

Abstract
Reverse transcription quantitative real-time polymerase chain reaction (RT-qPCR) is a widely used technique to investigate gene expression levels due to its high throughput, specificity, and sensitivity. An appropriate reference gene is essential for RT-qPCR analysis to obtain accurate and reliable results. To date, no reliable reference gene has been validated for the economically tropical tree, sandalwood (Santalum album L.). In this study, 13 candidate reference genes, including 12 novel putative reference genes selected from a large set of S. album transcriptome data, as well as the currently used β-actin gene (ACT), were validated in different tissues (stem, leaf, root and callus), as well as callus tissue under salicylic acid (SA), jasmonic acid methyl ester (MeJA), and gibberellin (GA) treatments using geNorm, NormFinder, BestKeeper, Delta Ct and comprehensive RefFinder algorithms. Several novel candidate reference genes were much more stable than the currently used traditional gene ACT. ODD paired with Fbp1 for SA treatment, CSA and Fbp3 for MeJA treatment, PP2C and Fbp2 for GA treatment, as well as Fbp1 combined with Fbp2 for the total of three hormone treatments were the most accurate reference genes, respectively. FAB1A, when combined with PP2C, was identified as the most suitable reference gene combination for the four tissues tested, while the combination of HLMt, PPR and FAB1A were the most optimal reference genes for all of the experimental samples. In addition, to verify our results, the relative expression level of the SaSSy gene was evaluated by the validated reference genes and their combinations in the three S. album tissues and under MeJA treatment. The evaluated reference genes in this study will improve the accuracy of RT-qPCR analysis and will benefit S. album functional genomics studies in different tissues and under hormone stimuli in the future.
Introduction
Reverse transcription quantitative real-time PCR (RT-qPCR) is a popular technique used to monitor the level of mRNA because of its high sensitivity, accuracy, specificity and efficiency1. To interpret the expression profiles of a target gene accurately and reliably, normalization of gene expression data using reference genes is essential in relative quantification analysis by RT-qPCR. Ideally, excellent reference genes should have a constantly stable or minimal variable expression in experimental conditions2. A few reference genes such as 18 S rRNA (18 S ribosomal RNA), TUBA (α-tubulin), EF1A (elongation factor 1α), ACTB (β-actin), and GAPDH (glyceraldehyde-3-phosphate dehydrogenase)2, which show relatively high levels of expression, are frequently used for RT-qPCR analysis in plants3,4. However, increasing evidence has demonstrated that the expression levels of these traditional reference genes vary considerably in different samples and under different experimental conditions3,5,6. Therefore, it is necessary to select and validate reference genes according to specific samples and experimental conditions.
Santalum album L., commonly known as sandalwood, is a hemiparasitic tropical tree distributed in India, Indonesia, Malaysia, and Australia7. It is famous for its valuable essential oil extracted from aromatic heartwood and roots that are used in aromatherapy, perfumes, cosmetics, medicine and sacred unguents8,9. A number of functional genes and their expression levels have been characterized and studied in S. album in recent years7,10–13. As far as we known, the traditional housekeeping gene ACT (β-actin) was the only reference gene used to date10,12,14,15, and there has been no systematic validation and evaluation of reference genes for RT-qPCR analysis in S. album.
In this study, 13 candidate reference genes, including 12 novel genes selected from a large set of RNA-seq data in three different tissues (stem, leaf, and root) of S. album, as well as the currently used traditional housekeeping gene ACT, were assessed by RT-qPCR. Five statistical algorithms (geNorm16, NormFinder17, BestKeeper18, Delta Ct19 and RefFinder20) were used to evaluate the expression stability of these putative reference genes. Furthermore, the key gene SaSSy for synthesizing the main component of sandal oil was investigated to validate the suitability of the newly identified stable reference genes. This work validated a set of more stable novel reference genes and will facilitate, expand and fortify gene expression analysis in different tissues and under hormone treatment of S. album.
Results
Selection of candidate reference genes based on transcriptome datasets
Based on the RNA-seq expression data previously published12,13,15, 12 genes (Table 1, Supplementary Table S1) with a CV ranging between 9.75% and 11.95% were selected. All 12 selected genes were newly identified candidate reference genes, as follows: contained a FYVE domain necessary for FAB1 gene FAB1A, protein with unknown function (UK), F-box protein (Fbp1, Fbp2 and Fbp3), cytochrome c biogenesis protein CCS1 (CCS1), pentatricopeptide repeat-containing protein (PPR), coatomer subunit alpha-1 (CSA), probable 2-oxoglutarate-dependent dioxygenase (ODD), probable protein phosphatase 2 C (PP2C), probable histone-lysine N-methyltransferase ATXR3 (HLMt), and 40 S ribosomal protein S8 (S8). The currently widely used housekeeping ACT gene, although not in the rank of most stable genes, was also assessed in our study for comparison.
Table 1.
Selected candidate reference genes, primers, Tm and KS-test p values, and amplicon characteristics.
| Gene name | Description | GeneBank accession number | Primer sequence (5′-3′) forward/reverse | Tm (oC) | Amplicon length (bp) | Amplicon efficiency (%) | R2 | KS-test p value |
|---|---|---|---|---|---|---|---|---|
| FAB1A | With FYVE domain necessary for FAB1 gene | MG282422 | AGCAGTTCTCAAAGGAGCTAAA | 62 | 104 | 109.168 | 0.998 | 0.826 |
| ACCTTCGTGCGACAACTAAA | ||||||||
| UK | Function unknown protein | MG282423 | TTTGGCAGTGATCGGTATCC | 62 | 105 | 112.855 | 0.996 | 0.302 |
| CCTCTGTGTTAGGTAGCTTTGG | ||||||||
| Fbp1 | F-box protein | MG282425 | TGGCGTGTCCTGTTTCTATC | 62 | 84 | 106.237 | 0.995 | 0.178 |
| CGCACTCCATAGGTTTCTTCT | ||||||||
| CCS1 | Cytochrome c biogenesis protein CCS1 | MG282427 | GGCCCAATTGGATTTCTCTCTA | 62 | 103 | 108.210 | 0.995 | 0.410 |
| GCAAACTTACTTCTCCGCTTTC | ||||||||
| PPR | Pentatricopeptide repeat-containing protein | MG282429 | TGCTGAATAGTGCCGGTAAG | 62 | 126 | 108.001 | 0.998 | 0.310 |
| TCTCCTTCATCTCATCCCAAATC | ||||||||
| CSA | Coatomer subunit alpha-1 | MG282432 | GCCAATATACCGAGGACAGAAG | 62 | 104 | 106.05 | 0.999 | 0.505 |
| CAACCGCAAGATCACAAACAG | ||||||||
| Fbp3 | F-box protein | MG282433 | CCTCGTGTACTGGGAAATGG | 62 | 110 | 107.471 | 0.998 | 0.243 |
| GCAAGAACGCAATGCCTAAA | ||||||||
| ODD | Probable 2-oxoglutarate-dependent dioxygenase | MG282424 | TTTAGCATTGGGTGGGACTC | 62 | 110 | 94.75 | 0.998 | 0.639 |
| CTTGGCGATTTGCATTGGTTA | ||||||||
| PP2C | Probable protein phosphatase 2C | MG282426 | ACTGACCAGGCAATCCTTTC | 62 | 92 | 93.919 | 0.994 | 0.391 |
| ATCCATAACCTTCGGCCATTTA | ||||||||
| HLMt | Probable histone-lysine N-methyltransferase ATXR3 | MG282428 | TGCTGAGGAAGACCAGGATA | 62 | 110 | 105.477 | 0.998 | 0.242 |
| CACCAAGACCCTTCCGATAAG | ||||||||
| Fbp2 | F-box protein | MG282430 | CGAAGCCTGGTTCACTCTATG | 62 | 94 | 98.121 | 0.997 | 0.051 |
| AAGCTAAGCCTCTGCAATGT | ||||||||
| S8 | 40 S ribosomal protein S8 | MG282431 | CCCGAGGATGATCTGGATAAC | 62 | 88 | 110.782 | 0.995 | 0.266 |
| CATTACTGGTGAACCCAACAC | ||||||||
| ACT | actin | EF452617 | GGATCCACGAGACTACCTACA | 62 | 90 | 99.872 | 0.998 | 0.988 |
| GAGCCACACTGAGCACAATA | ||||||||
| SaSSy | SaSSy | JX826486.1 | CCTTCCTGATCTTCTGCACTAC | 62 | 91 | 105.997 | 0.993 | _ |
| ATTATCGCCTCTTGCCATCTC |
Primer specificity, amplification efficiency and expression profile of candidate reference genes
The specificity of primer pairs for each candidate reference gene was verified by 2% agarose gel electrophoresis with a single expected size product (Supplementary Fig. S1), and further demonstrated by melting curve analysis with a single peak (Supplementary Fig. S2). The cDNA-free template controls (ddH2O as template) showed no obvious melting curve products (data not shown). All these results confirmed the specificity of all primer pairs and the absence of DNA and other contaminating materials during RT-qPCR amplification.
The amplification efficiency for primer pairs of all candidate reference genes ranged from 93.919% (PP2C) to 112.855% (UK), and the R2 values lay between 0.994 (PP2C) and 0.999 (CSA) (Table 1).
The expression profiles of the 13 candidate reference genes in all experimental samples was assessed by RT-qPCR using the Cq value for each sample, after testing for normality using the KS-test in which a p value > 0.05 was considered as normal. All KS-test p values of each sample were greater than the 0.05 cut-off value (Table 1), the average Cq values ranged from 19.8 to 28.96 (Fig. 1), and the majority of average Cq values lay between 23 and 25, which indicates that the expression of all candidate reference genes fitted within a suitable reference gene expression level (15 < Cq < 30)21. As shown in Fig. 1, the gene with highest expression was ACT (with the lowest Cq value), and the gene with the lowest expression was UK (with the highest Cq value). The candidate reference gene names, GeneBank accession numbers, primer sequences, Tm values, amplicon lengths, amplification efficiencies, R2 values, and KS-test p values are listed in Table 1.
Figure 1.
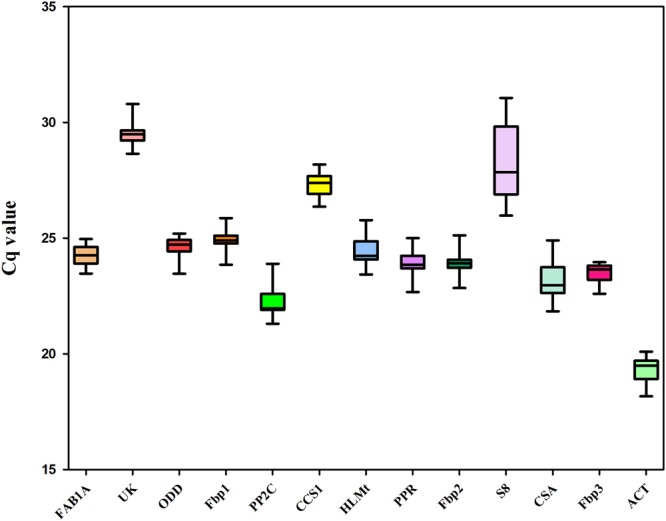
Distribution of Cq values of 13 candidate reference genes in all experimental samples. Boxplot graph showing maximum, minimum values, medians and 25/75 percentiles.
Expression stability of candidate reference genes in different tissues and under hormone treatment of santalum album
According to GeNorm analysis (Table 2), M values of all of the candidate reference genes tested were below 1.5, indicating that they all had relatively stable expression. Among all four tissues tested (Table 2), FAB1A and PPR were the most stable genes, while Fbp1 was the least stable gene. For salicylic acid (SA) treatment (Table 2), FAB1A and Fbp3 were the most stable reference genes. For jasmonic acid methyl ester (MeJA) (Table 2), PP2C and CSA were the top ranked genes. PP2C and CCS1 ranked as the most stable reference genes for gibberellin (GA) treatment (Table 2). PPR and Fbp2 (Table 2) were the most stable reference genes in all three hormone treatment sample sets. As for the total experimental samples (Table 2), HLMt and PPR were the most stable reference genes.
Table 2.
Expression stability of 13 candidate reference genes calculated by GeNorm, NormFinder, BestKeeper, Delta Ct and RefFinder.
| Group | Rank | GeNorm | NormFinder | BestKeeper | Delta Ct | RefFinder | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | MV | Gene | MV | Gene | CV ± SD | Gene | SV | Gene | SV | ||
| Total tissues | 1 | FAB1A | 0.45 | PP2C | 0.16 | ACT | 2.08 ± 0.43 | PP2C | 0.99 | FAB1A | 1.86 |
| 2 | PPR | 0.45 | FAB1A | 0.26 | PPR | 2.32 ± 0.6 | FAB1A | 1.03 | PP2C | 2.00 | |
| 3 | HLMt | 0.46 | HLMt | 0.37 | FAB1A | 3.12 ± 0.81 | HLMt | 1.08 | PPR | 2.51 | |
| 4 | PP2C | 0.48 | Fbp2 | 0.44 | PP2C | 3.46 ± 0.86 | PPR | 1.14 | HLMt | 3.41 | |
| 5 | Fbp3 | 0.52 | PPR | 0.48 | HLMt | 3.54 ± 0.93 | Fbp2 | 1.16 | ACT | 4.45 | |
| 6 | Fbp2 | 0.59 | Fbp3 | 0.49 | Fbp3 | 3.81 ± 0.99 | Fbp3 | 1.18 | Fbp2 | 5.38 | |
| 7 | ACT | 0.65 | ODD | 0.62 | Fbp2 | 4.04 ± 1.07 | ACT | 1.33 | Fbp3 | 5.73 | |
| 8 | ODD | 0.80 | ACT | 0.68 | CSA | 4.56 ± 1.12 | ODD | 1.33 | ODD | 7.97 | |
| 9 | S8 | 0.92 | CCS1 | 0.70 | ODD | 5.28 ± 1.4 | CCS1 | 1.44 | CCS1 | 9.49 | |
| 10 | CCS1 | 1.01 | S8 | 0.85 | CCS1 | 5.35 ± 1.52 | S8 | 1.55 | S8 | 9.97 | |
| 11 | UK | 1.15 | UK | 1.1 | S8 | 5.77 ± 1.66 | UK | 1.83 | CSA | 10.84 | |
| 12 | CSA | 1.27 | CSA | 1.19 | Fbp1 | 7.69 ± 2.23 | CSA | 1.93 | UK | 11.47 | |
| 13 | Fbp1 | 1.38 | Fbp1 | 1.20 | UK | 8.16 ± 2.26 | Fbp1 | 1.97 | Fbp1 | 12.74 | |
| SA | 1 | FAB1A | 0.17 | PPR | 0.14 | ODD | 0.97 ± 0.24 | ODD | 0.48 | ODD | 2.00 |
| 2 | Fbp3 | 0.17 | Fbp1 | 0.15 | Fbp1 | 1.18 ± 0.3 | Fbp1 | 0.49 | Fbp1 | 2.63 | |
| 3 | CCS1 | 0.18 | PP2C | 0.16 | CCS1 | 1.24 ± 0.34 | PP2C | 0.50 | Fbp3 | 3.20 | |
| 4 | ODD | 0.24 | ODD | 0.18 | Fbp3 | 1.36 ± 0.32 | PPR | 0.51 | PPR | 3.87 | |
| 5 | UK | 0.29 | Fbp2 | 0.22 | UK | 1.48 ± 0.44 | Fbp3 | 0.51 | FAB1A | 4.56 | |
| 6 | Fbp1 | 0.32 | HLMt | 0.32 | FAB1A | 1.66 ± 0.4 | Fbp2 | 0.54 | PP2C | 5.01 | |
| 7 | PP2C | 0.34 | Fbp3 | 0.35 | PPR | 1.68 ± 0.41 | UK | 0.55 | CCS1 | 5.89 | |
| 8 | PPR | 0.36 | UK | 0.35 | HLMt | 1.87 ± 0.46 | FAB1A | 0.56 | UK | 6.88 | |
| 9 | Fbp2 | 0.39 | FAB1A | 0.42 | ACT | 1.98 ± 0.39 | HLMt | 0.59 | Fbp2 | 7.38 | |
| 10 | HLMt | 0.41 | CCS1 | 0.48 | Fbp2 | 2.26 ± 0.55 | CCS1 | 0.59 | HLMt | 8.35 | |
| 11 | ACT | 0.44 | CSA | 0.50 | PP2C | 2.37 ± 0.54 | CSA | 0.71 | ACT | 9.43 | |
| 12 | CSA | 0.48 | ACT | 0.67 | CSA | 2.59 ± 0.62 | ACT | 0.73 | CSA | 11.49 | |
| 13 | S8 | 0.63 | S8 | 1.47 | S8 | 4.61 ± 1.37 | S8 | 1.48 | S8 | 13.00 | |
| MeJA | 1 | PP2C | 0.08 | CSA | 0.05 | Fbp3 | 0.26 ± 0.06 | CSA | 0.18 | CSA | 1.19 |
| 2 | CSA | 0.08 | Fbp3 | 0.08 | CSA | 0.27 ± 0.06 | Fbp3 | 0.19 | Fbp3 | 1.86 | |
| 3 | Fbp3 | 0.09 | ODD | 0.10 | Fbp2 | 0.28 ± 0.07 | PP2C | 0.20 | PP2C | 2.63 | |
| 4 | Fbp2 | 0.10 | PP2C | 0.11 | PPR | 0.37 ± 0.09 | ODD | 0.21 | Fbp2 | 4.16 | |
| 5 | ODD | 0.12 | Fbp2 | 0.12 | PP2C | 0.4 ± 0.09 | Fbp2 | 0.21 | ODD | 4.36 | |
| 6 | PPR | 0.12 | PPR | 0.13 | ODD | 0.42 ± 0.1 | PPR | 0.22 | PPR | 5.73 | |
| 7 | Fbp1 | 0.13 | Fbp1 | 0.16 | S8 | 0.44 ± 0.12 | Fbp1 | 0.23 | Fbp1 | 7.24 | |
| 8 | ACT | 0.15 | ACT | 0.17 | Fbp1 | 0.49 ± 0.12 | ACT | 0.24 | ACT | 7.74 | |
| 9 | S8 | 0.16 | S8 | 0.17 | ACT | 0.54 ± 0.11 | S8 | 0.25 | S8 | 9.00 | |
| 10 | CCS1 | 0.18 | CCS1 | 0.23 | UK | 0.56 ± 0.16 | CCS1 | 0.28 | CCS1 | 10.24 | |
| 11 | UK | 0.20 | UK | 0.25 | CCS1 | 0.61 ± 0.17 | UK | 0.31 | UK | 10.74 | |
| 12 | HLMt | 0.23 | HLMt | 0.32 | HLMt | 1.3 ± 0.32 | HLMt | 0.36 | HLMt | 12.00 | |
| 13 | FAB1A | 0.25 | FAB1A | 0.35 | FAB1A | 1.4 ± 0.34 | FAB1A | 0.39 | FAB1A | 13.00 | |
| GA | 1 | PP2C | 0.12 | Fbp2 | 0.09 | CCS1 | 0.6 ± 0.16 | Fbp2 | 0.23 | PP2C | 1.57 |
| 2 | CCS1 | 0.12 | UK | 0.13 | PP2C | 0.64 ± 0.14 | PP2C | 0.24 | Fbp2 | 2.59 | |
| 3 | HLMt | 0.16 | PP2C | 0.14 | S8 | 0.91 ± 0.24 | UK | 0.25 | CCS1 | 2.66 | |
| 4 | UK | 0.19 | HLMt | 0.16 | UK | 0.94 ± 0.28 | HLMt | 0.25 | UK | 3.60 | |
| 5 | Fbp2 | 0.19 | CCS1 | 0.17 | Fbp1 | 1.11 ± 0.27 | CCS1 | 0.26 | HLMt | 3.94 | |
| 6 | CSA | 0.21 | CSA | 0.19 | HLMt | 1.11 ± 0.27 | CSA | 0.27 | CSA | 6.82 | |
| 7 | PPR | 0.22 | PPR | 0.19 | FAB1A | 1.19 ± 0.29 | Fbp3 | 0.27 | S8 | 7.40 | |
| 8 | Fbp3 | 0.22 | Fbp3 | 0.19 | Fbp2 | 1.22 ± 0.29 | PPR | 0.27 | PPR | 8.10 | |
| 9 | Fbp1 | 0.23 | Fbp1 | 0.21 | ACT | 1.39 ± 0.26 | Fbp1 | 0.29 | Fbp1 | 8.13 | |
| 10 | S8 | 0.24 | S8 | 0.22 | CSA | 1.45 ± 0.33 | S8 | 0.29 | Fbp3 | 8.56 | |
| 11 | FAB1A | 0.25 | FAB1A | 0.25 | PPR | 1.54 ± 0.36 | FAB1A | 0.29 | ACT | 9.12 | |
| 12 | ACT | 0.26 | ACT | 0.28 | Fbp3 | 1.64 ± 0.38 | ACT | 0.33 | FAB1A | 10.16 | |
| 13 | ODD | 0.28 | ODD | 0.33 | ODD | 1.9 ± 0.46 | ODD | 0.37 | ODD | 13.00 | |
| Total hormone | 1 | PPR | 0.22 | Fbp2 | 0.12 | UK | 0.96 ± 0.28 | Fbp1 | 0.47 | Fbp1 | 1.73 |
| 2 | Fbp2 | 0.22 | PPR | 0.14 | Fbp1 | 1.09 ± 0.27 | Fbp2 | 0.48 | Fbp2 | 1.78 | |
| 3 | Fbp1 | 0.26 | Fbp1 | 0.16 | Fbp3 | 1.36 ± 0.32 | PPR | 0.49 | PPR | 2.55 | |
| 4 | HLMt | 0.30 | HLMt | 0.24 | ODD | 1.41 ± 0.35 | HLMt | 0.53 | UK | 4.53 | |
| 5 | UK | 0.35 | PP2C | 0.34 | FAB1A | 1.46 ± 0.35 | ODD | 0.54 | HLMt | 5.03 | |
| 6 | ODD | 0.37 | ODD | 0.35 | Fbp2 | 1.46 ± 0.35 | UK | 0.55 | ODD | 5.18 | |
| 7 | Fbp3 | 0.39 | UK | 0.37 | CCS1 | 1.49 ± 0.41 | CCS1 | 0.55 | Fbp3 | 6.24 | |
| 8 | CCS1 | 0.40 | CCS1 | 0.37 | PPR | 1.62 ± 0.39 | Fbp3 | 0.56 | CCS1 | 7.74 | |
| 9 | FAB1A | 0.41 | Fbp3 | 0.44 | HLMt | 1.86 ± 0.45 | PP2C | 0.58 | PP2C | 8.59 | |
| 10 | ACT | 0.42 | ACT | 0.46 | ACT | 2.15 ± 0.41 | ACT | 0.60 | FAB1A | 9.19 | |
| 11 | PP2C | 0.44 | CSA | 0.47 | PP2C | 2.23 ± 0.5 | FAB1A | 0.61 | ACT | 9.74 | |
| 12 | CSA | 0.48 | FAB1A | 0.49 | CSA | 2.77 ± 0.64 | CSA | 0.67 | CSA | 11.74 | |
| 13 | S8 | 0.61 | S8 | 1.34 | S8 | 4.63 ± 1.3 | S8 | 1.36 | S8 | 13.00 | |
| Total | 1 | HLMt | 0.37 | HLMt | 0.31 | CCS1 | 3.13 ± 0.86 | HLMt | 0.91 | HLMt | 1.50 |
| 2 | PPR | 0.37 | PPR | 0.35 | FAB1A | 3.16 ± 0.78 | PPR | 0.92 | PPR | 1.86 | |
| 3 | FAB1A | 0.48 | FAB1A | 0.41 | PPR | 3.52 ± 0.86 | FAB1A | 0.95 | FABIA | 2.71 | |
| 4 | Fbp2 | 0.53 | Fbp2 | 0.48 | HLMt | 3.59 ± 0.9 | Fbp2 | 0.97 | ACT | 4.45 | |
| 5 | Fbp3 | 0.55 | PP2C | 0.54 | ACT | 3.59 ± 0.71 | PP2C | 1.00 | Fbp2 | 4.76 | |
| 6 | PP2C | 0.57 | ODD | 0.54 | ODD | 3.87 ± 0.97 | Fbp3 | 1.03 | PP2C | 6.37 | |
| 7 | ACT | 0.61 | ACT | 0.63 | UK | 3.91 ± 1.13 | ODD | 1.04 | Fbp3 | 6.40 | |
| 8 | ODD | 0.66 | Fbp3 | 0.64 | Fbp3 | 4.43 ± 1.08 | ACT | 1.05 | ODD | 6.70 | |
| 9 | CCS1 | 0.73 | CCS1 | 0.68 | Fbp2 | 4.44 ± 1.1 | CCS1 | 1.14 | CCS1 | 7.35 | |
| 10 | CSA | 0.81 | CSA | 1.00 | CSA | 4.65 ± 1.1 | CSA | 1.32 | CSA | 9.74 | |
| 11 | Fbp1 | 0.94 | S8 | 1.38 | S8 | 5.44 ± 1.54 | S8 | 1.63 | S8 | 11.49 | |
| 12 | S8 | 1.05 | Fbp1 | 1.52 | PP2C | 5.51 ± 1.27 | Fbp1 | 1.70 | Fbp1 | 11.98 | |
| 13 | UK | 1.21 | UK | 1.98 | Fbp1 | 6.64 ± 1.74 | UK | 2.11 | UK | 12.17 | |
The geNorm program was also performed to determine the optimal number of reference genes for normalizing RT-qPCR data by calculating the pairwise variations Vn/Vn + 1. As shown in Fig. 2, the value of V2/3 was always below the cut-off value of 0.15 in different tissue samples and samples from all of the hormone treatments, indicating that the two most stable reference genes were sufficient to normalize expression data in these experiments. In the total experimental samples, the three most stable reference genes were ideal to normalize RT-qPCR data since the value of V3/4 (0.127) was below the cut off value of 0.15.
Figure 2.

Pairwise variation (V) analysis of 13 selected reference genes using geNorm software. The pairwise variations Vn/Vn + 1 were calculated by geNorm in different tissues and under hormone treatment samples.
The results calculated with NormFinder (Table 2) show that PP2C followed by FAB1A were the most stable genes in all tested tissues, Fbp1 was also considered to be a weakly stable gene in such a sample set. As for SA treatment, PPR and Fbp1 were the most stable reference genes. CSA and Fbp3 were the top ranked reference genes in MeJA treatment samples. Fbp2 and UK were the most stable reference genes for GA treatment. Fbp2 and PPR were the most highly ranked reference genes in all three hormone treatment samples. When assessing the total experimental samples, HLMt and PPR were the top stably expressed genes.
As shown in Table 2, when evaluated by the BestKeeper program, ACT followed by PPR were the most stable genes and UK was considered to be the least stable gene in the four tissue samples tested. ODD and Fbp1 for the SA treatment, Fbp3 and CSA for the MeJA treatment, CCS1 and PP2C for the GA treatment, as well as UK and Fbp1 for all three hormone treatments were the most stable reference genes. As for the total experimental samples, CCS1 and FAB1A were the top stably expressed reference genes.
According to the ranking orders generated by Delta Ct (Table 2), PP2C and FAB1A were the most stable genes and Fbp1 was the least stable gene in the total of four tissues tested. As for hormone treatment, ODD and Fbp1 for SA treatment, CSA and Fbp3 for MeJA treatment, Fbp2 and PP2C for GA treatment, and Fbp1 followed by Fbp2 for all three hormone treatments were the top ranking reference genes. HLMt and PPR were the most stable reference genes for the total of experimental samples.
Finally, RefFinder was used to comprehensively validate the stability of candidate reference genes. According to the results determined by RefFinder (Table 2) and geNorm (Fig. 2), the combination of FAB1A and PP2C for all four tissues tested, ODD and Fbp1 for SA treatment, CSA and Fbp3 for MeJA treatment, PP2C and Fbp2 for GA treatment, as well as Fbp1 and Fbp2 for the total of three hormone treatments were the most suitable reference genes. As for all of the experimental samples, the most suitable reference genes were the combination of HLMt, PPR and FAB1A.
Moreover, we also verified the stability of candidate reference genes in specific tissues and different tissue combinations using RefFinder. According to the comprehensive ranking recommended by RefFinder (Fig. 3), PPR and Fbp3 were the most stable reference genes in leaves, Fbp2 and Fbp3 were the most stable genes in roots, and Fbp3, CCS1 and CSA were the most stable genes in callus. In stems as well as the combination of leaf, stem and root (LSR), PP2C and PPR were the most stable reference genes. Fbp3 was also among the most stable reference genes in LSR.
Figure 3.

Comprehensive expression stability of 13 selected reference genes recommended by RefFinder in specific tissues and different tissue combinations.
Validation of Identified reference genes in different Tissues and under MeJA treatment
In order to validate the identified reference genes, the transcript profile of a key gene (SaSSy) was investigated in the reference genes, and evaluated in the three tissues and under MeJA treatment.
As shown in Fig. 4A, the expression level of the SaSSy gene was similar in the three tested tissues when using the two most stable reference genes (FAB1A and PP2C) to normalize RT-qPCR data. The combination of FAB1A + PP2C provides more accurate expression values for each tissue than a single reference gene. Although each of the reference genes (FAB1A, PP2C, ACT and Fbp1) and gene combination (FAB1A + PP2C) used for normalization provided a similar trend of SaSSy expression level (leaf < root < stem) (this trend was comparable with the RNA-seq result), when the least stable reference gene Fbp1 was used to normalize RT-qPCR data, the expression level of SaSSy was obviously over-estimated in tested tissues. Statistical analysis showed insignificantly different results in roots when normalized by Fbp1, so it generated inconsistent statistical results compared with the results normalized by more stable reference genes or their combination. These over-estimated results, especially in roots, did not match RNA-seq results. Furthermore, SaSSy expression level was considerably reduced in all tissues when the traditional housekeeping gene (ACT) was used for normalization.
Figure 4.
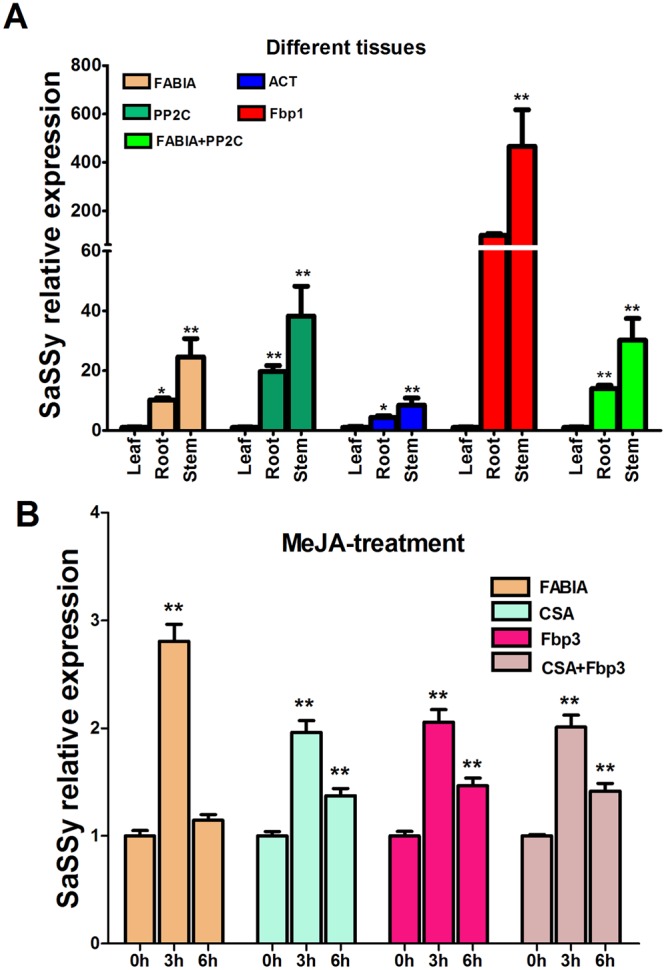
Relative expression levels of the SaSSy gene normalized by a validated reference gene alone or combination in different tissues (A) and under MeJA treatment (B) of Santalum album. Bars indicate standard deviation calculated from three biological replicates. Asterisk indicates significance at P < 0.05(*) or P < 0.01(**) using Duncan’s multiple range test.
Under the MeJA treatment, the expression of SaSSy at 3 h is about 2.0 times higher than no treatment control (0 h) when normalized by the most stable reference genes (CSA, Fbp3) and their combination (CSA + Fbp3). While using the least stable gene FAB1A, it was more than 2.8 times higher. The SaSSy expression at 6 h is about 1.4 times higher than at 0 h using the best reference genes (CSA, Fbp3, CSA + Fbp3). However, the least stable reference gene FAB1A produced an obviously reduced and statistically insignificant reduction of 1.15 times (Fig. 4B). This demonstrated an obviously effect of using different reference genes for normalization.
Discussion
A powerful technique, RT-qPCR, has been widely used for the detection and quantification of gene expression in plants. In order to interpret RT-qPCR data accurately and reliably, appropriate reference genes are essential. Many reports in several plant species such as Arabidopsis (Arabidopsis thaliana), tomato (Solanum lycopersicum) and grape (Vitis vinifera) have shown the importance and need to validate appropriate reference genes for normalizing RT-qPCR data in different tissues, developmental stages or experimental conditions22–27. To date, only the housekeeping gene ACT was used as a reference gene in S. album, and no systematic validation and evaluation of stable reference genes for RT-qPCR data normalization exists for the commercial S. album tree. In this study, we took advantage of previously published data12,13,15 to select stable reference genes in different tissues of S. album. A total of 12 novel genes were selected as candidate reference genes based on the CV value calculated from transcriptome data. These genes had an intermediate or low level of expression according to the mean expression values (MVs) (Supplementary Table S1), similar to Czechowski et al.22. In order to compare the stability of these 12 candidate reference genes, the currently used housekeeping gene ACT for S. album was also included in our analysis.
In general, multiple methods should be employed to validate the stability of reference genes to avoid artificial results. The most widespread methods used are geNorm, NormFinder and BestKeeper. In this study, the ranking of stability of the 13 candidate reference genes, as arranged by geNorm and NormFinder (Table 2), was rather similar. As can be seen in different tissues tested (Table 2), FAB1A was ranked in first position by geNorm and second by NormFinder, while Fbp1 was always ranked 13th based on both methods. However, BestKeeper generated a different ranking. For instance, ACT was ranked first by BestKeeper but in a medium position by geNorm and NormFinder in different tissues. Similar results could be found in other sample sets. Previous studies also demonstrated similar results in Taihangia rupestris flowers28, under hormone stress in Brassica napus29, and under heat stress as well as in all samples including various abiotic stresses, tissues and ages of Salicornia europaea30. This is because different algorithm models and statistical methods are used in each software. In order to obtain a global and comprehensive result, the integrative RefFinder web-based tool is widely used to arrange the final ranking of reference genes, such as in tissue development in kenaf (Hibiscus cannabinus)31, under abiotic stresses in creeping bentgrass (Agrostis stolonifera)32 as well as in Seashore paspalum (Paspalum vaginatum)33. Based on RefFinder analysis (Table 2), some novel genes were identified as the most stably expressed reference genes, i.e., FAB1A for all tissues tested, ODD for SA treatment, CSA for MeJA treatment, and HLMt for all experimental samples were the best stable reference genes. PP2C, a conserved serine/threonine protein phosphatase gene, was the most stable reference gene for GA treatment, and previous studies showed that another conserved serine/threonine protein phosphatase 2 A gene (PP2A) was the most stable reference gene in root tissue and hormone treatments of garden pea (Pisum sativum)34, both in diurnal and developmental time-course experiments in lettuce (Lactuca sativa)35, in total samples (including five different tissues and three different abiotic stresses) and under abiotic stress treatments in Isatis indigotica36, as well as under various stresses in rapeseed (Brassica napus)37. One of the best reference genes for all experimental samples, PPR, is one of the largest gene superfamilies and is essential in mitochondria and chloroplasts biogenesis, plastid gene expression, mitochondrial RNA editing, as well as early embryogenesis in plants38–41. Previous studies selected and validated several PPR superfamily genes as the most stable reference genes in B. napus29 and Arabidopsis thaliana22. The F-box family gene Fbp1, Fbp2 and Fbp3 were also among the most stable reference genes for SA and total hormone treatment, for GA treatment, and for MeJA treatment samples, respectively. In contrast, Fbp1 was also identified as the least stable gene among all four S. album tissues tested. A previous study demonstrated that the F-box gene was the most stable expression reference gene in B. napus37 and soybean (Glycine max)42, but was the least stably expressed gene in different tissues of licorice (Glycyrrhiza glabra) under drought stress43. This could be explained by the fact that the F-box protein is one of the largest and most heterogeneous superfamilies in plants, and plays a wide range of roles in plant growth and development44. Similar results were also found in our newly selected reference genes FAB1A and ODD (Table 2). Therefore, the expression of a reference gene within the same gene family can vary in different tissues and experimental conditions. For this reason, it is essential to select and validate reference genes for specific tissue samples and experimental conditions.
There is no universally stable reference gene for all experiments, and multiple reference genes should be used to obtain more accurate and reliable results2,3,16. In this study, we examined the interference of the most and least stable reference gene and their combination on the expression level of the SaSSy gene. In different tissues (Fig. 4A), the expression data of SaSSy normalized by the most stable genes FAB1A and PP2C was similar, but the data normalized by the combination of FAB1A and PP2C was more precise. When the least stable reference gene Fbp1 was used as the normalizer, the expression level of SaSSy was fully overestimated in tested tissues. Under MeJA treatment (Fig. 4B), the expression level of SaSSy presented a significant difference when the most and least stable reference genes were used for normalization. Results were identical when the two best reference genes alone or their combination were used for normalization. However, the least stable gene FAB1A not only overestimated SaSSy expression level at 6 h but also significantly reduced the expression at 3 h, and thus generated an incorrect interpretation of the results. Thus, for all four tissues, we recommend that the two most stable genes (FAB1A and PP2C) are ideal for normalization. As for hormone treatment, we suggest that one most stable reference gene could be sufficient to normalize RT-qPCR data.
There is increasing evidence that more stable reference genes can be selected using high-throughput transcriptomic data22,26,42,45. Our current study also demonstrates that several novel reference genes (FAB1A, CSA, ODD, PP2C, PPR and HLMt), which were selected from a set of RNA-seq data of S. album, performed better than the traditional housekeeping gene ACT. Indeed, when the RT-qPCR data was normalized by ACT, the expression level of SaSSy decreased and exhibited inconspicuous and inaccurate discrepancies among all tested tissue samples of S. album (Fig. 4A). Zhou et al.46 also demonstrated that unstably higher expression level of reference genes lowered the expression of the target gene and caused indistinguishable discrepancies in different species of oil-tea (Camellia sinensis). Therefore, our study confirmed the feasibility of reference gene selection using high-throughput transcriptome data. However, due to variation in the expression level of different samples and experimental conditions, reference genes selected based on RNA-seq data should be further validated when the samples and conditions are different from RNA-seq data. Furthermore, we recommend that transcriptome data be employed from diverse samples and conditions to obtain more universally stable reference genes.
In conclusion, for the first time in sandalwood research, this study has systematically selected and evaluated appropriate reference genes for RT-qPCR in four tissues (stem, root, leaf and callus) and the tissue of callus under three hormone treatments based on RNA-seq data. A total of 13 candidate reference genes were selected then verified using geNorm, NormFinder, BestKeeper, Delta Ct and RefFinder software, and the results was further validated by SaSSy gene expression analysis in different tissues and under MeJA treatment. The combination of FAB1A and PP2C was the optimal combination of reference genes for all tissues tested, while Fbp1 and Fbp2 were the most suitable reference genes under three hormone treatments. Our results demonstrated that the suitable reference genes for RT-qPCR normalization are not identical in different experimental samples, so optimal reference genes or their combinations should be selected according to specific experimental conditions. The stable reference genes obtained in this study will undoubtedly improve the accuracy of RT-qPCR data normalization and quantification under hormone treatment as well as in different tissues of S. album and facilitate functional gene analysis in sandalwood in the future.
Materials and Methods
Collection of plant materials and hormone treatments
The leaves, roots and stems were collected from three 5- to 7-year-old S. album tress growing in the South China Botanical Garden in April, 2017. The stem was prepared as shavings using a hand-driven drill at 20, 40 and 60 cm from the ground, and then pooled as the stem sample for each tree.
Callus was induced as follows: newly sprouting shoots from trees were selected as the explant, their surface was swabbed clean with cotton dipped in 75% alcohol, then immersed into 0.1% (w/v) mercuric chloride for 15–20 min, followed by five successive washes in sterile distilled water. Shoot segments with a single node were inoculated vertically on solid Murashige and Skoog (MS) basal medium47 to which 1.0 mg/L thidiazuron (TDZ) was added to induce callus. Cultures were incubated at 25 ± 1 °C under a 16-h photoperiod provided by cool white fluorescent lamps with a light intensity of approximately 50 µmol m−2 s−1. The resulting callus was proliferated on solid MS medium supplemented with 1.5 mg/L 2,4-dichlorophenoxyacetic acid (2,4-D) and 0.2 mg/L TDZ. After about 20 days, similarly good callus was collected as callus tissue samples and used for hormone treatment. Callus was frozen immediately in liquid nitrogen (N2) and stored at −80 °C until use. Each tissue (stem, leaf, root, callus clump) sample collected or generated from a tree was used as a biological replicate. Three biological replicates were used for each sample.
Similarly good callus proliferated after transfer into liquid Murashige and Skoog (MS) basal medium47 supplemented with 1.5 mg/L 2,4-D and 0.2 mg/L TDZ, and then placed on a shaker at 100 rpm/min in the same culture conditions mentioned above. After 24 h, dissolved SA, MeJA and GA were added separately into samples at a final concentration of 100 µM for each hormone treatment, and all samples were collected at 0, 3 and 6 h as triplicates. All samples were frozen immediately in N2 and stored at −80 °C until use.
RNA extraction and cDNA synthesis
RNA extraction and RT-qPCR experiments were carried out on the basis of the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guideline48 to ensure the reliability of results. Total RNA was isolated from all samples using a protocol reported for the isolation of RNA from woody plants49. To remove DNA, the total RNA of all samples was digested with RNase-free DNase I (Takara, Dalian, China) at 37 °C for 30 min according to the manufacturer’s instructions. Then, PCR of the ACT gene was conducted using RNA as the template in 40 cycles to determine DNA contamination with specific primers (F: AGGCTGTTCTTTCCCTTTA, R: TTCCTTGCTCATTCTATCG). The DNA-free total RNA was qualified and quantified using a NanoDrop ND-1000 spectrophotometer (Nanodrop Technologies, Wilmington, NC, USA). The RNA samples with an A260/A280 ratio between 1.9 and 2.1, and an A260/A230 ratio greater than 2.0 were used for subsequent analysis. RNA integrity was assessed by 1.0% (w/v) agarose gel electrophoresis, and confirmed with an Agilent 2100 Bioanalyzer. Total RNA (1 µg) was used to synthesize first strand cDNAs using an equivalent of oligo-(dT)15 and random primers with a GoScript™ Reverse Transcriptase system (Promega, Madison, WI, USA) in 20 µL volume according to the manufacturer’s protocols. Amplification was performed at 25 °C for 5 min, 42 °C for 60 min, and 70 °C for 15 min. The successfully synthesized cDNA samples were diluted 1:10 with nuclease-free water and stored at −20 °C until further use.
Selection of candidate reference genes using RNA-seq data and design of primers
The transcriptome sequencing data from wood in stems13 (GenBank Accession: PRJNA297453), roots12 (GenBank Accession: SRA150639) and leaves15 (GenBank Accession: SRR3731808, SRR3731809) of S. album were used to select the most stably expressed genes. To estimate the expression stability of every gene, we analyzed all the raw data for each gene using the method described by Wang et al.50, noted briefly as follows: (a) reads per kb per million reads (RPKM) values, mean expression values (MVs), and standard deviations (SDs) for each gene were calculated; (b) the coefficient of variation (CV) of each gene was calculated using the formula CV = SD/MV × 100%; (c) all genes were ranked based on their CV value. In general, gene expression is more stable while its CV value is much lower. Based on this principal and the method described by de Jonge et al.51, candidate reference genes that met the following requirements were selected: (a) maximum fold change (MFC) < 2; (b) mean value of FPKM of any gene pairs < (maximum expression value − 2 × SD) in the dataset; (c) a CV ≤ 12%.
RT-qPCR primers were designed using primer3 plus (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi). Primer design considered the following criteria: (a) primer size: 20–23 bp; (b) product size: 80–200 bp; (c) GC% content: 40–60% (primers are shown in Table 1). Moreover, primer accuracy and specificity were checked by 2.0% (w/v) agarose gel electrophoresis. The melting curve and no template control (NTC) were prepared to further validate the specificity and absence of primer dimer formation and DNA contamination for every primer pair.
Quantitative real-time PCR (qPCR) and amplification efficiency
RT-qPCR was performed in 96-well plates in an ABI 7500 Real-time system (ABI, Alameda, CA, USA) using the SYBR Premix Ex Taq™ Kit (Takara). The qPCR reaction in a total volume of 10 µL consisted of 5 µL SYBR Premix Ex Taq (1×), 500 nM of each forward and reverse primer, 100 ng of cDNA, and 3 µL of ddH2O. The cycling conditions were: 95 °C for 2 min, followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min. After 40 cycles, a melting curve analysis was performed ranging from 60 to 95 °C.
A standard curve was established by triplicate repeats of RT-qPCR amplification using serial dilutions (1:1, 1:10, 1:100, 1:1000, and 1:10000) of all tested cDNA sample pools. The correlation coefficient (R2) and amplification efficiency (E) for each gene were calculated based on the standard curve. The amplification efficiency of each gene was calculated using the equation E = (10–1/slope – 1) × 100%48. All RT-qPCR experiments were carried out using three biological replicates of each sample, as indicated above, and three technical replicates of each biological replicate.
Gene stability analysis
Five different programs, geNorm (version 3.5)16, NormFinder17, BestKeeper18, Delta Ct19 and RefFinder20, a web-based tool (http://150.216.56.64/referencegene.php), were used to determine the stability of candidate reference genes. For the geNorm and NormFinder algorithms, the raw Cq data from each sample was converted into relative quantity (RQ) using the formula 2ΔCq, where ΔCq = min Cq (of each gene) – sample Cq. The GeNorm program first calculates an expression stability (M) value for each gene and then calculates a pairwise variation (Vn/n + 1) value between genes. Genes with an M value below 1.5 are supposed to be stably expressed, and a lower M value indicates a more stable level of expression16. Moreover, optimal number of reference genes for normalization is indicated. The value of “n” is the optimal number of reference genes when the pairwise value of variation (Vn/Vn + 1) is below a cut-off value of 0.1516. NormFinder was used to rank the stability of candidate reference genes with an M value that took into account the value of inter- and intra-group variance. Genes with the lowest stability value indicate the most stable expression within the examined gene set17. The BestKeeper program examines the ranking of reference genes based on the calculation of the CV and SD values for each gene. Any gene with an SD value less than 1.0 was recommended as a gene with stable expression while genes with both the lowest CV and SD values represent the highest stability in the BestKeeper program18. The Delta Ct approach compares the difference in Cq values of reference genes pairwise and ranks the candidate reference genes using the variability of averaged SD19. RefFinder integrates the three classical algorithms (geNorm, NormFind and BestKeeper) and a comparative ΔCt method to comprehensively validate and rank the stability of candidate reference genes by calculating the geometric mean of their weights for the overall final ranking20.
Validation of reference genes
To validate the reliability of the selected reference genes (including the least and most stable reference genes and their combinations) recommended by the RefFinder tool, the expression level of SaSSy, a key gene for synthesizing α-, β-, and epi-β-santalene (the precursor of the main component of S. album oil (Z)-α-santalol and (Z)-β-santalol)9–11, was determined in stem, leaf, and root tissues and the tissue of callus under MeJA treatment at different times using the 2-ΔΔCq method52. The primer for SaSSy was designed according to the criteria mentioned above. Specificity was checked as described above (primer pairs are shown in Table 1).
Statistical analysis
The Kolmogorov-Smirnov test, which is used to verify data normality with a KS-test p value, was performed in SPSS13.0 (SPSS Inc., Chicago, IL, USA). Data with a KS-test p value > 0.05 was considered as normal. Following one-way analysis of variance (ANOVA), significant differences were assessed by Duncan’s multiple range test at P < 0.05 (*) and P < 0.01 (**).
Electronic supplementary material
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant numbers 31470685, 31270720, and 31100498), the Natural Science Foundation of Guangdong Province (S2012010009025), and the Guangdong Science and Technology project (2015B020231008).
Author Contributions
G.M. received and designed the experiment. H.Y., Y.Z., Y.X., and Q.C. performed the experiment. H.L., M.N., and B.G. contributed materials. H.Y., Y.L. and M.L. analyzed the data. H.Y., X.Z., and J.A.T.S. wrote the manuscript. J.A.T.S. provided critical analysis of the design and data. All authors read and proved the final manuscript.
Data Availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files).
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Xinhua Zhang, Email: xhzhang@scib.ac.cn.
Jaime A. Teixeira da Silva, Email: jaimetex@yahoo.com
Guohua Ma, Email: magh@scib.ac.cn.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-35883-6.
References
- 1.Bustin SA. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J. Mol. Endocrinol. 2002;29:23–29. doi: 10.1677/jme.0.0290023. [DOI] [PubMed] [Google Scholar]
- 2.Kozera B, Rapacz M. Reference genes in real-time PCR. J. Appl. Genet. 2013;54:391–406. doi: 10.1007/s13353-013-0173-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicot N, Hausman JF, Hoffmann L, Evers D. Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress. J. Exp. Bot. 2005;56:2907–2914. doi: 10.1093/jxb/eri285. [DOI] [PubMed] [Google Scholar]
- 4.Liu J, et al. Selection of reference genes for quantitative real-time PCR normalization in Panax ginseng at different stages of growth and in different organs. PLoS One. 2014;9:e112177. doi: 10.1371/journal.pone.0112177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martins PK, et al. Selection of reliable reference genes for RT-qPCR analysis during developmental stages and abiotic stress in Setaria viridis. Sci. Rep. 2016;6:28348. doi: 10.1038/srep28348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaramillo ML, et al. Identification and evaluation of reference genes for expression studies by RT-qPCR during embryonic development of the emerging model organism. Macrobrachium olfersii. Gene. 2017;598:97–106. doi: 10.1016/j.gene.2016.11.001. [DOI] [PubMed] [Google Scholar]
- 7.Jones CG, et al. Isolation of cDNAs and functional characterisation of two multi-product terpene synthase enzymes from sandalwood, Santalum album L. Arch. Biochem. Biophys. 2008;477:121–130. doi: 10.1016/j.abb.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 8.Kim TH, et al. Aromatic constituents from the heartwood of Santalum album L. Chem. Pharm. Bull. 2005;53:641–644. doi: 10.1248/cpb.53.641. [DOI] [PubMed] [Google Scholar]
- 9.Baldovini N, Delasalle C, Joulain D. Phytochemistry of the heartwood from fragrant Santalum species: a review. Flavour Frag. J. 2011;26:7–26. doi: 10.1002/ffj.2025. [DOI] [Google Scholar]
- 10.Rani A, Ravikumar P, Reddy MD, Kush A. Molecular regulation of santalol biosynthesis in Santalum album L. Gene. 2013;527:642–648. doi: 10.1016/j.gene.2013.06.080. [DOI] [PubMed] [Google Scholar]
- 11.Srivastava PL, et al. Functional characterization of novel sesquiterpene synthases from Indian sandalwood, Santalum album. Sci. Rep. 2015;5:10095. doi: 10.1038/srep10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang X, et al. RNA-Seq analysis identifies key genes associated with haustorial development in the root hemiparasite Santalum album. Front. Plant Sci. 2015;6:661. doi: 10.3389/fpls.2015.00661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Celedon JM, et al. Heartwood-specific transcriptome and metabolite signatures of tropical sandalwood (Santalum album) reveal the final step of (Z)-santalol fragrance biosynthesis. Plant J. 2016;86:289–299. doi: 10.1111/tpj.13162. [DOI] [PubMed] [Google Scholar]
- 14.Misra BB, Dey S. Developmental variations in sesquiterpenoid biosynthesis in East Indian sandalwood tree (Santalum album L.) Trees. 2013;27:1071–1086. doi: 10.1007/s00468-013-0858-0. [DOI] [Google Scholar]
- 15.Zhang X, et al. Physiological and transcriptomic analyses reveal a response mechanism to cold stress in Santalum album L. leaves. Sci. Rep. 2017;7:42165. doi: 10.1038/srep42165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, research 0034 (2002). [DOI] [PMC free article] [PubMed]
- 17.Andersen CL, Jensen JL, Ørntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64:5245–5250. doi: 10.1158/0008-5472.CAN-04-0496. [DOI] [PubMed] [Google Scholar]
- 18.Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper – Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004;26:509–515. doi: 10.1023/B:BILE.0000019559.84305.47. [DOI] [PubMed] [Google Scholar]
- 19.Silver N, Best S, Jiang J, Thein SL. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Biol. 2006;7:33. doi: 10.1186/1471-2199-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie F, Xiao P, Chen D, Xu L, Zhang B. miRDeepFinder: a miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol. Biol. 2012;80:75–84. doi: 10.1007/s11103-012-9885-2. [DOI] [PubMed] [Google Scholar]
- 21.Wan H, et al. Selection of appropriate reference genes for gene expression studies by quantitative real-time polymerase chain reaction in cucumber. Anal Biochem. 2010;399:257–261. doi: 10.1016/j.ab.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 22.Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible WR. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 2005;139:5–17. doi: 10.1104/pp.105.063743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borges AF, Fonseca C, Ferreira RB, Lourenço AM, Monteiro S. Reference gene validation for quantitative RT-PCR during biotic and abiotic stresses in Vitis vinifera. PLoS One. 2014;9:e111399. doi: 10.1371/journal.pone.0111399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Upadhyay A, Jogaiah S, Maske SR, Kadoo NY, Gupta VS. Expression of stable reference genes and SPINDLY gene in response to gibberellic acid application at different stages of grapevine development. Biol. Plant. 2015;59:436–444. doi: 10.1007/s10535-015-0521-2. [DOI] [Google Scholar]
- 25.Gonzalez-Aguilera KL, Saad CF, Chavez Montes RA, Alves-Ferreira M, de Folter S. Selection of reference genes for quantitative real-time RT-PCR studies in tomato fruit of the genotype MT-Rg1. Front. Plant Sci. 2016;7:1386. doi: 10.3389/fpls.2016.01386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pombo MA, Zheng Y, Fei Z, Martin GB, Rosli HG. Use of RNA-seq data to identify and validate RT-qPCR reference genes for studying the tomato-Pseudomonas pathosystem. Sci. Rep. 2017;7:44905. doi: 10.1038/srep44905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng Y, et al. Genome-wide identification and evaluation of reference genes for quantitative RT-PCR analysis during tomato fruit development. Front. Plant Sci. 2017;8:1440. doi: 10.3389/fpls.2017.01440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li W, et al. Selection and validation of appropriate reference genes for quantitative real-time PCR normalization in staminate and perfect flowers of andromonoecious Taihangia rupestris. Front. Plant Sci. 2017;8:729. doi: 10.3389/fpls.2017.00729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang H, et al. Selection and evaluation of novel reference genes for quantitative reverse transcription PCR (qRT-PCR) based on genome and transcriptome data in Brassica napus L. Gene. 2014;538:113–122. doi: 10.1016/j.gene.2013.12.057. [DOI] [PubMed] [Google Scholar]
- 30.Xiao X, et al. Validation of suitable reference genes for gene expression analysis in the halophyte Salicornia europaea by real-time quantitative PCR. Front. Plant Sci. 2014;5:788. doi: 10.3389/fpls.2014.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou B, et al. Candidate reference genes selection and application for RT-qPCR analysis in kenaf with cytoplasmic male sterility background. Front. Plant Sci. 2017;8:1520. doi: 10.3389/fpls.2017.01520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Y, et al. Selection of reference genes for quantitative real-time PCR normalization in creeping bentgrass involved in four abiotic stresses. Plant Cell Rep. 2015;34:1825–1834. doi: 10.1007/s00299-015-1830-9. [DOI] [PubMed] [Google Scholar]
- 33.Liu Y, et al. Identification and validation of reference genes for seashore paspalum response to abiotic stresses. Int. J. Mol. Sci. 2017;18:1322. doi: 10.3390/ijms18061322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Die JV, Román B, Nadal S, González-Verdejo CI. Evaluation of candidate reference genes for expression studies in Pisum sativum under different experimental conditions. Planta. 2010;232:145–153. doi: 10.1007/s00425-010-1158-1. [DOI] [PubMed] [Google Scholar]
- 35.Sgamma T, Pape J, Massiah A, Jackson S. Selection of reference genes for diurnal and developmental time-course real-time PCR expression analyses in lettuce. Plant Methods. 2016;12:21. doi: 10.1186/s13007-016-0121-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li T, et al. Selection and validation of appropriate reference genes for qRT-PCR analysis in Isatis indigotica Fort. Front. Plant Sci. 2017;8:1139. doi: 10.3389/fpls.2017.01139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z, et al. Selection of reference genes for quantitative reverse-transcription polymerase chain reaction normalization in Brassica napus under various stress conditions. Mol. Genet. Genomics. 2014;289:1023–1035. doi: 10.1007/s00438-014-0853-1. [DOI] [PubMed] [Google Scholar]
- 38.Lurin C, et al. Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell. 2004;16:2089–2103. doi: 10.1105/tpc.104.022236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding YH, Liu NY, Tang ZS, Liu J, Yang WC. Arabidopsis glutamine-rich protein 23 is essential for early embryogenesis and encodes a novel nuclear PPR motif protein that interacts with RNA polymerase II subunit III. Plant Cell. 2006;18:815–830. doi: 10.1105/tpc.105.039495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Verbitskiy D, Merwe JA, Zehrmann A, Hartel B, Takenaka M. The E-class PPR protein MEF3 of Arabidopsis thaliana can also function in mitochondrial RNA editing with an additional DYW domain. Plant Cell Physiol. 2012;53:358–367. doi: 10.1093/pcp/pcr182. [DOI] [PubMed] [Google Scholar]
- 41.Shikanai T, Fujii S. Function of PPR proteins in plastid gene expression. RNA Biol. 2013;10:1446–1456. doi: 10.4161/rna.25207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yim AK, et al. Using RNA-seq data to evaluate reference genes suitable for gene expression studies in soybean. PLoS One. 2015;10:e0136343. doi: 10.1371/journal.pone.0136343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maroufi A. Selection of reference genes for real-time quantitative PCR analysis of gene expression in Glycyrrhiza glabra under drought stress. Biol. Plant. 2016;60:645–654. doi: 10.1007/s10535-016-0601-y. [DOI] [Google Scholar]
- 44.Stefanowicz K, Lannoo N, Van Damme EJM. Plant F-box proteins – judges between life and death. Crit. Rev. Plant Sci. 2015;34:523–552. doi: 10.1080/07352689.2015.1024566. [DOI] [Google Scholar]
- 45.Demidenko NV, Logacheva MD, Penin AA. Selection and validation of reference genes for quantitative real-time PCR in buckwheat (Fagopyrum esculentum) based on transcriptome sequence data. PLoS One. 2011;6:e19434. doi: 10.1371/journal.pone.0019434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou CF, et al. Selection of reference genes for quantitative real-time PCR in six oil-tea camellia based on RNA-seq. Mol. Biol. 2013;47:836–851. doi: 10.1134/S0026893313060198. [DOI] [PubMed] [Google Scholar]
- 47.Murashige T, Skoog F. A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol. Plant. 1962;15:473–497. doi: 10.1111/j.1399-3054.1962.tb08052.x. [DOI] [Google Scholar]
- 48.Bustin SA, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 49.Kolosova N, et al. Isolation of high-quality RNA from gymnosperm and angiosperm trees. BioTechniques. 2004;35:821–824. doi: 10.2144/04365ST06. [DOI] [PubMed] [Google Scholar]
- 50.Wang H, Zhang X, Liu Q, Liu X, Ding S. Selection and evaluation of new reference genes for RT-qPCR analysis in Epinephelus akaara based on transcriptome data. PLoS One. 2017;12:e0171646. doi: 10.1371/journal.pone.0171646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Jonge HJ, et al. Evidence based selection of housekeeping genes. PLoS One. 2007;2:e898. doi: 10.1371/journal.pone.0000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files).