

Abstract
Assessing the molecular mechanism of a chemical-biological interaction and bonding stands as the ultimate goal of any modern quantitative structure-activity relationship (QSAR) study. To this end the present work employs the main chemical reactivity structural descriptors (electronegativity, chemical hardness, chemical power, electrophilicity) to unfold the variational QSAR though their min-max correspondence principles as applied to the Simplified Molecular Input Line Entry System (SMILES) transformation of selected uracil derivatives with anti-HIV potential with the aim of establishing the main stages whereby the given compounds may inhibit HIV infection. The bonding can be completely described by explicitly considering by means of basic indices and chemical reactivity principles two forms of SMILES structures of the pyrimidines, the Longest SMILES Molecular Chain (LoSMoC) and the Branching SMILES (BraS), respectively, as the effective forms involved in the anti-HIV activity mechanism and according to the present work, also necessary intermediates in molecular pathways targeting/docking biological sites of interest.
Keywords: anti-HIV activity; 1,3-disubstituted uracil derivatives; QSAR; SMILES; electronegativity; chemical hardness; chemical power; electrophilicity; chemical reactivity principles; lipophilicity
1. Introduction
There is a tremendous current demand for new materials and substances for the betterment of life, applications, health and the environment, but new synthesis cannot sufficiently guarantee the sustainability of the new compounds. In a global effort to diminish the toxicological and adverse effects of the multi-scale interaction and fate of chemicals in silico (computational) methods appear more and more as a viable alternative and prerequisite for any experimental endeavor, in vitro first, and then moving on to the final in vivo tests. Accordingly, an intimate relationship between the structure of a compound, in physicochemical terms, and the manifested reactivity (in the chemical realm), activity (in the bio-/eco-/pharmaco-logical realm) and functionality (in the nano-toxicology and technology realm) should be computationally established as a “road map” of expectations, conditions of use, prediction and prevention. In this context, the computational mathematical and statistical algorithms for modeling the chemical-biological interaction of a compound with organisms have become known as quantitative structure-activity relationships (QSAR) methods have come to the forefront. Especially in the last decade, they have evolved towards a regulatory framework, able to jointly address a variety of areas such as:
Toxicological dose (endpoint) response and risk for spatio-temporal multi-scale prediction [1,2];
Assessment of metabolic genotoxicity and screening of chemicals with bioaccumulation potential [3,4];
Modeling of nanomaterials [5], including the oxidative stress-potential [6] and the toxicity of nanoparticles [7];
Food and organic chemicals’ safety by computational analysis [8,9,10];
Computational toxicology [11];
Complex algebraic (networks) as well as simple arithmetic physiological activity and toxicity [12,13];
Quantifying the dynamics of environmental nutrients and contaminants [14] with a view toward nanochemistry [15] and nanomedicine [16];
Integrative structure-property and structure-activity computational workflows [17];
Interspecies toxicity analysis [18];
Design of safe drugs by employing structural similarity and computing toxicity predictions [19,20];
Guidance rules for the domain of applicability in QSAR approaches [21,22];
Considering, in relation to molecular structure, the molecular topology and quantum chemical descriptors among the basic causes of the observed toxicological properties, reactivity (or aromaticity) and activities [23,24,25,26];
The assessment of multilinear models for molecular classes and large sets of chemicals with environmental activity [27,28];
Establishing hierarchical models for the human health effects of toxicants [29,30];
The role of the hydrophobicity of new chemicals in relation with cells’ activity and associated mechanistic interactions [31,32].
Accordingly, in response to this increasing demand for benchmark principles to be followed by a reliable QSAR research project [33,34], the Organization for Economic Co-Operation and Development (OECD) had advanced a set of standard principles for the validation, for regulatory purposes, of (quantitative) structure-activity relationship models [35,36,37,38]. In short, these principles are:
QSAR-1: a defined endpoint;
QSAR-2: an unambiguous algorithm;
QSAR-3: a defined domain of applicability;
QSAR-4: appropriate measures of goodness-of–fit, robustness and predictivity;
QSAR-5: a mechanistic interpretation, if possible.
At this point one should distinguish between two main directions in which a QSAR study may be conducted, namely:
Drug design oriented, which is generated through extensive database screening [39,40], similarity and domain considerations [41,42], producing QSAR models which should be then validated by internal [43,44], external and read-across techniques [45] so that finally the molecules or molecular fragments predicted as most active or inhibitive depending on the endpoint target can be selected;
Mechanism oriented, which consists mainly in the identification of the fundamental types of interaction that happen at the chemical-to-biological scale so that the structural properties of a compound constitute the causes that can be related to the manifest and recorded effects at a biological site [46,47,48,49,50,51];
In phenomenological terms, while the first direction is more related to technology and to the prescriptions for new synthesis, the second QSAR route is more on the scientific side due to the fundamental approach it involves; nevertheless, they both are related since after all, drug design is based on the desired or assumed mechanism of action specific to a given class of compounds, so knowing or revealing the mechanism of action for a given chemical-biological interaction only based on QSAR models remains as the first and probably the most important stage in drug design process itself.
Then, one faces with the true challenge, namely how to extract from a single or from a collection of QSAR models the “first causes” of a chemical-biological interaction. Fortunately, one may rely on the (multi) linear form of QSAR models since, when considered in terms of physicochemical parameters with mechanistic interpretation at the nano-chemical scale, they provide just a manifestation of the quantum superposition principle [52]: while each structural parameter is associated with a given state or “chemical movement” specific to that state, their linear superposition combines into the macroscopic effect recorded as bio-/eco-/pharmaco-activity. Within this paradigm one has then the conceptual and computational freedom in establishing the “order” of the chemical states/movements toward the concerned endpoint. This direction has proven fruitful in assigning many useful QSAR tools thus enriching the related analysis and paving the way to mechanistic drug design through combination of various in cerebro (conceptual)—in silico (computational) approaches, such as:
Considering the elements of a QSAR model, i.e., both descriptors and activities as vectors in a multi-dimensional (chemical-biological) Banach-Hilbert (quantum) observable space [53,54,55,56];
Considering the descriptors of a QSAR model mainly with observable or physicochemical character, e.g., hydrophobicity for cellular wall transduction (the translation motion), the total energy for steric optimization (rotation motion), polarizability for molecular cloud deformation (vibrational motion) [57,58], or more recently, through the chemical reactivity indices (electronegativity, chemical hardness and related quantities) for gaining more insight into the subtle bonding description (binding movement)—leading to the so called chemical reactivity driven biological activity picture (which will be used also in the present work) [59];
Considering the systematical collection of QSAR models of descriptors in the previous entry along with their basic statistics, e.g., correlation factors, to be then employed either in an algebraic formulation of descriptor-activity correlations, proved to be always superior to the basic statistical one, or to entering in Euclidian paths among the computed endpoints [60], thus involving the square form of the correlation factor, to produce and compare minimum distances toward the most comprehensive (superior in correlation) QSAR model (in turn presumed to be the closest in the QSAR pool of models to the real/recorded activity). This approach, consecrated as Spectral-SAR [57,58,61,62,63], provides the mechanistic interpretation of biological action in terms of the hierarchy of structural causes (descriptors) along the least computed path across available QSAR mode;
Considering, more recently, the way of improving the previous entry by extensive use of the variational approach in all stages of Spectral-SAR, from screening (i.e., selecting the training set) from a set of toxicants, to assessing the minimum path by considering the molecular passage through cellular walls accompanied by the partial chemical bonds in molecules [64], according with the Simplified Molecular-Input Line-Entry System (SMILES) [65,66,67,68].
This last point is from where the present work continues the idea of fully considering the SMILES structure in the computational development of QSARs, by calculating the associated descriptors and involving them in the mechanistic analysis. Actually, it was found that when using SMILES forms only for screening purposes, as in the present case for modeling the anti-HIV activity of selected uracil derivatives [69], the output mechanism provides an activated chemical-biological bonding not properly indicating the finalization of the ligand-receptor coupling to explain the anti-HIV activity. Therefore, the present report takes this concept one step further in order to complete the chemical-bonding picture by fully using the SMILES structures not only as a graphical tool but also considering them as an intermediate reality in the mechanistic picture of chemical ligand-biological receptor interaction yielding the recorded effect in the organism. To this end, the above mechanistic-oriented framework will be unfolded, by applying the OECD-QSAR principles to the present purpose and conceptual-computational stages [70], by combining Spectral-SAR methodology with variational principles of chemical reactivity driving biological activity and with the recursive minimization of paths across systematic QSAR with SMILES molecular (chemical reactivity) descriptors, to recognize the preferred hierarchy and the “first causes” that eventually result in the envisaged chemical binding and resulting anti-HIV activity. This mechanism may be further used in a subsequent stage when extensive validation and drug design studies to recognize the molecular shape and structure [71] which best accords with a particular mechanism of action can be envisaged.
2. Results and Discussion
2.1. OECD-QSAR Principle 1: A Defined Endpoint
According to OECD guidance, “the intent of QSAR Principle 1 (defined endpoint) is to ensure clarity in the endpoint being predicted by a given model, since a given endpoint could be determined by different experimental protocols and under different experimental conditions. It is therefore important to identify the experimental system that is being modeled by the (Q)SAR”. Note that the actual endpoint is still the inhibitory effect predicted by a series of 1,3-disubstituted uracil-based anti-HIV compounds [69] on reverse transcriptase [72,73,74,75] in highly active antiretroviral therapy (HAART) [76,77,78]. It arises, in principle, with the same binding mechanism as binding/breaking DNA, through a group of non-necessarily similar structures, giving rise to the following updating QSAR end-point approaches [79,80,81]:
(Eco-) toxicological studies, having various end-points (such as inhibition, activation, death, sterility, irritations, etc.) yet produced by a group of similar molecules, i.e., the case of congeneric studies;
and carcinogenic studies, having essentially the same end-point as the exacerbated apoptosis that in principle diffuses in the organism no matter what the initial trigger point is, and may be initiated by highly structurally diverse molecules, being therefore classified as non-congeneric studies.
While the first case above is usually treated by ordinary (or direct) QSAR approaches, the second category is less frequently treated with the central QSAR dogma of congenericity. It therefore requires special approaches, such as the recently described residual-QSAR study [82]. This relies on the fact that if no direct high correlation can be found, then there is a high probability that the action is residual, complementary or indirect [83]. For this point one considers the working molecules under study the most likely form producing the considered end-point, namely the anti-HIV activity produced by uracil-based pyrimidines [69,84], along two aspects of their SMILES structure, as presented in Table 1:
Table 1.
Working molecules (IUPAC name and molecular weight MW are indicated ) and their corresponding SMILES topology, i.e. the longest SMILES molecular chain (LoSMoC) as upper entry and the Branching SMILES (BraS) as down entry, for each pyrimidine structure considered, along the common activity A = log10(1/EC50) employed from half maximal effective concentration (EC50, μM) antiviral activity of 1,3-disubstituted uracils against human immunodeficiency virus (HIV-1) [69], with AIDS code indicated [84], respectively. The solubility parameter of lipophilicity (LogP), and the chemical reactivity parameters such as electronegativity (χ) and chemical hardness (η), chemical power (π) and electrophilicity (ω) are considered within the semiempirical (AM1) framework (Polak-Ribiere conjugate gradient algorithm and geometry optimization till the root mean square RMS gradient was equal to or less than 0.01 kcal/Åmol) as provided by the Hyperchem 7.01 computational environment [140], while the chemical reactivity values were computed in terms of HOMO and LUMO from equations (14) and (15)—see text and Table 2, (7) and (10), respectively. SMILES legend is:  principal SMILES chain;
principal SMILES chain;  secondary SMILES branch;
secondary SMILES branch;  tertiary SMILES branch;
tertiary SMILES branch;  quaternary SMILES branch; = double bond; # triple bond; /,\ directional bonds; ( ) branch; C, N, F, S, I — atoms present in the molecule; c, n — atoms place in an aromatic ring; C1/2/3, N1/2, c1/2/3, n2 — connectivity points.
quaternary SMILES branch; = double bond; # triple bond; /,\ directional bonds; ( ) branch; C, N, F, S, I — atoms present in the molecule; c, n — atoms place in an aromatic ring; C1/2/3, N1/2, c1/2/3, n2 — connectivity points.
| No. | Structure 2D | SMILES configurations | A | LogP | χ (eV) | η (eV) | π | ω (eV) | |
|---|---|---|---|---|---|---|---|---|---|
| IUPAC name MW AIDS code |
LoSMoC | Code LoSMoC | ... LoSMoC ... | ||||||
| BraS | Code BraS | ... BraS ... | |||||||
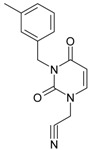
| 1 |
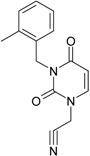
[3-(2-Methyl-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile 255.28 AIDS352092 |
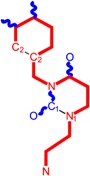
|
N#CCN1/C=C\C(=O)N(C1=O)Cc2ccc(C)c(C)c2 | 3.716698 | 0.91 | 23.107212 | 1.5817419 | 7.304356 | 168.78330 |
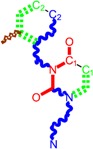
|
O=C1N(Cc(c(C)cc2)cc2)C(N(/C=C1\)CC#N)=O | 0.44 | 13.240955 | 2.8324015 | 2.3374078 | 30.949511 | |||
| 2 |
[3-(3-Methyl-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile 255.28 AIDS352093 |
|
N#CCN1/C=C\C(=O)N(C1=O)Cc2cccc(C)c2 | 5.173925 | 0.47 | 22.812517 | 1.5937610 | 7.156819 | 163.26505 |
|
O=C1N(Cc(cc(C)c2)cc2)C(N(/C=C1\)CC#N)=O | 0.44 | 13.043803 | 2.8273990 | 2.3066788 | 30.087865 | |||
| 3 |
[3-(4-Methyl-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile 255.28 AIDS352094 |
|
N#CCN1/C=C\C(=O)N(C1=O)Cc2ccc(C)cc2 | 4.023191 | 0.47 | 22.852718 | 1.5799314 | 7.232187 | 165.27512 |
|
O=C1N(Cc(ccc2C)cc2)C(N(/C=C1\)CC#N)=O | 0.88 | 13.149213 | 2.8323062 | 2.3212908 | 30.523148 | |||
| 4 |
[3-(2,4-Dimethyl-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile 269.30 AIDS352888 |
|
N#CCN1/C=C\C(=O)N(C1=O)Cc2ccc(C)cc2C | 3.943095 | 1.06 | 22.695343 | 1.4889604 | 7.621204 | 172.96584 |
|
O=C1N(Cc2c(cc(cc2)C)C)C(N(/C=C1\)CC#N)=O | 1.03 | 13.061603 | 2.7061581 | 2.4133112 | 31.521715 | |||
| 5 |
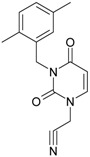
[3-(2,5-Dimethyl-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile 269.30 AIDS352889 |
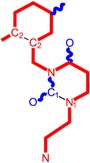
|
N#CCN1/C=C\C(=O)N(C1=O)Cc2cc(C)ccc2C | 4.610833 | 1.06 | 22.961910 | 1.5967679 | 7.190121 | 165.09891 |
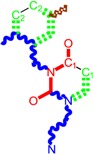
|
O=C1N(Cc(cc(C)c2)c(c2)C)C(N(/C=C1\)CC#N)=O | 0.6 | 13.344068 | 2.8843065 | 2.3132194 | 30.867758 | |||
| 6 |
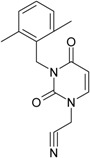
[3-(2,6-Dimethyl-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile 269.30 AIDS352890 |
|
N#CCN1/C=C\C(=O)N(C1=O)Cc2c(C)cccc2C | 3.707743 | 1.06 | 22.914792 | 1.5375402 | 7.45177 | 170.75577 |
|
O=C1N(Cc(c(C)cc2)c(C)c2)C(N(/C=C1\)CC#N)=O | 0.6 | 13.174123 | 2.7474378 | 2.3975289 | 31.585343 | |||
| 7 |
[3-(3,5-Dimethyl-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile 269.30 AIDS352095 |
|
N#CCN1/C=C\C(=O)N(C1=O)Cc2cc(C)cc(C)c2 | 6.229147 | 0.63 | 22.322613 | 1.3441469 | 8.303636 | 185.35884 |
|
O=C1N(Cc(cc(C)c2)cc2C)C(N(/C=C1\)CC#N)=O | 1.03 | 12.688503 | 2.5160717 | 2.5214906 | 31.993942 | |||
| 8 |
[3-(3,4-Dimethyl-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile 269.30 AIDS352891 |
|
N#CCN1/C=C\C(=O)N(C1=O)Cc2ccc(C)c(C)c2 | 5.425968 | 0.63 | 22.513298 | 1.4966364 | 7.521298 | 169.32923 |
|
O=C1N(Cc(cc(c2C)C)cc2)C(N(/C=C1\)CC#N)=O | 1.03 | 12.964034 | 2.7262701 | 2.3776137 | 30.823468 | |||
| 9 |
[3-(2,4,6-trimethyl-benzyl)- 2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile 283.33 AIDS352892 |
|
N#CCN1/C=C\C(=O)N(C1=O)Cc2c(C)cc(C)cc2C | 3.716698 | 1.22 | 22.436637 | 1.3498377 | 8.310865 | 186.46785 |
|
O=C1N(Cc2c(cc(cc2C)C)C)C(N(/C=C1\)CC#N)=O | 1.62 | 12.848802 | 2.5836971 | 2.4865149 | 31.948740 | |||
| 10 |
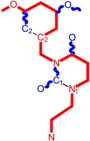
[3-(3-cyanophenyl)methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile 266.26 AIDS352893 |
|
N#CCN1/C=C\C(=O)N(C1=O)Cc2cccc(c2)C#N | 5.128427 | 0.04 | 22.981901 | 1.5807784 | 7.269172 | 167.05939 |
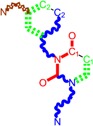
|
O=C1N(Cc(cc(C#N)c2)cc2)C(N(/C=C1\)CC#N)=O | 0.01 | 12.984607 | 2.7188679 | 2.3878703 | 31.00556 | |||
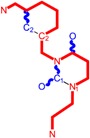
| 11 |
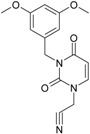
[3-(3,5-Dimethoxy-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile 301.30 AIDS352897 |
|
N#CCN1/C=C\C(=O)N(C1=O)Cc2cc(OC)cc(c2)OC | 5.248720 | -1.67 | 21.820275 | 1.0563595 | 10.32805 | 225.36097 |
|
O=C1N(Cc(cc2OC)cc(OC)c2)C(N(/C=C1\)CC#N)=O | -0.72 | 12.366078 | 2.2360288 | 2.7651875 | 34.194524 | |||
| 12 |
[3-(3,4,5-trimethoxy-benzyl)-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile 331.33 AIDS352898 |
|
N#CCN1/C=C\C(=O)N(C1=O)Cc2cc(OC)c(OC)c(c2)OC | 3.423658 | -2.66 | 21.365171 | 1.0625102 | 10.0541 | 214.80760 |
|
O=C1N(Cc2cc(c(OC)c(OC)c2)OC)C(N(/C=C1\)CC#N)=O | -2.26 | 12.143075 | 2.4593788 | 2.4687280 | 29.977950 | |||
| 13 |
|
|
N#CCN1/C=C\C(=O)N(C1=O)Cc3c2ccccc2ccc3 | 5.268411 | 1.16 | 25.868615 | 1.4726275 | 8.78315 | 227.20792 |
| (3-Naphthalen-1-ylmethyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl)-acetonitrile 291.31
AIDS352899 |
|
O=C1N(Cc(c(cc3)c(cc3)c2)cc2)C(N(/C=C1\)CC#N)=O | 0.25 | 14.682316 | 2.7628433 | 2.6571026 | 39.012422 | ||
| 14 |
(3-Naphthalen-2-ylmethyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl)-acetonitrile 291.31 AIDS352900 |
|
N#CCN1/C=C\C(=O)N(C1=O)Cc3cc2ccccc2cc3 | 4.435333 | 1.16 | 25.888824 | 1.3140309 | 9.850919 | 255.02871 |
|
O=C1N(Cc(cc(ccc3)c2c3)cc2)C(N(/C=C1\)CC#N)=O | 0.69 | 14.829177 | 2.6159392 | 2.8343888 | 42.031656 | |||
| 15 |
(3-Biphenyl-4-ylmethyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl)-acetonitrile 317.35 AIDS352901 |
|
N#CCN1/C=C\C(=O)N(C1=O)Cc2ccc(cc2)c3ccccc3 | 4.236572 | 1.25 | 27.000458 | 1.2990428 | 10.39244 | 280.60074 |
|
O=C1N(Cc(c2)ccc(c(cc3)ccc3)c2)C(N(/C=C1\)CC#N)=O | 0.79 | 15.020930 | 2.3806514 | 3.1547941 | 47.387942 | |||
| 16 |
1-Benzyl-3-phenyl-1H-pyrimidine-2,4-dione 278.31 AIDS352902 |
|
c1ccccc1CN2/C=C\C(=O)N(C2=O)c3ccccc3 | 3.665546 | 1.55 | 28.617336 | 1.4763650 | 9.691822 | 277.35413 |
|
O=C1N(c(cc2)ccc2)C(N(/C=C1\)Cc(ccc3)cc3)=O | 0.54 | 16.311764 | 2.7002385 | 3.0204302 | 49.268547 | |||
| 17 |
1,3-Dibenzyl-1H-pyrimidine-2,4-dione 292.34 AIDS352903 |
|
c1ccccc1CN2/C=C\C(=O)N(C2=O)Cc3ccccc3 | 4.954677 | 1.53 | 27.627131 | 1.4262804 | 9.685028 | 267.56953 |
|
O=C1N(Cc(ccc2)cc2)C(N(/C=C1\)Cc(ccc3)cc3)=O | 1.06 | 15.538736 | 2.6492805 | 2.9326332 | 45.569415 | |||
| 18 |
1-Benzyl-3-(3,5-dimethyl-benzyl)-1H-pyrimidine-2,4-dione 320.39 AIDS352096 |
|
c1ccccc1CN2/C=C\C(=O)N(C2=O)Cc3cc(C)cc(C)c3 | 6.630784 | 1.84 | 25.860489 | 0.7302591 | 17.70638 | 457.89563 |
|
O=C1N(Cc(cc(C)c2)cc2C)C(N(/C=C1\)Cc(ccc3)cc3)=O | 1.81 | 14.540931 | 1.5875011 | 4.5798175 | 66.594813 | |||
| 19 |
1-Benzyl-3-(4,6-dimethyl-pyridin-2-ylmethyl)-1H-pyrimidine-2,4-dione 321.38 AIDS352904 |
|
c1ccccc1CN2/C=C\C(=O)N(C2=O)Cc3nc(C)cc(C)c3 | 5.136082 | 0.41 | 26.114347 | 0.8253111 | 15.82091 | 413.15277 |
|
O=C1N(Cc(cc(C)c2)nc2C)C(N(/C=C1\)Cc(ccc3)cc3)=O | 0.15 | 14.748792 | 1.7122755 | 4.3067812 | 63.519822 | |||
| 20 |
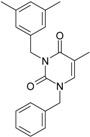
1-Benzyl-3-(3,5-dimethyl-benzyl)-5-methyl-1H-pyrimidine-2,4-dione334.42 AIDS352905 |
|
c1ccccc1CN2/C=C\(C)C(=O)N(C2=O)Cc3cc(C)cc(C)c3 | 5.841637 | 2.12 | 25.007275 | 1.0403700 | 12.01845 | 300.54873 |
|
O=C1N(Cc(cc(C)c2)cc2C)C(N(/C=C1\C)Cc(ccc3)cc3)=O | 2.39 | 14.063834 | 2.1272754 | 3.3055978 | 46.489379 | |||
| 21 |
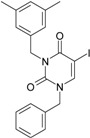
1-Benzyl-3-(3,5-dimethyl-benzyl)-5-iodo-1H-pyrimidine-2,4-dione 446.29 AIDS352906 |
|
c1ccccc1CN2/C=C\(I)C(=O)N(C2=O)Cc3cc(C)cc(C)c3 | 4.379863 | 2.48 | 25.393186 | 0.8931783 | 14.21507 | 360.96592 |
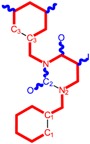
|
O=C1N(Cc(cc(C)c2)cc2C)C(N(/C=C1\I)Cc(ccc3)cc3)=O | 2.53 | 13.656576 | 1.4894424 | 4.5844594 | 62.608023 | |||
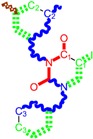
| 22 |
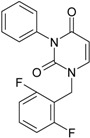
1-(2,6-Difluoro-benzyl)-3-phenyl-1H-pyrimidine-2,4-dione 314.29 AIDS352907 |
|
Fc1cccc(F)c1CN2/C=C\C(=O)N(C2=O)c3ccccc3 | 3.690369 | 1.08 | 28.610234 | 1.4786792 | 9.674253 | 276.78264 |
|
O=C1N(c(cc2)ccc2)C(N(/C=C1\)Cc(c(F)cc3)c(F)c3)=O | −0.66 | 16.175016 | 2.7665356 | 2.9233342 | 47.284980 | |||
| 23 |
1-(2,6-Difluoro-benzyl)-3-(3,5-dimethyl-benzyl)-1H-pyrimidine-2,4-dione 356.37 AIDS352908 |
|
Fc1cccc(F)c1CN2/C=C\C(=O)N(C2=O)Cc3cc(C)cc(C)c3 | 6.939302 | 1.37 | 25.844444 | 0.7517152 | 17.19032 | 444.27415 |
|
O=C1N(Cc(cc(C)c2)cc2C)C(N(/C=C1\)Cc(c(F)cc3)c(F)c3)=O | 0.6 | 14.486247 | 1.5713578 | 4.6094680 | 66.773895 | |||
| 24 |
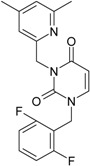
1-(2,6-Difluoro-benzyl)-3-(4,6-dimethyl-pyridin-2-ylmethyl)-1H-pyrimidine-2,4-dione357.36 AIDS352909 |
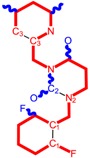
|
Fc1cccc(F)c1CN2/C=C\C(=O)N(C2=O)Cc3nc(C)cc(C)c3 | 5.193820 | −0.06 | 26.085800 | 0.8406863 | 15.51458 | 404.71036 |
|
O=C1N(Cc(cc(C)c2)nc2C)C(N(/C=C1\)Cc(c(F)cc3)c(F)c3)=O | −1.05 | 14.690744 | 1.6779412 | 4.3776098 | 64.310348 | |||
| 25 |
1-(2,6-Difluoro-benzyl)-3-(2,6-dimethyl-pyridin-4-ylmethyl)-1H-pyrimidine-2,4-dione 357.36 AIDS352910 |
|
Fc1cccc(F)c1CN2/C=C\C(=O)N(C2=O)Cc3cc(C)nc(C)c3 | 3.886056 | 0.57 | 26.493803 | 0.9063530 | 14.61561 | 387.22308 |
|
O=C1N(Cc(cc(C)n2)cc2C)C(N(/C=C1\)Cc(c(F)cc3)c(F)c3)=O | 0.77 | 14.950333 | 1.7825743 | 4.1934669 | 62.693730 | |||
| 26 |
1,3-Bis-(2,6-difluoro-benzyl)-1H-pyrimidine-2,4-dione 364.30 AIDS352911 |
|
Fc1cccc(F)c1CN2/C=C\C(=O)N(C2=O)Cc3c(F)cccc3F | 4.379863 | 0.59 | 27.958833 | 1.5546911 | 8.991764 | 251.39924 |
|
O=C1N(Cc(c(F)cc2)c(F)c2)C(N(/C=C1\)Cc(c(F)cc3)c(F)c3)=O | −1.34 | 15.611849 | 2.8690618 | 2.7207236 | 42.475527 | |||
| 27 |
3-(3,5-Dimethyl-benzyl)-1-phenethyl-1H-pyrimidine-2,4-dione334.42 AIDS352912 |
|
c1ccccc1CCN2/C=C\C(=O)N(C2=O)Cc3cc(C)cc(C)c3 | 5.206209 | 2.09 | 25.447501 | 0.8335692 | 15.26418 | 388.43520 |
|
O=C1N(Cc(cc(C)c2)cc2C)C(N(/C=C1\)CCc(cccc3)c3)=O | 2.06 | 14.410323 | 1.8477646 | 3.8993936 | 56.191522 | |||
| 28 |
3-(3,5-Dimethyl-benzyl)-1-prop-2-ynyl-1H-pyrimidine-2,4-dione 268.32 AIDS352913 |
|
C#CCN1/C=C\C(=O)N(C1=O)Cc2cc(C)cc(C)c2 | 5.966576 | 0.77 | 21.628890 | 1.4603086 | 7.405589 | 160.17466 |
|
O=C1N(Cc(cc(C)c2)cc2C)C(N(/C=C1\)CC#C)=O | 1.18 | 12.392809 | 2.5046350 | 2.4739751 | 30.659502 | |||
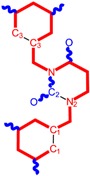
| 29 |
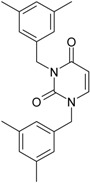
1,3-Bis-(3,5-dimethyl-benzyl)-1H-pyrimidine-2,4-dione348.44 AIDS352914 |
|
c1c(C)cc(C)cc1CN2/C=C\C(=O)N(C2=O)Cc3cc(C)cc(C)c3 | 6.283996 | 2.14 | 25.233546 | 0.8800182 | 14.33694 | 361.77196 |
|
O=C1N(Cc(cc(C)c2)cc2C)C(N(/C=C1\)Cc(cc(cc3C)C)c3)=O | 2.55 | 14.566107 | 1.9376961 | 3.7586149 | 54.748388 | |||
| 30 |
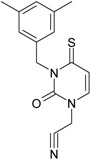
[3-(3,5-Dimethyl-benzyl)-2-oxo-4-thioxo-3,4-dihydro-2H-pyrimidin-1-yl]-acetonitrile 285.36 AIDS352915 |
|
N#CCN1/C=C\C(=S)N(C1=O)Cc2cc(C)cc(C)c2 | 7.309803 | 1.28 | 21.897722 | 1.6182386 | 6.765913 | 148.15807 |
|
S=C1N(Cc(cc(C)c2)cc2C)C(N(/C=C1\)CC#N)=O | 1.68 | 12.764862 | 3.0237637 | 2.1107572 | 26.943525 | |||
| 31 |
1-Benzyl-3-(3,5-dimethyl-benzyl)-4-thioxo-3,4-dihydro-1H-pyrimidin-2-one 336.45 AIDS352916 |
|
c1ccccc1CN2/C=C\C(=S)N(C2=O)Cc3cc(C)cc(C)c3 | 7.292429 | 2.49 | 25.217792 | 1.1471616 | 10.99139 | 277.17849 |
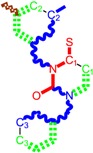
|
S=C1N(Cc(cc(C)c2)cc2C)C(N(/C=C1\)Cc(ccc3)cc3)=O | 2.45 | 14.289267 | 2.4197012 | 2.9526925 | 42.191813 | |||
| 32 |
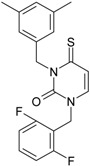
1-(2,6-Difluoro-benzyl)-3-(3,5-dimethyl-benzyl)-4-thioxo-3,4-dihydro-1H-pyrimidin-2-one 372.43 AIDS352917 |
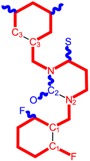
|
Fc1cccc(F)c1CN2/C=C\C(=S)N(C2=O)Cc3cc(C)cc(C)c3 | 7.229147 | 2.02 | 25.321304 | 1.0761564 | 11.76469 | 297.89740 |
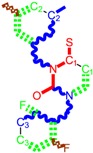
|
S=C1N(Cc(cc(C)c2)cc2C)C(N(/C=C1\)Cc(c(F)cc3)c(F)c3)=O | 1.25 | 14.434969 | 2.3806265 | 3.0317586 | 43.763344 | |||
the longest SMILES molecular chain (LoSMoC), when bonds are breaking on aromatic rings and moieties such that the resulting molecule displays a sort of 2D form of the original molecule along the “fractalic” chain, assumed to be the first stage in intermediary molecular defolding targeting the receptor. The maximum SMILES chains in LoSMoC are presumably responsible for best transport/transduction of ligand molecules through cellular (lipidic) walls, after which they may be released with a modified structure due to their further ionization resulting from interactions with cellular layers; accordingly, another SMILES form is generated and considered next, namely:
the Branching SMILES (BraS), representing the second phase of molecular defolding and providing ligand bond breakages such that many “bays” are formed, yet with consistent “arms” linking the short molecular “skeleton” aiming to favor the binding with a receptor in its pockets. Accordingly, the branching is not necessary in the same points of molecules through a series, but the maximum branching combined with equilibrium of branches is to be obtained in the final BraS. For instance, a long branch adjacent to a short one will not make a strong enough “float” to bind in a receptor pocket; therefore, the branching principle is to have the float-clefs balanced among themselves. To this end branching up to fourth order is performed for the molecules in Table 1.
However, one should note that the fact that the most drugs are ionized once immersed in a biological medium is in accordance with the present two-steps of SMILES conformations, since in each of them more nucleophilic compounds are considered due to the successive bonding breaking and the loss of pairs of electrons as the unfolding goes from the original to the LoSMoC to the BraS configuration. These SMILES metabolic intermediates acting as nucleophilic active sides are confirmed at least for fused and non-fused diazines [85], among which are also those based on pyrimidines, which have already demonstrated antiviral and anti-HIV activity [86,87,88,89] and antiinflammatory effects in general [90,91,92].
2.2. OECD-QSAR Principle 2: An Unambiguous Algorithm
According to the OECD guidance, the intent of QSAR-Principle 2 (unambiguous algorithm) is to ensure transparency in the predictive algorithm. In order to achieve this aim one needs reliable descriptors with physicochemical relevance. In this regard, the present QSAR modeling of ani-HIV activity employs the so called chemical orthogonal space–COS of chemical bonding [54,55], which is based on the main chemical reactivity indices and the principles of electronegativity (χ) and chemical hardness (η), alongside their related quantities such as chemical power index (π) and electrophilicity (ω) [93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135]. Their detailed description follows with the aim of better understanding the forthcoming QSAR- based mechanism of anti-HIV action for the present pool [136,137,138,139] of molecules.
2.2.1. Electronegativity and Its Principles
Electronegativity is viewed as an instantaneous variation of total (or valence) energy for a neutral or charged system with N-electrons [93]:
| (1) |
It may be also be related to frontier electronic behavior by performing the central finite difference development of equation (1) in terms of ionization potential (IP) and electronic affinity (EA), thus facilitating further connection with the highest occupied and lowest unoccupied molecular orbitals, (HOMO and LUMO), respectively, according to Koopmans’ frozen spin orbitals’ theorem [94]:
| (2) |
As such, in the course of a chemical reaction, or in chemical reactivity in general, electronegativity basically assures energetic stabilization through equalization of middle HOMO-LUMO levels among ligand (L) and receptor (R) active molecular structures; this is sustained by its inner definition [equation (1)] which identifies it with the negative of the chemical potential of a system, as according to Parr et al. [95], by the natural thermodynamic law of two fluids in contact its complex evolves towards equalization of the individual chemical potentials into a global one, while this principle, for the electronic fluid systems, was already consecrated from solid state physics [96], in chemistry it was coined by the so called electronegativity equalization (EE) principle ( ), as originally stated by Sanderson under the assumption that “for molecules in their fundamental state, the electronegativities of different electronic regions in the molecule—are equal” [97]; however, its variational form was recently clarified within the context of the double variational procedure [98], specific to chemical systems:
| (3) |
under the minimum electronegativity principle stating that: “a chemical reaction is promoted so as to minimize further charge transfer between atoms-in-molecules or between molecular fragments within a complex” [99,100,101]. Nevertheless it was firstly formulated by Parr and Yang as under the maximum form favoring chemical reactivity [102,103]: “given two different sites with generally similar disposition for reacting with a given reagent, the reagent prefers the one which on the reagent’s approach is associated with the maximum response of the system’s electronegativity. In short, is good for reactivity (n. a.)”. Yet, for assessing the chemical stability the reverse form of the latter idea will be considered, from where the minimum electronegativity principle immediately results. However, in order to not conflict with the equality of electronegativity, this principle should be seen as a quantum fluctuation remnant effects in system upon the EE was consumed, i.e., it needs to be minimized so that the system reaches stable equilibrium [104].
2.2.2. Chemical Hardness and Its Principles
Chemical hardness is viewed as the instantaneous electronegativity change with charge [105]:
| (4) |
It also supports the Koopmans’ frozen spin orbitals reformulation at the level of molecular frontier, i.e., there where chemical reactivity takes place, through the expression [106]:
| (5) |
At this point, while comparing Equations (2) and (5), it is clear that the electronegativity and chemical hardness may be viewed as the basis for an orthogonal space for chemical reactivity analysis since the conceptual and practical differences noted between the energetic level characterizing the “experimental” electronegativity and the energetic gap characterizing the “experimental” chemical hardness, respectively [107,108].
Like electronegativity, chemical hardness also supports two types of equations accompanying the chemical reactions and transformations. The first one promoting equalization of chemical hardness of the atoms in a molecule or between molecular fragments in a complex or between adducts in a chemical bond refers to the so called the hard and soft acids and bases (HSAB) principle [109,110,111]; it was initially formulated by Pearson and says that “the species with a high chemical hardness prefer the coordination with species that are high in their chemical hardness, and the species with low softness (the inverse of the chemical hardness) will prefer reactions with species that are low in their softness, respectively” [112]. This leads to numerous applications in both inorganic and organic chemistry, since it practically reshapes the basic Lewis and Brönsted qualitative theories of acids and bases [113] into a rigorous orbital-based rule of chemical reactivity and bonding quantification. Nevertheless, being of a quantum nature, chemical hardness inherently contains fluctuations leading to the inequality or variational form of its evolution towards bonding stabilization; as such, within the abovementioned double-variational variational formalism the actual maximum hardness principle is advanced [114,115,116]:
| (6) |
stating that the charge transfer during a chemical reaction or binding continues until the resulted bonded complex acquires maximum stability through hardness; i.e., maximizing the HOMO-LUMO energetic gap thus impeding further electronic transitions [117]. It was originally based on the Pearson observation according which “there seems to be a rule of nature that molecules (or the many-electronic systems in general; n. a.) arrange themselves (in their ground or valence states; n. a.) to be as hard as possible” [113]; it also leads to the practical application merely through its inverse formulation; the chemical softness is in turn related with the polarizability features of a system; i.e., as an observable quantity rooted in the quantum structure of the system; so that the minimum polarization principle was actually tested for various chemical systems [118]; e.g., to rotational barriers accounting for conformational properties and thus with the steric effects [119]; such that the actual chemical hardness variational principle of equation (6) is also indirectly validated.
2.2.3. Chemical Power and Its Principle
Since noting the opposition of electronegativity and chemical hardness, i.e., being the former associated with the tendency of the system to attract electrons and the latter with the tendency to inhibit the coordination and with the system stability, one may introduce the concept of chemical power, as the dynamic charge of atoms in a molecule, between molecular fragments or between adducts in a chemical bond, through the basic definition [59]:
| (7) |
Initially, expression (7) was recognized as maximum electronic uptake in a bonding [120], yet one actually realizes that it gives us a sort of “reduced” or “normalized” electronegativity when its inertial hardness also counts. Moreover, for establishing a quantitative meaning one considers the Cartesian system where the coordinates are the hardness (on abscise) and electronegativity (on ordinate), see Figure 1a; in this framework there follows that:
| (8) |
Figure 1.

(a) Orthogonal hardness-electronegativity (η – χ) representation for an electronic system with coordinate A (ηA, χA); (b) the “ABB” mechanism of frontier chemical reactivity driven by chemical power in A-B bonding complex.
The last identity in (8) follows from chemical hardness-to-electronegativity definition (4) and allows the practical interpretation of chemical power in the chemical reactivity and bonding realm, providing the electronic charge transfer released by the adduct “A” when in bonding in an “A-B” complex, see Figure 1b.
Accordingly, the original frontier orbital HOMOA is minimized to the HOMOB in bonding, through the intermediate LUMOB. In variational terms, the chemical power index is associated with minimizing HOMOs in bonding by means of charge transfer without spin changing:
| (9) |
While principle (9) is consistent with principles relating minimum electronegativity and inverse of maximum of chemical hardness, it also emphasizes the necessity of the double variational principle when combined with Equation (8), i.e., the released charge transfer of A in bonding is minimized so as to fit with the HOMO of bonding; in other terms, LUMO/HOMOA and LUMO/HOMOB levels also tend to equalize in bonding thus jointly fulfilling the conditions of equalization of electronegativity and chemical hardness.
2.2.4. Electrophilicity and Its Principle
Electrophilicity [120], further allows coupling of chemical power index with electronegativity to provide the energetic information of activation towards charge tunneling of the potential between adducts [59,64]:
| (10) |
Electrophilicity actually accounts for energy consumed by a system for manifesting its chemical power in a chemical orthogonal space see Figure 2a, essentially complementing it in bonding by electron transfer through tunneling between the bonding adducts, having the parent LUMO as an intermediate state, see Figure 2b as “orthogonal/complementary” to that of Figure 1b.
Figure 2.

(a) Orthogonal representation of chemical power-electrophilicity (π – ω) scheme for a parabolic form of total energy respecting the number of electrons for an elementary reactivity (accepting-donating) range; (b) the “AAB” mechanism of frontier chemical reactivity driven by electrophilicity in a A-B bonding complex as the complementary/orthogonal one respecting the “ABB” counterpart of Figure 1(b).
As a mixed reactive index electrophilicity was developed to characterize the electrophilic/ nucleophilic action of charge transfer through accepting/donating electrons, in modeling a variety of physical-chemical phenomena such as site selectivity [121,122], molecular vibrations and rotation [123], intramolecular and intermolecular reactivity patterns [124,125], solvent and external field effects [126,127,128] as well as biological activity and toxicity [59,64,129,130,131,132,133,134,135].
However, electrophilicity involves even stronger than the chemical power the double minimum character (through squaring of electronegativity and of its principle) which corresponds to charge penetration of the A-B energetic barrier towards fulfilling electronic pairing in a bonded complex. In practical circumstances, electrophilicity drives the electronic jump from HOMOA to LUMOA then relaxes to HOMOB in an “A-B” bond complex thus covering the “AAB” pathway in chemical reactivity and bonding, see Figure 2b; in this case it minimizes the LUMOB-HOMOB gap, as the inverse of chemical hardness as promoted by equation (10), that nevertheless leaves the bonding complex in an activated state which competes with minimization of electronegativity (through pairing) which tends to stabilizes the structure. For this reason the overall variational principle of electrophilicity assumes its minimization form:
| (11) |
yet whether this is a characteristic of a reactive or stabilized bonding system remains an open issue and should be assessed for each case under study.
These reactivity indices and principles are suited for analyzing the molecular interaction mechanism for a bonding complex chemically formed in a chemical-biological interaction, as is the present anti-HIV concerned action.
2.3. OECD-QSAR Principle 3: A Defined Domain of Applicability
OECD guidance justifies the need to define an applicability domain (Principle 3) by the fact that (Q)SARs are reductionist models with inevitable limitations. These include limitations in terms of the types of chemical structures, physicochemical properties and mechanisms of action for which the models can generate reliable predictions [136,137,138,139].
This principle is inherently linked with the first OECD-QSAR endpoint criterion but equally influences the final mechanism of action, the “OECD-QSAR fifth commandment”. However, in the present anti-HIV study, establishing the domain of applicability is associated with the SMILES screening in searching of the working (trial) test of molecules, among the molecules of Table 1, as follows.
The given chemical structures are employed via their highest occupied and lowest unoccupied molecular orbitals, HOMO and LUMO, respectively, to provide the basic chemical reactivity indices as such the electronegativity and chemical hardness, chemical power and electrophilicity, as previously described, since they are naturally interpreted in a successive and combined QSAR models by their associate chemical principles [59].
To accomplish such a goal, an original step was recently undertaken when for a QSAR series one considers also their counterpart Simplified Molecular Input Line Entry System (SMILES) transformations, which were assumed as being responsible for an intermediate stage in the molecular interaction mechanism targeting the receptor site [64]. However, the present endeavor continues the approach where the SMILES molecules were involved only in the screening for QSAR modeling, by effectively invoking also the SMILES structures in the QSAR models employed. This is because in the former stage the ligand-receptor binding mechanism remained unfinished at the level of activated 1,3-disubstituted uracil-reverse transcriptase complex, while the present ansatz is that the activated complex will be eventually relax and this can be studied by considering the computed structure parameters for the SMILES counterparts. Yet, as was pointed out in the previous paper [64], the mechanistic QSARs should be always driven and selected by the variational min-max principles, at all stages of conceptual and computational analysis. They will also be considered in the present analysis, having the additional SMILES molecular configurations as intermediates between the free molecules and the molecules binding to the biological receptor.
Computationally, this behavior is reflected in considering the SMILES forms of Table 1 in ionization [+2n] states, with “n” representing the number of broken bonds in the gas-phase molecule. The computational framework chosen was the semiempirical AM1 as executed in the Hyperchem code [140], with which help the respective HOMO and LUMO states were determined, beyond the first order of frontier orbitals used in “custom” chemical reactivity calculations; see equations (2) and (5) for electronegativity and chemical hardness, respectively. This approach is also consistent with the “branching” effect at the energetic level of SMILES structures. Fortunately, within Koopmans’ approximation, such formulations exist up to the third order of compact finite differences and they look like [54,106,141]:
| (12) |
| (13) |
When they are employed here under the spectral-like-resolution numerics [142], equations (12) and (13) reduce to the working ones [54,106,141]:
| (14) |
| (15) |
The analytical descriptors of equations (14) and (15) greatly help in considering the chain and branching modeling of actual molecules as being differentiated for LoSMoC and BraS intermediates also at the level of frontier chemical reactivity. As reported in Table 2:
Table 2.
The AM1 computed values (in electron-volts, eV) for the first three highest occupied and lowest unoccupied molecular orbitals in both variants as the longest SMILES molecular chain (LoSMoC, upper entry) and the Branching SMILES (BraS, lower entry), employed for computation of electronegativity (χ), chemical hardness (η), chemical power (π) and electrophilicity (ω), for the compounds of Table 1. Note that, in either LoSMoC or BraS forms, the overall compound was considered as carrying the [+2n] charge due to removed electronic pair out of each “broken bond” in SMILES configurations for compounds of Table 1. “X” indicates the truncation to the first order of HOMO and LUMO in LoSMoC calculations of electronegativity and chemical hardness of equations (14) and (15), respectively.
| No. | HOMO1 | LUMO1 | HOMO2 | LUMO2 | HOMO3 | LUMO3 |
|---|---|---|---|---|---|---|
| ... LoSMoC ... | ||||||
| ... BraS ... | ||||||
| 1 | −24.49903 | −19.06514 | X | X | X | X |
| −24.48801 | −19.03451 | −24.88237 | −18.05411 | −25.10602 | −15.57611 | |
| 2 | −24.24188 | −18.7667 | X | X | X | X |
| −24.23715 | −18.75946 | −24.69547 | −17.93489 | −24.84179 | −15.32821 | |
| 3 | −24.25602 | −18.82835 | X | X | X | X |
| −24.2567 | −18.82977 | −24.58191 | −17.70927 | −24.85183 | −15.37954 | |
| 4 | −23.95141 | −18.83626 | X | X | X | X |
| −23.95204 | −18.83621 | −24.28008 | −17.60903 | −24.88104 | −15.38259 | |
| 5 | −24.38787 | −18.90236 | X | X | X | X |
| −24.38787 | −18.90236 | −24.42277 | −17.3528 | −24.90982 | −15.43411 | |
| 6 | −24.24172 | −18.95968 | X | X | X | X |
| −24.23569 | −18.95218 | −24.49526 | −17.86779 | −25.04196 | −15.49452 | |
| 7 | −23.35131 | −18.73365 | X | X | X | X |
| −23.35188 | −18.73767 | −24.22514 | −17.79723 | −24.54057 | −15.31291 | |
| 8 | −23.79299 | −18.65147 | X | X | X | X |
| −23.79239 | −18.65293 | −24.13254 | −17.38683 | −24.7122 | −15.20959 | |
| 9 | −23.46857 | −18.83136 | X | X | X | X |
| −23.46979 | −18.83921 | −24.28395 | −17.49789 | −24.46192 | −15.37871 | |
| 10 | −24.37925 | −18.94867 | X | X | X | X |
| −24.38 | −18.9506 | −24.99142 | −18.7636 | −25.14861 | −15.67584 | |
| 11 | −22.38345 | −18.75445 | X | X | X | X |
| −22.38345 | −18.75445 | −23.79029 | −17.31787 | −24.21747 | −15.29094 | |
| 12 | −21.96501 | −18.31488 | X | X | X | X |
| −21.96844 | −18.31149 | −23.85501 | −16.16856 | −23.89945 | −14.89846 | |
| 13 | −26.91465 | −21.85561 | X | X | X | X |
| −26.91465 | −21.85561 | −27.7802 | −20.69252 | −28.33738 | −18.9827 | |
| 14 | −26.66128 | −22.14708 | X | X | X | X |
| −26.66128 | −22.14708 | −27.47999 | −20.30796 | −27.875 | −19.37649 | |
| 15 | −27.68342 | −23.22071 | X | X | X | X |
| −27.68553 | −23.22033 | −28.91865 | −22.96564 | −28.9519 | −21.82943 | |
| 16 | −29.51216 | −24.44028 | X | X | X | X |
| −29.52823 | −24.42876 | −29.75423 | −23.06013 | −30.9813 | −22.86666 | |
| 17 | −28.49271 | −23.59289 | X | X | X | X |
| −28.47523 | −23.5581 | −29.36592 | −22.66404 | −30.06262 | −21.75203 | |
| 18 | −25.63183 | −23.12311 | X | X | X | X |
| −25.62548 | −23.11217 | −26.88207 | −22.20886 | −27.55654 | −20.01182 | |
| 19 | −26.0344 | −23.19914 | X | X | X | X |
| −26.03953 | −23.19181 | −27.0533 | −22.2293 | −27.84065 | −20.10159 | |
| 20 | −25.36022 | −21.78615 | X | X | X | X |
| −25.36493 | −21.78792 | −26.68329 | −20.71338 | −27.06831 | −19.37157 | |
| 21 | −25.47117 | −22.40276 | X | X | X | X |
| −24.47218 | −22.40179 | −26.77381 | −21.92655 | −27.2391 | −19.67952 | |
| 22 | −29.50944 | −24.42961 | X | X | X | X |
| −29.5088 | −24.42942 | −30.31708 | −23.21535 | −30.95683 | −22.84796 | |
| 23 | −25.65356 | −23.07113 | X | X | X | X |
| −25.6511 | −23.06654 | −26.89824 | −22.43524 | −27.60502 | −19.97251 | |
| 24 | −26.0339 | −23.14582 | X | X | X | X |
| −26.03578 | −23.15325 | −27.02319 | −22.45168 | −27.88159 | −20.08076 | |
| 25 | −26.5313 | −23.41763 | X | X | X | X |
| −26.55279 | −23.43647 | −27.38203 | −22.60093 | −28.51525 | −20.85345 | |
| 26 | −29.02596 | −23.685 | X | X | X | X |
| −28.90689 | −23.43443 | −29.78576 | −22.74551 | −29.8298 | −21.98785 | |
| 27 | −25.41998 | −22.55635 | X | X | X | X |
| −25.42157 | −22.55341 | −26.66657 | −21.11333 | −27.34483 | −19.49103 | |
| 28 | −22.8969 | −17.88018 | X | X | X | X |
| −22.90148 | −17.87531 | −22.96332 | −17.29096 | −24.06666 | −14.20252 | |
| 29 | −25.29808 | −22.27488 | X | X | X | X |
| −25.30234 | −22.27582 | −25.91112 | −20.09649 | −26.56577 | −19.35705 | |
| 30 | −23.42159 | −17.86232 | X | X | X | X |
| −23.42258 | −17.86581 | −23.58074 | −15.8222 | −23.9511 | −15.31823 | |
| 31 | −25.7421 | −21.80116 | X | X | X | X |
| −25.74458 | −21.80102 | −27.02937 | −20.01162 | −27.54453 | −19.79035 | |
| 32 | −25.71771 | −22.0207 | X | X | X | X |
| −25.71681 | −22.02284 | −27.02077 | −19.81276 | −27.47808 | −19.75182 | |
We consider only first orders of HOMO and LUMO for the LoSMoC molecules of Table 1;
We consider all three orders of HOMO and LUMO for the BraS molecules of Table 1.
Values of χ & η, of Table 1 are based on the HOMO and LUMO entries of Table 2 combined with equations (14) and (15). They are further implemented in π & ω of equation (7) and (10) to provide the respective LoSMoC and BraS results in Table 1 as well.
Along with the different hydrophobicities for LoSMoC and BraS molecules, these chemical-physical descriptors are further employed by QSAR modeling to explain the chemical-biological binding of the actual series of pyrimidines to the reverse-transcriptase enzyme in HIV cells causing its inhibition for further action against the host organism’s cells. Actually, we have to explain by variational QSAR models how the anti-HIV mechanism of Figure 3 [143,144,145,146,147,148,149,150,151] is possible by means of SMILES chain and branching intermediates such as the LoSMoC and BraS conformations considered in Table 1 and based only on their chemical reactivity descriptors.
Figure 3.

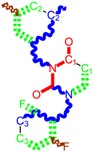
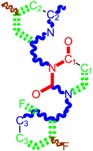
The mechanism of molecular interaction of the 1,3-disubstituted uracils, with prototype no. 31 of Table 1 (since belonging to all selected QSAR-SMILES criteria and cases of Table 3) against human immunodeficiency virus (HIV-1), after Ref. [143], through five stages: (A) the free molecular attack on the HIV viral envelope, after Ref. [144]; (B) the passage of the lipidic viral envelope of HIV under the form of longest SMILES molecular chain (LoSMoC) of Table 1, after Refs. [64,145,146]; (C) the transport though the protein layer of HIV capsid, after Refs. [147,148], yielding the Branching SMILES (BraS) configuration of Table 1 that further binds in (D) with the palm active region of p66 monomer of reverse transcriptase (RT), after Refs. [149,150], towards (E) the competitively inhibiting the RT by the formed ligand-receptor complex, after Ref. [151], by means of chemical reactivity frontier electronic transfer as detailed in the Figure 4.
QSAR analysis requires a preliminary screening such that out of the available pool of molecules the ones that further fulfill certain similarity criteria with an increased degree of correlation are retained.
This stage is presented in Table 3 separately for LoSMoC and BraS and for each such molecular defolding, and separately for the SMILES central chain case (i) as well as for the N3- pyrimidine atom neighbors case (ii) due to its central role in obtaining the spiroheterocyclic compounds and their reaction pathways [85], which are also presumably defolded in the chemical-biological interaction. Note that, consistent with the previous branching considerations the criteria for BraS are almost doubled with respect to the LoSMoC. The results of Table 3 leaves us with two sets of molecules for each SMILES intermediate, while they are not necessary selected based only upon the highest correlation factor recorded, but through a compromise between the correlation factor and the number of chemical reactivity variables and with the number of compounds employed in the correlation. As such, for each LoSMoC/BraS cases (i)/(ii) one should chose the molecular sets presenting the best combination between:
Table 3.
Case (i): screening based on SMILES central chain and Case (ii): screening based on SMILES central N-atom neighbors (N3 atom of the pyrimidine) for chain length and atomic neighboring in longest SMILES molecular chain (LoSMoC) in upper entry and the Branching SMILES (BraS) in down entry for various versions (V’s) of SMILES based screening criteria along the molecules of Table 1, respectively. The correlation factors are given for full dependency of parameters of Table 1, i.e., A = A (χ, η, π, ω, logP), and for statistical error tolerance of 0.0001, unless otherwise indicated for the best correlation’s combination such that the Topliss-Costello rule [152] for ratio molecule-to-descriptors ≥ 4 to be generally respected (at least for χ & η as the main QSAR descriptors); the marked correlation corresponds with selected criteria and implicitly with the working molecular pool of Table 1 for each SMILES configuration (LoSMoC and BraS) and screening case (i and ii) further considered (see text).
| Index | Criteria | CASE (i) | Case (ii) | ||
|---|---|---|---|---|---|
| Molecules | RQSAR | Molecules | RQSAR | ||
| V1 LoSMoC | Between 15–16 atoms LoSMoC | 1–4, 6–11, 28 | 0.90371960 (a) | 1–9, 28 | 0.92402295 (c) |
| V1 BraS | Main chain and secondary branch with maximum 14 atoms | 2–11, 13, 14, 16, 17, 22, 28 | 0.53158997 | 2, 3, 5–9, 13, 14, 16, 17, 22, 28, 29 | 0.70384894 |
| V2 LoSMoC | Between 18–21 atoms LoSMoC | 13–17, 19, 21, 22, 24, 26, 31, 32 | 0.75180080 | 15–18, 21–23, 27, 29, 31, 32 | 0. 95150144 (b) |
| V2 BraS | Main chain and secondary branch with minimum 14 atoms | 7, 11, 12, 15–17, 19, 22, 24–26, 28, 30–32 | 0.95109419 | 7, 15–17, 20, 21, 22, 27, 28, 29, 30–32 | 0.87354213 |
| V3 LoSMoC | At least one triple bond in the main chain LoSMoC | 1–7, 9–11, 13, 14, 28, 30 | 0.56411064 | 1–4, 6, 7, 9, 13, 15, 28, 30 | 0.49202776 |
| V3 BraS | Secondary and tertiary branches with maximum 14 atoms | 2–10, 13, 14, 28 | 0.62469181 | 1–7, 9, 13, 14, 28 | 0.75756597 |
| V4 LoSMoC | More than three branches in the main chain LoSMoC | 2–4, 6–11, 19, 21, 22, 24–26, 28, 30–32 | 0.43357261 | 2–4, 6, 7, 9, 15,20–23, 27–32 | 0.61510478 |
| V4 BraS | Secondary and tertiary branches with minimum 14 atoms | 11, 15–17, 19, 21–25, 31, 32 | 0.64694148 | 15–17, 20–23, 27, 29, 31, 32 | 0.94183439 |
| V5 LoSMoC | More than four branches in the main chain LoSMoC | 7–9, 11, 19, 21, 24–26, 28, 30–32 | 0.47454364 | 7, 8, 20, 23, 27–32 | 0.71500251 (d) |
| V5 BraS | Minimum 3 tertiary branches | 6, 11, 15–17, 19, 22–26, 31, 32 | 0.94899619 | 6, 15–17, 20–23, 27, 29, 31, 32 | 0.64718879 |
| V6 LoSMoC | Ramifications of LoSMoC main chain containing groups formed only carbon and hydrogen atoms (except common = O, C = O) | 2–4, 6–10, 19, 28, 30, 31 | 0.71050966 (b) | 2–4, 6, 7, 9, 15, 20,27–31 | 0.64508095 |
| V6 BraS | Minimum 1 quaternary branching | 1, 2, 4, 6–8, 10,13–15, 19, 21–25, 28, 30–32 | 0.48549586 | 1, 2, 4, 6–8,13, 14, 20–23, 27–29, 30–32 | 0.63906586 |
| V7 LoSMoC | Ramifications of LoSMoC main chain containing groups consisting of a single atom or –CH3 groups (except common = O, C = O) | 2–7, 9, 10, 19, 22,24–26, 28, 30–32 | 0.57636501 | 2–4, 6, 7, 9, 20–22,27– 32 | 0.61600596 (e) |
| V7 BraS | One of the secondary branches with minimum one triple bond | 1–7, 9–11, 13–15, 28 | 0.63904635 | 1–7, 9, 13–15, 28 | 0.73556023 (d) |
| V8 LoSMoC | At least one branch for the last 6 points main chain LoSMoC | 2–4, 6–11, 19, 23–25, 28, 30, 32 | 0. 51837657 | 2–4, 6, 7, 9, 20, 21, 27–32 | 0.69314160 (d) |
| V8 BraS | The secondary branch linked with C2 of pyrimidinic nucleus with minimum 2 heteroatoms | 1– 6, 8–11, 13– 15 | 0.58368204 | 1–6, 8, 9, 13–15 | 0.57765388 (f) |
| V9 LoSMoC | LoSMoC main chain contains after N3 atom of the pyrimidine nucleus (central main chain LoSMoC) a group –CH2– | 1–7, 9–11, 13–15, 19, 21, 24–26, 28, 30–32 | 0.37650771 | 1–8, 13–15, 20, 21, 27–32 | 0.63047473 |
| V9 BraS | The secondary branch linked with N3 of pyrimidinic nucleus contains only C and H atoms | 1– 8, 10, 11, 13–17, 25, 26, 28, 30–32 | 0.63881109 | 1–8, 13–17, 20, 21, 27–29, 30–32 | 0.72514327 |
| V10 BraS | The secondary branch linked with N3 of pyrimidinic nucleus contains 4 Carbon atoms | 2– 4, 6 8– 10, 13, 14, 16, 19, 22, 24, 26 | 0.61480396 | 2–6, 8, 9, 13, 14, 16, 17, 22 | 0.53480139 |
| V11 BraS | The secondary branch linked with N3 of pyrimidinic nucleus contains 5–6 Carbon atoms | 7, 12, 15, 18, 21, 23, 25, 28, 30–32 | 0.66627959 | 7, 15, 18, 20, 21, 23, 28, 29, 30–32 | 0.59914507 |
| V12 BraS | The tertiary branching are formed by maximum 3 atoms of C and H | 2, 4–10, 13, 16, 19, 21–25, 28, 30–32 | 0.38470862 | 2, 4, 6–9, 13, 16–18, 20, 21, 28–32 | 0.61909773 |
| V13 BraS | The tertiary branches are formed only by C and H atoms | 2– 10, 13, 14, 16, 17, 19, 28, 30, 31 | 0.56415743 | 2–9, 13–16, 20, 27–31 | 0.64691170 |
| V14 BraS | Quaternary branching are contains only one C atom or CH3 group | 1, 2, 5–7, 21–25, 28, 30–32 | 0.57731047 | 2, 5, 6, 20–23, 27, 28, 30–32 | 0.72850903 |
| V15 BraS | A single quaternary branching with maximum 2 atoms (C/O) and H | 1, 2, 5– 7, 19, 21, 22, 28, 30, 31 | 0.93051865 | 1, 2, 5, 6, 20–22, 27, 28, 30, 31 | 0.90565106 |
(a) A = A(χ, η, ω, logP); (b) within statistical error tolerance 0.00002; (c) within statistical error tolerance 1E−25; (d) within statistical error tolerance 0.00004; (e) within statistical error tolerance 0.00008; (f) within statistical error tolerance 0.00003;
higher correlation factors;
screening correlations having maxima of variables as descriptors;
almost equal sets of compounds producing the precedent points;
sets of compounds fulfilling the Topliss-Costello rule [152], or at least respecting the basic/independent descriptors of electronegativity and chemical hardness plus the hydrophobicity measure.
This way, the selected LoSMoC cases’ variants are:
the case (i)/V2 was chosen over V1 since it better fulfills the above criteria (e.g. being based on all variables and on 12 compounds and not on four variables and 11 compounds like V1);
the case (ii)/V6 was chosen despite the fact versions V1 and V2 have lesser compounds in the set, and to be closer to the previous case, for molecular sets’ cardinals.
On similar grounds, the selected BraS cases’ variants are:
the case (i)/V5 over variant V2 since it has a minimum of three tertiary branching instances, while being in the similar correlation range, so that it better fulfills the “spirit” of molecular branching;
the case (ii)/V2 over versions V4 and V15 (with lesser compounds in the set), being nevertheless in the same range of higher correlations and having the same cardinal of molecules in the set as its companion case (i)/V5
They are further used for integration in appropriate measures towards establishing the anti-HIV mechanism of action.
2.4. OECD-QSAR Principle 4: Appropriate Measures of Goodness-of-Fit, Robustness and Predictivity
OECD-QSAR principle 4 makes a distinction between the internal performance of a model as represented by goodness-of-fit and robustness or the correlation within the trial set of molecules and the predictivity of a model as determined by external validation on a test set of molecules [153,154].
However, in the present work we are considering internal measures of the present QSAR models (unfolded in Table 4) by their minimal search–formally written as:
| (16) |
where are the actual various computed endpoints, by means of the Euclidean paths across the available QSAR models, according with the rule [64]:
| (17) |
with the results presented in Table 5.
Table 4.
Statistical correlation results obtained for cases V2/(i) and V6/(ii) for longest SMILES molecular chain (LoSMoC) and respectively for the cases V5/(i) and V2/(ii) for branching SMILES (BraS) selected compounds’ sets form Table 3 with respective molecules of Table 1 (detailed respective QSAR models dependencies on chemical reactivity parameters are provided in Supplementary Material—Table S1).
| No. | A(x) | LoSMoC | BraS | ||
|---|---|---|---|---|---|
| RCase V2/(i) | RCase V6/(ii) | RCase V5/(i) | RCase V2/(ii) | ||
| I1 | A(logP) | 0.36160241 | 0.43043863 | 0.45645057 | 0.51687516 |
| I2 | A(χ) | 0.70875308 | 0.04142206 | 0.32832072 | 0.63329686 |
| I3 | A(η) | 0.3850668 | 0.27082157 | 0.3694801 | 0.10466918 |
| I4 | A(π) | 0.20001171 | 0.23419593 | 0.23910446 | 0.36217604 |
| I5 | A(ω) | 0.0679732 | 0.21014 | 0.12316764 | 0.52996859 |
| II1 | A(logP, χ) | 0.72462236 | 0.54711991 | 0.54563771 | 0.68322871 |
| II2 | A(logP, η) | 0.53462981 | 0.45498598 | 0.58822038 | 0.78078563 |
| II3 | A(logP, π) | 0.4587341 | 0.47447182 | 0.53086816 | 0.8624387 |
| II4 | A(logP, ω ) | 0.40635079 | 0.49281211 | 0.48406183 | 0.85830581 |
| II5 | A(χ, η) | 0.72042921 | 0.34882836 | 0.44147923 | 0.65793015 |
| II6 | A(χ, π) | 0.72662887 | 0.32861178 | 0.42540934 | 0.67176394 |
| II7 | A(χ, ω) | 0.72663277 | 0.33323936 | 0.41607475 | 0.67165975 |
| II8 | A(η, π) | 0.74023092 | 0.31232276 | 0.46816571 | 0.69205634 |
| II9 | A(η, ω) | 0.74918964 | 0.3278778 | 0.47282745 | 0.6980058 |
| II10 | A(π, ω) | 0.72422189 | 0.31072122 | 0.4687647 | 0.66987725 |
| III1 | A(logP, χ, η ) | 0.72946153 | 0.54741756 | 0.62478127 | 0.83591477 |
| III2 | A(logP, χ, π) | 0.73229267 | 0.54735654 | 0.62197159 | 0.86508134 |
| III3 | A(logP, χ, ω) | 0.73214282 | 0.5471543 | 0.61493693 | 0.86624574 |
| III4 | A(logP, η, π) | 0.74609564 | 0.48854915 | 0.62416978 | 0.87096819 |
| III5 | A(logP, η, ω ) | 0.751297 | 0.51239927 | 0.63374038 | 0.86007179 |
| III6 | A(logP, π, ω) | 0.72648755 | 0.52806785 | 0.65025857 | 0.86552207 |
| III7 | A(χ, η, π) | 0.75053661 | 0.35028746 | 0.4752325 | 0.7019648 |
| III8 | A(χ, η, ω) | 0.74939285 | 0.34885082 | 0.52544907 | 0.70077495 |
| III9 | A(χ, π, ω ) | 0.72663285 | 0.35789332 | 0.83429197 | 0.67179626 |
| III10 | A(η, π, ω) | 0.74919138 | 0.33193549 | 0.47362344 | 0.70165085 |
| V | A(logP, χ, η, π, ω) | 0.7518008 | 0.64508095 | 0.94899619 | 0.87354213 |
Table 5.
Endpoint paths and their lengths (δ) considered for the best/relevant QSAR’s correlations’ models of Table 4, in cases V2/(i) and V6/(ii) for longest SMILES molecular chain (LoSMoC) and in cases V5/(i) and V2/(ii) for branching SMILES (BraS) selected compounds’ sets from Table 3, upon the Euclidian metrics of Equation (17) applied on the first four shortest intermediary QSAR models of Table 4; the overall first three shortest path-lengths are identified in each configuration case by bolding and labeling as alpha (α), beta (β) and gamma (γ) superscripts, respectively.
| LoSMoC | BraS | ||||||
|---|---|---|---|---|---|---|---|
| Path | δV2/(i) | Path | δV6/(ii) | Path | δV5/(i) | Path | δV2/(ii) |
| I1-II1-III5-V | 0.363999003 | I1-II1-III1-V | 0.15216027 | I1-II1-III5-V | 0.339267818 | I1-II1-III3-V | 0.247430746 |
| I1-II1-III7-V | 0.363945917 | I1-II1-III2-V | 0.15219933 | I1-II1-III6-V | 0.328852605 γ | I1-II1-III4-V | 0.250851034 |
| I1-II1-III8-V | 0.363872037 | I1-II1-III3-V | 0.15232909 | I1-II1-III9-V | 0.323160465 β | I1-II1-III6-V | 0.246918396 |
| I1-II7-III5-V | 0.36586301 | I1-II2-III1-V | 0.13669055 | I1-II2-III5-V | 0.344705061 | I1-II2-III3-V | 0.277498475 |
| I1-II7-III7-V | 0.365814373 | I1-II2-III2-V | 0.13669292 | I1-II2-III6-V | 0.332349493 | I1-II2-III4-V | 0.27890546 |
| I1-II7-III8-V | 0.365747157 | I1-II2-III3-V | 0.13670114 | I1-II2-III9-V | 0.301780663 α | I1-II2-III6-V | 0.277296451 |
| I1-II8-III5-V | 0.378790523 | I1-II3-III1-V | 0.12960764 | I1-II3-III5-V | 0.339863056 | I1-II3-III3-V | 0.345661527 |
| I1-II8-III7-V | 0.378770846 | I1-II3-III2-V | 0.12961931 | I1-II3-III6-V | 0.330206319 | I1-II3-III4-V | 0.345678373 |
| I1-II8-III8-V | 0.378746997 | I1-II3-III3-V | 0.12965837 | I1-II3-III9-V | 0.332807819 | I1-II3-III6-V | 0.345670347 |
| I1-II9-III5-V | 0.387593286 | I1-II4-III1-V | 0.12810286 α | I1-II4-III5-V | 0.350074672 | I1-II4-III3-V | 0.341600891 |
| I1-II9-III7-V | 0.387591632 | I1-II4-III2-V | 0.1281234 β | I1-II4-III6-V | 0.342969246 | I1-II4-III4-V | 0.341675065 |
| I1-II9-III8-V | 0.387594763 | I1-II4-III3-V | 0.12819186 γ | I1-II4-III9-V | 0.369568114 | I1-II4-III6-V | 0.34160106 |
| I2-II1-III5-V | 0.031042298 | I2-II1-III1-V | 0.51504227 | I2-II1-III5-V | 0.392905816 | I2-II1-III3-V | 0.189846412 |
| I2-II1-III7-V | 0.030413493 | I2-II1-III2-V | 0.51505381 | I2-II1-III6-V | 0.383948387 | I2-II1-III4-V | 0.194283111 |
| I2-II1-III8-V | 0.029516257 β | I2-II1-III3-V | 0.51509217 | I2-II1-III9-V | 0.379084442 | I2-II1-III6-V | 0.18917817 |
| I2-II7-III5-V | 0.030467382 | I2-II2-III1-V | 0.43487567 | I2-II2-III5-V | 0.411103551 | I2-II2-III3-V | 0.170615371 β |
| I2-II7-III7-V | 0.029877668 γ | I2-II2-III2-V | 0.43487642 | I2-II2-III6-V | 0.40080012 | I2-II2-III4-V | 0.172894351 γ |
| I2-II7-III8-V | 0.029043119 α | I2-II2-III3-V | 0.434879 | I2-II2-III9-V | 0.37584055 | I2-II2-III6-V | 0.17028659 α |
| I2-II8-III5-V | 0.033370142 | I2-II3-III1-V | 0.44987922 | I2-II3-III5-V | 0.388579959 | I2-II3-III3-V | 0.229289585 |
| I2-II8-III7-V | 0.033146038 | I2-II3-III2-V | 0.44988258 | I2-II3-III6-V | 0.38016273 | I2-II3-III4-V | 0.22931498 |
| I2-II8-III8-V | 0.032872383 | I2-II3-III3-V | 0.44989384 | I2-II3-III9-V | 0.382424544 | I2-II3-III6-V | 0.229302881 |
| I2-II9-III5-V | 0.04049457 | I2-II4-III1-V | 0.46505147 | I2-II4-III5-V | 0.382158589 | I2-II4-III3-V | 0.225267191 |
| I2-II9-III7-V | 0.040478734 | I2-II4-III2-V | 0.46505713 | I2-II4-III6-V | 0.375660505 | I2-II4-III4-V | 0.225379654 |
| I2-II9-III8-V | 0.040508702 | I2-II4-III3-V | 0.46507599 | I2-II4-III9-V | 0.400091867 | I2-II4-III6-V | 0.225267448 |
| I3-II1-III5-V | 0.340602068 | I3-II1-III1-V | 0.29305119 | I3-II1-III5-V | 0.371725449 | I3-II1-III3-V | 0.606860446 |
| I3-II1-III7-V | 0.340545335 | I3-II1-III2-V | 0.29307147 | I3-II1-III6-V | 0.36224466 | I3-II1-III4-V | 0.608262992 |
| I3-II1-III8-V | 0.340466377 | I3-II1-III3-V | 0.29313888 | I3-II1-III9-V | 0.357085205 | I3-II1-III6-V | 0.606651729 |
| I3-II7-III5-V | 0.342455676 | I3-II2-III1-V | 0.22803128 | I3-II2-III5-V | 0.386400836 | I3-II2-III3-V | 0.681535121 |
| I3-II7-III7-V | 0.342403714 | I3-II2-III2-V | 0.2280327 | I3-II2-III6-V | 0.375420048 | I3-II2-III4-V | 0.682109209 |
| I3-II7-III8-V | 0.342331902 | I3-II2-III3-V | 0.22803762 | I3-II2-III9-V | 0.34864824 | I3-II2-III6-V | 0.681452889 |
| I3-II8-III5-V | 0.355336832 | I3-II3-III1-V | 0.23734499 | I3-II3-III5-V | 0.368802149 | I3-II3-III3-V | 0.75781421 |
| I3-II8-III7-V | 0.355315856 | I3-II3-III2-V | 0.23735136 | I3-II3-III6-V | 0.359922688 | I3-II3-III4-V | 0.757821894 |
| I3-II8-III8-V | 0.355290432 | I3-II3-III3-V | 0.23737269 | I3-II3-III9-V | 0.362310878 | I3-II3-III6-V | 0.757818233 |
| I3-II9-III5-V | 0.364129287 | I3-II4-III1-V | 0.24859544 | I3-II4-III5-V | 0.367313037 | I3-II4-III3-V | 0.753713772 |
| I3-II9-III7-V | 0.364127526 | I3-II4-III2-V | 0.24860602 | I3-II4-III6-V | 0.360547493 | I3-II4-III4-V | 0.753747392 |
| I3-II9-III8-V | 0.364130859 | I3-II4-III3-V | 0.24864131 | I3-II4-III9-V | 0.385936759 | I3-II4-III6-V | 0.753713849 |
| I4-II1-III5-V | 0.525288611 | I4-II1-III1-V | 0.32781038 | I4-II1-III5-V | 0.448453944 | I4-II1-III3-V | 0.369625875 |
| I4-II1-III7-V | 0.525251826 | I4-II1-III2-V | 0.32782851 | I4-II1-III6-V | 0.440627193 | I4-II1-III4-V | 0.371924125 |
| I4-II1-III8-V | 0.525200637 | I4-II1-III3-V | 0.32788877 | I4-II1-III9-V | 0.436395432 | I4-II1-III6-V | 0.369283099 |
| I4-II7-III5-V | 0.527198557 | I4-II2-III1-V | 0.25851495 | I4-II2-III5-V | 0.472588851 | I4-II2-III3-V | 0.42730628 |
| I4-II7-III7-V | 0.527164806 | I4-II2-III2-V | 0.2585162 | I4-II2-III6-V | 0.463653781 | I4-II2-III4-V | 0.428221331 |
| I4-II7-III8-V | 0.527118165 | I4-II2-III3-V | 0.25852055 | I4-II2-III9-V | 0.442255821 | I4-II2-III6-V | 0.42717511 |
| I4-II8-III5-V | 0.540332774 | I4-II3-III1-V | 0.26942851 | I4-II3-III5-V | 0.441695569 | I4-II3-III3-V | 0.500330351 |
| I4-II8-III7-V | 0.54031898 | I4-II3-III2-V | 0.26943412 | I4-II3-III6-V | 0.434308982 | I4-II3-III4-V | 0.500341989 |
| I4-II8-III8-V | 0.540302262 | I4-II3-III3-V | 0.26945292 | I4-II3-III9-V | 0.436290182 | I4-II3-III6-V | 0.500336444 |
| I4-II9-III5-V | 0.549182204 | I4-II4-III1-V | 0.281784 | I4-II4-III5-V | 0.426373085 | I4-II4-III3-V | 0.496246943 |
| I4-II9-III7-V | 0.549181037 | I4-II4-III2-V | 0.28179334 | I4-II4-III6-V | 0.420558718 | I4-II4-III4-V | 0.496298005 |
| I4-II9-III8-V | 0.549183247 | I4-II4-III3-V | 0.28182447 | I4-II4-III9-V | 0.44251816 | I4-II4-III6-V | 0.49624706 |
| I5-II1-III5-V | 0.657190923 | I5-II1-III1-V | 0.3508471 | I5-II1-III5-V | 0.534442949 | I5-II1-III3-V | 0.238824486 |
| I5-II1-III7-V | 0.657161522 | I5-II1-III2-V | 0.35086404 | I5-II1-III6-V | 0.52789265 | I5-II1-III4-V | 0.242366256 |
| I5-II1-III8-V | 0.657120609 | I5-II1-III3-V | 0.35092035 | I5-II1-III9-V | 0.524365617 | I5-II1-III6-V | 0.238293632 |
| I5-II7-III5-V | 0.65912139 | I5-II2-III1-V | 0.27934081 | I5-II2-III5-V | 0.563677521 | I5-II2-III3-V | 0.265077074 |
| I5-II7-III7-V | 0.659094395 | I5-II2-III2-V | 0.27934197 | I5-II2-III6-V | 0.556207653 | I5-II2-III4-V | 0.266549633 |
| I5-II7-III8-V | 0.659057091 | I5-II2-III3-V | 0.27934599 | I5-II2-III9-V | 0.53850008 | I5-II2-III6-V | 0.264865576 |
| I5-II8-III5-V | 0.672348982 | I5-II3-III1-V | 0.29108509 | I5-II3-III5-V | 0.52553652 | I5-II3-III3-V | 0.332571954 |
| I5-II8-III7-V | 0.672337897 | I5-II3-III2-V | 0.29109028 | I5-II3-III6-V | 0.519343768 | I5-II3-III4-V | 0.332589464 |
| I5-II8-III8-V | 0.672324461 | I5-II3-III3-V | 0.29110768 | I5-II3-III9-V | 0.521001709 | I5-II3-III6-V | 0.332581122 |
| I5-II9-III5-V | 0.681219886 | I5-II4-III1-V | 0.30401219 | I5-II4-III5-V | 0.502030388 | I5-II4-III3-V | 0.328514246 |
| I5-II9-III7-V | 0.681218945 | I5-II4-III2-V | 0.30402085 | I5-II4-III6-V | 0.497101738 | I5-II4-III4-V | 0.328591374 |
| I5-II9-III8-V | 0.681220726 | I5-II4-III3-V | 0.30404971 | I5-II4-III9-V | 0.515812781 | I5-II4-III6-V | 0.328514423 |
Note that the Euclidean distance itself employs the square of the correlations factors, i.e., a higher order statistical framework, which nevertheless may be further enriched with other statistical outputs and factors, although all directly or indirectly depend on the correlation factor [155].
In is also worth remarking that in the present uracil-derivative anti-HIV analysis, the four-descriptors’ dependency is not necessary in equation (17) since it is not needed in assessing the structural/reactivity parameters hierarchy in the minimum variational path principle of (16) by being absorbed in the rest of correlations by means of the transitivity chain rule:
whenever two descriptors are common for adjacent activities’ correlations—they will be considered as a single common influence in chemical causes for the observed biological activity.
This way, the redundancies or double counting of models are avoided, even at the cost of “jumping” some intermediate models, like the four-descriptors’ endpoints. The results are displayed in Table 5. They are interpreted in the sense of establishing the minimum of three path hierarchies, and then compared at the global level; note that more than three minimum paths will produce redundant information. Accordingly, the minimum paths, for LoSMoC and BraS cases (i)/(ii) separately, are:
- For case LoSMoC/V2/(i):
(α): I2-II7-III8-V δ[α]=0.029043119
(β): I2-II1-III8-V δ[β]=0.029516257
(γ): I2-II7-III7-V δ[γ]=0.029877668
(18) - For case LoSMoC/V6/(ii):
(α): I1-II4-III1-V δ[α]=0.12810286
(β): I1-II4-III2-V δ[β]=0.1281234
(γ): I1-II4-III3-V δ[γ]=0.12819186
(19) - For case BraS/V5/(i):
(α): I1-II2-III9-V δ[α]=0.301780663
(β): I1-II1-III9-V δ[β]=0.323160465
(γ): I1-II1-III6-V δ[γ]=0.328852605
(20) - For case BraS/V2/(ii):
(α): I2-II2-III6-V δ[α]=0.17028659
(β): I2-II2-III3-V δ[β]=0.170615371
(γ): I2-II2-III4-V δ[γ]=0.172894351
(21)
The variational results of Table 5 summarized by equations (19)–(21) are most involved in ensuring the reliability of the present approach because:
All the LoSMoC least path lengths are shorter than those of BraS, this way confirming that the chain based SMILES intermediates are prior to those displaying branching SMILES conformations, i.e., in accordance with the steps [A] →[B] of Figure 3 in pyrimidine-related uracil attack onreserve transcriptase;
- While passing from LoSMoC to BraS configurations in the chemical-biological interaction of uracil derivatives–reverse transcriptase binding phenomenology one notes the maintenance of the same criteria variants, namely V2 of Table 3:
meaning that the chain-to-branching passage seems to require the same features of the principal chain and of the secondary branch alike;(22) Looking now to the cases interchanged in the transformation of equation (22) one also notes that the passage from case (i) based on longest chain in the SMILES configuration to the case (ii) based on the pyrimidinic N3 atom’s neighbors, happens consistently. The mechanism of interaction is described as involving the trans-membrane transduction by means of the longest chain of SMILES configuration; it is followed by the bonding stage centered on the N3 atom of the pyrimidine ring nuclei as already proved to be specific for spirodiazine derivatives in their transformations towards recorded anti-inflammatory activities, anti-HIV activity included [126].
With these we exposed the pre-final stage of ligand-receptor interaction explained by variational/spectral-QSAR analysis. It assumes the linking of the LoSMoC and BraS least paths’ models to mirror the successive SMILES transformations of the free molecule inside the HIV cell, by passing its lipidic walls and plasmidic environment hitting the reverse transcriptase palm-p66 pocket, see Figure 3E.
2.5. OECD-QSAR Principle 5: A Mechanistic Interpretation
The intent of OECD QSAR Principle 5 is not to reject models that have no apparent mechanistic basis, but to ensure that some consideration is given to the possibility of a mechanistic association between the descriptors used in a model and the endpoint being predicted and to ensure that this association is documented. Since the physicochemical QSAR parameters were chosen in this study, a mechanistic interpretation of the models is possible. This nevertheless follows specific steps integrated in the previously discussed OECD-QSAR principles.
Accordingly, on the concrete study of actual uracils’ anti-HIV action, the transformation (22) is projected on the structural or chemical reactivity descriptors it encompasses for the shortest path lengths so that it concludes the variational QSAR modeling:
| αLoSMoC/V2/(i) → αBraS/V2/(ii) | (23) |
which is equivalently rewritten with the help of Equations (18) and (21):
| [I2-II7-III8-V] → [I2-II2-III6-V] | (24) |
and even more with the help of endpoint identifications of Table 4, respectively as:
| [(χ)→(χ,ω)→(χ,ω,η)→(χ,ω,η,π,logP)] → [(χ)→(η,logP)→(logP,π, ω)→(χ,η,logP,π,ω)] | (25) |
Now, the solution of the structural/reactivity causes driving the ligand receptor binding mechanism in the present 1,3-disubstituted uracils against human immunodeficiency virus (HIV-1) action is given by combining the two variational principles noted before:
Transitivity chain rule, and
Minimization of redundancies
in structural/chemical reactivity dependencies. As such, the first alpha spectral-SAR hierarchy in (25) solves the first three causes:
| χ→ω→η→(π,logP) | (26a) |
while the second alpha path hierarchy of (25) solves the last degeneracy of (26a) as explained next: one considers the already solved structural/reactivity causes of (26a) from where it results that η follows χ; with this ordering back in (25) one yields that logP follows η; this should finally applied also in (26a); all in all, the ordered causes of structural/reactivity influences in actual anti-HIV mechanism look like:
| χ→ω→η→logP→π | (26b) |
Equation (26b) may be represented by the orbital based scheme of chemical reactivity driving biological (anti-HIV) activity as provided in Figure 4. It is explained in the light of chemical reactivity principles, (see Section 2.2) within the “time-space” framework fixed by the chemical reactivity-biological activity interaction:
Figure 4.

Representation of mechanistic molecular orbital interaction and bonding between the the uracil derivative compounds and HIV through binding the SMILES (essentially the BraS) molecule (the ligand, L) with the molecular pocket of the receptor (R) site, see the stages (D) & (E) of Figure 3, through variational principles of chemical reactivity of 1. electronegativity (χ), 2. electrophilicity (ω), 3. chemical hardness (η), 4. lipophilicity (logP) and 5. chemical power (π), according with the Spectral-QSAR analysis of equations (16)–(26).
The development time is not the physical one but an internal one related with the reaction coordinates, so that the reactivity-driven-activity steps are phenomenological ordered through being interrelated and inter-conditioned during the entire physical time of the binding (on a nano-second scale);
The described interaction is spatially placed between the ligand (L) represented by the SMILES branched molecule resulted upon the HIV cell’s transduction (at least of the viral envelope) and the receptor–the palm region of the p66 region of the reverse transcriptase.
In these conditions the found mechanism for uracil derivatives’ anti-HIV activity goes as follows:
The first step is triggered by electronegativity (χ) and of its principle of minimization difference between ligand (L) and receptor (R) HOMO-LUMO middle-levels, as provided by equation (3). In this stage the ligand and receptor are energetically aligned around a common electronegativity; it also associates with “preparation” of HOMO and LUMO states for ceding and accepting electrons by the accompanying interchanging charge;
The second step accompanies the first one through the electrophilicity (ω) by putting into action the charge transfer by tunneling of the L-R barrier for one electron of the HOMOL level passing to the LUMOL and then down to the HOMOR state by means of the LLR mechanism, see Figure 2b; the minimization principle for electrophilicity, equation (11), further allows the relaxation of the transferred electron from the HOMOR to the HOMOR* level;
The third step appears naturally “called” by the second one: the R to R* actually corresponds with the expansion of the HOMOR-LUMOR gap to HOMOR*-LUMOR* to be equal with HOMOL-LUMOL one, in accordance with the maximum hardness principle, equation (6), being this step driven by chemical hardness;
The fourth step converts spatially the energetic HOMO-LUMO coupling of ligand-receptor by hydrophobicity/lipophilicity (logP) action eventually assuring also the capsid penetration; note that the previous charge transfer was realized through (quantum) tunneling, in accordance with electrophilicity driving action, thus being consistent with the earlier (second step) long-range action of the pyrimidines in the plasmidic region of HIV cell against its reverse transcriptase enzyme inside of the capsid, see Figure 3;
The fifth and the last step is accomplished by chemical power (π) which assures the effective ligand-receptor binding (now also spatial in nature) by transferring the remaining electron of HOMOL to LUMOL and then to LUMOR* by means of the LRR mechanisms of Figure 1b; it nevertheless fulfils the minimization principle, equation (9), by undergoing the final LUMOR* to HOMOR* relaxation, when it pairs with the electron arrived from the electrophilicity step above.
Overall, the presented molecular mechanism fully explains the ligand-receptor binding in all respects:
Spatially (the molecule is placed in the pocket of HIV’s reverse transcriptase);
Energetically (all transitions compensate each other);
By electronic pairing (assured by electrophilicity and chemical power actions);
By bonding on the relaxed HOMOR* level
This way the presented variational QSAR anti-HIV mechanism assures the stabilization of pyrimidine complex with the enzyme transcriptase receptor towards the concerned apoptosis of the HIV cells through inhibiting his enzyme activities for further actions (and replications) in the host organism. This study complements the previous one [64], by effectively employing the various forms of SMILES configuration for the ligand molecules, with the satisfactory result that the proposed molecular anti-HIV mechanism appears to be reliable and self-consistent, aiding us to envisage ligand-receptor binding. However, while being aware of the importance the branching SMILES procedure has played in the actual endeavor, further study may be directed towards employing the topological branching information of the involved molecules, being this field equally rich and promising in QSAR chemical systems with high complexity [156,157,158,159]. Moreover, when the actual mechanistic analysis is envisaged to be further used in drug design, i.e., in searching for new anti-HIV agents, one should employ the resulting minimum path, namely the path (α) in Equation (18), and the intermediate QSAR models contained along this path, i.e., A(χ), A(χ,ω), and A(χ,ω,η), respectively, to further identify uracil derivative shapes best fulfilling the synergistic needs of all these models, finally tested also for external robustness. This step is under our purview in achieving the self-consistent mechanistic drug design in an in-cerebro-in silico framework.
3. Conclusions
Chemical bonding and reactivity were at the forefront of modern chemistry in the last century, described through various qualitative theories (viz. Lewis’ theory of atoms and molecules [160] or the resonance theory of Pauling [161,162,163,164]) as well as through quantitative ones (e.g., Heitler-London homopolar theory [165], Hückel and extended Hückel heteropolar theories [166,167,168], or the Bader-Gillespie Atoms in Molecules–AIM [169,170,171] and Valence Shell Electron Pair Repulsion—VSEPR formulations [172,173], just to name a few), before finally being united within the conceptual Density Functional Theory [174,175] leading to the the recent bonding-by-reactivity scenario within the so- called chemical orthogonal space [54,55] of electronegativity [95] and chemical hardness [105]. The next step was made when chemical-biology binding interactions and binding were considered as a superior phenomenological level of ordinary chemical bonding. To treat it, however, the descriptors’ orthogonality feature turns out to be of prime importance so that the quantitative structure-activity relationship QSAR approach, while incorporating it, establishes itself as the current paradigm in modeling biological activity. Eventually it may fully employ the fundamental chemical reactivity concepts such as the electronegativity and chemical hardness along their second generation of descriptors such as chemical power [59,64] and electrophilicity [120], and their associated variational principles, while assuming a given (parabolic) electronic total energy vs. number of electrons E = E(N) shape dependency [117,176,177].
In this chemical reactivity-driven biological activity context, the present work has succeeded in clarifying the mechanism of molecular-cellular action by means of chemical reactivity indices and of their variational principles viewed as descriptors in a QSAR context, while studying available uracil derivatives’ anti-HIV action.
This way, one is left with the variational QSAR recipe generally summarized following the Organization for Economic Co-Operation and Development (OECD) related principles (see Introduction):
For QSAR-OECD Principle 1 (a defined endpoint): considering SMILES longest chain (LoSMoC)- and branching (BraS)-based counterparts of envisaged molecules as the actual molecular ansatz for modeling the envisaged anti-HIV activity by the end-point of half maximal effective concentration (EC50, μM) antiviral activity of 1,3-disubstituted uracils against human immunodeficiency virus (HIV-1)—see Table 1;
For QSAR-OECD Principle 2 (an unambiguous algorithm): implementing QSAR orthogonal descriptors with associate min-max principles of chemical reactivity: electronegativity and chemical hardness, and of their mixed forms under electrophilicity and chemical power indices; the first two descriptors were also considered with “branching” working forms for BraS molecules up to the third order in HOMO and LUMO, within Koopmans theorem and spectral like resolution frameworks; the last two descriptors are merely associated with chemical charge transfer at the molecular frontier (HOMO and LUMO). Together, they all assure the chemical reactivity-driving-biological activity and provide the molecular mechanism linking structural causes with recorded biological effects (anti-HIV in the present application), while being accompanied by the hydrophobicity/lipophilicity index (logP) modeling the transduction through cellular HIV membranes;
For QSAR-OECD Principle 3 (a defined domain of applicability): selecting the appropriate QSAR correlation through the screening based on chain (LoSMoC) and branching (BraS) SMILES molecular structures; this stage allows further application of transitivity and minimum redundancy rules for the QSAR descriptors as they are present in the various multi-linear computed endpoints;
For QSAR-OECD Principle 4 (appropriate measures of goodness-of–fit, robustness and predictivity): ordering the multi-descriptor dependencies with the help of spectral-path length hierarchy for chain (LoSMoC) and branching (BraS) SMILES molecular interaction, and globally in between them, with the aim of Euclidian path measure and of their systematic minimum search across all QSAR models and of their combinations;
For QSAR-OECD Principle 5 (a mechanistic interpretation, if possible): constructing the molecular (orbital/frontier) diagram describing the mechanism of ligand-receptor interaction based on correlating the least alpha paths of LoSMoC and BraS QSAR analyses with the chemical reactivity descriptors’ electronic manifestations and principles.
All these steps and algorithm were applied and directed for establishing the general molecular mechanism whereby 1,3-disubstituted uracils act against human immunodeficiency virus (HIV-1) by inhibiting its reverse transcriptase enzyme by means of ligand-receptor binding. Results were satisfactory and show reliability in all steps, while complementing the recent work where the resulting ligand-complex ended in an activated state [64], with the actual fully predicted bonding behavior; however, for future works, it would be interesting to research the biological effect of a mixture between a marine drug and a pyrimidine derivate with anti-HIV activity, as well as extending the branching study from SMILES to topological characterization of molecules aiming to identifying the best molecular shape responding to the best/minimal path in providing the ligand-receptor interaction and of its mechanism by the synergetic mechanistic drug design.
Acknowledgments
This work was supported by the Romanian National Council of Scientific Research (CNCS-UEFISCDI) through project TE16/2010-2013 within the PN II-RU-TE-2010-1 framework. MVP thanks Ionel I. Mangalagiu from “Al. I. Cuza” University of Iasi (Romania) for vivid interest in the present research direction and for furnishing supporting references that further stimulated the actual work. We also thank the referees for their convergent and constructive appreciations on our approach motivating its improvement to the final actual form as well to Molecules’ editors for professionally handling of our manuscript.
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/18/8/9061/s1.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Not available.
References and Notes
- 1.Péry A.R.R., Brochot C., Zeman F.A., Mombelli E., Desmots S., Pavan M., Fioravanzo E., Zaldivar J.M. Prediction of dose-hepatotoxic response in humans based on toxicokinetic/ toxicodynamic modeling with or without in vivo data: A case study with acetaminophen. Toxicol. Lett. 2013;220:26–34. doi: 10.1016/j.toxlet.2013.03.032. [DOI] [PubMed] [Google Scholar]
- 2.Diaz Ochoa J.G., Bucher J., Péry A.R.R., Zaldivar J.M., Niclas J., Mauch K. A multi-scale modeling framework for individualized, spatiotemporal prediction of drug effects and toxicological risk. Front. Pharmacol. 2013;3:204. doi: 10.3389/fphar.2012.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Worth A.P., Lapenna S., Serafimova R. QSAR and metabolic assessment tools in the assessment of genotoxicity. Method. Mol. Biol. 2012;930:125–162. doi: 10.1007/978-1-62703-059-5_6. [DOI] [PubMed] [Google Scholar]
- 4.Tonnelier A., Coecke S., Zaldivar J.M. Screening of chemicals for human bioaccumulative potential with a physiologically based toxicokinetic model. Arch. Toxicol. 2012;86:393–403. doi: 10.1007/s00204-011-0768-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burello E., Worth A.P. QSAR modeling of nanomaterials. WIREs Nanomed. Nanobiotechnol. 2011;3:298–306. doi: 10.1002/wnan.137. [DOI] [PubMed] [Google Scholar]
- 6.Burello E., Worth A.P. A theoretical framework for predicting the oxidative stress potential of oxide nanoparticles. Nanotoxicology. 2011;5:228–235. doi: 10.3109/17435390.2010.502980. [DOI] [PubMed] [Google Scholar]
- 7.Burello E., Worth A.P. Computational nanotoxicology: Predicting toxicity of nanoparticles. Nat. Nanotechnol. 2011;6:138–139. doi: 10.1038/nnano.2011.27. [DOI] [PubMed] [Google Scholar]
- 8.Lo Piparo E., Worth A., Manibusan M., Yang C., Schilter B., Mazzatorta P., Jacobs M.N., Steinkellner H., Mohimont L. Use of computational tools in the field of food safety. Regul. Toxicol. Pharm. 2011;60:354–362. doi: 10.1016/j.yrtph.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 9.Raevsky O.A., Liplavskaya E.A., Yarkova A.V., Raevskaya O.E., Worth A.P. Linear and nonlinear QSAR models of acute intravenous toxicity of organic chemicals for mice. Biochem. (Moscow) Suppl. Ser. B Biomed. Chem. 2011;5:213–225. doi: 10.1134/S1990750811030103. [DOI] [Google Scholar]
- 10.Worth A.P., Mostrag-Szlichtyng A. Towards a common regulatory framework for computational toxicology: current status and future perspectives. In: Wilson A.G.E., editor. New Horizons in Predictive Toxicology: Methods and Applications. The Royal Society of Chemistry; Cambridge, UK: 2011. pp. 38–69. [Google Scholar]
- 11.Mostrag-Szlichtyng A., Zaldivar J.M., Worth A.P. Computational toxicology at the European Commission`s Joint Research Centre. Expert Opin. Drug Met. 2010;6:785–792. doi: 10.1517/17425255.2010.489551. [DOI] [PubMed] [Google Scholar]
- 12.Novellino A., Zaldivar J.M. Recurrence quantification analysis of spontaneous electrophysiological activity during development: Characterization of in vitro neuronal networks cultured on multi electrode array chips. Adv. Artificial Intelligence. 2010;2010 doi: 10.1155/2010/209254. [DOI] [Google Scholar]
- 13.Raevsky O.A., Grigor'ev V.J., Modina E.A., Worth A.P. Prediction of acute toxicity to mice by the Arithmetic Mean Toxicity (AMT) modelling approach. SAR QSAR Environ. Res. 2010;21:265–275. doi: 10.1080/10629361003771025. [DOI] [PubMed] [Google Scholar]
- 14.Bacelar F.S., Dueri S., Hernandez-Garcia E., Zaldivar J.M. Joint effects of nutrients and contaminants on the dynamics of a food chain in marine ecosystems. Math. Biosci. 2009;218:24–32. doi: 10.1016/j.mbs.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 15.Poater A., Gallegos A., Sola M., Cavallo L., Worth A.P. Computational methods to predict the reactivity of nanoparticles through structure-property relationships. Expert Opin. Drug Del. 2009;7:1–11. doi: 10.1517/17425240903508756. [DOI] [PubMed] [Google Scholar]
- 16.Poater A., Gallegos A., Carbo-Dorca R., Poater J., Sola M., Cavallo L., Worth A.P. Modeling the structure-property relationships of nanoneedles: A journey toward nanomedicine. J. Comput. Chem. 2009;30:275–284. doi: 10.1002/jcc.21041. [DOI] [PubMed] [Google Scholar]
- 17.Bassan A., Worth A.P. The integrated use of models for the properties and effects of chemicals by means of a structured workflow. QSAR Comb. Sci. 2008;27:6–20. doi: 10.1002/qsar.200710119. [DOI] [Google Scholar]
- 18.Kahn I., Maran U., Benfenati E., Netzeva T.I., Schultz T.W., Cronin M.T.D. Comparative quantitative structure-activity-activity relationships for toxicity to Tetrahymena pyriformis and Pimephales promelas. Altern. Lab. Anim. ATLA. 2007;35:15–24. doi: 10.1177/026119290703500112. [DOI] [PubMed] [Google Scholar]
- 19.Gallegos A. Mini-review on chemical similarity and prediction of toxicity. Curr. Comp.-Aid. Drug. 2006;2:105–122. doi: 10.2174/157340906777441681. [DOI] [Google Scholar]
- 20.Patlewicz G. Computational methods to predict drug safety. Curr. Comp.-Aid. Drug. 2006;2:151–168. doi: 10.2174/157340906777441735. [DOI] [Google Scholar]
- 21.Dimitrov S., Dimitrova G., Pavlov T., Dimitrova N., Patlewicz G., Niemela J., Mekenyan O. A stepwise approach for defining the applicability domain of SAR and QSAR models. J. Chem. Inf. Model. 2005;45:839–849. doi: 10.1021/ci0500381. [DOI] [PubMed] [Google Scholar]
- 22.Gallegos A., Carbó-Dorca R., Lodier F., Cancès E., Savin A. Maximal probability domains in linear molecules. J. Comput. Chem. 2005;26:455–460. doi: 10.1002/jcc.20180. [DOI] [PubMed] [Google Scholar]
- 23.Gallegos A., Gironés X. Topological quantum similarity indices based on fitted densities: Theoretical background and QSPR application. J. Chem. Inf. Model. 2005;45:321–326. doi: 10.1021/ci049721y. [DOI] [PubMed] [Google Scholar]
- 24.Gallegos A., Gironés X. Topological quantum similarity measures: Applications in QSAR. J. Mol. Struct. (THEOCHEM) 2005;727:97–106. doi: 10.1016/j.theochem.2004.11.062. [DOI] [Google Scholar]
- 25.Netzeva T.I., Aptula A.O., Benfenati E., Cronin M.T.D., Gini G., Lessigiarska I., Maran U., Vracko M., Schuurmann G. Description of the electronic structure of organic chemicals using semiempirical and ab initio methods for development of toxicological QSARs. J. Chem. Inf. Model. 2005;45:106–114. doi: 10.1021/ci049747p. [DOI] [PubMed] [Google Scholar]
- 26.Ponec R., Bultinck P., Gallegos A. Multicenter bond indices as a new means for the quantitative characterization of homoaromaticity. J. Phys. Chem. A. 2005;109:6606–6609. doi: 10.1021/jp052179b. [DOI] [PubMed] [Google Scholar]
- 27.Schultz T.W., Netzeva T.I., Roberts D.W., Cronin M.T.D. Structure-Toxicity relationships for the effects to Tetrahymena pyriformis of aliphatic, α,β-carbonyl-containing, unsaturated chemicals. Chem. Res. Toxicol. 2005;18:330–341. doi: 10.1021/tx049833j. [DOI] [PubMed] [Google Scholar]
- 28.Lessigiarska I., Worth A.P., Sokull-Klüttgen B., Jeram S., Dearden J.C., Netzeva T.I., Cronin M.T.D. QSAR investigation of a large data set for fish, algae and Daphnia toxicity. SAR QSAR Environ. Res. 2004;15:413–431. doi: 10.1080/10629360412331297416. [DOI] [PubMed] [Google Scholar]
- 29.Cronin M.T.D., Dearden J.C., Walker J.D., Worth A.P. Quantitative structure-activity relationships for human health effects: Commonalities with other endpoints. Environ. Toxicol. Chem. 2003;22:1829–1843. doi: 10.1897/01-274. [DOI] [PubMed] [Google Scholar]
- 30.Worth A.P., Cronin M.T.D. The use of discriminant analysis, logistic regression and classification tree analysis in the development of classification models for human health effects. J. Mol. Struct. (Theochem) 2003;622:97–111. doi: 10.1016/S0166-1280(02)00622-X. [DOI] [Google Scholar]
- 31.Cronin M.T.D., Dearden J.C., Duffy J.C., Edwards R., Manga N., Worth A.P., Worgan A.D.P. The importance of hydrophobicity and electrophilicity descriptors in mechanistically-based QSARs for toxicological endpoints. SAR QSAR Environ. Res. 2002;13:167–176. doi: 10.1080/10629360290002316. [DOI] [PubMed] [Google Scholar]
- 32.Cronin M.T.D., Aptula A.O., Dearden J.C., Duffy J.C., Netzeva T.I., Patel H., Rowe P.H., Schultz T.W., Worth A.P., Voutzoulidis K., et al. Structure-based classification of antibacterial activity. J. Chem. Inf. Comp. Sci. 2002;42:869–878. doi: 10.1021/ci025501d. [DOI] [PubMed] [Google Scholar]
- 33.Worth A.P., van Leeuwen C.J., Hartung T. The prospects for using (Q)SARs in a changing political environment — high expectations and a key role for the Commission's Joint Research Centre. SAR QSAR Environ. Res. 2004;15:331–343. doi: 10.1080/10629360412331297371. [DOI] [PubMed] [Google Scholar]
- 34.Worth A.P., Cronin M.T.D., van Leeuwen C.J. A framework for promoting the acceptance and regulatory use of (Quantitative) Structure-Activity Relationships. In: Cronin M.T.D., Livingstone D., editors. Predicting Chemical Toxicity and Fate. CRC Press; Boca Raton, FL, USA: 2004. pp. 427–438. [Google Scholar]
- 35.OECD. OECD; Paris, France: 2004. [(accessed on 3 March 2011)]. Report from the Expert Group on (Quantitative) Structure-Activity Relationships [(Q)SARs] on the Principles for the Validation of (Q)SARs. (Series on Testing and Assessment, No. 49). Available online: http://www.oecd.org/document/30/0,2340,en_2649_34365_1916638_1_1_1_1,00.html. [Google Scholar]
- 36.OECD. OECD; Paris, France: 2005. [(accessed on 3 March 2011)]. Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. (Series on Testing and Assessment, No. 34). Available online: http://www.oecd.org/document/30/0,2340,en_2649_34365_1916638_1_1_1_1,00.html. [Google Scholar]
- 37.OECD. OECD; Paris, France: 2006. [(accessed on 3 March 2011)]. Report on the Regulatory Uses and Applications in OECD Member Countries of (Quantitative) Structure-Activity Relationship [(Q)SAR] Models in the Assessment of New and Existing Chemicals, Series on Testing and Assessment, No. 58. Available online: http://www.oecd.org/document/30/0,2340,en_2649_34365_1916638_1_1_1_1,00.html. [Google Scholar]
- 38.OECD. Vol. 154. OECD; Paris, France: 2006. [(accessed on 3 March 2011)]. Guidance Document on the Validation of (Quantitative) Structure-Activity Relationship [(Q)SAR] Models. (Series on Testing and Assessment, No. 69.). Available online: http://www.oecd.org/document/30/0,2340,en_2649_34365_1916638_1_1_1_1,00.html. [Google Scholar]
- 39.Patlewicz G., Jeliazkova N., Gallegos A., Worth A.P. Toxmatch—A new software tool to aid in the development and evaluation of chemically similar groups. SAR QSAR Environ. Res. 2008;19:397–412. doi: 10.1080/10629360802083848. [DOI] [PubMed] [Google Scholar]
- 40.Todeschini R., Ballabio D., Consonni V., Mauri A., Pavan M. CAIMAN (Classification And Influence Matrix Analysis): A new approach to the classification based on leverage-scaled functions. Chemom. Intell. Lab. 2007;87:3–17. doi: 10.1016/j.chemolab.2005.11.001. [DOI] [Google Scholar]
- 41.Patlewicz G., Jeliazkova N., Safford R.J., Worth A.P., Aleksiev B. An evaluation of the implementation of the Cramer classification scheme in the Toxtree software. SAR QSAR Environ. Res. 2008;19:495–524. doi: 10.1080/10629360802083871. [DOI] [PubMed] [Google Scholar]
- 42.Gallegos A., Poater A., Jeliazkova N., Patlewicz G., Worth A.P. Toxmatch—A chemical classification and activity prediction tool based on similarity measures. Regul. Toxicol. Pharm. 2008;52:77–84. doi: 10.1016/j.yrtph.2008.05.012. [DOI] [PubMed] [Google Scholar]
- 43.Patlewicz G., Aptula A.O., Uriarte E., Roberts D.W., Kern P.S., Gerberick G.F., Kimber I., Dearman R.J., Ryan C.A., Basketter D.A. An evaluation of selected global (Q)SARs/expert systems for the prediction of skin sensitisation potential. SAR QSAR Environ. Res. 2007;18:515–541. doi: 10.1080/10629360701427872. [DOI] [PubMed] [Google Scholar]
- 44.Jaworska J., Nikolova-Jeliazkova N, Aldenberg T. QSAR applicability domain estimation by projection of the training set descriptor space: a review. Altern. Lab. Anim. 2005;33:445–459. doi: 10.1177/026119290503300508. [DOI] [PubMed] [Google Scholar]
- 45.Jeliazkova N., Jaworska J., Worth A.P. Open source tools for read across and category formation. In: Cronin M.T.D., Madden J., editors. In Silico Toxicology. Principles and Applications. Royal Society of Chemistry; Cambridge, UK: 2010. pp. 408–445. [Google Scholar]
- 46.Pavan M., Todeschini R. Multi-criteria decision making methods. In: Brown S., Walczak B., Tauler R., editors. Comprehensive Chemometrics. Elsevier; Amsterdam, The Netherlands: 2009. pp. 591–629. [Google Scholar]
- 47.Pavan M., Todeschini R., editors. Science and Technology. 1st ed. Volume 27 Elsevier; Oxford, UK: 2008. Scientific data ranking methods: theory and applications. Data handling. [Google Scholar]
- 48.Roberts D.W., Aptula A.O., Patlewicz G., Pease C. Chemical reactivity indices and mechanism-based read-across for non-animal based assessment of skin sensitisation potential. J. Appl. Toxicol. 2008;28:443–454. doi: 10.1002/jat.1293. [DOI] [PubMed] [Google Scholar]
- 49.Spycher S., Netzeva T.I., Worth A.P., Escher B.I. Mode of action-based classification and prediction of activity of uncouplers for the screening of chemical inventories. SAR QSAR Environ. Res. 2008;19:433–463. doi: 10.1080/10629360802348803. [DOI] [PubMed] [Google Scholar]
- 50.Benigni R., Bossa C., Netzeva T.I., Rodomonte A., Tsakovska I. Mechanistic QSAR of aromatic amines: New models for discriminating between homocyclic mutagens and nonmutagens, and validation of models for carcinogens. Environ. Mol. Mutagen. 2007;48:754–771. doi: 10.1002/em.20355. [DOI] [PubMed] [Google Scholar]
- 51.Vracko M., Bandelj V., Barbieri P., Benfenati E., Chaudhry Q., Cronin M.T.D., Devillers J., Gallegos A., Gini G., Gramatica P., et al. Validation of counter propagation neural network models for predictive toxicology according to the OECD principles: A case study. SAR QSAR Environ. Res. 2006;17:265–284. doi: 10.1080/10659360600787650. [DOI] [PubMed] [Google Scholar]
- 52.Dirac P.A.M. The Principles of Quantum Mechanics. 2nd ed. Clarendon Press; Oxford, UK: 1947. [Google Scholar]
- 53.Putz M.V., editor. QSAR & SPECTRAL-SAR in Computational Ecotoxicology. Apple Academics; Ontario, Canada: 2012. [Google Scholar]
- 54.Putz M.V. Chemical orthogonal spaces; Mathematical Chemistry Monographs. Volume 14 Publisher; Kragujevac, Serbia: 2012. [Google Scholar]
- 55.Putz M.V. Chemical Orthogonal Spaces (COSs): From structure to reactivity to biological activity. Int. J. Chem. Model. 2013;5 in press. [Google Scholar]
- 56.Putz M.V., Putz A.M., Lazea M., Ienciu L., Chiriac A. Quantum-SAR extension of the Spectral-SAR algorithm. Application to polyphenolic anticancer bioactivity. Int. J. Mol. Sci. 2009;10:1193–1214. doi: 10.3390/ijms10031193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Putz M.V., Lacrămă A.M. Introducing spectral structure activity relationship (S-SAR) Analysis. application to ecotoxicology. Int. J. Mol. Sci. 2007;8:363–391. doi: 10.3390/i8050363. [DOI] [Google Scholar]
- 58.Putz M.V., Putz A.M. Timisoara Spectral–Structure Activity Relationship (Spectral-SAR) Algorithm: From statistical and algebraic fundamentals to quantum consequences. In: Putz M.V., editor. Quantum Frontiers of Atoms and Molecules. NOVA Science Publishers, Inc.; New York, NY, USA: 2011. pp. 539–580. (Series, Chemistry Research and Applications). [Google Scholar]
- 59.Putz M.V., Putz A.M. DFT chemical reactivity driven by biological activity: Applications for the toxicological fate of chlorinated PAHs. Struct. Bond. 2013;150:181–232. doi: 10.1007/978-3-642-32750-6_6. [DOI] [Google Scholar]
- 60.Putz M.V., Putz A.M., Lazea M., Chiriac A. Spectral vs. statistic approach of Structure-Activity Relationship. Application on ecotoxicity of aliphatic amines. J. Theor. Comput. Chem. 2009;8:1235–1251. doi: 10.1142/S0219633609005453. [DOI] [Google Scholar]
- 61.Lacrămă A.M., Putz M.V., Ostafe V. A Spectral-SAR model for the anionic-cationic interaction in ionic liquids: application to Vibrio fischeri ecotoxicity. Int. J. Mol. Sci. 2007;8:842–863. doi: 10.3390/i8080842. [DOI] [Google Scholar]
- 62.Putz M.V., Lacrămă A.M., Ostafe V. Spectral-SAR ecotoxicology of ionic liquids. The Daphnia magna Case. Int. J. Ecology (former Res. Lett. Ecology) 2007;2007 doi: 10.1155/2007/12813. [DOI] [Google Scholar]
- 63.Putz M.V., Putz A.M., Ostafe V., Chiriac A. Spectral-SAR Ecotoxicology of Ionic Liquids-Acetylcholine Interaction on E. Electricus Species. Int. J. Chem. Model. 2010;2:85–96. [Google Scholar]
- 64.Putz M.V., Dudaş N.A. Variational principles for mechanistic quantitative structure–activity relationship (QSAR) studies: Application on uracil derivatives’ anti-HIV action. Struct. Chem. 2013 doi: 10.1007/s11224-013-0249-6. [DOI] [Google Scholar]
- 65.Weininger D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Model. 1988;28:31–36. doi: 10.1021/ci00057a005. [DOI] [Google Scholar]
- 66.Weininger D., Weininger A., Weininger J.L. SMILES. 2. Algorithm for generation of unique SMILES notation. J. Chem. Inf. Model. 1989;29:97–101. doi: 10.1021/ci00062a008. [DOI] [Google Scholar]
- 67.Weininger D. SMILES. 3. DEPICT. Graphical depiction of chemical structures. J. Chem. Inf. Model. 1990;30:237–243. doi: 10.1021/ci00067a005. [DOI] [Google Scholar]
- 68.Helson H.E. Structure Diagram Generation. In: Lipkowitz K.B., Boyd D.B., editors. Reviews in Computational Chemistry. Volume 13. John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2007. pp. 313–398. [Google Scholar]
- 69.Maruyama T., Kozai S., Yamasaki T., Witvrouw M., Pannecouque C., Balzarini J., Snoeck R., Andrei G., De Clercq E. Synthesis and antiviral activity of 1,3-disubstituted uracils against HIV-1 and HCMV. Antivir. Chem. Chemoth. 2003;14:271–279. doi: 10.1177/095632020301400506. [DOI] [PubMed] [Google Scholar]
- 70.Putz M.V., Putz A.M., Barou R. Spectral-SAR realization of OECD-QSAR principles. Int. J. Chem. Model. 2011;3:173–190. [Google Scholar]
- 71.Putz M.V., Duda-Seiman C., Duda-Seiman D.M., Putz A.M. Turning SPECTRAL-SAR into 3D-QSAR analysis. Application on H+K+-ATPase inhibitory activity. Int. J. Chem. Model. 2008;1:45–62. [Google Scholar]
- 72.Garg R., Gupta S.P., Gao H., Babu M.S., Debnath A.K., Hansch C. QSAR studies on anti HIV-1 drugs. Chem. Rev. 1999;99:3525–3601. doi: 10.1021/cr9703358. [DOI] [PubMed] [Google Scholar]
- 73.Mehellou Y., De Clercq E. Twenty-six years of anti-HIV drug discovery: Where do we stand and where do we go? J. Med. Chem. 2010;53:521–538. doi: 10.1021/jm900492g. [DOI] [PubMed] [Google Scholar]
- 74.Esposito F., Corona A., Tramontano E. HIV-1 reverse transcriptase still remains a new drug target: structure, function, classical inhibitors, and new inhibitors with innovative mechanisms of actions. Mol. Biol. Int. 2012;2012 doi: 10.1155/2012/586401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Quashie P.K., Sloan R.D., Wainberg M.A. Novel therapeutic strategies targeting HIV integrase. BMC Medicine. 2012;10:34. doi: 10.1186/1741-7015-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kaufmann G.R., Cooper D.A. Antiretroviral therapy of HIV-1 infection: established treatment strategies and new therapeutic options. Curr. Opin. Microbiol. 2000;3:508–514. doi: 10.1016/S1369-5274(00)00131-4. [DOI] [PubMed] [Google Scholar]
- 77.De Clercq E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int. J. of Antimicrob. Agents. 2009;33:307–320. doi: 10.1016/j.ijantimicag.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 78.Krausslich H.G., Bartenschlager R., editors. Handbook of Experimental Pharmacology. Volume 189 Springer-Verlag; Berlin-Heidelberg, Germany: 2010. Antiviral strategies. [Google Scholar]
- 79.Benigni R., Netzeva T.I., Benfenati E., Bossa C., Franke R., Helma C., Hulzebos E., Marchant C., Richard A., Woo Y.T., et al. The expanding role of predictive toxicology: An update on the (Q)SAR models for mutagens and carcinogens. J. Environ. Sci. Health C. 2007;25:53–97. doi: 10.1080/10590500701201828. [DOI] [PubMed] [Google Scholar]
- 80.Benigni R., Bossa C., Jeliazkova N., Netzeva T., Worth A. The Benigni/Bossa Rules for Mutagenicity and CarcInogenicity—A Module of Toxtree. IdeaConsult Ltd.; Sofia, Bulgaria: 2008. [(accessed on 3 August 2011)]. European Commission report EUR 23241 EN. Available online: http://toxtree.sourceforge.net/carc.html. [Google Scholar]
- 81.Lessigiariska I., Worth A.P., Cronin M.T.D. Structure-Activity Relationships for Pharmacotoxicological Endpoints. Lambert Academic Publishing; Saarbrucken, Germany: 2010. [Google Scholar]
- 82.Putz M.V. Residual-QSAR. Implications for Genotoxic Carcinogenesis. Chem. Cent. J. 2011;5:29–40. doi: 10.1186/1752-153X-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tarko L., Putz M.V. On Quantitative Structure-Toxicity Relationships (QSTR) using high chemical diversity molecules group. J. Theor. Comput. Chem. 2012;11:265–272. doi: 10.1142/S0219633612500174. [DOI] [Google Scholar]
- 84.Chemical Identifier Resolver beta 4. [(accessed on 23 February 2013)]. Available online: http://cactus.nci.nih.gov/chemical/structure.
- 85.Moldoveanu C.C., Jones P.G., Mangalagiu I.I. Spiroheterocyclic compounds: Old stories with new outcomes. Tetrahedron Lett. 2009;50:7205–7208. doi: 10.1016/j.tetlet.2009.10.044. [DOI] [Google Scholar]
- 86.Gammon D.B., Snoeck R., Fiten P., Krecmerova M., Holy A., De Clercq E., Opdenakker G., Evans D.H., Andrei G. Mechanism of antiviral drug resistance of Vaccinia virus: Identification of residues in the viral DNA polymerase conferring differential resistance to Antipoxvirus drugs. J. Virol. 2008;82:12520–12534. doi: 10.1128/JVI.01528-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fan S.Y., Zheng Z.B., Mi C.L., Zhou X.B., Yan H., Gong Z.H., Li S. Synthesis and evaluation of novel chloropyridazine derivatives as potent human rhinovirus (HRV) capsid-binding inhibitors. Bioorg. Med. Chem. 2009;17:621–624. doi: 10.1016/j.bmc.2008.11.061. [DOI] [PubMed] [Google Scholar]
- 88.De Clercq E. New approaches toward anti-HIV chemotherapy. J. Med. Chem. 2005;48:1297–1313. doi: 10.1021/jm040158k. [DOI] [PubMed] [Google Scholar]
- 89.Muhanji C.I., Hunter R. Current developments in the synthesis and biological activity of HIV-1 double-drug inhibitors. Curr. Med. Chem. 2007;14:1207–1222. doi: 10.2174/092986707780597952. [DOI] [PubMed] [Google Scholar]
- 90.Butnariu R., Mangalagiu I.I. New pyridazine derivatives: Synthesis, Chemistry and biological activity. Bioorg. Med. Chem. 2009;17:2823–2829. doi: 10.1016/j.bmc.2009.02.028. [DOI] [PubMed] [Google Scholar]
- 91.Balan A.M., Florea O., Moldoveanu C., Zbancioc G., Iurea D., Mangalagiu I.I. Diazinium salts with dihydroxyacetophenone skeleton: syntheses and antimicrobial activity. Eur. J. Med. Chem. 2009;44:2275–2279. doi: 10.1016/j.ejmech.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 92.Butnariu R., Caprosu M., Bejan V., Ungureanu M., Poiata A., Tuchilus C., Florescu M., Mangalagiu I.I. Pyridazine and phthalazine derivatives with potential antimicrobial activity. J. Heterocycl. Chem. 2007;44:1149–1152. doi: 10.1002/jhet.5570440528. [DOI] [Google Scholar]
- 93.Parr R.G., Yang W. Density-Functional Theory Of Atoms And Molecules. Oxford University Press; New York, NY, USA: 1989. [Google Scholar]
- 94.Koopmans T. Uber die Zuordnung von Wellen Funktionen und Eigenwerter zu den einzelnen Elektronen eines Atom. Physica. 1934;1:104–113. doi: 10.1016/S0031-8914(34)90011-2. [DOI] [Google Scholar]
- 95.Parr R.G., Donnelly R.A., Levy M., Palke W.E. Electronegativity: The density functional viewpoint. J. Chem. Phys. 1978;68:3801–3808. doi: 10.1063/1.436185. [DOI] [Google Scholar]
- 96.Mortier W.J., Genechten K.V., Gasteiger J. Electronegativity equalization: Application and parametrization. J. Am. Chem. Soc. 1985;107:829–835. doi: 10.1021/ja00290a017. [DOI] [Google Scholar]
- 97.Sanderson R.T. Principles of electronegativity Part I. General nature. J. Chem. Edu. 1988;65:112–119. doi: 10.1021/ed065p112. [DOI] [Google Scholar]
- 98.Putz M.V. Chemical action concept and principle. MATCH Commun. Math. Comput. Chem. 2011;66:35–63. [Google Scholar]
- 99.Tachibana A. Density functional rationale of chemical reaction coordinate. Int. J. Quantum Chem. 1987;21:181–190. doi: 10.1002/qua.560320721. [DOI] [Google Scholar]
- 100.Tachibana A., Parr R.G. On the redistribution of electrons for chemical reaction systems. Int. J. Quantum. Chem. 1992;41:527–555. doi: 10.1002/qua.560410402. [DOI] [Google Scholar]
- 101.Tachibana A., Nakamura K., Sakata K., Morisaki T. Application of the regional density functional theory: The chemical potential inequality in the HeH+ system. Int. J. Quantum Chem. 1999;74:669–679. doi: 10.1002/(SICI)1097-461X(1999)74:6<669::AID-QUA8>3.0.CO;2-O. [DOI] [Google Scholar]
- 102.Parr R.G., Yang W. Density functional approach to the frontier electron theory of chemical reactivity. J. Am. Chem. Soc. 1984;106:4049–4050. doi: 10.1021/ja00326a036. [DOI] [Google Scholar]
- 103.Yang W., Parr R.G., Pucci R. Electron density, Kohn-Sham frontier orbitals, and Fukui functionS. J. Chem. Phys. 1984;81:2862–2863. doi: 10.1063/1.447964. [DOI] [Google Scholar]
- 104.Putz M.V. Contributions within Density Functional Theory with Applications in Chemical Reactivity Theory and Electronegativity. Dissertation.com; Parkland, FL, USA: 2003. [Google Scholar]
- 105.Pearson R.G. Chemical Hardness. Wiley-VCH; Weinheim, Germany: 1997. [Google Scholar]
- 106.Putz M.V. Electronegativity and chemical hardness: different patterns in quantum chemistry. Curr. Phys. Chem. 2011;1:111–139. doi: 10.2174/1877946811101020111. [DOI] [Google Scholar]
- 107.Putz M.V. Electronegativity: Quantum observable. Int. J. Quantum Chem. 2009;109:733–738. doi: 10.1002/qua.21957. [DOI] [Google Scholar]
- 108.Putz M.V. Chemical hardness: Quantum observable? Studia Univ. Babeş-Bolyai-Ser. Chem. 2010;55:47–50. [Google Scholar]
- 109.Chattaraj P.K., Schleyer P.V.R. An ab initio study resulting in a greater understanding of the HSAB principle. J. Am. Chem. Soc. 1994;116:1067–1071. doi: 10.1021/ja00082a031. [DOI] [Google Scholar]
- 110.Chattaraj P.K., Maiti B. HSAB principle applied to the time evolution of chemical reactions. J. Am. Chem. Soc. 2003;125:2705–2710. doi: 10.1021/ja0276063. [DOI] [PubMed] [Google Scholar]
- 111.Putz M.V., Russo N., Sicilia E. On the application of the HSAB principle through the use of improved computational schemes for chemical hardness evaluation. J. Comput. Chem. 2004;25:994–1003. doi: 10.1002/jcc.20027. [DOI] [PubMed] [Google Scholar]
- 112.Pearson R.G. Hard and soft acids and bases—the evolution of a chemical concept. Coord. Chem. Rev. 1990;100:403–425. doi: 10.1016/0010-8545(90)85016-L. [DOI] [Google Scholar]
- 113.Pearson R.G. Absolute electronegativity and absolute hardness of Lewis acids and bases. J. Am. Chem. Soc. 1985;107:6801–6806. doi: 10.1021/ja00310a009. [DOI] [Google Scholar]
- 114.Chattaraj P.K., Lee H., Parr R.G. Principle of maximum hardness. J. Am. Chem. Soc. 1991;113:1854–1855. doi: 10.1021/ja00005a072. [DOI] [Google Scholar]
- 115.Chattaraj P.K., Liu G.H., Parr R.G. The maximum hardness principle in the Gyftpoulos-Hatsopoulos three-level model for an atomic or molecular species and its positive and negative ions. Chem. Phys. Lett. 1995;237:171–176. doi: 10.1016/0009-2614(95)00280-H. [DOI] [Google Scholar]
- 116.Putz M.V. Maximum hardness index of quantum acid-base bonding. MATCH Commun. Math. Comput. Chem. 2008;60:845–868. [Google Scholar]
- 117.Ayers P.W., Parr R.G. Variational principles for describing chemical reactions: The Fukui function and chemical hardness revisited. J. Am. Chem. Soc. 2000;122:2010–2018. doi: 10.1021/ja9924039. [DOI] [PubMed] [Google Scholar]
- 118.Mineva T., Sicilia E., Russo N. Density functional approach to hardness evaluation and its use in the study of the maximum hardness principle. J. Am. Chem. Soc. 1998;120:9053–9058. doi: 10.1021/ja974149v. [DOI] [Google Scholar]
- 119.Torrent-Sucarrat M., Solà M. Gas-phase structures, rotational barriers, and conformational properties of hydroxyl and mercapto derivatives of cyclohexa-2,5-dienone and cyclohexa-2,5-dienthione. J. Phys. Chem. A. 2006;110:8901–8911. doi: 10.1021/jp060664g. [DOI] [PubMed] [Google Scholar]
- 120.Parr R.G., Szentpaly L.V., Liu S. Electrophilicity Index. J. Am. Chem. Soc. 1999;121:1922–1924. doi: 10.1021/ja983494x. [DOI] [Google Scholar]
- 121.Pérez P., Toro-Labbé A., Aizman A., Contreras R. Comparison between experimental and theoretical scales of electrophilicity in benzhydryl cations. J. Org. Chem. 2002;67:4747–2752. doi: 10.1021/jo020255q. [DOI] [PubMed] [Google Scholar]
- 122.Chamorro E., Chattaraj P.K., Fuentealba P. Variation of the electrophilicity index along the reaction path. J. Phys. Chem. A. 2003;107:7068–7072. doi: 10.1021/jp035435y. [DOI] [PubMed] [Google Scholar]
- 123.Parthasarathi R., Elango M., Subramanian V., Chattaraj P.K. Variation of electrophilicity during molecular vibrations and internal rotations. Theor. Chem. Acc. 2005;113:257–266. doi: 10.1007/s00214-005-0634-3. [DOI] [Google Scholar]
- 124.Domingo L.R., Asensio A., Arroyo P. Density functional theory study of the Lewis acid-catalyzed Diels–Alder reaction of nitroalkenes with vinyl ethers using aluminum derivatives. J. Phys. Org. Chem. 2002;15:660–666. doi: 10.1002/poc.529. [DOI] [Google Scholar]
- 125.Domingo L.R., Aurell M.J., Pérez P., Contreras R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron. 2002;58:4417–4423. doi: 10.1016/S0040-4020(02)00410-6. [DOI] [Google Scholar]
- 126.Pérez P., Toro-Labbé A., Contreras R. Solvent effects on electrophilicity. J. Am. Chem. Soc. 2001;123:5527–5531. doi: 10.1021/ja004105d. [DOI] [PubMed] [Google Scholar]
- 127.Meneses L., Fuentealba P., Contreras R. On the variations of electronic chemical potential and chemical hardness induced by solvent effects. Chem. Phys. Lett. 2006;433:54–57. doi: 10.1016/j.cplett.2006.10.124. [DOI] [Google Scholar]
- 128.Parthasarathi R., Subramanian V., Chattaraj P.K. Effect of electric field on the global and local reactivity indices. Chem. Phys. Lett. 2003;382:48–56. doi: 10.1016/j.cplett.2003.09.160. [DOI] [Google Scholar]
- 129.Parthasarathi R., Padmanabhan J., Subramanian V., Maiti B., Chattaraj P.K. Chemical reactivity profiles of two selected polychlorinated biphenyls. J. Phys. Chem. A. 2003;107:10346–10352. doi: 10.1021/jp035620b. [DOI] [Google Scholar]
- 130.Parthasarathi R., Subramanian V., Roy D.R., Chattaraj P.K. Electrophilicity index as a possible descriptor of biological activity. Bioorg. Med. Chem. 2004;12:5533–5543. doi: 10.1016/j.bmc.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 131.Padmanabhan J., Parthasarathi R., Subramanian V., Chattaraj P.K. Molecular structure, reactivity, and toxicity of the complete series of chlorinated benzenes. J. Phys. Chem. A. 2005;109:11043–11049. doi: 10.1021/jp0538621. [DOI] [PubMed] [Google Scholar]
- 132.Padmanabhan J., Parthasarathi R., Subramanian V., Chattaraj P.K. Chemical reactivity indices for the complete series of chlorinated benzenes: solvent effect. J. Phys. Chem. A. 2006;110:2739–2745. doi: 10.1021/jp056630a. [DOI] [PubMed] [Google Scholar]
- 133.Padmanabhan J., Parthasarathi R., Subramanian V., Chattaraj P.K. Group philicity and electrophilicity as possible descriptors for modeling ecotoxicity applied to chlorophenols. Chem. Res. Toxicol. 2006;19:356–364. doi: 10.1021/tx050322m. [DOI] [PubMed] [Google Scholar]
- 134.Rong C., Lian S., Yin D., Zhong A., Zhang R., Liu S. Effective simulation of biological systems: choice of density functional and basis set for heme-containing complexes. Chem. Phys. Lett. 2007;434:149–154. doi: 10.1016/j.cplett.2006.11.092. [DOI] [Google Scholar]
- 135.Roy D.R., Pal N., Mitra A., Bultinck P., Parthasarathi R., Subramanian V., Chattaraj P.K. An atom counting strategy towards analyzing the biological activity of sex hormones. Eur. J. Med. Chem. 2007;42:1365–1369. doi: 10.1016/j.ejmech.2007.01.028. [DOI] [PubMed] [Google Scholar]
- 136.Schultz T.W., Hewitt M., Netzeva T.I., Cronin M.T.D. Assessing applicability domains of toxicological QSARs: Definition, Confidence in predicted values, And the role of mechanisms of action. QSAR Comb. Sci. 2007;26:238–254. doi: 10.1002/qsar.200630020. [DOI] [Google Scholar]
- 137.Netzeva T.I., Gallegos A., Worth A.P. Comparison of the applicability domain of a QSAR for estrogenicity with a large chemical inventory. Environ. Toxicol. Chem. 2006;25:1223–1230. doi: 10.1897/05-367R.1. [DOI] [PubMed] [Google Scholar]
- 138.Roberts D.W., Aptula A.O., Patlewicz G. Mechanistic applicability domains for non-animal based prediction of toxicological endpoints. QSAR analysis of the Schiff base applicability domain for skin sensitization. Chem. Res. Toxicol. 2006;19:1228–1233. doi: 10.1021/tx060102o. [DOI] [PubMed] [Google Scholar]
- 139.Aptula A.O., Patlewicz G., Roberts D.W. Skin sensitization: Reaction mechanistic applicability domains for Structure-Activity Relationships. Chem. Res. Toxicol. 2005;18:1420–1426. doi: 10.1021/tx050075m. [DOI] [PubMed] [Google Scholar]
- 140.Program Package, HyperChem 7.01. Hypercube, Inc.; Gainesville, FL, USA: 2002. [Google Scholar]
- 141.Putz M.V. On absolute aromaticity within electronegativity and chemical hardness reactivity pictures. MATCH Commun. Math. Comput. Chem. 2010;64:391–418. [Google Scholar]
- 142.Lele S.K. Compact finite difference schemes with spectral-like resolution. J. Comput. Phys. 1992;103:16–42. doi: 10.1016/0021-9991(92)90324-R. [DOI] [Google Scholar]
- 143.ViiV Healthcare Group of Companies. The biology of HIV, 2012. [(accessed on 4 May 2013)]. Available online: http://www.apositivelife.com/forasos/biology-of-hiv.html.
- 144.Genetic Engineering & Biotechnology News. Pfizer Inks Deal with K.U. Leuven for HIV drugs with new mechanism of action. 2010. [(accessed on 5 April 2013)]. Available online: http://www.genengnews.com/gen-news-highlights/pfizer-inks-deal-with-k-u-leuven-for-hiv-drugs-with-new-mechanism-of-action/81243931/
- 145.Aksimentiev A., Heng J.B., Timp G., Schulten K. Microscopic kinetics of DNA translocation through synthetic nanopores. Biophys. J. 2004;87:2086–2097. doi: 10.1529/biophysj.104.042960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Heng J.B., Ho C., Kim T., Timp R., Aksimentiev A., Grinkova Y.V., Sligar S., Schulten K., Timp G. Sizing DNA using a nanometer-diameter pore. Biophys. J. 2004;87:2905–2911. doi: 10.1529/biophysj.104.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Perilla J.R., Zhao G., Chandler D., Gronenborn A., Zhang P., Schulten K. Refinement of Atomic Models of HIV-1 Oligomers. [(accessed on 5 April 2013)]. Available online: http://www.ks.uiuc.edu/
- 148.Ganser-Pornillos B.K., Yeager M., Sundquist W.I. The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 2008;18:203–217. doi: 10.1016/j.sbi.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Picado M.J. Avances en la diseminación del VIH. Apr 30, 2012. [(accessed on 5 April 2013)]. La Ciencia y sus Demonios. Available online: http://lacienciaysusdemonios.com/2012/04/30/avances-en-la-diseminacion-del-vih/#more-24038.
- 150.C.H.A.N.G.E.—Counteracting HIV/AIDS through new gene enhancement. [(accessed on 5 April 2013)]. Available online: http://dev.nsta.org/evwebs/577/Present_Technology_Page.html.
- 151.Madrid M., Jacobo-Molina A., Ding J., Arnold E. Major subdomain rearrangement in HIV-1 reverse transcriptase simulated by molecular dynamics. Proteins Struct. Funct. Bioinf. 1999;35:332–337. doi: 10.1002/(SICI)1097-0134(19990515)35:3<332::AID-PROT7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 152.Topliss J.G., Costello J.D. Chance correlation in structure-activity studies using multiple regression analysis. J. Med. Chem. 1972;15:1066–1069. doi: 10.1021/jm00280a017. [DOI] [PubMed] [Google Scholar]
- 153.Puzyn T., Mostrag-Szlichtyng A., Gajewicz A., Skrzynski M., Worth A.P. Investigating the influence of data splitting on the predictive ability of QSAR/QSPR models. Struct. Chem. 2011;22:795–804. doi: 10.1007/s11224-011-9757-4. [DOI] [Google Scholar]
- 154.Pavan M., Netzeva T.I., Worth A.P. Validation of a QSAR Model for Acute Toxicity. SAR QSAR Environ. Res. 2006;17:147–171. doi: 10.1080/10659360600636253. [DOI] [PubMed] [Google Scholar]
- 155.Putz M.V., Lazea M., Putz A.M., Seiman-Duda C. Introducing Catastrophe-QSAR. Application on Modeling Molecular Mechanisms of Pyridinone Derivative-Type HIV Non-Nucleoside Reverse Transcriptase Inhibitors. Int. J. Mol. Sci. 2011;12:9533–9569. doi: 10.3390/ijms12129533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Balaban A.T. Chemical Applications of Graph Theory. Academic Press; London, UK: 1976. [Google Scholar]
- 157.Balaban A.T. Highly discriminating distance-based topological index. Chem. Phys. Lett. 1982;89:399–404. doi: 10.1016/0009-2614(82)80009-2. [DOI] [Google Scholar]
- 158.Basak S.C., Mills D.R., Balaban A.T., Gute B.D. Prediction of mutagenicity of aromatic and heteroaromatic amines from structure: a hierarchical QSAR approach. J. Chem. Inf. Comput. Sci. 2011;41:671–678. doi: 10.1021/ci000126f. [DOI] [PubMed] [Google Scholar]
- 159.Balaban A.T. From chemical topology to 3D geometry. J. Chem. Inf. Comput. Sci. 1997;37:645–650. doi: 10.1021/ci960168x. [DOI] [Google Scholar]
- 160.Lewis G.N. The atom and the molecule. J. Am. Chem. Soc. 1916;38:762–785. doi: 10.1021/ja02261a002. [DOI] [Google Scholar]
- 161.Pauling L., Wheland G.W. The nature of the chemical bond. V. The quantum-mechanical calculation of the resonance energy of benzene and naphthalene and the hydrocarbon free radicals. J. Chem. Phys. 1933;1:362–375. doi: 10.1063/1.1749304. [DOI] [Google Scholar]
- 162.Pauling L., Sherman J. The nature of the chemical bond. VI. The calculation from thermochemical data of the energy of resonance of molecules among several electronic structures. J. Chem. Phys. 1933;1:606–618. doi: 10.1063/1.1749335. [DOI] [Google Scholar]
- 163.Pauling L., Wilson E.B. Introduction to Quantum Mechanics with Applications to Chemistry. McGraw-Hill; New York, NY, USA: 1935. [Google Scholar]
- 164.Wheland G.W. The Theory of Resonance and its Application to Organic Chemistry. Wiley; New York, NY, USA: 1944. [Google Scholar]
- 165.Heitler W., London F. Wechselwirkung neutraler Atome und homöopolare Bindung nach der Quantenmechanik. Z. Phys. 1927;44:455–472. doi: 10.1007/BF01397394. [DOI] [Google Scholar]
- 166.Hückel E. Quantentheoretische Beiträge zum Benzolproblem. I. Z. Physik. 1931;71:204–286. doi: 10.1007/BF01339530. [DOI] [Google Scholar]
- 167.Hückel E. Quantentheoretische Beiträge zum Benzolproblem. II. Z. Physik. 1931;72:310–337. doi: 10.1007/BF01341953. [DOI] [Google Scholar]
- 168.Hoffmann R. An extended Hückel theory. I. Hydrocarbons. J. Chem. Phys. 1963;39:1397–1412. doi: 10.1063/1.1734456. [DOI] [Google Scholar]
- 169.Bader R.F.W. Atoms in Molecules - A Quantum Theory. Oxford University Press; Oxford, UK: 1990. [Google Scholar]
- 170.Bader R.F.W. Principle of stationary action and the definition of a proper open system. Phys. Rev. B. 1994;49:13348–13356. doi: 10.1103/PhysRevB.49.13348. [DOI] [PubMed] [Google Scholar]
- 171.Bader R.F.W. A bond path: a universal indicator of bonded interactions. J. Phys. Chem. A. 1998;102:7314–7323. doi: 10.1021/jp981794v. [DOI] [Google Scholar]
- 172.Gillespie R.J., Nyholm R.S. Inorganic stereochemistry. Quart. Rev. Chem. Soc. 1957;11:339–380. doi: 10.1039/qr9571100339. [DOI] [Google Scholar]
- 173.Gillespie R.J., Hargittai I. The VSEPR Model of Molecular Geometry. Allyn and Bacon; Boston, MA, USA: 1991. [Google Scholar]
- 174.Kohn W., Becke A.D., Parr R.G. Density functional theory of electronic structure. J. Phys. Chem. 1996;100:12974–12980. doi: 10.1021/jp960669l. [DOI] [Google Scholar]
- 175.Putz M.V. Chemical action and chemical bonding. J. Mol. Structure THEOCHEM. 2009;900:64–70. doi: 10.1016/j.theochem.2008.12.026. [DOI] [Google Scholar]
- 176.Putz M.V. Quantum parabolic effects of electronegativity and chemical hardness on carbon π-systems. In: Putz M.V., editor. Carbon Bonding And Structures: Advances in Physics and Chemistry. Springer Verlag; London, UK: 2011. pp. 1–32. [Google Scholar]
- 177.Putz M.V. Quantum Theory: Density, Condensation, and Bonding. Apple Academics; Ontario, New Jersey, NJ, USA: 2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.