

Abstract
The concept of sequence-definition in the sense of polymer chemistry is introduced to conjugated, rod-like oligo(phenylene ethynylene)s via an iterative synthesis procedure. Specifically, monodisperse sequence-defined trimers and pentamers were prepared via iterative Sonogashira cross-coupling and deprotection. The reaction procedure was extended to tetra- and pentamers for the first time yielding a monodisperse pentamer with 18% and a sequence-defined pentamer with 3.2% overall yield. Furthermore, three novel trimers with a 9H-fluorene building block at predefined positions within the phenylene ethynylene chain were synthesised in 23–52% overall yields. Hence, it was confirmed that a functionality of interest can be incorporated selectively at a pre-defined position of these monodisperse oligomers. All respective intermediate structures were fully characterised by proton and carbon NMR, mass spectrometry, size-exclusion chromatography, and IR spectroscopy. Additionally, thermal and optical transitions are reported for the different oligomers.
Introduction
The synthesis of monodisperse macromolecules with a defined monomer sequence came into the focus of polymer science lately1–3. In nature, sequence-defined macromolecules or oligomers, such as peptides and proteins, are produced in a highly precise manner4. The latter are based on a building set of more than 20 amino acids, enabling diverse secondary and tertiary structures5. Theoretically, an infinite number of monomer units can be utilised in polymer chemistry, but the extent of perfection observed in nature is not yet achieved. Several synthetic methods to sequence-defined macromolecules are described, including solution phase6–9, solid phase10–13, as well as fluorous-14,15 and polymer-tethered16,17 approaches. Procedures based on solution phase chemistry are often associated with complex purification procedures, but usually offer high yields and the possibility to characterise and optimise every step. For solid phase procedures, the purification process is drastically facilitated, but the yields as well as scales are rather restricted. Fluorous-tethered approaches use a fluorous tag for a simplified purification process with perfluorinated columns and the polymer-tethered options are purified by simple precipitation2.
In the field of conjugated polymers, monodisperse oligomers are investigated as model systems for potential applications18. For rod-like molecules, such as oligo(1,4-phenylene ethynylene)s, various procedures to monodisperse oligomers were pursued. In a recent approach, Bunz et al. synthesised water-soluble oligo(1,4-phenylene ethynylene)s (monomer to tetramer) by non-iterative procedures19. Many iterative procedures, which in principle allow to obtain sequence-defined macromolecules, were published by the group of Tour, for example a divergent approach to a 16mer with dodecyl side groups or a bidirectional approach to a molecule exhibiting seven benzene units20–22. Non-iterative as well as divergent or bidirectional procedures, however, do not provide full molecular control over each repeating unit. Only via iterative synthesis procedures, the structure and more specifically the sequence of building blocks can be controlled. Therefore, gradual changes in the electronic level of the conjugated oligomer backbone can be achieved23. So far, only few studies focus on the incorporation of diverse monomers17,23–26. For instance, Lutz et al. prepared oligo(1,4-arylene ethynylene)s iteratively with the aid of a soluble polystyrene support by Sonogashira cross-coupling and subsequent deprotection17. Unfunctionalised benzene and pyridine building blocks enabled the positioning within the sequence. UV/Vis and fluorescence spectra of the polymers exhibiting one to four aromatic units were measured and revealed a bathochromic shift with increasing length and pyridine content. The Tour group established another linear synthesis procedure to oligo(1,4-phenylene ethynylene)s up to the trimer stage25.
The Sonogashira reaction is also the method of choice for preparing rod-like polymers in form of poly(arylene ethynylene)s by converting aryl diiodides or dibromides with aromatic diynes27–29. Poly(arylene ethynylene)s are mainly investigated in applications for molecular wires and sensors, but also as transistors27,30–32. Usually, these polymers exhibit structural defects due to butadiyne formation related to Glaser coupling33,34. Monodisperse oligo(1,4-phenylene ethynylene)s are free from structural defects, since chromatographic procedures are applied for purification.
Our approach towards sequence-defined oligo(1,4-phenylene ethynylene)s is based on the Sonogashira cross-coupling as well29. Although short monodisperse rod-like oligomers obtained by Sonogashira cross-coupling were published before (see above), an extension to sequence-defined pentamers was not achieved yet. Furthermore, novel molecules with a targeted incorporation of a fluorene unit were synthesised, demonstrating the possibility to systematically tune the structure of such highly defined macromolecules. All oligomers are molecularly fully characterised and demonstrate the success as well as limitations of this approach (see electronic supplementary information with all details). Furthermore, optical and thermal properties are reported to demonstrate the influence of the sequence on the properties of the obtained macromolecules.
Results and Discussion
Synthesis strategy
For the synthesis of sequence-defined rod-like oligomers, five building blocks were synthesised starting from hydroquinone. By applying Williamson ether synthesis with 1-bromopropane, 2-bromopropane, bromocyclohexane and 1-bromooctane, four 1,4-dialkoxybenzenes were obtained35. An iodination of 1,4-dimethoxybenzene and the obtained 1,4-dialkoxybenzenes with periodic acid yielded the 1,4-diiodo-2,5-dialkoxybenzenes36. These were converted with trimethylsilylacetylene into protected bifunctional building blocks via Sonogashira mono-coupling37. First, building block 1 with propoxy side groups was used for establishing the synthesis procedure in a solution and a solid phase organic synthesis (SPOS) approach (Fig. 1). For SPOS, a triazene linker was applied38.
Figure 1.

Synthesis procedure towards monodisperse oligo(1,4-phenylene ethynylene)s through Sonogashira cross-coupling with the building block 1 and subsequent deprotection. The approach in solution yields the protected monomer 2, the deprotected monomer 3, the respective versions of the dimers 4 and 5, the trimers 6 and 7, the tetramers 8 and 9 as well as the pentamers 10 and 11. Cleavage from the resin in SPOS results in the azide functionalised monomer 12.
The synthesis in solution started with a Sonogashira reaction of the building block 1 with phenylacetylene and proceeded smoothly to yield the respective monomer 2 in 99% yield. The deprotection was performed with potassium carbonate and the deprotected monomer 3 was received with 97%. After ten iterative steps of Sonogashira reaction with 1 and subsequent deprotection, the deprotected monodisperse pentamer 11 was obtained in an overall yield of 18% and in a scale of 116 mg. All intermediate steps are fully characterised in the Supplementary Information (1H and 13C NMR, SEC, and MS), unambiguously confirming the success of the synthesis. The purification was challenging, since Glaser coupling due to traces of oxygen could not be suppressed completely. Thus, this side reaction constitutes the major limiting factor for better yields in the Sonogashira cross-coupling.
The SPOS approach was initially performed in analogy to the solution approach, however product formation was not observed. The acquired conditions for the solution approach are therefore not directly transferable to SPOS and the above-mentioned Glaser coupling side reaction is even more problematic in this case, as it leads to further cross-linking of the resin. Further investigations enabled the cleavage of azide functionalised monomer 12 (Fig. 1) in an overall yield of 7 mg (60%). However, the initial concept of a fast and direct synthesis strategy via SPOS could not sustain, and the yield and scale are significantly lower as for the solution approach to deprotected monomer 3 with 3.15 g (96%). Therefore, SPOS was not further investigated here.
Polycondensation with chain stopper
A further direct oligomerisation approach in solution was performed as depicted in Fig. 2. A deprotected version of 1 exhibiting an unprotected tripled bond served as monomer (13, Fig. 2a), phenylacetylene and building block 1 act as starting unit and chain stopper, respectively. By applying the ratio 1:3:1 (starting unit, monofunctional monomer 13, chain stopper 1), short chain oligomers like the tetramer 8 were expected to form preferentially within the oligomer mixture. The comparative size exclusion chromatography (SEC) traces (Fig. 2b) confirm the formation of short chain oligomers, but also clearly demonstrate the high dispersity within the product mixture. Thus, the SEC trace clearly outlines the necessity of the herein described iterative approach towards defined oligomers (Fig. 1).
Figure 2.

The polycondensation approach and its comparison with monodisperse oligomers. (a) Schematic representation of the polycondensation approach with starting unit (phenylacetylene), monomer 13 and chain stopper 1. (b) SEC traces of a step-growth approach with monofunctional monomers (brown) in comparison to the monodisperse protected oligomers - monomer 2 (black), dimer 4 (red), trimer 6 (blue), tetramer 8 (pink) and pentamer 10 (green) and the building block 1 (grey). The trimer 4 is most abundant in the mixture. (c) Absorption (dotted) and photoluminescence (dotted) spectra of the polycondensation approach (brown), in comparison to the oligomers.
Furthermore, absorption and emission properties of the oligomer approach in comparison to the monodisperse representatives were investigated (Fig. 2c). The absorption maximum for the trimer 6 is observed at 402 nm, for the tetramer 8 at 410 nm and for the oligomer mixture at 406 nm. Therefore, the most abundant oligomer in the mixture should be the trimer 6 or the tetramer 8, which is in accordance with the SEC trace (Fig. 2b). Generally, the maximum of the photoluminescence intensity increases from the monomer 2 (400 nm) to the pentamer 10 (465 nm) and the oligomer mixture exhibits its maximum at 471 nm. Thus, in order to achieve defined photophysical properties, monodisperse oligomers offer unique advantages.
A sequence-defined rod-like pentamer
With the previously synthesised building blocks in hand and the experience gained in the synthesis of pentamer 10, subsequently sequence-defined oligomers were synthesised starting from deprotected monomer 3. The protected, sequence-defined dimer 14 exhibits propoxy side groups at position 1 and isopropoxy side groups at position 2. Deprotection yields deprotected, sequence-defined dimer 15 (Fig. 3).
Figure 3.
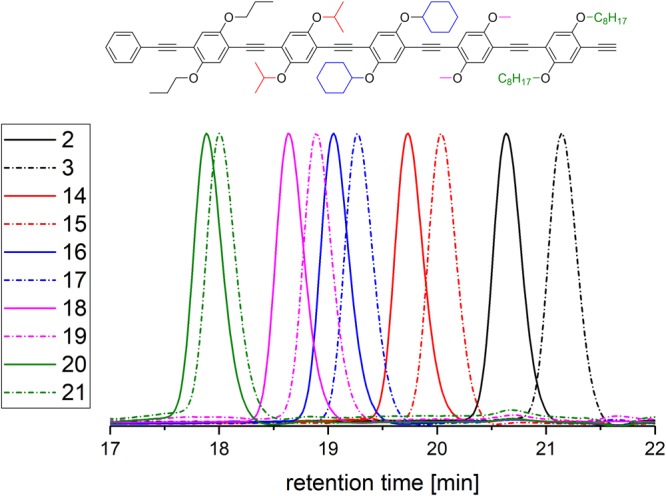
SEC traces of the sequence-defined oligomers. The protected versions are displayed as bold, the deprotected representatives as dotted lines: protected monomer 2 (black, bold), deprotected monomer 3 (black, dotted), the respective versions of the dimers 14 and 15 (red), trimers 16 and 17 (blue), tetramers 18 and 19 (pink) as well as pentamers 20 and 21 (green). The colour code of the SEC traces corresponds to the coloured side chains of the structure displayed above.
Eventually, sequence-defined deprotected pentamer 21 (compare structure depicted in Fig. 3) was synthesised iteratively according to Fig. 1 in a lower overall yield of 3.2% and in a scale of 73.6 mg. The sterically demanding moieties (i.e. cyclohexyl) restricted better yields in the Sonogashira reaction. Also, the side reaction is favoured as the respective Glaser coupling products are sterically less hindered compared to the desired respective oligomers. In Fig. 3, the SEC traces of the sequence-defined oligomers and their respective protected precursors are depicted confirming the high purities of the compounds and intermediates. All other characterisation data can be found in the supporting information.
The absorption and photoluminescence spectra of the sequence-defined oligomers in comparison to the monodisperse compounds differ only slightly, since the respective electronic properties do not change significantly. The thermal properties differ however, since the monodisperse and the sequence-defined oligomers exhibit distinct molecular weight and side groups (Table 1). The sequence-defined representatives have higher melting points (apart from the dimers) and higher glass transition temperatures compared to the monodisperse oligomers. For instance, the monodisperse trimer 6 shows a Tg of 34.4 °C and a Tm of 125.5 °C, whereas the sequence-defined trimer 16 shows a Tg of 52.7 °C and a Tm of 189.2 °C. Higher melting points result from the higher molecular weight of the molecules and the higher glass transition temperatures from the sterically more demanding side groups (especially cyclohexyl), which leads to high rotation barriers39.
Table 1.
Overview of melting points (Tm) and glass transition temperatures (Tg). The monodisperse trimer 6, tetramer 8 and pentamer 10 exhibit lower melting points and glass transition temperatures compared to the sequence-defined representatives
| Monodisperse oligomers | T g | T m | Sequence-defined oligomers | T g | T m |
|---|---|---|---|---|---|
| dimer 4 | 19.3 °C | 129.2 °C | dimer 14 | 22.9 °C | 123.2 °C |
| trimer 6 | 34.4 °C | 125.5 °C | trimer 16 | 52.7 °C | 189.2 °C |
| tetramer 8 | 45.6 °C | 160.5 °C | tetramer 18 | 67.7 °C | 176.6 °C |
| pentamer 10 | — | 186.8 °C | pentamer 20 | — | — |
Novel trimers with pre-defined incorporation of a fluorene moiety
The confirmation of the synthesis strategy inspired us to synthesise a 9H-fluorene building block 22 derived from 2,7-diiodofluorene. This novel building block, exhibiting again an iodine moiety as well as a trimethylsilyl-protected triple bond, was applied for the sequence-specific incorporation into oligo(1,4-phenylene ethynylene)s. In this way, three trimers (see Fig. 4 for respective structures) with systematically altered position of the fluorene unit were obtained (23, 24, 25). The overall yields and scales amounted to 21% and 188 mg (23), 36% and 319 mg (24), as well as 52% and 426 mg (25). All characterisation data of these trimers is provided in the supporting information, confirming their structure and high purity. In order to investigate the influence of the fluorene position, differential scanning calorimetry (DSC) was performed for the three trimers and the monodisperse (1,4-phenylene ethynylene) trimer 6 as depicted in Fig. 4.
Figure 4.
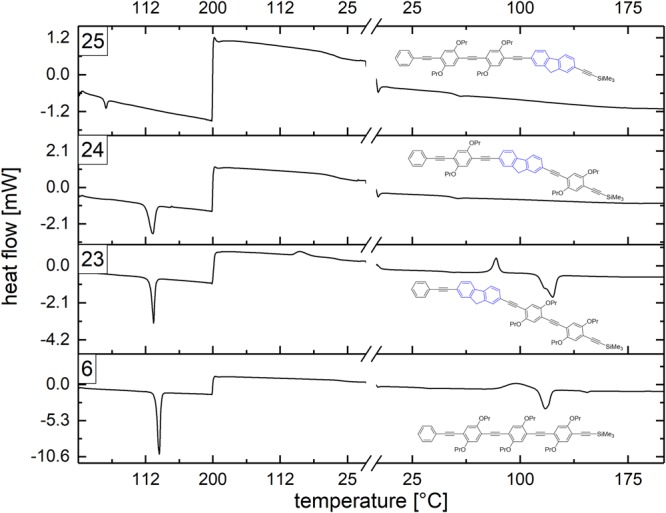
DSC traces of the sequence-defined trimers 23, 24 and 25 and the monodisperse trimer 6 with the respective molecular structures. The molecules were heated up to 200 °C and then cooled to 0 °C (left). The temperature was retained at 0 °C for 10 minutes (not depicted) and then a further heating cycle to 200 °C was performed (right).
Interestingly, the melting points of the trimers incorporating the fluorene units at position 1 (23) with 118.3 °C and at position 2 (24) with 112.6 °C resemble the melting point of monodisperse trimer 6 with 125.5 °C. Trimer 25 does not exhibit a melting point, but a glass transition (Tg) at 57.3 °C. Performing the DSC measurement of trimer 24 again, no melting point, but a Tg at 54.2 °C is detected as well. In further measurements, exclusively the Tg was detectable. Annealing attempts at 75 °C for 1 hour and altered heating and cooling rates did not lead to crystallisation of 24; the same applies to trimer 25. The monodisperse trimer 6 with its rod-like structure can obviously arrange comparably easy into a crystal structure, also 23 is able to recrystallise. Both representatives exhibit a solid-solid transition in the second heating cycle40. Trimer 24 and 25 do not recrystallise easily proving that the position of the fluorene unit has a relevant impact on the thermal properties.
According to SEC, 24 with the fluorene unit in the middle exhibits the largest hydrodynamic volume (Fig. 5a), probably since the different bond angle in the middle of the molecule has the largest influence within the series of stiff trimers. However, one would assume that 23 and 25 elute similar, but 23 exhibits a relatively smaller hydrodynamic radius. Thus, the interchanged phenyl and trimethylsilyl groups lead to a significantly altered hydrodynamic volume.
Figure 5.

Properties of novel sequence-defined trimers with one fluorene unit and monodisperse trimer 6. (a) SEC traces of trimer 23 with the fluorene unit at position 1 (orange), 24 with the fluorene unit at position 2 (green) and 25 with the fluorene unit at position 3 (blue) in comparison to monodisperse trimer 6 (red, dotted). (b) Absorption spectra (dotted) of monodisperse and sequence-defined trimers 6 (red), 23 (orange) 24 (green) 25 (blue) and photoluminescence spectra of 6, 23–25 (bold).
Furthermore, the maximum absorption and photoluminescence intensities differed (Fig. 5b): 24 displayed the lowest wavelengths. Moreover, the Stokes shift is significantly smaller for 24 compared to the other trimers. The exact wavelengths of the trimers are depicted in Table 2. Trimers 23 and 25 exhibit similar wavelengths of maximum absorption intensities, maximum photoluminescence intensities and similar Stokes shifts. Monodisperse trimer 6 exhibits the highest wavelength of maximum absorption and photoluminescence intensities. The Stokes shift is similar to 23 and 25. The latter results indicate that the conjugated system especially in trimer 24 is slightly disturbed if compared to the non-fluorene containing trimer 6. This effect seems to be less pronounced for fluorene units placed at positions 1 and 3 of trimers 23 and 25, respectively.
Table 2.
Overview of maximum absorption and photoluminescence intensities and the Stokes shift. The monodisperse trimer 6 exhibits the highest wavelengths of maximum absorption and photoluminescence intensities. The Stokes shift is similar to fluorene-containing trimers 23 and 25. Trimer 24 with the fluorene unit at position 2 has lower maximum absorption and photoluminescence intensity wavelengths; also the Stokes-shift is significantly smaller
| Oligomer | λmax absorption | λmax photoluminescence | Stokes-shift |
|---|---|---|---|
| 6 | 404 nm | 446 nm | 42 nm |
| 23 | 395 nm | 437 nm | 42 nm |
| 24 | 392 nm | 425 nm | 33 nm |
| 25 | 398 nm | 442 nm | 44 nm |
Additionally, time-resolved emission spectra were recorded, and the transient characteristic of the photoluminescence was fitted by a biexponential decay function. All transients are strongly dominated by the shorter-lived decay component with a lifetime of 1.15 ns for the monodisperse trimer 6 and only 0.97 ns for trimer 24. The trimers 23 and 25 show similar decay times of 1.03 ns and 1.01 ns, respectively. Photoluminescence quantum yield (PLQY) experiments at an excitation wavelength of 405 nm showed that trimer 25 exhibits the lowest PLQY of 83.3%. Trimers 6, 23 and 24 exhibit a similar PLQY of 88.5%, 88.7% and 89.3%, respectively. Longer lifetimes usually relate to lower quantum yields41, which cannot explain the significantly lower PLQY value for trimer 25 in this case.
Conclusion
To conclude, a monodisperse as well as a sequence-defined pentamer were synthesised in solution. So far, only trimers have been synthesised in an iterative fashion, thus significantly extending existing concepts. In parallel to the solution approach, SPOS and a direct oligomerisation were tested as fast alternatives but could not compete. The concept of sequence-definition was transferred to the synthesis of three novel sequence-defined trimers incorporating one fluorene moiety at a pre-defined position. These molecules were not only analysed by 1H and 13C NMR, MS, IR, and SEC, but also with DSC and several photophysical methods. The photophysical properties of the monodisperse and sequence-defined trimers differed only slightly, but the sequence had an impact on the thermal properties and the hydrodynamic volume. However, no clear trend of the properties according to the sequence could be observed. In the future, iterative synthesis procedures could be applied for the investigation and understanding of structure-property relationships by synthesising further structures with more distinct sequences.
Methods
General Synthetic Procedures
General procedure for the Williamson ether synthesis to 1,4-dialkoxybenzenes according to H. Meier et al.35.
Hydroquinone (1.00 eq., typically 275 mmol) was dissolved in 250 mL absolute ethanol. Potassium hydroxide (2.50 eq.) was added and the mixture was stirred for 30 minutes under reflux. Subsequently, the bromoalkane (2.20 eq.) was slowly added over a 1 h time period and stirred under reflux for another 2 h. Ethanol was removed with a rotary evaporator and the residue was taken up in dichloromethane. The organic phase was washed with water three times and once more with saturated NaHCO3 solution. It was then dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The crude product was either recrystallised from methanol (if solid) or column chromatography was performed (cyclohexane/ethyl acetate 20:1). Typical yields: 28–76%.
General procedure for the iodination to 1,4-diiodo-2,5-dialkoxybenzenes after Park et al.36.
Periodic acid (0.636 eq., typically 14.0 mmol) was dissolved in 25 mL methanol and stirred for 10 minutes. Subsequently, iodine (1.23 eq.) was added and after an additional stirring time of 10 minutes, 1,4-dialkoxybenzene (1.00 eq.) was added. The reaction mixture was stirred at 70 °C for 4 h. The residue was carefully poured into 50 mL water containing potassium disulfite. The precipitate was washed with methanol and dissolved in dichloromethane. The solution was filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by recrystallisation from methanol or ethanol. Typical yields: 64–92%).
General procedure for the Sonogashira mono-coupling to the dialkoxy-substituted building blocks (e.g. 1) after Tour et al.37.
All Sonogashira reactions were performed under argon atmosphere.
1,4-Diiodo-2,5-dialkoxybenzene (1.00 eq., typically 22.4 mmol), 2.5 mol% bis(triphenylphosphine)palladium(II) dichloride and 5 mol% copper(I) iodide were placed into a Schlenk flask and degassed. Under continuous argon flow, 400 mL dry THF and 30 mL dry triethylamine were added, and the mixture was stirred for 10 minutes. Subsequently, trimethylsilylacetylene (1.10 eq.) with 5 mL dry THF was added dropwise with a syringe. The reaction mixture was stirred for 20 h at room temperature, taken up in dichloromethane and washed with saturated NH4Cl solution. The aqueous phase was extracted three times with dichloromethane. The combined organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica column chromatography (cyclohexane/dichloromethane 9:1 or 5:1). Typical yields: 33–46%.
Procedure for the Sonogashira mono-coupling to the 9H-fluorene building block 22.
2,7-Diiodo-9H-fluorene (1.00 eq., typically 12.0 mmol), 2.5 mol% bis(triphenylphosphine)palladium(II) dichloride and 5 mol% copper(I)iodide were placed into a Schlenk flask and degassed. Under continuous argon flow, 200 mL dry THF and 17 mL dry triethylamine were added, and the mixture was stirred for 10 minutes. Subsequently, trimethylsilylacetylene (0.603 eq) was added dropwise with a syringe. The reaction mixture was stirred for 20 h at room temperature, taken up in dichloromethane and washed with saturated NH4Cl solution. The aqueous phase was extracted three times with dichloromethane. The combined organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica column chromatography (cyclohexane/dichloromethane 30:1) to yield the product as a white solid (43%).
General procedure for the Sonogashira cross-coupling to monomers (2 and fluorene-containing monomer).
1 or 22 (1.00 eq., typically 12.0 mmol), 2.5 mol% bis(triphenylphosphine) palladium(II) dichloride and 5 mol% copper(I) iodide were placed into a Schlenk flask and degassed three times. Under continuous argon flow, 150 mL dry THF and dry triethylamine (10.0 eq.) were added and the mixture was stirred for 10 minutes. Subsequently, phenylacetylene (3.00 eq.) in 5 mL THF was added dropwise with a syringe. The reaction mixture was stirred for 48 h at room temperature, taken up in dichloromethane and washed with saturated NH4Cl solution. The aqueous phase was extracted three times with dichloromethane. The combined organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica column chromatography twice (cyclohexane/dichloromethane 4:1 and cyclohexane/ethyl acetate 20:1). Typical yields: 92–99%.
General procedure for the deprotection of the trimethylsilyl group (3, 5, 7, 9, 11, 13, 15, 17, 19, 21).
The protected oligomer (1.00 eq., typically 10 mmol) and two equivalents of potassium carbonate were placed into a Schlenk flask and degassed three times. Under continuous argon flow, 200 mL dry dichloromethane and 200 mL dry methanol were added. The reaction mixture was stirred overnight at room temperature under argon atmosphere and quenched with distilled water. The aqueous phase was extracted three times with dichloromethane, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash silica column chromatography (either cyclohexane/ethyl acetate or dichloromethane/cyclohexane). Typical yields: 85–100%.
General procedure for the Sonogashira cross-coupling to dimers and higher molecular weight oligomers (4, 6, 8, 10, 12, 14, 16, 18, 20, 23, 24, 25).
The building block (e.g. 1 or 22) (3.00–5.00 eq.), 5 mol% bis(triphenylphosphine) palladium(II) dichloride and 5 mol% copper(I) iodide were placed into a Schlenk flask and degassed three times. Under continuous argon flow, 100 mL dry THF and dry triethylamine (10.0 eq.) were added and the mixture was stirred for 10 minutes. Subsequently, rod-like oligomer (e.g. 3, 5, 7, 9, 13, 15, 17, 19) (1.00 eq.) in 20 mL dry THF was added dropwise with a syringe. The reaction mixture was stirred for 72 h (3 d) at 45 °C, taken up in dichloromethane and washed with saturated NH4Cl solution. The aqueous phase was extracted three times with dichloromethane. The combined organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica column chromatography (cyclohexane/dichloromethane) and a flash silica column (cyclohexane/ethyl acetate) or by recrystallisation from cyclohexane/ethyl acetate. Typical yields: 33–84%.
Electronic supplementary material
Acknowledgements
We kindly thank N. Wippert for the (E)-3-benzyl-3-M*-1-(4-iodophenyl)triaz-1-ene resin. R. Schulte and T. Sattelberger are thanked for experimental support. This work was supported by the DFG in the framework of the collaborative research centre 1176 (SFB 1176, project A4).
Author Contributions
R.V.S., K.A.W. and R.S. synthesised the molecules in solution and M.L. conducted the solid phase approaches. A.P.A. performed the photophysical analysis, D.B. the PLQY measurements and R.V.S. the thermal analysis. R.V.S. wrote the paper, K.A.W. and M.A.R.M. reviewed the manuscript. M.A.R.M., U.L. and S.B. designed the project.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-35933-z.
References
- 1.Lutz J-F, Ouchi M, Liu DR, Sawamoto M. Sequence-controlled polymers. Science. 2013;341:1238149. doi: 10.1126/science.1238149. [DOI] [PubMed] [Google Scholar]
- 2.Solleder SC, Schneider RV, Wetzel KS, Boukis AC, Meier MAR. Recent Progress in the Design of Monodisperse, Sequence-Defined Macromolecules. Macromol. Rapid Commun. 2017;38:1600711. doi: 10.1002/marc.201600711. [DOI] [PubMed] [Google Scholar]
- 3.Lutz J-F, Lehn J-M, Meijer EW, Matyjaszewski K. From precision polymers to complex materials and systems. Nat. Rev. Mater. 2016;1:16024–16037. doi: 10.1038/natrevmats.2016.24. [DOI] [Google Scholar]
- 4.Lucas-Lenard J, Lipmann F. Protein biosynthesis. Annu. Rev. Biochem. 1971;40:409–448. doi: 10.1146/annurev.bi.40.070171.002205. [DOI] [PubMed] [Google Scholar]
- 5.Ambrogelly A, Palioura S, Söll D. Natural expansion of the genetic code. Nat. Chem. Biol. 2007;3:29–35. doi: 10.1038/nchembio847. [DOI] [PubMed] [Google Scholar]
- 6.Takizawa K, Tang C, Hawker CJ. Molecularly defined caprolactone oligomers and polymers. Synthesis and characterization. J. Am. Chem. Soc. 2008;130:1718–1726. doi: 10.1021/ja077149w. [DOI] [PubMed] [Google Scholar]
- 7.Foarta F, Landis CR. Condensation Oligomers with Sequence Control but without Coupling Reagents and Protecting Groups via Asymmetric Hydroformylation and Hydroacyloxylation. J. Org. Chem. 2016;81:11250–11255. doi: 10.1021/acs.joc.6b02210. [DOI] [PubMed] [Google Scholar]
- 8.Konrad W, et al. A Combined Photochemical and Multicomponent Reaction Approach to Precision Oligomers. Chem. Eur. J. 2018;24:3413–3419. doi: 10.1002/chem.201705939. [DOI] [PubMed] [Google Scholar]
- 9.Solleder SC, Zengel D, Wetzel KS, Meier MAR. A Scalable and High-Yield Strategy for the Synthesis of Sequence-Defined Macromolecules. Angew. Chem. Int. Ed. 2016;55:1204–1207. doi: 10.1002/anie.201509398. [DOI] [PubMed] [Google Scholar]
- 10.Al Ouahabi A, Charles L, Lutz J-F. Synthesis of non-natural sequence-encoded polymers using phosphoramidite chemistry. J. Am. Chem. Soc. 2015;137:5629–5635. doi: 10.1021/jacs.5b02639. [DOI] [PubMed] [Google Scholar]
- 11.Young JK, Nelson JC, Moore JS. Synthesis of Sequence Specific Phenylacetylene Oligomers on an Insoluble Solid Support. J. Am. Chem. Soc. 1994;116:10841–10842. doi: 10.1021/ja00102a082. [DOI] [Google Scholar]
- 12.Grate JW, Mo K-F, Daily MD. Triazine-Based Sequence-Defined Polymers with Side-Chain Diversity and Backbone-Backbone Interaction Motifs. Angew. Chem. Int. Ed. 2016;55:3925–3930. doi: 10.1002/anie.201509864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martens S, van den Begin J, Madder A, Du Prez FE, Espeel P. Automated Synthesis of Monodisperse Oligomers, Featuring Sequence Control and Tailored Functionalization. J. Am. Chem. Soc. 2016;138:14182–14185. doi: 10.1021/jacs.6b07120. [DOI] [PubMed] [Google Scholar]
- 14.Jian H, Tour JM. Preparative fluorous mixture synthesis of diazonium-functionalized oligo(phenylene vinylene)s. J. Org. Chem. 2005;70:3396–3424. doi: 10.1021/jo048051s. [DOI] [PubMed] [Google Scholar]
- 15.Kanasty RL, et al. Sequence-Defined Oligomers from Hydroxyproline Building Blocks for Parallel Synthesis Applications. Angew. Chem. Int. Ed. 2016;55:9529–9533. doi: 10.1002/anie.201602748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Solleder SC, Meier MAR. Sequence control in polymer chemistry through the Passerini three-component reaction. Angew. Chem. Int. Ed. 2014;53:711–714. doi: 10.1002/anie.201308960. [DOI] [PubMed] [Google Scholar]
- 17.Szweda R, Chendo C, Charles L, Baxter PNW, Lutz J-F. Synthesis of oligoarylacetylenes with defined conjugated sequences using tailor-made soluble polymer supports. Chem. Commun. 2017;53:8312–8315. doi: 10.1039/C7CC03633G. [DOI] [PubMed] [Google Scholar]
- 18.Müllen, K. & Wegner, G. Electronic materials. The oligomer approach (Wiley-VCH, 1998).
- 19.Hergert M, Bender M, Seehafer K, Bunz UHF. Molecular Wire Effects in Phenyleneethynylene Oligomers. Surprising Insights. Chem. Eur. J. 2018;24:3132–3135. doi: 10.1002/chem.201705604. [DOI] [PubMed] [Google Scholar]
- 20.Huang S, Tour JM. Rapid bi-directional synthesis of oligo(1, 4-phenylene ethynylene)s. Tetrahedron Lett. 1999;40:3447–3450. doi: 10.1016/S0040-4039(99)00463-3. [DOI] [Google Scholar]
- 21.Schumm JS, Pearson DL, Tour JM. Iterative Divergent/Convergent Approach to Linear Conjugated Oligomers by Successive Doubling of the Molecular Length: A Rapid Route to a 128-Long Potential Molecular Wire. Angew. Chem. Int. Ed. 1994;33:1360–1363. doi: 10.1002/anie.199413601. [DOI] [Google Scholar]
- 22.Jones L, Schumm JS, Tour JM. Rapid Solution and Solid Phase Syntheses of Oligo(1,4-phenylene ethynylene)s with Thioester Termini: Molecular Scale Wires with Alligator Clips. Derivation of Iterative Reaction Efficiencies on a Polymer Support. J. Org. Chem. 1997;62:1388–1410. doi: 10.1021/jo962336q. [DOI] [Google Scholar]
- 23.Jørgensen M, Krebs FC. Stepwise unidirectional synthesis of oligo phenylene vinylenes with a series of monomers. Use in plastic solar cells. J. Org. Chem. 2005;70:6004–6017. doi: 10.1021/jo0506783. [DOI] [PubMed] [Google Scholar]
- 24.Elliott EL, Ray CR, Kraft S, Atkins JR, Moore JS. Solid-phase synthesis of m-phenylene ethynylene heterosequence oligomers. J. Org. Chem. 2006;71:5282–5290. doi: 10.1021/jo0607212. [DOI] [PubMed] [Google Scholar]
- 25.Hwang J-J, Tour JM. Combinatorial synthesis of oligo(phenylene ethynylene)s. Tetrahedron. 2002;58:10387–10405. doi: 10.1016/S0040-4020(02)01409-6. [DOI] [Google Scholar]
- 26.Norris BN, et al. Sequence Matters: Modulating Electronic and Optical Properties of Conjugated Oligomers via Tailored Sequence. Macromolecules. 2013;46:1384–1392. doi: 10.1021/ma400123r. [DOI] [Google Scholar]
- 27.Bunz UHF, Seehafer K, Bender M, Porz M. Poly(aryleneethynylene)s (PAE) as paradigmatic sensor cores. Chem. Soc. Rev. 2015;44:4322–4336. doi: 10.1039/C4CS00267A. [DOI] [PubMed] [Google Scholar]
- 28.Giesa R, Schulz RC. Soluble poly(1,4‐phenyleneethynylene)s. Makromol. Chem. 1990;191:857–867. doi: 10.1002/macp.1990.021910413. [DOI] [Google Scholar]
- 29.Sonogashira K, Tohda Y, Hagihara N. A convenient synthesis of acetylenes: catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines. Tetrahedron Lett. 1975;16:4467–4470. doi: 10.1016/S0040-4039(00)91094-3. [DOI] [Google Scholar]
- 30.Dong H, et al. Nanowire crystals of a rigid rod conjugated polymer. J. Am. Chem. Soc. 2009;131:17315–17320. doi: 10.1021/ja907015p. [DOI] [PubMed] [Google Scholar]
- 31.Hu W, et al. A self-assembled nano optical switch and transistor based on a rigid conjugated polymer, thioacetyl-end-functionalized poly(para-phenylene ethynylene) J. Am. Chem. Soc. 2005;127:2804–2805. doi: 10.1021/ja0433929. [DOI] [PubMed] [Google Scholar]
- 32.Kim I-B, Erdogan B, Wilson JN, Bunz UHF. Sugar-poly(para-phenylene ethynylene) conjugates as sensory materials. Efficient quenching by Hg2+ and Pb2+ ions. Chem. Eur. J. 2004;10:6247–6254. doi: 10.1002/chem.200400788. [DOI] [PubMed] [Google Scholar]
- 33.Glaser C. Beiträge zur Kenntniss des Acetenylbenzols. Ber. Dtsch. Chem. Ges. 1869;2:422–424. doi: 10.1002/cber.186900201183. [DOI] [Google Scholar]
- 34.Bunz, U. H. F. Poly(arylene ethynylene)s (ed. Weder, C.) 1–52 (Springer, 2005).
- 35.Meier H, et al. Preparation and Nonlinear Optics of Monodisperse Oligo(1,4-phenyleneethynylene)s. Eur. J. Org. Chem. 2001;2001:4431–4443. doi: 10.1002/1099-0690(200112)2001:23<4431::AID-EJOC4431>3.0.CO;2-J. [DOI] [Google Scholar]
- 36.Ko S-B, Cho A-N, Kim M-J, Lee C-R, Park N-G. Alkyloxy substituted organic dyes for high voltage dye-sensitized solar cell. Effect of alkyloxy chain length on open-circuit voltage. Dyes. Pigm. 2012;94:88–98. doi: 10.1016/j.dyepig.2011.10.014. [DOI] [Google Scholar]
- 37.Morin J-F, Sasaki T, Shirai Y, Guerrero JM, Tour JM. Synthetic routes toward carborane-wheeled nanocars. J. Org. Chem. 2007;72:9481–9490. doi: 10.1021/jo701400t. [DOI] [PubMed] [Google Scholar]
- 38.Bräse S, Enders D, Köbberling J, Avemaria F. A Surprising Solid-Phase Effect. Development of a Recyclable “Traceless” Linker System for Reactions on Solid Support. Angew. Chem. Int. Ed. 1998;37:3413–3415. doi: 10.1002/(SICI)1521-3773(19981231)37:24<3413::AID-ANIE3413>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 39.Mark, J. E. et al. Physical properties of polymers. (American Chemical Society, 1993).
- 40.Perrenot B, Widmann G. Polymorphism by differential scanning calorimetry. Thermochim. Acta. 1994;234:31–39. doi: 10.1016/0040-6031(94)85133-6. [DOI] [Google Scholar]
- 41.Greenham NC, et al. Measurement of absolute photoluminescence quantum efficiencies in conjugated polymers. Chem. Phys. Lett. 1995;241:89–96. doi: 10.1016/0009-2614(95)00584-Q. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.