

Abstract
YARS2 variants have previously been described in patients with myopathy, lactic acidosis and sideroblastic anemia 2 (MLASA2). YARS2 encodes the mitochondrial tyrosyl-tRNA synthetase, which is responsible for conjugating tyrosine to its cognate mt-tRNA for mitochondrial protein synthesis. Here we describe 14 individuals from 11 families presenting with sideroblastic anemia and YARS2 variants that we identified using a sideroblastic anemia gene panel or exome sequencing. The phenotype of these patients ranged from MLASA to isolated congenital sideroblastic anemia. As in previous cases, inter- and intra-familial phenotypic variability was observed, however, this report includes the first cases with isolated sideroblastic anemia and patients with biallelic YARS2 variants that have no clinically ascertainable phenotype. We identified ten novel YARS2 variants and three previously reported variants. In vitro amino-acylation assays of five novel missense variants showed that three had less effect on the catalytic activity of YARS2 than the most commonly reported variant, p.(Phe52Leu), associated with MLASA2, which may explain the milder phenotypes in patients with these variants. However, the other two missense variants had a more severe effect on YARS2 catalytic efficiency. Several patients carried the common YARS2 c.572 G>T, p.(Gly191Val) variant (minor allele frequency =0.1259) in trans with a rare deleterious YARS2 variant. We have previously shown that the p.(Gly191Val) variant reduces YARS2 catalytic activity. Consequently, we suggest that biallelic YARS2 variants, including severe loss-of-function alleles in trans of the common p.(Gly191Val) variant, should be considered as a cause of isolated congenital sideroblastic anemia, as well as the MLASA syndromic phenotype.
Introduction
Sideroblastic anemia is defined by the presence of bone marrow ringed sideroblasts, which are erythroblasts containing pathological intramitochondrial iron deposits.1 Congenital sideroblastic anemias (CSAs) are caused by a growing list of genetic variants that affect mitochondrial pathways, including heme synthesis, iron-sulfur cluster biogenesis, mitochondrial protein synthesis, and oxidative phosphorylation.2,3 Variants in YARS2 have been associated with myopathy, lactic acidosis, and sideroblastic anemia 2 (MLASA2; OMIM #613561),4–8 and recently cases of YARS2-related myopathy in the absence of sideroblastic anemia have been reported.9 YARS2 encodes the mitochondrial tyrosyl-tRNA synthetase, YARS2, which is responsible for the ATP-dependent conjugation of tyrosine to its cognate tRNA, required to support mitochondrial protein synthesis.10 YARS2 catalyses this reaction in a two-step process. In the first step, tyrosine and ATP bind to the catalytic domain to form the tyrosyl-adenylate intermediate. In the second step, cognate tRNATyr binds the synthetase and the tyrosyl moiety is transferred to the tRNA CCA-end. The resulting tyrosyl-tRNATyr will be delivered to the ribosome.
The most commonly reported YARS2 variant, p.(Phe52Leu), prevalent in patients of Lebanese Christian descent, has been shown to reduce YARS2 amino-acylation catalytic efficiency by approximately 9-fold, and leads to a reduction in mitochondrial protein synthesis in patients with MLASA2.4 Here we report YARS2 variants, some of which were associated with milder effects on amino-acylation, in patients with isolated CSA, or CSA with mild myopathy and lactic acidosis. In addition, we describe two pairs of genotypically identical siblings with divergent, affected and unaffected, clinical phenotypes. Importantly, some patients carry a common YARS2 c.572 G>T, p.(Gly191Val), that we and others have previously shown has a mild effect on amino-acylation activity,5,11 and suggest that these milder alleles may be the basis of the reduced penetrance and expressivity.
Methods
Clinical data
The patients and their immediate family members were referred to MMH, MDF, NS or LA for clinical consultation. Written informed consent was obtained from participants in the study, as approved by the Institutional Review Boards of Boston Children’s Hospital, USA, the Radboud University Medical Center, the Netherlands, and the Hospital Germans Trias i Pujol, Badalona, Spain. In each case, CSA was ascertained by complete blood counts (CBCs), and peripheral blood or bone marrow morphology. Detailed clinical histories are provided in the Online Supplementary Appendix.
Variant detection
Targeted sequencing of nuclear encoded CSA genes,12 and the mitochondrial genome as well as mitochondrial DNA deletion analysis was performed on the probands of families 1-3 and 5-9. Genomic DNA was isolated from peripheral blood or skin fibroblasts, using the Puregene DNA Purification Kit (Qiagen, Valencia, CA, USA). DNA templates for sequencing were amplified from genomic DNA by PCR, enzymatically cleaned, bidirectionally sequenced using fluorescent dye termination sequencing chemistry, and analyzed with the Sequencher 5.3 DNA sequence assembly software (Gene Codes, Ann Arbor, MI, USA), as previously described.12
Exome sequencing for Patient 4 was performed on genomic DNA isolated from whole blood. The experimental workflow was performed at BGI Europe (Bejing Genome Institute Europe, Copenhagen, Denmark) using an Illumina Hiseq (Illumina, CA, USA) platform. Variants in genes previously associated with Mendelian diseases (OMIM), including CSAs, were analyzed bioinformatically.
Patient 10 DNA was analyzed using a targeted gene panel for congenital and acquired sideroblastic anemias, including ABCB7, ALAS2, GLRX5, PUS1, SF3B1, SLC19A2, SLC25A38, STEAP3, TRNT1 and YARS2. The library was constructed using the Custom HaloPlex™ Target Enrichment System (Agilent, Santa Clara, CA, USA) and sequenced on a MiSeq platform (Illumina, San Diego, CA, USA). Data were analyzed with SureCall software (Agilent, Santa Clara, CA, USA).
Patient 11 DNA was analyzed using a targeted gene panel for sideroblastic anemia (ABCB7, ALAS2, GLRX5, HSCB, HSPA9, PUS1, SLC25A38, STEAP3, YARS2) and ion semiconductor sequencing as developed by Ion Torrent systems.13
In silico predictions of variant pathogenicity were performed using the Alamut Visual suite of genetic analysis software (Interactive Biosoftware, Rouen, France), and linking externally to the PolyPhen2 and SIFT analytical tools.14,15 Minor allele frequencies are reported as in gnomAD (gnomad.broadinstitute.org) current as of September 2017.16
Amino-acylation assays
Recombinant wild-type and the p.(Leu61Val), p.(Met195Ile), p.(Ser203Ile), p.(Tyr236Cys) and p.(Gly244Ala) YARS2 variants were expressed in E. coli, purified to homogeneity and assayed for tyrosylation activity as previously described.10 Apparent kinetic parameters were determined from Lineweaver-Burk plots in the presence of 4.8 to 6.5 nM YARS2 and 0.28 to 1.12 mM native E. coli tRNATyr (Sigma, St. Louis, MO, USA). Experimental errors on kcat and Km varied at most by 20%. Numerical values are averages of at least two independent experiments.
Results
Phenotypic spectrum
Eleven probands with CSA were identified with potentially pathogenic YARS2 variants by targeted gene sequencing panels or exome sequencing (Table 1A and 1B). The majority of these families were derived from a group of more than 200 probands with CSA referred to SSB, MDF and MMH, in which approximately 4% of cases were attributed to YARS2 variants. YARS2 variants have previously been identified in patients with myopathy, lactic acidosis and sideroblastic anemia 2 (MLASA2);4 however, some patients in this study did not have overt clinical features of MLASA2 other than CSA, and several individuals with biallelic variants had no phenotype whatsoever. In two families, the proband had moderate sideroblastic anemia (P8a and P9a), while a sibling with the same YARS2 genotype was not anemic and was otherwise asymptomatic (P8b and P9b) (Table 1B). In a third family (P2a and P2b) (Table 1A), the proband was identified with a severe, new onset anemia at six years of age, and, subsequent to her brother’s diagnosis, the younger sibling was found to be anemic. Four of the probands presented within the first two years of life (P5, P6, P7, P9a), and 4 presented in adolescence (P1, P4, P8a, P11). Two patients have died (P1, P5), both from multi-organ failure, one of these following two unsuccessful hematopoietic stem cell transplantations (HSCTs). One patient (P4) has undergone successful HSCT.
Table 1A.
Clinical data.

Table 1B.
Clinical data.

The 11 probands all had moderate to severe normocytic to macrocytic anemia. In nine probands, the presence of ringed sideroblasts, ranging from 10% to over 50% of bone marrow erythroblasts, was documented on bone marrow aspiration; marrows were not examined in 3 other patients and 2 clinically unaffected siblings (Table 1A and B). Eight patients required transfusion; however, one patient spontaneously became transfusion independent at 16 months of age (P7), and 3 patients had periods of hematologic remission (P4, P5, P9a), transiently becoming RBC transfusion independent. In addition to anemia, 3 probands had variable neutropenia and/or thrombocytopenia (P1, P6, P8a). Four patients were treated with pyridoxine with no improvement in their anemia (P4, P5, P6, P11).
Two patients had severe lactic acidosis (P1, P3), but the remaining cases in which it was studied had mild or no lactic acidosis (Table 1A and B). Two patients had elevated blood lactate upon light exercise (P4, P8a); those with mild lactic acidosis also tended to have mild myopathy, although one patient with no reported lactic acidosis had moderate myopathy (P7). Patient 1 (P1) with severe lactic acidosis and myopathy had combined respiratory chain deficiency in skeletal muscle, and the muscle biopsy showed histopathological features typical of a mitochondrial myopathy, including ragged red fibers on trichrome stain and “parking lot” inclusions and whorled arrays of mitochondrial cristae by transmission electron microscopy (data not shown). In one family, the proband (P9a) and his clinically unaffected, but genotypically identical sibling (P9b), had distinctive “triangular” faces, unlike their parents or genotypically normal sibling, which has not previously been reported in association with YARS2 variants, but has been described in mitochondrial myopathy with lactic acidosis and sideroblastic anemia 1 (MLASA1; OMIM #600462) due to pseudouridine synthase 1 (PUS1) variants.17
YARS2 variants in patients with congenital sideroblastic anemia
We identified three previously described YARS2 variants and ten novel variants in patients with CSA: the Lebanese Christian founder variant, p.(Phe52Leu),4 was in the homozygous state in 4 patients; the p.(Asp311Glu) variant8 homozygous in one patient; and a novel variant, p.(Leu61Val) homozygous in one patient. The remaining six families had compound heterozygous variants including four novel missense variants: p.(Met195Ile), p.(Ser203Ile), p.(Tyr236Cys), p.(Gly244Ala); a novel nonsense variant p.(Ser33*); three novel indels, p.(Thr197_Leu208del), p.(Leu389Cysfs*6), p.(Ile454Serfs*10); one novel splicing variant, c.1104-1G>A; and two previously reported missense variants, p.(Gly191Val) and p.(Asp311Glu).5,8 No patient had two indel or splicing variants.
The five novel missense variants all lie in the catalytic domain of YARS2 (Figure 1A) and are rare in the gnomAD database (gnomad.broadinstitute.org) (Table 2). In silico predictions of pathogenicity for p.(Leu61Val), p.(Met195Ile) and p.(Tyr236Cys) vary between the SIFT and PolyPhen2 prediction programs while p.(Ser203Ile) and p.(Gly244Ala) are predicted to be damaging to the YARS2 protein by both algorithms (Table 2 and Figure 1B). Conservation among species for each missense variant is shown in Online Supplementary Figure S1.
Figure 1.

Representation of mutated YARS2 proteins. (A) Schematic view of YARS2 domains: MTS: mitochondrial targeting sequence; ACB: anticodon binding domain; S4-Like: S4 ribosomal protein-like domain. Amongst all the variants identified, only those tested in this study are shown in cyan. Note that the recombinant YARS2 used in the amino-acylation assays is deprived of the MTS. (B) Model of YARS2 p.(Thr197-Leu208del), built with I-TASSER.28 The structural domains from (A) are shown with the same color code. The locations of the variants, which have the weakest effects on amino-acylation [p.(Leu61Val), p.(Met195Ile), p.(Tyr236Cys)] are shown in cyan. (C) Crystal structure of YARS2 catalytic domain19 with the tyrosyl-adenylate analog (TyrAMS, magenta) bound to the active site. The locations of variants p.(Ser203Ile) and p.(Gly244Ala), characterized by the strongest effects on amino-acylation, are indicated in cyan.
Table 2.
In silico predictions of pathogenicity for YARS2 missense variants.

The nonsense variant, the splicing variant and three novel indels are likely to be deleterious. The splicing variant c.1104-1G>A alters a canonical position in the 3ʹ splice acceptor site of intron 3 and it is predicted to result in skipping of exon 4. The YARS2 c.98C>A, p.(Ser33*) nonsense variant and the c.1165_1166insG, p.(Leu389Cysfs*6) frameshift variant both lie greater than 55 nucleotides upstream of the last exon-exon junction and are most likely targeted for nonsense mediated decay.18 The p.(Thr197_Leu208) in frame deletion results in loss of 12 residues in α-helical regions of the catalytic domain, and more precisely of cluster 1, which is important for tRNA acceptor end recognition19 (Figure 1B). The c.1360_1361insG, p.(Ile454Serfs*10) variant lies in the last exon of YARS2 and is not predicted to be targeted for nonsense mediated decay.18 This variant would cause a frameshift at position 454 in the S4-like domain, which is found in all prokaryotic and organellar tyrosyl-tRNA synthetases, and is thought to stabilize the interaction between the tRNA and YARS2.19,20
Amino-acylation activity of YARS2 missense variants
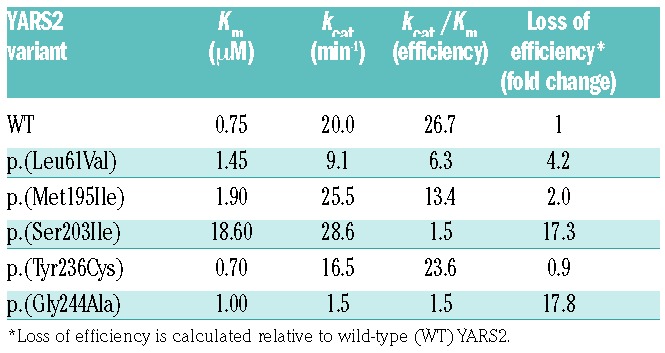
Amino-acylation assays are commonly used to evaluate the effect of variants on aminoacyl-tRNA synthetase activity, with reduced activity being a strong predictor of pathogenicity.21 Consequently, the effect of the five missense variants, p.(Leu61Val), p.(Met195Ile), p.(Ser203Ile), p.(Tyr236Cys) and p.(Gly244Ala) on amino-acylation activity was measured by the incorporation of [14C]-tyrosine into an E. coli tRNATyr substrate and compared to wild-type YARS2 activity. In vitro studies of the YARS2 variants revealed that amino-acylation efficiency was mildly reduced for p.(Leu61Val) and, p.(Met195Ile), while p.(Tyr236Cys) was not affected as compared to the wild-type enzyme (Table 3). YARS2 p.(Ser203Ile) and p.(Gly244Ala) demonstrated a 17-fold loss in catalytic efficiency. The reduced activity of YARS2 p.(Ser203Ile) is a consequence of an increased Km, indicating that its affinity for tRNATyr was reduced. On the other hand, the YARS2 p.(Gly244Ala) is characterized by a 13-fold lower kcat suggesting that the variant hinders efficient transfer of the tyrosyl moiety from the active site to the tRNA.
Table 3.
Kinetic parameters for tyrosylation of E. coli tRNATyr by YARS2 wild-type and novel missense variant recombinant proteins.
Discussion
Here we expand the clinical spectrum associated with YARS2 variants and describe patients with milder phenotypes who do not display all the features of MLASA2. Rather, most of the patients we describe presented principally with a normo- or macrocytic CSA; they are mostly non-syndromic and unlike the most common forms of non-syndromic sideroblastic anemia (e.g. ALAS2 or SLC25A38 deficiency), the anemia is not microcytic. Nevertheless, in addition to ringed sideroblasts, some of these patients had vacuolization of marrow precursors and/or other cytopenias that are often seen in the syndromic sideroblastic anemias (e.g. Pearson syndrome), which may be a diagnostic clue.
Patients 1 and 3 had all the typical features of MLASA2, whereas Patients 2a, and 2b, who share homozygosity for the YARS2 Lebanese founder allele, p.(Phe52Leu), had only anemia. Patient 1 also had other features not typically associated with MLASA2, including neutropenia, thrombocytopenia, pericardial effusion, and premature ovarian failure. Neutropenia and pericardial effusion have each been reported in one other patient homozygous for the p.(Phe52Leu) variant.5,22 Two other patients in the current series with other genotypes also had mild or intermittent neutropenia. Premature ovarian failure is associated with variants in several mitochondrial aminoacyl-tRNA synthetase-encoding genes including HARS2, LARS2 and AARS2,23–25 and thus may be a feature common to mitochondrial protein synthesis defects. There are now 10 reported individuals homozygous for the YARS2 p.(Phe52Leu) variant5,22 and all have been symptomatic, supporting complete penetrance of this allele. However, the great range of phenotypic severity strongly suggests the presence of other genetic and environmental influences that can modify the effects of YARS2 deficiency.
Patient 4 presented in late adolescence with sideroblastic anemia without myopathy and has a homozygous p.(Leu61Val) variant that diminished the amino-acylation catalytic efficiency 4-fold. Leu61 is located in a region of the catalytic domain specific to mitochondrial YARSs that was proposed to contact the tRNATyr acceptor helix (Figure 1B).19 In this case, HSCT appeared to be an effective treatment, restoring the patient’s hemoglobin levels to normal.
Patient 5 presented in infancy with CSA and was transfusion dependent other than a remission occurring between three and six years of age; she had no myopathy until her post-HSCT terminal illness. This patient had a YARS2 c.1165_1166insG variant predicted to result in a null allele, and a novel p.(Met195Ile) variant which lies within cluster 1, in a region involved in recognition of the tyrosine accepting arm of tRNATyr (Figure 1B).19 Some YARS proteins (e.g. yeast) have an isoleucine (Ile) at this position, suggesting that it might be a milder allele. Indeed, in vitro this mutant had little effect on YARS2 catalytic efficiency.
Patient 10 is a compound heterozygote for a splicing mutation (c.1104-1G>A) predicted to cause skipping of exon 4, and a missense variant p.(Ser203Ile), also located in cluster 1. YARS2 (p.Ser203Ile) led to a reduced affinity for tRNATyr, resulting in a 17-fold loss in catalytic efficiency (Figure 1C). Patient 10 has no lactic acidosis or myopathy, and presented with isolated normocytic anemia and asthenia, and has not required transfusion.
Patient 7 presented with anemia in infancy requiring two transfusions within the first 16 months of life and then became transfusion independent. She has moderate myopathy and no lactic acidosis and a compound heterozygous genotype: a missense variant, p.(Gly244Ala), occurring in cis with p.(Gly191Val) and in trans with the p.(Asp311Glu) variant. Gly244 is a critical residue for tyrosyl-adenylate binding.19 YARS2 p.(Gly244Ala) only affected the kcat indicating that, as predicted, this variant hinders binding of the tyrosyl-adenylate in the active site (Figure 1C). YARS2 Asp311 is involved in the recognition of anti-codon residue G34 of tRNATyr.19 The p.(Asp311Glu) variant is respiratory deficient in a yeast model, and patients homozygous for this allele also have transfusion-dependent sideroblastic anemia in the first year of life; however, in contrast to patient 7, they have lactic acidosis but no myopathy.8 Further phenotypic variability for the p.(Asp311Glu) variant was observed in Patient 11 who was homozygous for p.(Asp311Glu), with transfusion-dependent MLASA2.
In two families in this study (Families 6 and 8), affected patients have the common p.(Gly191Val) allele (MAF =0.1259) in trans of a predicted null allele. Importantly, all of the unaffected carriers of predicted null alleles in these and other families, where probands had the ancestral p.Gly191 variant in trans, were asymptomatic (data not shown). Patient 6 presented in infancy with CSA requiring transfusions every three weeks. She has mild lactic acidosis, no myopathy and intermittent neutropenia. She has a c.590_645del variant resulting in a 12 amino acid deletion in the catalytic domain (Figure 1A), which would almost certainly lead to a completely dysfunctional protein, in trans with p.(Gly191Val). Individuals 8a and 8b also carry p.(Gly191Val) in trans with a predicted null or severe loss-of-function allele, c.1360_1360insG, p.(Ile454Serfs*10). This variant truncates the S4-like domain which is thought to stabilize the interaction with tRNATyr, and the deletion of the YARS2 S4-like domain leads to a 100-fold reduced amino-acylation activity in vitro.20 Patient 8a had sideroblastic anemia and edema. Lactate was elevated only on exertion and the patient did not have myopathy. Her sister (P8b) is asymptomatic. Patient 8a also had a somatic mutation in SF3B1 p.(Lys700Glu) that is strongly associated with myelodysplastic syndromes with ringed sideroblasts.26 Based on the childhood presentation of her anemia and exercise intolerance that was exacerbated significantly decades later, and the fact that a mutation in SF3B1 would be exceptional in a patient under 30 years of age, we infer the YARS2 mutations to be the primary cause of her anemia with the SF3B1 mutation occurring as a secondary somatic event, which exacerbated her anemia, bringing her to clinical attention. In addition to the reduced activity in vitro,5 support of the notion that YARS2 p.(Gly191Val) contributes to the disease phenotype in these patients comes from the observation that this variant is a disease modifier in Leber Hereditary Optic Neuropathy (LHON); the three common LHON mitochondrial DNA mutations have incomplete penetrance. However, all patients who carry both the LHON m.11778G>A mtDNA disease-associated variant in combination with a homozygous YARS2 p.(Gly191Val) genotype were symptomatic.11
Patients 9a and 9b carried the YARS2 c.98C>A, p.(Ser33*) nonsense variant, which would result in a null allele, and the p.(Tyr236Cys) variant (Figure 1A and B) that did not alter amino-acylation activity in vitro. In addition, in silico analysis using Alamut did not predict that this variant would lead to alteration of an exonic splicing enhancer site. Patient 9a presented in infancy with sideroblastic anemia that has come and gone throughout his life. He has no lactic acidosis or myopathy. He and his unaffected brother have some dysmorphic features, which have not previously been reported in association with YARS2 variants, but are typical of MLASA1 patients with pseudouridine synthase 1 (PUS1) mutations.17,27 His genotypically concordant fraternal twin (P9b) has only mild anemia and similar facial dysmorphology, once again highlighting the potential for decreased penetrance and/or expressivity of the disorder.
Interestingly, some YARS2 patients with myopathy, but no sideroblastic anemia, have recently been reported by Sommerville et al.9 They report siblings with a homozygous YARS2 p.(Leu392Ser) variant who had MLASA2, while another individual homozygous for the same variant had myopathy without sideroblastic anemia or lactic acidosis.
To summarize, the inter- and intra-familial phenotypic variability, intermittent transfusion dependence of some YARS2 cases, and the association of a common variant with disease, suggest that all MLASA2 phenotypes may be susceptible to subtle changes in YARS2 function, which may in turn be influenced by genetic and/or environmental modifiers. This study shows that YARS2 variants can result in CSA in the absence of clinically significant myopathy or lactic acidosis. Thus, we recommend that YARS2 variants be considered as a cause of isolated sideroblastic anemia as well as MLASA2 or mitochondrial myopathy.
Supplementary Material
Acknowledgments
We thank Katinka Redert for her help in data collection. We thank Beatriz Cadenas from Josep Carreras Leukaemia Research Institute (IJC) and Whole Genix, S.L. for excellent technical and bioinformatic assistance, and Dr. Carme Pedro and Dr. Sara Montesdeoca from IMIM Hospital del Mar for medical assistance for P10. We also gratefully acknowledge donations to JC by the Crane and Perkins families as well as the participation of the research subjects. The research conducted at the Murdoch Children’s Research Institute was supported by the Victorian Government’s Operational Infrastructure Support Program.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/103/12/2008
Funding
This research was supported by Australian NHMRC grant 1026891 to JC, NIH DK087992 to MDF, and grants SAF2015-70412-R from the Spanish Secretary of Research, Development and Innovation (MINECO), DJCLS R14/04 from Deutsche José Carreras Leukämie Stiftung, 2014 SGR225 (GRE) from Generalitat de Catalunya and economical support from Fundació Internacional Josep Carreras and from Obra Social “la Caixa” Spain to MS.
References
- 1.Cartwright GE, Deiss A. Sideroblasts, siderocytes, and sideroblastic anemia. N Engl J Med. 1975;292(4):185–193. [DOI] [PubMed] [Google Scholar]
- 2.Bottomley SS, Fleming MD. Sideroblastic anemia: diagnosis and management. Hematol Oncol Clin North Am. 2014;28(4):653–700. [DOI] [PubMed] [Google Scholar]
- 3.Fleming MD. Congenital sideroblastic anemias: iron and heme lost in mitochondrial translation. Hematology Am Soc Hematol Educ Program. 2011;2011:525–531. [DOI] [PubMed] [Google Scholar]
- 4.Riley LG, Cooper S, Hickey P, et al. Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia--MLASA syndrome. Am J Hum Genet. 2010;87(1):52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Riley LG, Menezes MJ, Rudinger-Thirion J, et al. Phenotypic variability and identification of novel YARS2 mutations in YARS2 mitochondrial myopathy, lactic acidosis and sideroblastic anaemia. Orphanet J Rare Dis. 2013;8:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sasarman F, Nishimura T, Thiffault I, Shoubridge EA. A novel mutation in YARS2 causes myopathy with lactic acidosis and sideroblastic anemia. Hum Mut. 2012;33(8):1201–1206. [DOI] [PubMed] [Google Scholar]
- 7.Nakajima J, Eminoglu TF, Vatansever G, et al. A novel homozygous YARS2 mutation causes severe myopathy, lactic acidosis, and sideroblastic anemia 2. J Hum Genet. 2014;59(4):229–232. [DOI] [PubMed] [Google Scholar]
- 8.Ardissone A, Lamantea E, Quartararo J, et al. A Novel Homozygous YARS2 Mutation in Two Italian Siblings and a Review of Literature. JIMD Rep. 2015;20:95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sommerville EW, Ng YS, Alston CL, et al. Clinical Features, Molecular Heterogeneity, and Prognostic Implications in YARS2-Related Mitochondrial Myopathy. JAMA Neurol. 2017;74(6):686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonnefond L, Fender A, Rudinger-Thirion J, Giegé R, Florentz C, Sissler M. Toward the full set of human mitochondrial aminoacyl- tRNA synthetases: characterization of AspRS and TyrRS. Biochemistry. 2005;44(12):4805–4816. [DOI] [PubMed] [Google Scholar]
- 11.Jiang P, Xiaofen J, Peng Y, et al. The exome sequencing identified the mutation in YARS2 encoding the mitochondrial tyrosyl-tRNA synthetase as a nuclear modifier for the phenotypic manifestation of Leber’s hereditary optic neuropathy-associated DNA mutation. Hum Mol Genet. 2016;25(3):584–596. [DOI] [PubMed] [Google Scholar]
- 12.Bergmann AK, Campagna DR, McLoughlin EM, et al. Systematic molecular genetic analysis of congenital sideroblastic anemia: evidence for genetic heterogeneity and identification of novel mutations. Pediatr Blood Cancer. 2010;54(2):273–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rusk N. Torrents of sequence. Nat Methods. 2011;8:44. [Google Scholar]
- 14.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;Chapter 7:Unit7 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40(Web Server issue):W452–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeharia A, Fischel-Ghodsian N, Casas K, et al. Mitochondrial myopathy, sideroblastic anemia, and lactic acidosis: an autosomal recessive syndrome in Persian Jews caused by a mutation in the PUS1 gene. J Child Neurol. 2005;20(5):449–452. [DOI] [PubMed] [Google Scholar]
- 18.Popp MW, Maquat LE. Leveraging rules of nonsense-mediated mRNA decay for genome engineering and personalized medicine. Cell. 2016;165(6):1319–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonnefond L, Frugier M, Touzé E, et al. Crystal structure of human mitochondrial tyrosyl-tRNA synthetase reveals common and idiosyncratic features. Structure. 2007;15(11):1505–1516. [DOI] [PubMed] [Google Scholar]
- 20.Paukstelis P, Chari N, Lambowitz A, Hoffman D. NMR structure of the C-terminal domain of a tyrosyl-tRNA synthetase that functions in group I intron splicing. Biochemistry. 2011;50(18):3816–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oprescu SN, Griffin LB, Beg AA, Antonellis A. Predicting the pathogenicity of aminoacyl-tRNA synthetase mutations. Methods. 2017;113:139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shahni R, Wedatilake Y, Cleary MA, Lindley KJ, Sibson KR, Rahman S. A distinct mitochondrial myopathy, lactic acidosis and sideroblastic anemia (MLASA) phenotype associates with YARS2 mutations. Am J Med Genet Part A. 2013;161a(9):2334–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dallabona C, Diodato D, Kevelam SH, et al. Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology. 2014;82(23):2063–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pierce SB, Chisholm KM, Lynch ED, et al. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc Natl Acad Sci USA. 2011;108(16):6543–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Solda G, Caccia S, Robusto M, et al. First independent replication of the involvement of LARS2 in Perrault syndrome by whole-exome sequencing of an Italian family. J Hum Genet. 2016;61(4):295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64–69. [DOI] [PubMed] [Google Scholar]
- 27.Bykhovskaya Y, Casas K, Mengesha E, Inbal A, Fischel-Ghodsian N. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am J Hum Genet. 2004;74(6):1303–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: Protein structure and function prediction. Nat Methods. 2015;12(1):7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.