

Abstract
The synthesis and structure-activity relationship (SAR) studies of praziquantel derivatives with activity against adult Schistosoma japonicum are described. Several of them showed better worm killing activity than praziquantel and could serve as leads for further optimization.
Keywords: praziquantel derivatives, antischistosomal activity, Schistosoma japonicum, neglected tropical disease
1. Introduction
Schistosomiasis is a neglected tropical disease (NTD) caused by blood-dwelling trematodes belonging to the genus Schistosoma. Schistosoma haematobium, Schistosoma japonicum, and Schistosoma mansoni are the main species parasitizing humans [1]. It has been estimated that some 779 million people are at risk for schistosomiasis transmission, with 207 million infected in 76 countries and territories [1,2]. In China, the disease caused by Schistosoma japonicum remains a major public health concern with more than 280 thousand people infected [3]. In the absence of a vaccine, praziquantel (PZQ) has been the only drug recommended by the World Health Organization for the treatment and control of schistosomiasis through mass drug administration (MDA) programs for almost three decades [4]. However, PZQ does not prevent reinfection, and it is inactive against juvenile schistosomes. Furthermore, it has only a limited effect on developed liver and spleen lesions, as well as on the emergent schistosome phenotypes that are resistant to PZQ chemotherapy [5,6]. Therefore, it is imperative to develop new antischistosomal agents. A number of recent studies were directed towards exploration of new chemical entities from natural products [7,8,9,10] and towards the identification of additional drug targets for schistosomiasis [11,12,13,14,15].
In addition, there are two other major issues with regard to PZQ: (1) the mechanism of action of PZQ is uncertain [16] and (2) as currently administered, PZQ is a racemic mixture [16]. It is a great challenge to prepare (−)-PZQ inexpensively and stereoselectively [17]. The less active (+)-PZQ enantiomer has well been documented as having a bitter and disgusting taste [18]. Recently, many synthetic routes for PZQ and its derivatives have been developed [19,20], providing more opportunities to make novel PZQ derivatives as potential drugs for the treatment of schistosomiasis.
Ever since the discovery of PZQ, there has been interest in the elucidation of the structure–activity relationships (SAR) [21], mostly from data generated using S. mansoni, and to some extent with data for S. japonicum. There are five positions amenable for chemical modification in the PZQ molecular structure [16,22] (Figure 1). Among these, only position R1 has been heavily investigated [21]. Recently, more variants at the R1 position as well as PZQ-ozonide hybrids were synthesized and tested against juvenile and adult stages of S. mansoni in vitro and in vivo. Unfortunately, all the variants showed decreased activity compared to the parent compound PZQ [23]. Patra and co-workers reported PZQ analogues that replaced the cyclohexyl group at position 2 with different ferrocenyl moieties. Unfortunately, the Fc-PZQ derivatives were considerably less active than PZQ when tested against S. mansoni in vitro [22]. Recently, Gasser and coworkers reported Cr-PZQ derivatives with comparable antischistosomal activities to PZQ when tested against adult S. mansoni [24]. In 2007, the Todd group first reported several derivatives through nitration and amination of the C10 position of the aromatic ring on PZQ. However, these compounds showed significantly decreased worm killing activity against S. mansoni in vitro [25]. In 2012, the Qiao group synthesized eight PZQ/peroxide conjugates, with some compounds showing superior worm-killing activity against S. japonicum in in vitro assays compared to PZQ [26]. Recently, the Qiao group reported aromatic ring-modified praziquantel derivatives with activity against both juvenile and adult S. japonicum, and a compound with a bromide atom at C8 position showed higher potency against adult S. japonicum than PZQ in vitro and comparable antischistosomal activity to PZQ in vivo. However, none of them showed superior worm-killing ability to PZQ [27]. The Rao group developed an efficient synthetic route to provide structurally diverse analogues for SAR studies. Several PZQ analogues with an indol ring were synthesized and tested against adult S. mansoni. Unfortunately, no compounds showed better activity than PZQ in vitro [28]. Taken together, all this data indicates that although much work has been done on PZQ, it is still necessary to further expand the range of PZQ analogues in order to obtain chemical probes to elucidate the real biological mechanism of PZQ, and to generate alternative new chemotherapies. Therefore, we became interested in the synthesis of new PZQ analogues to find new potential antischistosomal agents against S. japonicum.
Figure 1.

Chemical structure of praziquanel (PZQ) and some reported derivatives.
2. Results and Discussion
2.1. Chemistry
Due to the lack of SAR details from S. japonicum, we initially resynthesized some PZQ analogues with position 2 substitution and evaluated their worm-killing activities against S. japonicum (Scheme 1). The racemic PZQ from Aldrich was hydrolyzed by 2 N HCl to yield praziquanamine (1) using the method described by the Vennerstrom group [23], followed by coupling with acyl chlorides to yield compounds 2–12. Reductive amination reactions of appropriate aldehydes with praziquanamine (1) were performed with sodium triacetoxyborohydride in dichloroethane at room temperature to afford compounds 13–15 in yields ranging from 27% to 35%. Praziquanamine (1) was reduced by lithium aluminum hydride to yield compound 16, followed by acylation to yield compounds 17–22. To extend the SAR studies, the aromatic moiety was replaced by a thiophene ring, and compound 23 was synthesized using the method described by Frehel and his coworkers [29]. Compound 23 was hydrolyzed with 2N HCl to yield amine 24, followed by acylation with appropriate acyl chlorides to yield compounds 25–33 (Scheme 2). As in previous reports [22], we also observed the major isomer in mixtures with its minor enantiomer with respect to the 11b position on the ring system for compounds 1–33. The results were also in agreement with the nature of the commercially available PZQ [19,30] and its thiophene analogue 23 [20].
Scheme 1.
Synthesis of PZQ analogues 1–22.

Reagents and conditions: (a) 2 N HCl, reflux, overnight, 62%; (b) TFAA, Et3N, DCM, 62% or RCOCl, Et3N, DCM, 29%–50%; (c) RCHO, NaBH4, HOAc, MeOH, 0 °C to 60 °C, 27%–35%; (d) LiAlH4, THF, 0 °C, 35%; (e) RCOCl, Et3N, DCM, 32%–69%.
Scheme 2.
Synthesis of PZQ analogues 24–33.

Reagents and conditions: (f) 2 N HCl, reflux, overnight, 78%; (g) RCOCl, Et3N, DCM, 40%–76%.
2.2. Biological Activities
The biological results are listed in Table 1. Among compounds with an aliphatic cyclic ring (2–4 and PZQ), the worm-killing activity was improved by increasing the ring size. Compounds with an acetyl group (i.e., 5) and with an isobutyryl group (i.e., 6) lacked worm-killing activity. Among compounds with a halogen atom, compound 7 with the chloroacetyl group killed 100% of the worms at the concentration of 5 μM, showing better activity than PZQ. However, compound 8 with a 2,2,2-trifluoroacetyl group lost worm killing activity. Replacement of the cyclohexyl ring with a heterocyclic ring, (compound 11 with a thiophene-2-carbonyl group) led to a similar activity to that of PZQ. However, compound 10 with an isonicotinoyl group and compound 12 with a furan-2-carbonyl group did not show any worm-killing activity below the concentration of 100 μM. Compound 9, with a benzoyl group, showed similar activity as PZQ. Replacement of the carbonyl group at position 2 with CH2 gave compounds 13–15, which showed no worm-killing activity. The results indicated that the amide bond at position 2 is essential for activity against S. japonicum. The SAR results displayed similarities between compounds 17–22 and compounds containing amide (3, 4, PZQ, 9, 11, and 7), showing decreased activity. The results indicated that the amide bond at position 4 was very important to maintain the activity of PZQ derivatives. The SAR results from S. japonicum adult worms were similar with those from S. mansoni [21]. However, it still gave us valuable information to explore novel PZQ derivatives as antischistosomal agents.
Table 1.
Worm-killing activity on S. japonicum adult worms in vitro by compounds 1–33.
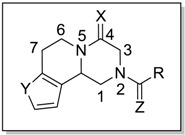
| Compound | R | X | Y | Z | Killing activity a | |||
|---|---|---|---|---|---|---|---|---|
| Conc (μM) b | 24 h | 48 h | 72 h | |||||
| Vehicle | n.e | n.e | n.e | |||||
| PZQ | 10 | 25.0%D | 25.0%D | 37.5%D | ||||
| 25 | 25.0%D | 25.0%D | 62.5%D | |||||
| 50 | 25.0%D | 37.5%D | 62.5%D | |||||
| 100 | 37.5%D | 50.0%D | 87.5%D | |||||
| 1 d, e | None | O | CH=CH | None | 100 | n.e. | n.e. | n.e. |
| 2 d, e | 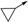 |
O | CH=CH | O | 100 | sluggish | 25.0% | 75.0% |
| 3 e | 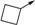 |
O | CH=CH | O | 10 | n.e. | n.e. | sluggish |
| 25 | n.e. | n.e. | 10.0%D | |||||
| 50 | n.e. | sluggish | 75.0%D | |||||
| 100 | sluggish | sluggish | 87.5%D | |||||
| 4 e | 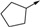 |
O | CH=CH | O | 10 | sluggish | sluggish | 37.5%D |
| 25 | sluggish | sluggish | 50.0%D | |||||
| 50 | sluggish | sluggish | 100%D | |||||
| 100 | 12.5%D | 12.5%D | 75.0%D | |||||
| 5 d, e | CH3 | O | CH=CH | O | 100 | n.e. | n.e. | n.e. |
| 6 d, e | 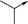 |
O | CH=CH | O | 100 | n.e. | n.e. | 20.0%D |
| 7 c, e | ClCH2 | O | CH=CH | O | 1 | n.e. | n.e. | n.e. |
| 3 | n.e. | 25.0%D | 25.0%D | |||||
| 5 | 70.0%D | 90.0%D | 100%D | |||||
| 8 | 100%D | 100%D | 100%D | |||||
| 10 | 100%D | 100%D | 100%D | |||||
| 25 | 100%D | 100%D | 100%D | |||||
| 50 | 100%D | 100%D | 100%D | |||||
| 100 | 100%D | 100%D | 100%D | |||||
| 8 d, e | 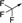 |
O | CH=CH | O | 100 | n.e. | n.e. | n.e. |
| 9 e | 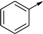 |
O | CH=CH | O | 10 | n.e. | n.e. | 55.6%D |
| 25 | sluggish | 25.0%D | 87.5%D | |||||
| 50 | 18.2%D | 36.4%D | 45.5%D | |||||
| 100 | 45.5%D | 45.5%D | 81.8%D | |||||
| 10 d, e | 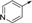 |
O | CH=CH | O | 100 | n.e. | n.e. | sluggish |
| 11 e | 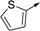 |
O | CH=CH | O | 10 | n.e. | n.e. | 62.5%D |
| 25 | n.e. | n.e. | 62.5%D | |||||
| 50 | 25.0%D | 37.5%D | 87.5%D | |||||
| 100 | 14.3%D | 28.6%D | 87.5%D | |||||
| 12 d, e | 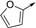 |
O | CH=CH | O | 100 | n.e. | n.e. | n.e. |
| 13 d | 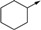 |
O | CH=CH | H2 | 100 | n.e. | n.e. | n.e. |
| 14 d, e | 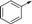 |
O | CH=CH | H2 | 100 | n.e. | n.e. | n.e. |
| 15 d | 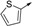 |
O | CH=CH | H2 | 100 | n.e. | n.e. | n.e. |
| 16 d, e | None | H2 | CH=CH | None | 100 | n.e. | n.e. | n.e. |
| 17 d | 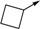 |
H2 | CH=CH | O | 100 | n.e. | n.e. | n.e. |
| 18 | 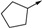 |
H2 | CH=CH | O | 10 | sluggish | sluggish | sluggish |
| 25 | sluggish | 18.2%D | 18.2%D | |||||
| 50 | 10.0%D | 20.0%D | 50.0%D | |||||
| 100 | 25.0%D | 25.0%D | 75.0%D | |||||
| 19 | 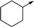 |
H2 | CH=CH | O | 10 | n.e. | n.e. | n.e. |
| 25 | n.e. | 14.3%D | 57.1%D | |||||
| 50 | sluggih | 33.3%D | 66.7%D | |||||
| 100 | sluggish | sluggish | 85.7%D | |||||
| 20 d, e | 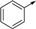 |
H2 | CH=CH | O | 100 | n.e. | n.e. | n.e. |
| 21 d | 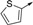 |
H2 | CH=CH | O | 100 | n.e. | n.e. | n.e. |
| 22 | ClCH2 | H2 | CH=CH | O | 10 | n.e. | sluggish | 50.0%D |
| 25 | 87.5%D | 100%D | 100%D | |||||
| 50 | 100%D | 100%D | 100%D | |||||
| 100 | 100%D | 100%D | 100%D | |||||
| 23 e | 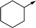 |
O | S | O | 10 | sluggish | 75.0%D | 75.0%D |
| 25 | sluggish | 87.5%D | 87.5%D | |||||
| 50 | 25.0% | 75.0%D | 87.5%D | |||||
| 100 | 28.6%D | 71.4%D | 71.4%D | |||||
| 24 d | None | O | S | None | 100 | n.e. | n.e. | n.e. |
| 25 d |  |
O | S | O | 100 | n.e. | 12.5%D | 37.5%D |
| 26 d | 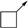 |
O | S | O | 100 | n.e. | sluggish | sluggish |
| 27 d | 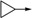 |
O | S | O | 100 | n.e. | n.e. | sluggish |
| 28 d |  |
O | S | O | 100 | n.e. | n.e. | n.e. |
| 29 d |  |
O | S | O | 100 | sluggish | sluggish | sluggish |
| 30 e | 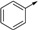 |
O | S | O | 10 | n.e. | n.e. | n.e. |
| 25 | n.e. | sluggish | sluggish | |||||
| 50 | n.e. | 14.3%D | 14.3%D | |||||
| 100 | sluggish | 37.5%D | 75.0%D | |||||
| 31 d, e | 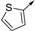 |
O | S | O | 100 | n.e. | n.e. | n.e. |
| 32 d, e | 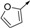 |
O | S | O | 100 | n.e. | n.e. | n.e. |
| 33 c | ClCH2 | O | S | O | 1 | n.e. | n.e. | n.e. |
| 3 | n.e. | n.e. | n.e. | |||||
| 5 | sluggish | 25%D | 37.5%D | |||||
| 8 | 75%D | 87.5%D | 87.5%D | |||||
| 10 | 100%D | 100%D | 100%D | |||||
| 25 | 100%D | 100%D | 100%D | |||||
| 50 | 100%D | 100%D | 100%D | |||||
| 100 | 100%D | 100%D | 100%D | |||||
a Data collected by visual examination of worm movement and shape (the values were the averages of two tests); n.e. (no effect): all worms are scored as active in culture with typical appearance; sluggish: worm movement is significantly reduced; % D = number of dead worms/total number of worms observed, and dead worms judged by lack of movement within 2 minutes in addition to morphological and tegumental alterations; b The concentration of the chemicals on S. japonicum adult worms in vitro; c Starting concentration of 1 μM when the chemicals killed 100% of the worms at the concentration of 10 μM; d Data only showed the concentration of 100 μM when the chemicals have no effect on the worms at 10, 25 and 50 μM; e Reported in References [20,21,29] and references therein.
To enlarge the chemical space of potential drug candidates, replacement of the phenyl ring on PZQ with a thiophene ring gave compounds 23–33. Among compounds with an alkyl group (i.e., 23, 25–29), compound 23 with a cyclohexanecarbonyl group showed the best worm-killing activity, and it showed similar activity to PZQ. However, the worm-killing activity of compounds 24–29 was dramatically decreased. The amine-containing compound 24 had no activity. Among compounds with an aromatic ring, compound 30 with a benzoyl group showed slight worm-killing activity, while compound 31 with a thiophene-2-carbonyl group and compound 32 with a furan-2-carbonyl group had no activity. Interestingly, compound 33 with a chloroacetyl group showed good worm killing activity at 10 μM, similar to PZQ derivative 7. Among the compounds reported here, compounds 7, 22 and 33 with a chloroacetyl group showed better antischistosomal activity than PZQ. The evaluation of their cytotoxity is in progress. There have been many compounds with a chloroacetyl group under development [31,32,33]. For example, TNP470 [O-(chloroacetylcarbamoyl)fumagillol], which was derived from the natural product fumagillin, was in clinical trials for Kaposi’s sarcoma [34], as well as renal, brain, breast, cervical and prostate cancers [35].
3. Experimental
3.1. Chemistry
All chemicals were reagent grade and used as purchased. All reactions were performed under an inert atmosphere of dry argon or nitrogen using distilled dry solvent. 1H-NMR spectra (400 MHz) were recorded on a Bruker AVⅢ 400 MHz spectrometer. The chemical shifts were reported in (ppm) using the 7.26 signal of CDCl3 (1H-NMR) as internal standards. Mass spectra (MS) were obtained on a Waters Micromass Platform LCZ Mass Spectrometer.
3.1.1. Procedure for the Preparation of Compound 1
A mixture of PZQ (10 g, 32 mmol) and 2 N HCl (50 mL) was refluxed overnight. After PZQ was consumed, the reaction solution was cooled to room temperature, neutralized with NaHCO3 (aq.) and then extracted with DCM/MeOH (V/V 10/1, 100 mL × 3). The organic layer was washed with water and brine, dried with anhydrous Na2SO4 and concentrated to get the crude product. The crude product was washed with petroleum ether/EtOAc (V/V 10/1) to yield compound 1 (4 g, 62%) as a yellow solid. 1H-NMR (CDCl3) δ: 2.73–3.03 (m, 4H), 3.48–3.77 (m, 3H), 4.79–4.89 (m, 2H), 7.13–7.27 (m, 4H); MS (ESI): m/z calcd for C12H15N2O [M+H]+: 203.1, found: 203.4.
3.1.2. Procedure for the Preparation of Compound 8
A stirred solution of compound 1 (500 mg, 2.5 mmol) in DCM (20 mL) was added to trifluoroacetic anhydride (275 μL, 2 mmol) and triethylamine (275 μL, 2 mmol). After the addition, the mixture was stirred at room temperature overnight. The reaction residue was poured into water, and extracted with EtOAc (50 mL × 3). The organic phases were then processed in the usual way and chromatographed (1:1 petroleum ether/EtOAc) to afford compound 8 (450 mg, 62%) as a white solid. 1H-NMR (CDCl3) δ:2.96 (m), 3.38 (dd, J = 10.8 Hz, 14.0 Hz) (total 4H), 4.00 (d, J = 18.4 Hz), 4.26 (d, J = 17.6 Hz) (total 1H), 4.61 (dd, J = 14.0Hz, 18.0 Hz, 1H), 4.84-5.10 (m, 3H), 7.16–7.34 (m, 4H); MS (ESI): m/z calcd. for [M+H]+: 299.1, found: 299.2.
3.1.3. General Procedure for the Preparation of Derivatives 2–7 and 9–12
A stirred solution of compound 1 (500 mg, 2.5 mmol) in DCM (50 mL) was added to cyclopropanecarbonyl chloride (420 μL, 3.7 mmol) at 0 °C slowly. After the addition, the mixture was stirred at room temperature overnight. The reaction residue was poured into NaHCO3 (aq.), extracted with DCM (250 mL), washed with water and brine/dried over Na2SO4. The organic phases were then processed in the usual way and chromatographed (1:1 petroleum ether/EtOAc) to afforded compound 2 (350 mg, 49%). 1H-NMR (CDCl3) δ: 0.89 (m, 2H), 1.04-1.15 (m, 2H), 1.73 (m, 1H), 2.79–3.02 (m, 4H), 4.23 (d, J = 17.2 Hz, 1H), 4.71 (d, J = 17.6 Hz, 1H), 4.84-4.87 (m, 2H), 5.15 (d, J = 12.8 Hz, 1H), 7.20–7.28 (m, 4H); MS (ESI): m/z calcd. for [M+H]+: 271.1, found: 271.2. The following compounds were similarly prepared.
2-(Cyclobutanecarbonyl)-2,3,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-4(11bH)-one (3). Yield = 40%; 1H-NMR (CDCl3) δ: 1.88-2.11 (m, 2H), 2.19–2.27 (m, 2H), 2.32–2.50 (m, 2H), 2.78–3.03 (m, 4H), 3.15–3.42 (m, 1H), 3.85 (d, J = 18.8 Hz), 3.99 (d, J = 17.2 Hz) (total 1H), 4.27 (d, J = 17.6 Hz, 1H), 4.74–4.91 (m, 2H), 5.14 (m, 1H), 7.17–7.33 (m, 4H); MS (ESI): m/z calcd. for [M+H]+: 285.2, found: 285.3.
2-(Cyclopentanecarbonyl)-2,3,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-4(11bH)-one (4). Yield = 43%; 1H-NMR (CDCl3) δ: 1.62–1.96 (m, 8H), 2.78–3.02 (m, 5H), 3.54–3.90 (m, 1H), 4.10 (d, J = 17.2 Hz), 4.51 (d, J = 17.2 Hz) (total 1H), 4.82–4.4.91 (m), 5.18 (dd, J = 2.8 Hz, 13.2 Hz)(total 3H), 7.16–7.30 (m, 4H); MS (ESI): m/z calcd. for [M+H]+: 299.2, found: 299.4 [M+H]+.
2-Acetyl-2,3,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-4(11bH)-one (5). Yield = 43%; 1H-NMR (CDCl3) δ: 2.19 (s), 2.25 (s) (total 3H), 2.79–3.01 (m), 3.29–3.35 (m) (total 4H), 3.92 (d, J = 18.4 Hz), 4.11 (d, J = 17.6 Hz) (total 1H), 4.31–4.47 (m), 4.38 (d, J = 17.6 Hz) (total 1H), 4.77–4.89 (m, 2H), 5.13–5.18 (m, 1H), 7.20–7.33 (m, 4H); MS (ESI): m/z calcd. for [M+H]+: 245.1, found: 245.1.
2-Isobutyryl-2,3,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-4(11bH)-one (6). Yield = 43%; 1H- NMR (CDCl3) δ: 1.10 (d, J = 6.8 Hz), 1.13 (d, J = 6.8Hz) (total 6H), 2.71–2.97 (m), 3.21 (m) (total 5H), 3.81 (d, J = 18.4 Hz), 4.01–4.09 (m) (total 1H), 4.41(d, J = 17.2 Hz, 1H), 4.76 (m, 2H), 5.11 (dd, J = 2.4 Hz, 12.8 Hz, 1H), 7.12–7.23 (m, 4H); MS (ESI): m/z calcd. for [M+H]+: 273.2, found: 273.0.
2-(2-Chloroacetyl)-2,3,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-4(11bH)-one (7). Yield = 44%; 1H-NMR (CDCl3) δ: 2.78–2.96 (m), 3.28–3.34 (m) (total 4H), 3.89 (d, J = 18.4 Hz), 4.19–4.26 (m) (total 1H), 4.19 (s), 4.15 (s) (total 2H), 4.42–4.46 (m, 1H), 4.77–4.88 (m, 2H), 5.02–5.08 (m, 1H), 7.19–7.28 (m, 4H); MS (ESI): m/z calcd. for [M+H]+: 279.1/281.1, found: 279.2/281.3.
2-Benzoyl-2,3,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-4(11bH)-one (9). Yield = 34%; 1H-NMR (CDCl3) δ: 2.77–3.10 (m, 4H), 4.06–4.11 (m, 1H), 4.25–4.39 (m, 1H), 4.82–4.99 (m, 2H), 5.25 (m, 1H), 7.19–7.49 (m, 9H); MS (ESI): m/z calcd for [M+H]+: 307.1, found: 307.2.
2-Isonicotinoyl-2,3,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-4(11bH)-one (10). Yield = 29%; 1H-NMR (CDCl3) δ: 2.80–3.30 (m, 4H), 4.10–4.27 (m, 2H), 4.81–5.24 (m, 3H), 6.75 (m), 7.23–7.39 (m) (total 6H), 8.79 (2H); MS (ESI): m/z calcd. for [M+H]+: 308.1, found: 308.4.
2-(Thiophene-2-carbonyl)-2,3,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-4(11bH)-one (11). Yield = 29%; 1H-NMR (CDCl3) δ: 2.80–2.85 (m, 1H), 2.90–3.04 (m, 2H), 3.12–3.18 (m, 1H), 4.27 (d, J = 17.6 Hz, 1H), 4.86 (m, 2H), 5.03 (dd, J = 4.0 Hz, 10.8 Hz, 1H), 5.11 (d, J = 11.2 Hz, 1H), 7.14 (dd, J = 3.6 Hz, 4.8 Hz, 1H), 7.22–7.30 (m, 4H), 7.46 (d, J = 3.6 Hz, 1H), 7.57 (d, J = 4.4 Hz, 1H); MS (ESI): m/z calcd. for [M+H]+: 313.1, found: 313.2.
2-(Furan-2-carbonyl)-2,3,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-4(11bH)-one (12). Yield = 50%; 1H-NMR (CDCl3) δ: 2.80–3.19 (m, 4H), 4.29 (m, 1H), 4.87 (d, J = 11.2 Hz, 1H), 5.01 (d, J = 15.2 Hz, 2H), 5.19 (d, J = 11.2 Hz, 1H), 6.56 (d, J = 3.2 Hz, 1H), 7.19 (d, J = 3.6 Hz, 1H), 7.22–7.31 (m, 4H), 7.58 (s, 1H); MS (ESI): m/z calcd. for [M+H]+: 297.1, found: 297.2.
3.1.4. General Procedure for the Preparation of Derivatives 13–15
To a solution of compound 1 (305 mg, 1.5 mmol) in methanol (10 mL), cyclohexanecarbaldehyde (186 μL, 1.5 mmol) was added at 0 °C, followed by acetic acid (170 μL, 3.0 mmol) addition. The mixture was thus maintained at 0 °C for 1 h. Then it was heated at 60 °C for 2 h. After cooled to 0 °C, NaBH4 (0.45 g, 12.0 mmol) was added by portions. The reaction mixture was stirred at 60–70 °C for 12 h, followed by evaporation to remove methanol. The residue was diluted with water (30 mL) and extracted with ethyl acetate (30 mL × 3). The organic phases were then processed in the usual way and chromatographed (2:1 petroleum ether/EtOAc) to afforded compound 13 (140 mg, 32%) as a white solid. 1H-NMR (CDCl3) δ: 0.89–0.95 (m, 4H), 1.59–1.83 (m, 6H), 2.23–2.35 (m, 3H), 2.75–2.99 (m, 4H), 3.46–3.60 (m, 3H), 4.80–4.90 (m, 2H), 7.16–7.28 (m, 4H); MS (ESI): m/z calcd. for [M+H]+: 299.2, found: 299.3. The following compounds were similarly prepared.
2-Benzyl-2,3,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-4(11bH)-one (14). Yield = 35%; 1H-NMR (CDCl3) δ: 2.37 (t, J = 10.8 Hz, 1H), 2.75 (d, J = 14.8 Hz, 1H), 2.86–2.97 (m, 3H), 3.49–3.59 (m, 2H), 3.64 (AB, J = 13.2 Hz, 2H), 4.78 (m, 1H), 4.87 (m, 1H), 6.97-7.36 (m, 9H); MS (ESI): m/z calcd. for [M+H]+: 293.2, found: 293.4.
2-(Thiophen-2-ylmethyl)-2,3,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-4(11bH)-one (15). Yield = 27%; 1H-NMR (CDCl3) δ: 2.37 (t, J = 10.4 Hz, 1H), 2.75 (d, J = 14.8 Hz, 1H), 2.89–3.02 (m, 3H), 3.49–3.63 (m, 2H), 3.86 (s, 2H), 4.79 (m, 1H), 4.89(m, 1H), 6.97–7.29 (m, 7H). MS (ESI): m/z calcd. for [M+H]+: 299.1, found: 299.0.
3.1.5. Procedure for the Preparation of Compound 16
Compound 1 (3.03 g, 15 mmol) was added to a solution of lithium aluminum hydride (1.14 g, 30 mmol) in tetrahydrofuran (63 mL) and the mixture was refluxed for 10 h After cooled to 0 °C, the reaction was quenched with aqueous sodium hydroxide (15%, 1.2 mL), and the precipitate was filtrated. The organic phases were then processed in the usual way and chromatographed (10:1 CH2Cl2/MeOH) to afforded compound 16 (1 g, 35%) as red oil. 1H-NMR (CDCl3) δ: 2.39–2.56 (m, 2H), 2.62–2.68 (m, 2H), 2.83–2.91 (m, 2H), 2.97 (dd, J = 2.8 Hz, 8.4 Hz, 2H), 3.07–3.15 (m, 1H), 3.25 (d, J = 10.4 Hz, 1H), 3.52 (dd, J = 2.8 Hz, 12.0 Hz, 1H), 4.58 (brs, 1H), 7.04–7.13 (m, 4H); MS (ESI): m/z calcd. for [M+H]+: 189.1, found:189.3.
3.1.6. General Procedure for the Preparation of Derivatives 17–22
To a stirred solution of compound 16 (500 mg, 2.7 mmol) in DCM (50 mL), cyclohexanecarbonyl chloride (532 μL, 4.0 mmol) was added at 0 °C. The mixture was stirred at room temperature overnight. The reaction was quenched with NaHCO3 (aq.), extracted with DCM (50 mL × 3). The organic phases were then processed in the usual way and chromatographed (1:1 petroleum ether/ EtOAc) to afforded compound 19 (350 mg, 43%) as white solid. 1H-NMR (CDCl3) δ:1.24–1.87 (10H), 2.50 (m, 1H), 2.65 (m, 2H), 2.78 (m, 1H), 2.90–3.13 (m, 3H), 3.28 (m, 2H), 3.44 (m, 1H), 3.92 (d, J = 12.4 Hz), 4.37 (d, J =12.8 Hz) (total 1H), 4.65 (d, J = 13.2 Hz), 5.19 (d, J = 12.4 Hz) (total 1H), 7.13–7.33 (4H); MS (ESI): m/z calcd. for [M+H]+: 299.2, found: 299.1. The following compounds were similarly prepared.
Cyclobutyl(3,4,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-2(11bH)-yl)methanone (17). Yield = 35%; 1H-NMR (CDCl3) δ: 1.92–2.01 (m, 2H), 2.15–2.25 (m, 2H), 2.33–2.50 (m, 3H), 2.54–2.64 (m, 2H), 2.75 (m, 1H), 2.87–3.04 (m, 3H), 3.15–3.46 (m, 3H), 3.68 (d, J = 12.8 Hz), 4.11-4.18 (m) (total 1H), 4.60 (d, J = 12.0 Hz), 5.16 (d, J = 12.4 Hz) (total 1H), 7.14–7.33 (m, 4H); MS (ESI): m/z calcd. for [M+H]+: 271.2, found: 271.3.
Cyclopentyl(3,4,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-2(11bH)-yl)methanone (18). Yield = 32%; 1H-NMR (CDCl3) δ: 1.58–1.98 (8H), 2.46 (m, 1H), 2.61 (m, 2H), 2.76 (d, J = 17.2 Hz, 1H), 2.91–3.10 (m, 3H), 3.23 (m, 2H), 3.40 (m, 1H), 3.97 (d, J = 12.4 Hz), 4.45 (d, J = 12.8 Hz) (total 1H), 4.66 (d, J = 12.8 Hz), 5.21 (d, J = 12.8 Hz) (total 1H), 7.13–7.33 (m, 4H); MS (ESI): m/z calcd. for [M+H]+: 285.2, found: 285.1.
Phenyl(3,4,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-2(11bH)-yl)methanone (20). Yield = 69%; 1H-NMR (CDCl3) δ: 2.40–2.60 (m, 2H), 2.66–2.71 (m, 1H), 2.79 (m, 1H), 2.97 (m, 2H), 3.13–3.33 (m, 3H), 3.67 (m), 4.18 (m) (total 1H), 4.64 (m), 5.21 (m) (total 1H), 6.58–7.38 (9H); MS (ESI): m/z calcd. for [M+H]+: 293.2, found: 293.2.
(3,4,6,7-Tetrahydro-1H-pyrazino[2,1-a]isoquinolin-2(11bH)-yl)(thiophen-2-yl)methanone (21). Yield = 44%; 1H-NMR (CDCl3) δ: 2.61 (m, 2H), 2.77 (m, 1H), 3.00–3.08 (m, 3H), 3.18–3.43 (m, 3H), 4.44 (brs, 1H), 5.04 (brs, 1H), 7.12 (t, J = 4.4 Hz, 1H), 7.15–7.21 (m, 4H), 7.40 (d, J = 2.8 Hz, 1H), 7.51 (d, J = 5.2 Hz, 1H); MS (ESI): m/z calcd. for [M+H]+: 299.1, found: 299.3.
2-Chloro-1-(3,4,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-2(11bH)-yl)ethanone (22). Yield = 36%; 1H-NMR (CDCl3) δ: 2.52–2.78 (m, 3H), 2.95–3.51 (m, 6H), 3.86 (m), 4.34 (m) (total 1H), 4.15 (AB, J = 12.0 Hz), 4.22 (AB, J = 12.0 Hz) (total 2H), 4.57 (m, ), 5.10 (m) (total 1H), 7.15–7.31 (m, 4H); MS (ESI): m/z calcd. for [M+H]+: 265.1/267.1, found: 265.2/267.1.
3.1.7. The Procedure for the Preparation of Compound 24
A stirred solution of compound 23 (1.0 g, 3.1 mmol) in HCl (2 N, 10 mL) was heated to 110 °C overnight. The reaction mixture was neutralized with solid NaHCO3, and extracted with DCM/MeOH (V/V 5/1, 30 mL × 3). The organic phases were then processed in the usual way and chromatographed (10:1 CH2Cl2/MeOH) to afforded compound 24 (500 mg, 78%). 1H-NMR (CDCl3) δ: 2.80–2.88 (m, 3H), 2.95–3.01 (m, 1H), 3.54 (d, J = 17.6 Hz, 1H), 3.66 (m, 1H), 3.67 (d, J = 17.2 Hz, 1H), 4.71 (m, 1H), 5.12 (m, 1H), 6.80 (d, J = 5.2 Hz, 1H), 7.19 (d, J = 5.2 Hz, 1H); MS (ESI): m/z calcd. for [M+H]+: 209.1, found: 209.2.
3.1.8. General Procedure for the Preparation of Compound 25–33
To a stirred solution of compound 24 (208 mg, 1 mmol) in DCM (10 mL), cyclobutanecarbonyl chloride (136 μL, 1.2 mmol) was added and stirred at room temperature overnight. The reaction was quenched with NaHCO3 (aq.), extracted with DCM. The organic phases were then processed in the usual way and chromatographed (1:1 petroleum ether/EtOAc) to afforded compound 26 (150 mg, 52%). 1H-NMR (CDCl3) δ: 1.92–2.37 (m, 6H), 2.75 (t, J = 11.2 Hz, 1H), 2.84–2.96 (m, 3H), 3.27 (m, 1H), 3.75 (d, J = 18.8 Hz) 3.94 (d, J = 17.6 Hz) (total 1H), 4.18 (d, J = 12.8 Hz), 4.25 (d, J = 17.6 Hz) (total 1H), 4.71 (d, J = 10.4 Hz, 1H), 4.91 (d, J = 19.2 Hz), 5.04 (dd, J = 3.2 Hz, 12.8 Hz) (total 1H), 5.10 (dd, J = 2.8 Hz, 13.2 Hz, 1H), 6.81 (d, J = 5.2 Hz), 6.91 (d, J = 5.2 Hz) (total 1H), 7.19 (d, J = 4.8 Hz), 7.23 (d, J = 4.8 Hz) (total 1H); MS (ESI): m/z calcd. for [M+H]+: 291.1, found: 291.2.
2-(Cyclopentanecarbonyl)-2,3,6,7-tetrahydro-1H-thieno[3',2':3,4]pyrido[1,2-a]pyrazin-4(10bH)-one (25). Yield = 52%; 1H-NMR (CDCl3) δ: 1.61–1.91 (m, 8H), 2.74 (t, J = 11.2 Hz, 1H), 2.86–2.97 (m, 4H), 3.76 (d, J = 18.4 Hz), 4.04 (d, J = 17.6 Hz) (total 1H), 4.50 (d, J = 17.6 Hz, 1H), 4.71 (d, J = 9.2 Hz, 1H), 4.93 (d, J = 19.2 Hz), 5.04 (dd, J = 5.2 Hz, 12.8 Hz) (total 1H), 5.15 (dd, J = 2.4 Hz, 13.2 Hz, 1H), 6.90 (d, J = 4.8 Hz, 1H), 7.19 (d, J = 5.2 Hz, 1H); MS (ESI): m/z calcd. for [M+H]+: 305.1, found: 305.1.
2-(Cyclopropanecarbonyl)-2,3,6,7-tetrahydro-1H-thieno[3',2':3,4]pyrido[1,2-a]pyrazin-4(10bH)-one (27). Yield = 57%; 1H-NMR (CDCl3) δ: 1.01 (m, 2H), 1.12 (m, 2H), 1.70 (m, 1H), 2.72–2.98 (m, 4H), 4.18 (d, J = 17.6 Hz, 1H), 4.72 (m, 2H), 5.04–5.14 (m, 2H), 6.90 (d, J = 4.8 Hz, 1H), 7.19 (d, J = 4.8 Hz, 1H); MS (ESI): m/z calcd. for [M+H]+: 277.1, found: 276.9.
2-Isobutyryl-2,3,6,7-tetrahydro-1H-thieno[3',2':3,4]pyrido[1,2-a]pyrazin-4(10bH)-one (28). Yield = 46%; 1H-NMR (CDCl3) δ: 1.15 (d, J = 6.8 Hz), 1.17 (d, J = 6.8 Hz), (total 6H), 2.70–2.80 (m, 1H), 2.86–2.98 (m), 3.16–3.25 (m) (4H), 3.77 (d, J = 18.4 Hz), 4.06 (d, J = 17.6 Hz) (total 1H), 4.34–4.40 (m), 4.48 (d, J = 17.6 Hz) (total 1H), 4.71–4.78 (m, 1H), 4.89–5.06 (m, 1H), 5.15 (d, J = 13.2 Hz, 1H), 6.90 (d, J = 5.2 Hz, 1H), 7.20 (d, J = 5.2 Hz, 1H); MS (ESI): m/z calcd. for [M+H]+: 279.1, found: 279.2.
2-Pivaloyl-2,3,6,7-tetrahydro-1H-thieno[3',2':3,4]pyrido[1,2-a]pyrazin-4(10bH)-one (29). Yield = 76%; 1H-NMR (CDCl3) δ: 1.34 (s, 9H), 2.81–2.97 (m, 4H), 3.96 (d, J = 18.0 Hz, 1H), 4.76 (m, 1H), 4.82 (d, J = 17.6 Hz, 1H), 5.04(dd, J = 4.8 Hz, 12.4Hz, 2H), 6.87 (d, J = 5.2 Hz, 1H), 7.21 (d, J = 5.2 Hz, 1H); MS (ESI): m/z calcd. for [M+H]+: 293.1, found: 293.1[M+H]+.
2-Benzoyl-2,3,6,7-tetrahydro-1H-thieno[3',2':3,4]pyrido[1,2-a]pyrazin-4(10bH)-one (30). Yield = 51%; 1H-NMR (CDCl3) δ: 2.86–3.02 (m, 4H), 4.08 (m, 1H), 4.36–4.38 (m, 1H), 4.88 (m, 1H), 5.01 (m, 1H), 5.19–5.25 (m, 1H), 6.98–7.62 (m, 6H), 8.10 (d, J = 7.6 Hz, 1H); MS (ESI): m/z calcd. for [M+H]+: 313.1, found: 313.2.
2-(Thiophene-2-carbonyl)-2,3,6,7-tetrahydro-1H-thieno[3',2':3,4]pyrido[1,2-a]pyrazin-4(10bH)-one (31). Yield = 40%; 1H-NMR (CDCl3) δ: 2.87–2.97 (m, 4H), 4.20–4.24 (m, 1H), 4.85–5.10 (m, 4H), 6.90–7.88 (m, 5H); MS (ESI): m/z calcd. for [M+H]+: 319.1, found: 319.0.
2-(Furan-2-carbonyl)-2,3,6,7-tetrahydro-1H-thieno[3',2':3,4]pyrido[1,2-a]pyrazin-4(10bH)-one (32). Yield = 46%; 1H-NMR (CDCl3) δ: 2.85–3.03 (m, 4H), 4.21–4.25 (m, 1H), 4.88 (m, 1H), 5.01–5.07 (m, 2H), 5.15 (m, 1H), 6.55 (d, J = 2.8 Hz, 1H), 6.94 (brs, 1H), 7.17 (d, J = 3.2 Hz, 1H), 7.22 (d, J = 5.2 Hz, 1H), 7.56 (s, 1H); MS (ESI): m/z calcd. for [M+H]+: 303.1, found: 303.2 [M+H]+.
2-(2-Chloroacetyl)-2,3,6,7-tetrahydro-1H-thieno[3',2':3,4]pyrido[1,2-a]pyrazin-4(10bH)-one (33). Yield = 76%; 1H-NMR (CDCl3) δ: 2.82–2.98 (m), 3.24–3.29 (m) (total 4H), 3.81 (d, J = 18.8 Hz), 4.23 (d, J = 12.4 Hz) (total 1H), 4.13 (s), 4.17 (s) (total 2H), 4.38–4.46 (m, 1H), 4.75–5.06 (m, 3H), 6.87 (d, J = 5.2 Hz), 6.90 (d, J = 5.2 Hz) (total 1H), 7.22 (d, J = 5.2 Hz), 7.24 (d, J = 5.2 Hz) (total 1H); MS (ESI): m/z calcd. for [M+H]+: 285.0/287.0, found: 285.1/287.1 [M+H]+.
3.2. Killing Activity of Compounds 1–33 on S. Japonicum Adult Worms in Vitro [36]
Stock solutions of compounds 1–33 and praziquantel were prepared by dissolving 1 mg of the drugs in 0.4 ml dimethyl sulfoxide (DMSO) and adding 0.6 mL RPMI 1640 medium. S. japonicum worms obtained from mice (C57BL/6, female, 22–24 g, each infected with 50 cercariae) were washed in RPMI 1640 medium, kept at pH 7.5 with HEPES 20 mM and supplemented with penicillin (100 UI/mL), streptomycin (100 mg/mL) and 10% fetal bovine serum (FBS, Gibco) . After washing, 8–15 adult worms were transferred to each well of a 24-well culture plate containing 2 mL of the same medium. The worms were cultured for 30 to 60 min at 37 °C in a humid atmosphere containing 5% CO2, and then different concentrations of compounds 1–33 (10, 25, 50, 100 µM) diluted with RPMI 1640 medium were added. Control worms were treated with equal volumes of RPMI 1640 or DMSO, and worms treated with 10, 25, 50, 100 µM praziquantel were also observed. The worm mobility, tegumental alterations and parasite survival were monitored under an inverted microscope (Leica, Wetzlar, Germany) at 24, 48 and 72 h. Parasite death was defined as having no motor activity during 2 min of continuous observation as well as morphological and tegumental alterations. The tests were repeated two times when compouds showed worm killing activity below the concentration of 100 μM.
4. Conclusions
Given the widespread morbidity and mortality derived from schistosomiasis and the possible emergence of PZQ-resistant parasites in the near future, it is undoubtedly urgent to develop new chemotherapy for the disease. In this work, we first reinvestigated the SAR of PZQ analogues at positions 2 and 4, and we identified that the introduction of a chloroacetyl group at position 2 led to compound 7 with higher worm-killing activity against adult S. japonicum than PZQ in vitro. Second, we extended the SAR studies by replacing the aromatic moiety with a thiophene ring, which displayed similar activity as PZQ derivatives, further confirming the respective SAR. Notably, compound 33 with the chloroacetyl group killed 100% of the worms at concentrations as low as 10 μM, which represents a more potent compound than PZQ. The study of these structurally diverse compounds will likely provide meaningful information for the design of new PZQ analogues. After the initial in vitro screening, in vivo activity and toxicity will have to be tested. Design and synthesis of more potent antischistosomal agents are currently undergoing in our laboratory.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 30972581), the Natural Science Foundation of Jiangsu Province (No. BK2008110), the Health Promotion Project Foundation of Jiangsu Province (No. ZX201108), the National S & T Major Program (No. 2012ZX 10004-220), and the Fundamental Research Funds for the Central Universities (JUSRP1040). We also thank Sergio C. Chai at St. Jude Children’s Research Hospital for his suggestions.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 1–33 are available from the authors.
References
- 1.Danso-Appiah A., Olliaro P.L., Donegan S., Sinclair D., Utzinger J. Drugs for treating Schistosoma mansoni infection. Cochrane. Database. Syst. Rev. 2013;2:CD000528. doi: 10.1002/14651858.CD000528.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gryseels B., Polman K., Clerinx J., Kestens L. Human schistosomiasis. Lancet. 2006;368:1106–1118. doi: 10.1016/S0140-6736(06)69440-3. [DOI] [PubMed] [Google Scholar]
- 3.Zheng H., Zhang L.J., Zhu R., Xu J., Li S.Z., Guo J.G., Xiao N., Zhou X.N. Schistosomiasis situation in People's Republic of China in 2011. Chin. J. Schisto. Control. 2012;24:621–626. [PubMed] [Google Scholar]
- 4.Caffrey C.R., Secor W.E. Schistosomiasis: From drug deployment to drug development. Curr. Opin. Infect. Dis. 2011;24:410–417. doi: 10.1097/QCO.0b013e328349156f. [DOI] [PubMed] [Google Scholar]
- 5.Stelma F.F., Talla I., Sow S., Kongs A., Niang M., Polman K., Deelder A.M., Gryseels B. Efficacy and side effects of praziquantel in an epidemic focus of Schistosoma mansoni. Am. J. Trop. Med. Hyg. 1995;53:167–170. doi: 10.4269/ajtmh.1995.53.167. [DOI] [PubMed] [Google Scholar]
- 6.Wang W., Wang L., Liang Y.S. Susceptibility or resistance of praziquantel in human schistosomiasis: a Review. Parasitol. Res. 2012;111:1871–1877. doi: 10.1007/s00436-012-3151-z. [DOI] [PubMed] [Google Scholar]
- 7.Caixeta S.C., Magalhaes L.G., de Melo N.I., Wakabayashi K.A., Aguiar Gde P., Aguiar Dde P., Mantovani A.L., Alves J.M., Oliveira P.F., Tavares D.C., et al. Chemical composition and in vitro schistosomicidal activity of the essential oil of Plectranthus neochilus grown in Southeast Brazil. Chem. Biodivers. 2011;8:2149–2157. doi: 10.1002/cbdv.201100167. [DOI] [PubMed] [Google Scholar]
- 8.Moraes J., Almeida A.A., Brito M.R., Marques T.H., Lima T.C., Sousa D.P., Nakano E., Mendonca R.Z., Freitas R.M. Anthelmintic activity of the natural compound (+)-limonene epoxide against Schistosoma mansoni. Planta Med. 2013;79:253–258. doi: 10.1055/s-0032-1328173. [DOI] [PubMed] [Google Scholar]
- 9.Porto T.S., da Silva Filho A.A., Magalhaes L.G., dos Santos R.A., Furtado N.A., Arakawa N.S., Said S., de Oliveira D.C., Gregorio L.E., Rodrigues V., et al. Fungal transformation and schistosomicidal effects of pimaradienoic acid. Chem. Biodivers. 2012;9:1465–1474. doi: 10.1002/cbdv.201100336. [DOI] [PubMed] [Google Scholar]
- 10.Ndjonka D., Rapado L.N., Silber A.M., Liebau E., Wrenger C. Natural Products as a Source for Treating Neglected Parasitic Diseases. Int. J. Mol. Sci. 2013;14:3395–3439. doi: 10.3390/ijms14023395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jao S.C., Chen J., Yang K., Li W.S. Design of potent inhibitors for Schistosoma japonica glutathione S-transferase. Bioorg. Med. Chem. 2006;14:304–318. doi: 10.1016/j.bmc.2005.07.077. [DOI] [PubMed] [Google Scholar]
- 12.Rai G., Sayed A.A., Lea W.A., Luecke H.F., Chakrapani H., Prast-Nielsen S., Jadhav A., Leister W., Shen M., Inglese J., et al. Structure Mechanism Insights and the Role of Nitric Oxide Donation Guide the Development of Oxadiazole-2-Oxides as Therapeutic Agents against Schistosomiasis. J. Med. Chem. 2009;52:6474–6483. doi: 10.1021/jm901021k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rai G., Thomas C.J., Leister W., Maloney D.J. Synthesis of oxadiazole-2-oxide analogues as potential antischistosomal agents. Tetrahedron Lett. 2009;50:1710–1713. doi: 10.1016/j.tetlet.2009.01.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sayed A.A., Simeonov A., Thomas C.J., Inglese J., Austin C.P., Williams D.L. Identification of oxadiazoles as new drug leads for the control of schistosomiasis. J. Nat. Med. 2008;14:407–412. doi: 10.1038/nm1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu J., Dyer D., Wang J., Wang S., Du X., Xu B., Zhang H., Wang X., Hu W. 3-Oxoacyl-ACP Reductase from Schistosoma japonicum: Integrated In Silico-In Vitro Strategy for Discovering Antischistosomal Lead Compounds. PLoS One. 2013;8:e64984. doi: 10.1371/journal.pone.0064984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Domling A., Khoury K. Praziquantel and schistosomiasis. Chemmedchem. 2010;5:1420–1434. doi: 10.1002/cmdc.201000202. [DOI] [PubMed] [Google Scholar]
- 17.Woelfle M., Seerden J.P., de Gooijer J., Pouwer K., Olliaro P., Todd M.H. Resolution of praziquantel. PLoS. Negl. Trop. Dis. 2011;5:e1260. doi: 10.1371/journal.pntd.0001260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyer T., Sekljic H., Fuchs S., Bothe H., Schollmeyer D., Miculka C. Taste, a new incentive to switch to (R)-praziquantel in schistosomiasis treatment. PLoS Negl. Trop. Dis. 2009;3:e357. doi: 10.1371/journal.pntd.0000357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao H., Liu H., Domling A. Efficient multicomponent reaction synthesis of the schistosomiasis drug praziquantel. Chemistry. 2010;16:12296–12298. doi: 10.1002/chem.201002046. [DOI] [PubMed] [Google Scholar]
- 20.Liu H., William S., Herdtweck E., Botros S., Domling A. MCR synthesis of praziquantel derivatives. Chem. Biol. Drug. Des. 2012;79:470–477. doi: 10.1111/j.1747-0285.2011.01288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andrews P., Thomas H., Pohlke R., Seubert J. Praziquantel. Med. Res. Rev. 1983;3:147–200. doi: 10.1002/med.2610030204. [DOI] [PubMed] [Google Scholar]
- 22.Patra M., Ingram K., Pierroz V., Ferrari S., Spingler B., Keiser J., Gasser G. Ferrocenyl derivatives of the anthelmintic praziquantel: Design, synthesis, and biological evaluation. J. Med. Chem. 2012;55:8790–8798. doi: 10.1021/jm301077m. [DOI] [PubMed] [Google Scholar]
- 23.Dong Y.X., Chollet J., Vargas M., Mansour N.R., Bickle Q., Alnouti Y., Huang J.G., Keiser J., Vennerstrom J.L. Praziquantel analogs with activity against juvenile Schistosoma mansoni. Bioorg. Med. Chem. Lett. 2010;20:2481–2484. doi: 10.1016/j.bmcl.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 24.Patra M., Ingram K., Pierroz V., Ferrari S., Spingler B., Gasser R.B., Keiser J., Gasser G. [(eta(6)-Praziquantel)Cr(CO)3] derivatives with remarkable in vitro anti-schistosomal activity. Chemistry. 2013;19:2232–2235. doi: 10.1002/chem.201204291. [DOI] [PubMed] [Google Scholar]
- 25.Ronketti F., Ramana A.V., Chao-Ming X., Pica-Mattoccia L., Cioli D., Todd M.H. Praziquantel derivatives I: Modification of the aromatic ring. Bioorg. Med. Chem. Lett. 2007;17:4154–4157. doi: 10.1016/j.bmcl.2007.05.063. [DOI] [PubMed] [Google Scholar]
- 26.Duan W.W., Qiu S.J., Zhao Y., Sun H., Qiao C., Xia C.M. Praziquantel derivatives exhibit activity against both juvenile and adult Schistosoma japonicum. Bioorg. Med. Chem. Lett. 2012;22:1587–1590. doi: 10.1016/j.bmcl.2011.12.133. [DOI] [PubMed] [Google Scholar]
- 27.Wang Z.X., Chen J.L., Qiao C. Praziquantel Derivatives with Anti-Schistosomal Activity: Aromatic ring Modification. Chem. Biol. Drug. Des. 2013;82:216–225. doi: 10.1111/cbdd.12153. [DOI] [PubMed] [Google Scholar]
- 28.Sadhu P.S., Kumar S.N., Chandrasekharam M., Pica-Mattoccia L., Cioli D., Rao V.J. Synthesis of new praziquantel analogues: Potential candidates for the treatment of schistosomiasis. Bioorg. Med. Chem. Lett. 2012;22:1103–1106. doi: 10.1016/j.bmcl.2011.11.108. [DOI] [PubMed] [Google Scholar]
- 29.Frehel D., Maffrand J.P. Synthesis of the new tricyclic system thieno[3',2':3,4]pyrido[1,2-a]pyrazin-4-one. Heterocycles. 1984;22:143–149. doi: 10.3987/R-1984-01-0143. [DOI] [Google Scholar]
- 30.Todd M.H., Ndubaku C., Bartlett P.A. Amino acid derived heterocycles: Lewis acid catalyzed and radical cyclizations from peptide acetals. J. Org. Chem. 2002;67:3985–3988. doi: 10.1021/jo010990m. [DOI] [PubMed] [Google Scholar]
- 31.Bissinger E.M., Heinke R., Spannhoff A., Eberlin A., Metzger E., Cura V., Hassenboehler P., Cavarelli J., Schule R., Bedford M.T., et al. Acyl derivatives of p-aminosulfonamides and dapsone as new inhibitors of the arginine methyltransferase hPRMT1. Bioorg. Med. Chem. 2011;19:3717–3731. doi: 10.1016/j.bmc.2011.02.032. [DOI] [PubMed] [Google Scholar]
- 32.Kim D.J., Reddy K., Kim M.O., Li Y., Nadas J., Cho Y.Y., Kim J.E., Shim J.H., Song N.R., Carper A., et al. (3-Chloroacetyl)-indole, a Novel allosteric AKT inhibitor, Suppresses colon cancer growth in vitro and in vivo. Cancer Prev. Res. 2011;4:1842–1851. doi: 10.1158/1940-6207.CAPR-11-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nam K.N., Koketsu M., Lee E.H. 5-Chloroacetyl-2-amino-1,3-selenazoles attenuate microglial inflammatory responses through NF-kappa B inhibition. Eur. J. Pharmacol. 2008;589:53–57. doi: 10.1016/j.ejphar.2008.03.034. [DOI] [PubMed] [Google Scholar]
- 34.Dezube B.J., Von Roenn J.H., Holden-Wiltse J., Cheung T.W., Remick S.C., Cooley T.P., Moore J., Sommadossi J.P., Shriver S.L., Suckow C.W., et al. Fumagillin analog in the treatment of Kaposi’s sarcoma: A phase I AIDS Clinical Trial Group Study. J. Clin. Oncol. 1998;16:1444–1449. doi: 10.1200/JCO.1998.16.4.1444. [DOI] [PubMed] [Google Scholar]
- 35.Kruger E.A., Figg W.D. TNP-470: An Angiogenesis inhibitor in clinical development for cancer. Expert Opin. Invest. Drug. 2000;9:1383–1396. doi: 10.1517/13543784.9.6.1383. [DOI] [PubMed] [Google Scholar]
- 36.Song L.J., Li J.H., Xie S.Y., Qian C.Y., Wang J., Zhang W., Yin X.R., Hua Z.C., Yu C.X. Thioredoxin Glutathione Reductase as a Novel Drug Target: Evidence from Schistosoma japonicum. PLoS One. 2012;7:e31456. doi: 10.1371/journal.pone.0031456. [DOI] [PMC free article] [PubMed] [Google Scholar]