

Abstract
Naturally occurring anthraquinones, damnacanthal (1) and nordamnacanthal (2) were synthesized with modified reaction steps and investigated for their cytotoxicity against the MCF-7 and K-562 cancer cell lines, respectively. Intermediate analogues 2-bromomethyl-1,3-dimethoxyanthraquinone (5, IC50 = 5.70 ± 0.21 and 8.50 ± 1.18 μg/mL), 2-hydroxymethyl-1,3-dimethoxyanthraquinone (6, IC50 = 12.10 ± 0.14 and 14.00 ± 2.13), 2-formyl-1,3-dimethoxyantharquinone (7, IC50 = 13.10 ± 1.02 and 14.80 ± 0.74), 1,3-dimethoxy-2-methylanthraquinone (4, IC50 = 9.40 ± 3.51 and 28.40 ± 2.33), and 1,3-dihydroxy-2-methylanthraquinone (3, IC50 = 25.60 ± 0.42 and 28.40 ± 0.79) also exhibited moderate cytotoxicity against MCF-7 and K-562 cancer cell lines, respectively. Other structurally related compounds like 1,3-dihydroxyanthraquinone (13a, IC50 = 19.70 ± 0.35 and 14.50 ± 1.28), 1,3-dimethoxyanthraquinone (13b, IC50 = 6.50 ± 0.66 and 5.90 ± 0.95) were also showed good cytotoxicity. The target compound damnacanthal (1) was found to be the most cytotoxic against the MCF-7 and K-562 cancer cell lines, with IC50 values of 3.80 ± 0.57 and 5.50 ± 1.26, respectively. The structures of all compounds were elucidated with the help of detailed spectroscopic techniques.
Keywords: Anthraquinone, damnacanthal, nordamnacanthal, MCF-7, K-562, cytotoxic effects
1. Introduction
Anthraquinone compounds, especially anthracyclines, have long been used as effective anticancer drugs. Depending on their chemical structure, anthraquinone drugs can kill tumor cells by diverse mechanisms, involving different initial intracellular targets that normally contribute to drug-induced toxicity [1,2,3]. Anthraquinones are known as “multipotent antioxidants”, as they are molecules that besides antioxidant activity possess additional pharmacological activities such as inhibition of platelet-aggregation or display antineoplastic and anticancer activities [4,5]. Many anthraquinones also display various biological activities such as antimicrobial, antifungal, hypotensive, analgesic, antimalarial [6,7,8,9,10,11], antileukemic, mutagenicity and anti-inflammatory properties [12,13,14]. Natural anthraquinones from Damnacanthus subspinosus and Morinda parvifolia have long been used in traditional medicine for the treatment of cancer [15]. The discovery of new compounds with antitumor activity has become one of the most important challenges in medicinal chemistry. The detail study on anthraquinones has revealed that a range of DNA-recognizing molecules that act as antitumor agents, including groove binders, alkylating and intercalator compounds. DNA intercalators have attracted particular attention because of their antitumor activity. For example, a number of acridine and anthracycline compounds are excellent DNA intercalators that are now on the market as chemotherapeutic agents [15,16]. Substituted anthraquinones such as rubiadin, subspinosin and morindaparvin are widely distributed in nature and are known to display various pharmacological activities [17,18,19,20,21]. Previously, we have reported the antitumor and anti-oxidant activities of anthraquinones isolated from Morinda elliptica [22].Recently, we have also reported the cytotoxic and immunomodulatory effects of damnacanthal and nordamnacanthal against different cell lines [23,24]. Damnacanthal and nordamnacanthal were originally isolated from Damnacanthus major [25]. Previous studies on the synthesis of damnacanthal and nordmnacanthal were reported by Hirose, Roberts and Saha [26,27,28]. The current study describes the total synthesis of damnacanthal (1) and nordamnacanthal (2) with modified reaction steps, their cytotoxic activities of against MCF-7 and K-562 cancer cell lines and their structure activity relationships (SARs).
2. Results and Discussion
Anthraquinone skeletons are generally synthesized by Friedel-Crafts acylation condensation between phthalic anhydride and benzene derivatives [29]. 1,3-Dihydroxy-2-methylanthraquinone (3) was used as the common precursor for the synthesis of damnacanthal (1) and nordamnacanthal (2). This was synthesized by mixing of phthalic anhydride and 1,3-dihydroxy-2-methylbenzene in a molten mixture of AlCl3/NaCl [29,30].
The synthesis of nordamnacanthal (2) was accomplished by first acetylating the precursor compound 3 with acetic anhydride and potassium carbonate to afford the monoacetylated intermediate 8. Upon methylation of compound 8 with K2CO3/(CH3)2SO4 in dry acetone to afforded 1,3-dimethoxy-2-methylathraquinone (4), which was then brominated with Wohl-Ziegler’s reagent (N-bromosuccinimide) in dry CCl4 to yield 2-bromomethyl-1,3-dimethoxyathraquinone (5) [25,31,32] and structure was confirmed by single X-ray diffraction (Figure 1). It is noteworthy that the use of a catalytic amount of benzoyl peroxide in this reaction gave 2-dibromomethyl-1,3-dimethoxyanthraquinone [26]. Compound 5 was hydrolyzed by refluxing it in acetic acid-water (8:2) to give the desired 2-hydroxymethyl-1,3-dimethoxyanthraqunone (6) in quantitative yield [26,31]. Compound 6, which contains a hydroxymethyl moiety, was converted into the corresponding aldehyde 7 in 92.3% yield using a mild oxidizing agent [pyridinium chlorochromate (PCC) in dry CH2Cl2 at 20–25 °C]. The use of an excess amount (1.5 equiv.) of PCC also gave the undesired 1-hydoxy-3-methoxyanthraquinone-2-carboxylic acid. Upon treating compound 7 with AlCl3/CH2Cl2, nordamnacanthal (2) was obtained in 28% yield. The detailed reaction conditions are shown in Scheme 1.
Figure 1.

The ORTEP diagram of 2-bromomethyl-1,3-dimethoxy-9,10-athraquinone (5).
Scheme 1.
Reactions pathway for synthesis of nordamnacanthal (2).

Reagents and conditions: (a) anhydrous AlCl3, NaCl, 165–175 °C, 1 h; (b) Anhydrous K2CO3 (CH3)2SO4, dry acetone, refluxed, 22 h; (c) NBS/CCl4 refluxed for 30 h; (d) 80% acetic acid refluxed 24 h; (e) PCC, CH2Cl2 stir. 2–4 h. rt; (f) AlCl3 /CH2Cl2, HCl.
The synthesis of damnacanthal (1) was accomplished by first acetylated the precursor compound 3 with (CH3)2SO4 and potassium carbonate to afforded monoacetylated derivative 8. Upon methylation of compound 8 with K2CO3/CH3I compound 9 was obtained, which then brominated with NBS to yield 1-methoxy-3-aectoxy-2-bromomethyl-1-methoxyathraquinone (10) [25,32]. The bromo derivative 10 was converted into ethoxymethyl derivative 11 by dissolving it in a mixture of aq. NaOH/ethanol and followed by reflux in acidic media. Compound 11 was acetylated using acetic anhydride and K2CO3 to afforded 12, which on oxidation with PCC in CH2Cl2 to give damnacanthal (1) in good yield. Thus the synthesis of damnacanthal (1) was achieved through modified steps as shown in Scheme 2.
Scheme 2.
Reactions pathway for synthesis of damnacanthal (1).

Reagents and conditions: (a) anhydrous K2CO3 /Ac2O, acetone; (b) anhydrous K2CO3/(CH3)2SO4 acetone, refluxed 22 h; (c) NBS/CCl4 refluxed 30 h; (d) 10% aq. NaOH/EtOH, aq. HCl; (e) Ac2O/ K2CO3 stirred for 24 h; (f) PCC, CH2Cl2 stir 2–4 h. rt.
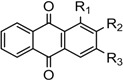
For the SARs additional anthraquinone analogous 13a–h were synthesized by Friedel-Crafts acylation of phthalic anhydride with resorcinol or catechol to yielded respective anthraquinones [29,30] as shown in Figure 2. All compounds showed significant in vitro cytotoxicity against two cancer cell lines, indicating that anthraquinone is an interesting class of compounds for cancer therapy. The compound damnacanthal (1) contains methoxy, formyl and hydroxyl groups at the 1, 2 and 3 positions that might be important for the cytotoxicity since the standard drug doxorubicin [33] also possesses an anthraquinone moiety. However, nordamnacanthal (2), 3, 8, 13a, 13c, 13f and 13h exhibited less cytotoxic effects on both the MCF-7 and K-562 cell lines, suggesting that a protected OH group at position 1 and 3 increase the solubility and might make it easy for the compounds to diffuse across the cellular membrane as investigated by a theoretical study of 188 drug-like compounds by MI-QSAR analysis [34,35], while for poorly soluble drugs dissolution could be the rate limiting step in the absorption process. The methylated compound 13b (IC50 = 24.25 ± 2.46 μM and 22.01 ± 3.54 μM) showed the stronger cytotoxicity than 13a (IC50 = 82.08 ± 1.46 and 60.42 ± 5.33 μM) and 13c (IC50 = 87.80 ± 2.01 and 64.96 ± 1.57μM). Other derivatives 13e (IC50 = 43.64 ± 2.12 and 46.61 ± 12.37) and 2-hydroxymethylanthraquinone (13d) (IC50 = 49.58 ± 12.61and 50.84±14.12) showed poor cytotoxicity as shown in Table 1. However, previously synthesized compounds 13f–h with mono or dihydroxyl groups at the 1 and 2 positions exhibited moderate cytotoxic activity [36]. Further derivatives as exemplified by compounds 13c–h indicated that this series of compounds only exhibited moderate activities not exceeding that of damnacanthal (1), suggesting that functional groups such as formyl at C-2, methoxy at C-1, hydroxyl groups at C-3 (damnacanthal) and bromomethyl at C-2 are important pharmacophores for the anticancer activity. However, the active compound 5 ((IC50 = 15.83 ± 0.58 and 23.61 ± 3.28 μM) might involve another mechanism like perfusion. The overall cytotoxic compounds (1, 5, 13b) showed more selectivity towards cancer cell lines because of their side chains and might fulfill the Lipinski rule-of-5 and drug-like properties.
Figure 2.

Structure of compounds 13a to 13h.
Table 1.
IC50 values of compounds 1 to 13h against MCF-7 and K-562 cell lines.
| Compound No. | IC50 value (μM) | ||
|---|---|---|---|
| Chemical Structure | MCF-7 | K562 | |
| 1 | 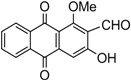 |
13.48 ± 2.02 | 19.50 ± 4.47 |
| 2 | 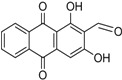 |
60.07 ± 6.46 | 32.46 ± 8.73 |
| 3 | 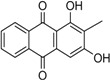 |
90.78 ± 1.49 | 100.71 ± 2.80 |
| 4 | 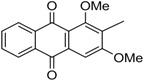 |
37.01 ± 13.82 | 87.81 ± 9.17 |
| 5 |  |
15.83 ± 0.58 | 23.61 ± 3.28 |
| 6 | 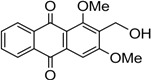 |
43.54 ± 0.48 | 47.62 ± 7.24 |
| 7 | 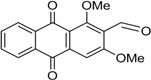 |
43.96 ± 3.42 | 49.66 ± 2.48 |
| 8 | 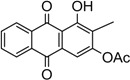 |
28.38 ± 7.94 | 69.59 ± 1.62 |
| 9 | 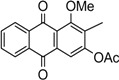 |
59.35 ± 8.23 | 46.77 ± 3.81 |
| 10 | 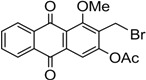 |
39.69 ± 6.24 | 54.90 ± 7.11 |
| 11 | 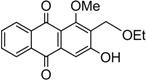 |
59.13 ± 11.59 | 42.06 ± 6.39 |
| 12 | 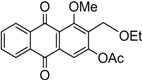 |
43.62 ± 5.35 | 89.36 ± 3.83 |
 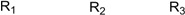
|
|||
| 13a | 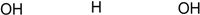 |
82.08 ± 1.46 | 60.42 ± 5.33 |
| 13b | 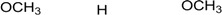 |
24.25 ± 2.46 | 22.01 ± 3.54 |
| 13c |  |
87.80 ± 2.01 | 64.96 ± 1.57 |
| 13d | 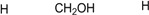 |
49.58 ± 12.61 | 50.84 ± 14.12 |
| 13e |  |
43.64 ± 2.12 | 46.61 ± 12.37 |
| 13f |  |
45.98 ± 2.95 | 107.14 ± 0.54 |
| 13g |  |
63.84 ± 12.46 | 70.54 ± 1.56 |
| 13h |  |
52.50 ± 2.63 | 94.17 ± 1.88 |
| Doxorubicin | 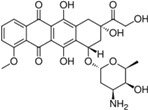 |
0.94 ± 0.20 | 0.24 ± 0.07 |
3. Experimental
3.1. General
Melting points were determined on a XSP-12 500X hot-stage melting point apparatus, and are uncorrected. UV spectra were recorded on a CARY 100 Conc. UV-visible spectrophotometer in MeOH. IR spectra were recorded on a Perkin–Elmer RXI Fourier Transform Infrared spectrometer (FTIR) as KBr discs. Mass spectra were measured on a Finnigan Mat SSQ 710 Spectrometer with ionization induced by electron impact at 70 eV. NMR spectra were recorded in CDCl3 and acetone-d6 using a Varian 500 MHz NMR spectrometer. Column chromatography was performed on silica gel (60 Merck 9385, 230–400 mesh, ASTM). The molecular structure of the intermediate 1,3-dimethoxy-2-bromomethylanthraquinone (5) was determined by X-ray crystallography. The data were collected at 100K using a Bruker APEXII with a CCD area-detector X-ray diffractometer. The structure was solved by direct method with SHELXS97 program and refined on F2 by full-matrix least-squares methods with anisotropic non-hydrogen atoms. The compound crystallizes in the monoclinic P2(1)/c space group with the crystallographic detail given in Table 2. The molecule exists as a planar molecule with r.m.s deviation of 0.018Å (Figure 1).
Table 2.
Crystal data and structure refinement for 2-bromomethyl-1,3-dimethoxyanthraquinone (5).
| Empirical formula | C17H13O4Br |
| Formula weight | 361.18 |
| Temperature | 100(2) K |
| Wavelength | 0.71073 Å |
| Crystal system | Monoclinic |
| Space group | P2(1)/c |
| Unit cell dimensions | a = 4.7401(8)Å, α = 90° |
| b = 17.364(3)Å, β = 94.543(2)° | |
| c = 16.825(3)Å, γ = 90° | |
| Volume | 1380.5(4) Å3 |
| Z | 4 |
| Calculated density | 1.738 Mgm−3 |
| Absorption coefficient | 2.994 mm−1 |
| F(000) | 728 |
| Crystal size | 0.20 × 0.10 × 0.10 mm |
| θ range for data collection | 1.69–28.26° |
| Limiting indices | −6 ≤ h ≤6, −14 ≤ k ≤23, −22 ≤ l ≤ 16 |
| Reflections collected | 3404 |
| Independent reflections | 2522 [R (int) = 0.0494] |
| Completeness to θ = 26.00º | 99.40% |
| Absorption correction | Multi-scan |
| Max. and min. transmission | 0.5973 and 0.7457 |
| Refinement method | Full-matrix least-squares on F2 |
| Data / restraints / parameters | 3404/0/201 |
| Goodness-of-fit on F2 | 1.038 |
| Final R indices [I>2σ(I)] | R1 = 0.0417, wR2 = 0.0984 |
| R indices (all data) | R1 = 0.0633, wR2 = 0.1124 |
| Largest difference peak and hole | 0.521 and −0.782 e.Å−3 |
3.2. General Procedure for Anthraquinone Synthesis
A mixture of anhydrous aluminium chloride (30 g, 225.0 mmol) and sodium chloride (12 g, 205.0 mmole) were melted at 125–130 °C. Phthalic anhydride (6.7 g, 45.0 mmole) and 2-methyl resorcinol (40.5 mmole) were mixed well and added slowly into the molten mixture of aluminum chloride and sodium chloride. The reaction temperature was raised till to 165–175 °C and maintained for 45–60 min. Another portion of anhydrous aluminum chloride (30 g, 225.0 mmole) was added into the hot mixture of the reactants [28]. After being cooled, the deep red solid product was decomposed by adding a mixture of ice water (250 mL) and conc. hydrochloric acid (250 mL). The crude mixture was dissolved in distilled water and organic layer was extracted with ethyl acetate, washed with brine and dried over anhydrous sodium sulfate. The crude products were purified by flash silica gel column chromatography with elution of the ethyl acetate /hexane as yellow needles.
1,3-Dihydroxy-2-methylanthraquinone (3). Yellow needles: 65% yield: m.p. 280–282 °C; {lit. [27] 280–283 °C}. UV (nm) in MeOH: 408, 279, 251. IR (cm−1): 3402 (OH), 2930, (C-H), 1661 (C=O non-chelated), 1628, 1591 (C=C< aromatic), 1338, 1310, 1122, 712. 1H-NMR (acetone-d6): δ 13.22 (s, 1H, 1-OH), 8.13 (dd, 1H, J = 1.5, 7.5 Hz, H-8), 8.23 (dd, 1H, J = 1.5, 7.5 Hz, H-5), 7.95–7.90 (m, 2H, H-6 & H-7), 7.37 (1H, s, H-4), 2.19 (3H, s, CH3). MS m/z (rel.int.): 254 (M+, 100), 226, 152 (10), 128 (21), 105 (11).
2-Bromomethyl-1,3-dimethoxyanthraqinone (5). To a solution of compound 4 (400 mg. 1.4 mmole) in dry CCl4 (40 mL), N-bromosuccinimide (500 mg, 3.0 mmole) was added portionwise. The whole mixture was stirred for 30 h. Compound 5 was extracted with ethyl acetate, filtered and purified by passing through a short column containing anhydrous sodium sulfate. The compound was recrystallized in a mixture of EtOAc and drops of MeOH as yellow crystals. Yield 90%; m.p.: 154–155 °C; {lit [27] 159–160 °C}. IR (cm−1): 2938 (CH), 1670 (C=O non-chelated), 1589 (C=C< aromatic), 1330, 1288, 1231, 1161, 730. 1H-NMR (CDCl3): δ 8.30 (d, 1H, J = 8.0 HZ, H-8), 8.25 (d, 1H, J = 8.0 Hz, H-5), 7.80–7.75 (m, 2H, H-6 & H-7), 7.26 (s, 1H, H-4), 4.76 (s, 2H, -CH2Br), 4.05 (s, 3H, OCH3), 3.97 (s, 3H, OCH3). MS m/z (rel.int.): 361 (M++H, 38) 362 (M++2, 37), 281 (100), 267 (35), 253 (36), 236 (42), 221 (7), 105 (11), 223 (41), 77 (21).
1,3-Dihydroxy-2-formylanthraquinone (nordamnacanthal) (2). Compound 7 (150 mg, 0.5 mmole) was suspended in dry CH2Cl2 (10 mL), stirred for 10 min. at rt and anhydrous AlCl3 (500 mg, 3.7 mmole) was added and stirred for 1h and then reflux for 4–6 h. The reaction mixture was cooled and added mixture of ice HCl (250 mL) and water (250 mL). The products was extracted by EtOAc and purified by CC and followed by recrystallized from a mixture of MeOH/EtOAc as orange crystals. Yield 28%; m.p.: 217–218 °C; {lit. [25] 220–221 °C}. UV (nm) in CHCl3: 422, 292, 263, 250. IR (cm−1): 3436 (OH), 2928, 2927, (CH), 1630 (C=O), 1560 (>C=C< aromatic), 1331, 1108, 786, 715. 1H-NMR (MeOD): δ 10.34 (s, 1H, CHO), 8.29 (d, 1H, J = 7.0 Hz, H-8), 8.25 (d, 1H, J = 7.0 Hz, H-5), 7.85–7.82 (m, 2H, H-6 & H-7), 7.29 (s, 1H, H-4). MS m/z (rel.int.): 268 (M+, 65) (10), 281 (100), 249 (100), 212 (34), 184 (32), 1197(24), 155 (7), 138 (12), 128 (21), 77 (12).
2-Formyl-3-hydroxy-1-methoxyanthraquinone (1). Compound 12 (200 mg 0.56 mmole) was dissolved in dry dichloromethane (30 mL) and PCC (125 × 2 mg, 1.39 mmole) was introduced slowly in a stirred solution. The reaction mixture was stirred at room temperature for 4–6 h. A black brown solid was obtained, dissolved in aqueous solution and extracted with EtOAc, solvent removed in vacuo on a rotary evaporator and dried over anhydrous sodium sulfate. The product was purified by column chromatography using EtOAc/hexane as solvent system. The product was obtained as orange crystals. Yield 64.5%; m.p.: 209–211 °C; (lit. (25) 211–212 °C. IR (cm−1): 3434 (OH), 2957, 2927, (CH), 1670 (non-chelated C=O), 1648 (C=O non-chelated), 1566 (C=C< aromatic), 1344, 1260, 1231, 1132, 7310. UV (nm) in CHCl3: 412, 284, 264, 254. 1H-NMR (acetone-d6): δ 10.45 (s , 1H, CHO), 8.32 (dd, 1H, J = 1.5, 7.5 Hz, H-8), 8.26 (dd, 1H, J = 1.5, 7.5 Hz, H-5), 7.87–7.78 (m, 2H, H-6 & H-7) 7.69 (s, 1H, H-4), 4.11 (s, 3H, OCH3). MS m/z (rel.int.): 282 (M+, 100) 267 (35), 254 (100), 237 (16), 225 (7), 197 (24), 180 (31), 168 (6) 152 (9), 139 (41).
3.3. X-ray Crystallography of Compound 5
A single crystal of compound 5 was obtained by slow evaporation at room temperature from a mixture of ethyl acetate-MeOH (drops). The crystal structure was solved by direct method with SHELXS97 programe and refined on F2 by full-matrix least-squares methods with anisotropic non-hydrogen bond atoms. The compound crystallized as a monoclinic P2(1)/c space group with the crystallographic data is given in Table 2. The molecule exists as a planar molecule with r.m.s deviation of 0.018 Å (Figure 1). There is no evidence of any hydrogen bonding interaction or π–π stacking present in the crystal packing of the compound. Full crystallographic data for structures 5 has been deposited at the Cambridge Crystallographic Data Center (CCDC) as supplementary publication number CCDC870566. Copies of the data can be obtained, free of charge, on application to CCDC 12 Union Road, Cambridge CB2 1EZ, UK [Fax: þ44 1223 336033. email: deposit@ccdc.cam.ac.uk or at www.ccdc.cam.ac.uk].
4. Cytotoxicity Assay
4.1. Preparation of Compounds
The compounds were dissolved in dimethylsulfoxide (DMSO, Sigma, St. Louis., MO, USA) to get a stock solution of 1 mg/mL and stored at 4 °C.
4.2. Preparation of Cell Lines
Adherent human estrogen dependent breast carcinoma MCF-7 and suspension human chronic myelogenic leukemia K-562 cell lines were obtained from ATCC, USA. Both MCF-7 and K562 were cultured in RPMI-1640 supplemented with 10% FBS and incubated at 37 °C, 5% CO2 and 90% humidity throughout the study. Cell viability was assessed by trypan blue exclusion method where both MCF-7 and K562 cell lines were collected, washed with PBS and mixed with 1:1 ratio of trypan blue dye and the cell number and viability were evaluated using haemacytometer under inverted light microscope. Only cell with viability higher than 95% will be subjected to MTT cell viability assay.
4.3. Cell Viability Assay
The effect of all compounds on cell viability of MCF-7 and K-562 cell lines were determined using the colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay [37]. MCF-7 cell were seeded on a 96 well microtiter plate with each well containing 200 µL of culture media with 10% of FBS per well at a density of 1 × 105 cells/mL and incubated overnight at 37 °C, 5% CO2 at humidified atmosphere of 95% air. When cells were reached 70% confluence then subjected to the cell viability assay. Medium containing the seeded MCF-7 cell was discarded. Treatment of both cultures were carried out by first adding 100 µL of RPMI-1640 medium supplemented with 10% of FBS into all wells except row A of the 96 well plates (TPP, Zollstrasse, Trasadingen, Switzerland). Then, 100 µL of each diluted compound (60 µg/mL) was added into row A and row B. A series of two-fold dilutions of compounds were carried out from row B to row G to give concentrations of 30, 15, 7.5, 3.75, 1.375 and 0.687 µg/mL, while row H was left as untreated control cell. For K-562 culture plate, 100 µL of cells (2 × 105 cells/mL) was added into all wells. For MCF-7 culture plate, 100 µL of RPMI-1640 medium was added into all wells. The plates were incubated in 37 °C, 5% CO2 and 90% humidity incubator for 72 h. After incubation period, 20 µL of MTT (Sigma, 5 mg/mL) was added into all wells and the plates were further incubated for 4 h. Then, 170 µL of supernatant was aspirated from every well plate. The plates containing K-562 culture were centrifuged at 200 × g for 5 min prior to medium aspiration. The resulting formazan crystals in each well were solubilized by 100 µL of DMSO (Fisher Scientific, Waltham, MA, USA) followed by incubation for 20 min. Finally, the plates were read at 570 nm and 630 nm as reference wavelength by using µQuant ELISA Reader (Bio-tech Instruments, Winooski, VT, USA). The results of the compound-treated cells were compared with the standard doxorubicin. Each compound and control was assayed in triplicates in three independent experiments. The percentage of inhibitions were calculated by using Graph pad and expressed in µg/mL or µM and listed in Table 1.
5. Conclusions
The synthesis of damnacanthal (1) and nordamnacanthal (2) was achieved. The key difference from the previously synthesis was the conversion of hydroxymethylanthraquinones 7 or 12 into the corresponding aldehyde, which was achieved by the use of optimized mild conditions with PCC as oxidant. The synthesis in good yield of 2-ethoxymethyl-3-hydroxy-1-methoxyanthraquinone (11) was achieved through a short sequence. The structure activity relationship suggested that methoxy, formyl, bromomethyl and hydroxyl groups are important for the cytotoxicity and selectivity in these substituted anthraquinones. These side chain modifications give an idea for further efforts to increase the therapeutic potential of this class of compounds.
Acknowledgments
This project was supported by the Universiti Putra Malaysia, (RUGS research grant no. 05-03-10-0967 RU) and University Malaysia Pahang (internal grant No. RDU 120373). For EI-MS analysis HEJ Research Institute of Chemistry, Karachi, Pakistan is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 1–13h are available from the authors.
References
- 1.Murphy G.P., Lawrence W.J., Lenhard R.E. American Society Textbook of Clinical Oncology. 2nd ed. American Cancer Society; Atlanta, GA, USA: 1995. [Google Scholar]
- 2.Doroshow J.H. Anthracyclines and anthracenediones. In: Chabner B.A., Longo D.L., editors. Cancer Chemotherapy and Biotherapy: Principles and Practice. 2nd ed. Lippincott-Raven Publishers; Philadelphia, PA, USA: 1996. pp. 409–434. [Google Scholar]
- 3.Faulds D., Balfour J.A., Chrisp P., Langtry H.D. Mitoxantrone, a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in the chemotherapy of cancer. Drugs. 1991;3:400–449. doi: 10.2165/00003495-199141030-00007. [DOI] [PubMed] [Google Scholar]
- 4.Kosalec I., Kremer D., Locatelli M., Epifano F., Genovese S., Carlucci G., Randic M., Koncic Z.M. Anthraquinone profile, antioxidant and antimicrobial activity of bark extracts of Rhamnus alaternus, R. fallax, R. intermedia and R. pumila. Food Chem. 2013;136:335–341. doi: 10.1016/j.foodchem.2012.08.026. [DOI] [PubMed] [Google Scholar]
- 5.Shi Y.Q., Fukai T., Sakagami H., Kuroda J., Miyaoka R., Tamura M., Yoshida N., Nomura T. Cytotoxic and DNA damage-inducing activities of low molecular weight phenols from rhubarb. Anticancer Res. 2001;21:2847–2853. [PubMed] [Google Scholar]
- 6.Sittie A.A., Lemmich E., Olsen C.E., Hviid L., Kharazmi A., Nkrumah F.K., Christensen S.B. Structure-activity studies: in vitro antileishmanial and antimalarial activities of anthraquinones from Morinda lucida. Planta Med. 1999;65:259–261. doi: 10.1055/s-2006-960473. [DOI] [PubMed] [Google Scholar]
- 7.Rath G., Ndonzao M., Hostettmann K. Antifungal anthraquinones from Morinda lucida. Int. J. Pharmacogol. 1995;33:107–114. doi: 10.3109/13880209509055208. [DOI] [Google Scholar]
- 8.Younos C., Rolland A., Fleurentin J., Lanhers M., Misslin R., Mortier F. Analgesic and behavioral effects of Morinda citrifolia. Planta Med. 1990;56:430–434. doi: 10.1055/s-2006-961004. [DOI] [PubMed] [Google Scholar]
- 9.Adwankar M.K., Chitnis M.P. In vivo Anti-Cancer Activity of RC-18. Chemotherapy. 1982;28:291–293. doi: 10.1159/000238092. [DOI] [PubMed] [Google Scholar]
- 10.Koumaglo K., Gbeassor M., Nikabu O., de Souza C., Werner W. Effects of three compounds extracted from Morinda lucida on Plasmodium falciparum. Planta Med. 1992;58:533–434. doi: 10.1055/s-2006-961543. [DOI] [PubMed] [Google Scholar]
- 11.Tripathi Y.B., Sharma M., Manickam M. Rubiadin, a new antioxidant from Rubia cordifolia. Indian J. Biochem. Biophys. 1997;34:302–306. [PubMed] [Google Scholar]
- 12.Chang P., Chen C. Isolation and Characterization of Antitumor Anthraquinones from Morinda umbellate. Chin. Pharm. J. 1995;47:347–353. (Taipei) [Google Scholar]
- 13.Ismail N.H., Ali A.M., Aimi N., Kitajima M., Takayama H., Lajis N.H. Anthraquinones from Morinda elliptica. Phytochemistry. 1997;45:1723–1725. doi: 10.1016/S0031-9422(97)00252-5. [DOI] [Google Scholar]
- 14.Di Marco A., Gaetani M., Orezzi P., Scarpinato B. M., Silvestrini R., Soldati M., Dasdia T., Valentini L. Daunomycin, A New Antibiotic of the Rhodomycin Group. Nature. 1964;201:706–707. doi: 10.1038/201706a0. [DOI] [PubMed] [Google Scholar]
- 15.Kim S.J., Kim M.C., Lee B.J., Park D.H., Hong S.H., Um J.Y. Anti-Inflammatory Activity of Chrysophanol through the Suppression of NF-κB/Caspase-1 Activation in Vitro and in Vivo. Molecules. 2010;15:6436–6451. doi: 10.3390/molecules15096436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Preobrazhenskaya M.N., Shchekotikhin A.E., Shtil A.A., Huang H.S. Antitumor Anthraquinone Analogues for Multidrug Resistant Tumor Cells. J. Med. Sci. 2006;26:1–4. [Google Scholar]
- 17.Teng C.H., Won S.J., Lin C.N. Design, synthesis and cytotoxic effect of hydroxy- and 3-alkylaminopropoxy-9,10-anthraquinone derivatives. Bioorg. Med. Chem. 2005;13:3439–3435. doi: 10.1016/j.bmc.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 18.Jin G.Z., Song G.Y., Zheng X.G., Kim Y., Sok D.E., Ahn B.Z. 2-(1-Oxyalkyl)-1,4-dioxy-9,10-anthraquinone: synthesis and Evaluation of Antitumor Activity. Arch. Pharm. Res. 1998;21:198–206. doi: 10.1007/BF02974028. [DOI] [PubMed] [Google Scholar]
- 19.Cairns D., Michalitsi E., Jenkins T.C., Mackay S.P. Molecular Modelling and Cytotoxicity of Substituted Anthraquinones as Inhibitors of Human Telomerase. Bioorg. Med. Chem. 2002;10:803–817. doi: 10.1016/S0968-0896(01)00337-6. [DOI] [PubMed] [Google Scholar]
- 20.Murdock K.C., Child R.C., Fabio P.F., Angier R.B., Wallace R.E., Durr F.E., Chella R.V. Antitumor Agents.1.1,4-Bis[(Aminoalkyl)amino]-9,19-Anthracenediones. J. Med. Chem. 1979;22:1024–1030. doi: 10.1021/jm00195a002. [DOI] [PubMed] [Google Scholar]
- 21.Chang P., Lee K.H., Shingu T., Hirayama T., Hall I.H., Huang H.C. Antitumor agents 50. Morindaparvin-A, a New Antileukemic Anthraquinone, and Alizarin-1-methyl ether from Morinda parvifolia, and the Antileukemic Activity of the Related Derivatives. J. Nat. Prod. 1982;45:206–210. doi: 10.1021/np50020a017. [DOI] [PubMed] [Google Scholar]
- 22.Ali A.M., Ismail N.H., Mackeen M.M., Yazan L.S., Mohammad S.M., Ho A.S.H., Lajis N.H. Antiviral, Cytotoxic and Antimicrobial Activities of Anthraquinones isolated from the roots of Morinda elliptca. Pharm. Biol. 2000;38:298–301. doi: 10.1076/1388-0209(200009)3841-AFT298. [DOI] [PubMed] [Google Scholar]
- 23.Alitheen N.B., Mashitoh A.R., Yeap S.K., Shuhaimi M., Manaf A.A., Lajis N.H. Cytotoxic effect of damnacanthal, nordamnacanthal, zerumbone and betulinic acid isolated from Malaysian plant sources. Int. Food Res. J. 2010;17:711–719. [Google Scholar]
- 24.Noorjahan B.M.A., Ali A.M., Yeap S.K., Suhaimi M., Lajis N.H., Mashitoh A.R., Ho W.Y., Ismail N.H. Cytotoxicity and Immunomodulatory Effects of Damnacanthal and Nordamnacanthal Isolated from Roots of Morinda elliptica. J. Agrobiotec. 2010;1:29–42. [Google Scholar]
- 25.Hirose Y. Syntheses of Damnacanthal, Damnacanthol, Norjuzunal and Norjuzunol, the Coloring Matters of Damnacanthus Spp. Chem. Pharm. Bull. 1960;8:417–426. doi: 10.1248/cpb.8.417. [DOI] [Google Scholar]
- 26.Roberts J.L., Rutledge P.S., Trebilcock M.J. Experiments Directed Towards the Synthesis of Anthracyclinones.1 Synthesis of 2-formylmethoxyanthraquinones. Aust. J. Chem. 1977;30:1553–1560. doi: 10.1071/CH9771553. [DOI] [Google Scholar]
- 27.Roberts J.L., Rutledge P.S. Experiments Directed Towards the Synthesis of Anthracyclinones. 111 P-Formylation of Hydroxyanthraquinonesby the Claisen Rearrangement. Aust. J. Chem. 1977;30:1743–1760. doi: 10.1071/CH9771743. [DOI] [Google Scholar]
- 28.Saha K., Lam K.W., Abas F., Hamzah A.S., Stanslas J., Hui L.S., Lajis N.H. Synthesis of damnacanthal, a naturally occurring 9,10-anthraquinone and its analogues, and its biological evaluation against five cancer cell lines. Med. Chem. Res. 2013;22:2093–2104. doi: 10.1007/s00044-012-0197-5. [DOI] [Google Scholar]
- 29.Wei B.L., Wu S.H., Chung M.I., Won S.J., Lin C.N. Synthesis and cytotoxic effect of 1,3-dihydroxy-9,10-anthrquinone derivatives. Eur. J. Med. Chem. 2000;35:1089–1098. doi: 10.1016/S0223-5234(00)01190-9. [DOI] [PubMed] [Google Scholar]
- 30.Horii Z., Hakusui H., Momose T. Synthetic Studies on Anthracyclinones. VII. Synthesis of Bisanhydroaklavinone. Chem. Pharm. Bull. 1968;16:1262–1265. doi: 10.1248/cpb.16.1262. [DOI] [PubMed] [Google Scholar]
- 31.Castonguay A., Brassard P. Total synthesis of (+)-averufin and (+)-bipolarin. Can. J. Chem. 1977;55:1324–1332. doi: 10.1139/v77-183. [DOI] [Google Scholar]
- 32.Ma H.M., Liu Z.Z., Chen S.Z. Regioselective Bromination of 3, 4-Dimethoxytoluene with N-Bromosuccinimide. Chin. Chem. lett. 2003;14:371–374. [Google Scholar]
- 33.Abraham S.A., Waterhouse D.N., Mayer L.D., Cullis P.R., Madden T.D., Bally M.B. The liposomal formulation of doxorubicin. Meth. Enzymol. 2005;391:71–97. doi: 10.1016/S0076-6879(05)91004-5. [DOI] [PubMed] [Google Scholar]
- 34.Iyer M., Tseng Y.J., Senese C.L., Liu J., Hopfinger A.J. Prediction and Mechanistic Interpretation of Human Oral Drug Absorption Using MI-QSAR Analysis. Mol. Pharm. 2007;4:218–231. doi: 10.1021/mp0600900. [DOI] [PubMed] [Google Scholar]
- 35.Zhao Y.H., Le J., Abraham M.H., Hersey A., Eddershaw P.J., Luscombe C.N., Butina D., Beck G., Sherborne B., Cooper I., Platts J.A. Evaluation of human intestinal absorption data foruse in QSAR studies and quantitative relationship obtained with the Abraham descriptors. J. Pharm. Sci. 2001;90:749–784. doi: 10.1002/jps.1031. [DOI] [PubMed] [Google Scholar]
- 36.Saha K., Lajis N.H., Abas F., Naji N.A., Hamzah A.S., Shaari K. Rearrangement in Lewis Acid-Catalyzed Friedel-Crafts Conditions: Evidence of Competitive Initial Protonation and Acylation. Aust. J. Chem. 2008;61:821–825. doi: 10.1071/CH08084. [DOI] [Google Scholar]
- 37.Mossman T. Rapid calorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Meth. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]