol Commun. 2017;5:78. doi: 10.1186/s40478-017-0479-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Louis DN, Ohgaki H, Wiestler OD, et al., editors. WHO Classification of Tumours of the Central Nervous System. International Agency for Research on Cancer; Lyon, France: 2016. Revised 4th Edition. [Google Scholar]
- 14.Rodriguez FJ, Scheithauer BW, Burger PC, Jenkins S, Giannini C. Anaplasia in pilocytic astrocytoma predicts aggressive behavior. Am J Surg Pathol. 2010;34:147–60. doi: 10.1097/PAS.0b013e3181c75238. [DOI] [PubMed] [Google Scholar]
- 15.Heaphy CM, Subhawong AP, Hong SM, et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Pathol. 2011;179:1608–15. doi: 10.1016/j.ajpath.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopes MB, Wiestler B, Stemmer-Rachamimov AO, et al. Subependymal giant cell astrocytoma. In: Louis DN, Ohgaki H, Wiestler B, et al., editors. WHO Classification of Tumours of the Central Nervous System. Revised. 4. International Agency for Research on Cancer; Lyon: 2016. pp. 90–3. [Google Scholar]
- 19.Franz DN, Belousova E, Sparagana S, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2013;381:125–32. doi: 10.1016/S0140-6736(12)61134-9. [DOI] [PubMed] [Google Scholar]
- 20.Bollo RJ, Berliner JL, Fischer I, et al. Extraventricular subependymal giant cell tumor in a child with tuberous sclerosis complex. J Neurosurg Pediatr. 2009;4:85–90. doi: 10.3171/2009.3.PEDS08225. [DOI] [PubMed] [Google Scholar]
- 21.Ritchie K, Seah C, Moulin J, et al. Loss of ATRX leads to chromosome cohesion and congression defects. J Cell Biol. 2008;180:315–24. doi: 10.1083/jcb.200706083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Danussi C, Bose P, Parthasarathy PT, et al. Atrx inactivation drives disease-defining phenotypes in glioma cells of origin through global epigenomic remodeling. Nat Commun. 2018;9:1057. doi: 10.1038/s41467-018-03476-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, Allis CD. Daxx is an H3. 3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci U S A. 2010;107:14075–80. doi: 10.1073/pnas.1008850107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen DN, Heaphy CM, de Wilde RF, et al. Molecular and morphologic correlates of the alternative lengthening of telomeres phenotype in high-grade astrocytomas. Brain Pathol. 2013;23:237–43. doi: 10.1111/j.1750-3639.2012.00630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodriguez EF, Scheithauer BW, Giannini C, et al. PI3K/AKT pathway alterations are associated with clinically aggressive and histologically anaplastic subsets of pilocytic astrocytoma. Acta Neuropathol. 2011;121:407–20. doi: 10.1007/s00401-010-0784-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reinhardt A, Stichel D, Schrimpf D, et al. Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol. 2018 doi: 10.1007/s00401-018-1837-8. [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez FJ, Brosnan-Cashman JA, Vizcaino MA, et al. USCAP. Vol. 31. Vancouver, Canada: 2018. Alternative lengthening of telomeres, ATRX loss and H3 p.K27M mutations in Anaplastic Pilocytic Astrocytoma; pp. 655–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Sullivan RJ, Arnoult N, Lackner DH, et al. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat Struct Mol Biol. 2014;21:167–74. doi: 10.1038/nsmb.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia-Exposito L, Bournique E, Bergoglio V, et al. Proteomic Profiling Reveals a Specific Role for Translesion DNA Polymerase eta in the Alternative Lengthening of Telomeres. Cell Rep. 2016;17:1858–71. doi: 10.1016/j.celrep.2016.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Root H, Larsen A, Komosa M, et al. FANCD2 limits BLM-dependent telomere instability in the alternative lengthening of telomeres pathway. Hum Mol Genet. 2016;25:3255–68. doi: 10.1093/hmg/ddw175. [DOI] [PubMed] [Google Scholar]
- 31.Mukherjee J, Johannessen TA, Ohba S, et al. Cancer Res. 2018. Mutant IDH1 cooperates with ATRX loss to drive the alternative lengthening of telomere (ALT) phenotype in glioma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Croteau DL, Popuri V, Opresko PL, Bohr VA. Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem. 2014;83:519–52. doi: 10.1146/annurev-biochem-060713-035428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sidorova JM, Monnat RJ., Jr . Encyclopedia of Life Sciences. John Wiley & Sons; 2010. DNA Helicase-deficiency Disorders. [Google Scholar]
- 34.Croteau DL, Singh DK, Hoh Ferrarelli L, Lu H, Bohr VA. RECQL4 in genomic instability and aging. Trends Genet. 2012;28:624–31. doi: 10.1016/j.tig.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mo D, Zhao Y, Balajee AS. Human RecQL4 helicase plays multifaceted roles in the genomic stability of normal and cancer cells. Cancer Lett. 2018;413:1–10. doi: 10.1016/j.canlet.2017.10.021. [DOI] [PubMed] [Google Scholar]
- 36.Henson JD, Hannay JA, McCarthy SW, et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin Cancer Res. 2005;11:217–25. [PubMed] [Google Scholar]
- 37.Ulaner GA, Hoffman AR, Otero J, et al. Divergent patterns of telomere maintenance mechanisms among human sarcomas: sharply contrasting prevalence of the alternative lengthening of telomeres mechanism in Ewing’s sarcomas and osteosarcomas. Genes Chromosomes Cancer. 2004;41:155–62. doi: 10.1002/gcc.20074. [DOI] [PubMed] [Google Scholar]
- 38.Woo LL, Futami K, Shimamoto A, Furuichi Y, Frank KM. The Rothmund-Thomson gene product RECQL4 localizes to the nucleolus in response to oxidative stress. Exp Cell Res. 2006;312:3443–57. doi: 10.1016/j.yexcr.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 39.Ferrarelli LK, Popuri V, Ghosh AK, et al. The RECQL4 protein, deficient in Rothmund-Thomson syndrome is active on telomeric D-loops containing DNA metabolism blocking lesions. DNA Repair (Amst) 2013;12:518–28. doi: 10.1016/j.dnarep.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goodwin LR, Picketts DJ. The role of ISWI chromatin remodeling complexes in brain development and neurodevelopmental disorders. Mol Cell Neurosci. 2018;87:55–64. doi: 10.1016/j.mcn.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 41.Banerjee S, Crouse NR, Emnett RJ, Gianino SM, Gutmann DH. Neurofibromatosis-1 regulates mTOR-mediated astrocyte growth and glioma formation in a TSC/Rheb-independent manner. Proc Natl Acad Sci U S A. 2011;108:15996–6001. doi: 10.1073/pnas.1019012108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vizcaino MA, Caccamo DV, Fox E, Rodriguez FJ. Pleomorphic xanthoastrocytoma: report of two cases with unconventional clinical presentations. Clin Neuropathol. 2014;33:380–7. doi: 10.5414/NP300766. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.