

Abstract
The development of novel antimicrobial agents represents a timely research topic. Eighteen salicylanilide 4-(trifluoromethyl)benzoates were evaluated against Mycobacterium tuberculosis, M. avium and M. kansasii, eight bacterial strains including methicillin-resistant Staphylococcus aureus (MRSA) and for the inhibition of mycobacterial isocitrate lyase. Some compounds were further screened against drug-resistant M. tuberculosis and for their cytotoxicity. Minimum inhibitory concentrations (MICs) for all mycobacterial strains were within 0.5–32 μmol/L, with 4-chloro-2-[4-(trifluoromethyl)phenylcarbamoyl]phenyl 4-(trifluoromethyl)benzoate superiority. Gram-positive bacteria including MRSA were inhibited with MICs ≥ 0.49 μmol/L, while Gram-negative ones were much less susceptible. Salicylanilide 4-(trifluoromethyl)benzoates showed significant antibacterial properties, for many strains being comparable to standard drugs (isoniazid, benzylpenicillin) with no cross-resistance. All esters showed mild inhibition of mycobacterial isocitrate lyase and four compounds were comparable to 3-nitropropionic acid without a direct correlation between in vitro MICs and enzyme inhibition.
Keywords: antibacterial activity, antimycobacterial activity, cytotoxicity, isocitrate lyase inhibitor, multidrug-resistant tuberculosis, salicylanilide ester, 4-(trifluoromethyl)benzoic acid ester
1. Introduction
The inappropriate use or misuse of established antimicrobial drugs has led to the increasing emergence of microbial resistance. The possible loss of the global control of infectious diseases represents a serious threat for public health, since resistance has been described for many important human pathogens.
Tuberculosis (TB) is responsible for millions of human deaths and about 1.4 million people died from TB in 2010 [1]. The drug-resistant TB forms, especially multidrug-resistant and extensively drug-resistant tuberculosis (MDR- and XDR-TB), bring grave problems. MDR-TB consists in a resistance to at least isoniazid (INH) and rifampicin (RIF), the most effective first-line oral agents, while XDR-TB is MDR-TB in combination with both resistance to any fluoroquinolone and at least one second-line injectable drug (kanamycin, amikacin, capreomycin) [2]. Latent TB and also coincidence of TB with HIV-infection signify other principal impetuses for novel antimycobacterial drugs.
Contemporary research trends are focused, i.e., on the identification of unique mycobacterial pathways. Isocitrate lyase (ICL), an essential enzyme for the metabolism of fatty acids which splits isocitrate to succinate and glyoxylate, is one of two glyoxylate shunt enzymes. This pathway replenishes tricarboxylic acids cycle intermediates during growth on C2 substrates and is lacking in vertebrates. Disruption of icl gene attenuated bacterial persistence and virulence without affecting the bacterial growth during the acute phase of infection. ICL might contribute to mycobacterial adaptation to hypoxia. Conventional anti-tuberculosis drugs exhibit mostly an insufficient activity against slow- or non-growing (latent) mycobacteria. This is thought to be an important reason for the length of anti-tuberculosis therapy [3]. Novel ICL inhibitors may bring the shortening of TB treatment course and targeting of ICL represents one of the up-to-date research topics [4,5].
Nosocomial infections are a major challenge because of the high rates of morbidity and mortality. Worldwide, the state of resistance increased considerably in both Gram-positive and Gram-negative pathogens. Methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus spp., Klebsiella pneumoniae, Escherichia coli, Pseudomonas spp. or Acinetobacter baumannii producing extended spectrum β-lactamases (ESBL) represent such a most common and problematic drug-resistant bacteria, which have been rapidly spread throughout the world [6].
Consequently, novel drugs and strategies to control microbial spread are of critical importance, especially molecules with unique mechanism of action and no structural similarity. They can replace or combine with conventional antibiotics to combat problematic strains. The question if a new drug should be broad-spectral rather than have a narrow-spectrum of antimicrobial activity is controversial [7].
Salicylanilides (2-hydroxy-N-phenylbenzamides) and their derivatives have been reported to share a significant antibacterial activity, especially against atypical mycobacteria, Mycobacterium tuberculosis (Mtb.) including drug-resistant strains, Gram-positive cocci including MRSA with minimum inhibitory concentrations (MICs) in micromolar range [8,9,10,11,12,13,14]. Additionally, salicylanilides inhibit moderately mycobacterial isocitrate lyase and methionine aminopeptidase [4], but they affect multiple targets within the cells of pathogens [4,8,14,15].
Salicylanilide scaffold represents still an investigated group of compounds with many interesting pharmacological properties. Recently, they have been reported, e.g., as potential agents with a significant activity against Onchocerca volvulus [15], Toxoplasma gondii parasite [16] or with anticancer features [17,18,19,20].
Salicylanilides derived from 4-(trifluoromethyl)aniline have exhibited predominantly a high antimycobacterial activity; in general, the introduction of CF3 moiety into the salicylanilide scaffold enhanced antimicrobial properties [4,9,10,11,12,13]. Salicylanilide benzoates revealed a notable activity especially toward MDR-TB [13] as well as antibacterial one [11]. Aromatic esters of 4-(trifluoromethyl)benzoic acid have been reported as a mild antibacterial agents against Pseudomonas fluorescens and Bacillus subtilis, since inactive for S. aureus and E. coli [21], and some salicylanilide 4-(trifluoromethyl)benzoates exhibited antifungal activity, especially against moulds with MICs ≥ 0.49 μmol/L [22]. Additionally, an increased lipophilicity may ameliorate passing through cell walls and biomembranes, that is why we selected highly lipophilic salicylanilide 4-(trifluoromethyl)benzoates for the searching of a potential antibacterial and antimycobacterial agents with expected lower MIC.
2. Results and Discussion
Salicylanilide 4-(trifluoromethyl)benzoates 1 were evaluated for their in vitro antimycobacterial and antibacterial activity, cytotoxicity and inhibition of mycobacterial isocitrate lyase.
2.1. Chemistry
The synthesis and characterization of the salicylanilide 4-(trifluoromethyl)benzoates (Scheme 1) were published recently [22]. Yield of esters synthesized via N,N'-dicyclohexylcarbodiimide coupling ranged from 49% up to 86%.
Scheme 1.
Synthesis of salicylanilides SAL and corresponding 4-(trifluoromethyl)benzoates 1 (R1 for esters: 1 = 4-Cl, 5-Cl, 4-Br; R2 = 3-Cl, 4-Cl, 3,4-diCl, 3-Br, 4-Br, 3-F, 4-F, 3-CF3, 4-CF3).

Abbreviations: MW: microwave irradiation (530 W, 600 rpm, 20 min); Ph-Cl: chlorobenzene; DCC: N,N'-dicyclohexylcarbodiimide; DMF: N,N-dimethylformamide.
2.2. Antimycobacterial Evaluation
Table 1 summarizes the antimycobacterial activity of esters 1, isoniazid and para-aminosalicylic acid, two comparative drugs. All of the 4-(trifluoromethyl)benzoates displayed a significant antimycobacterial activity, with MICs of 0.5–32 μmol/L against all strains, with 1o and 1r showing superiority (1–4 μmol/L). Mycobacterium avium showed the highest MIC values, up to 32 μmol/L. All esters displayed a better in vitro activity against four mycobacterial strains than PAS, a second-line oral drug sharing a structural similarity. Additionally, salicylanilide derivatives exhibited lower MICs than INH for M. kansasii 235/80 and M. avium; MIC values against M. kansasii 6509/96 were comparable or a slightly better in the case of esters. Only three esters (1e, 1o and 1r) showed equal activity like INH against M. tuberculosis (1 μmol/L). 4-(Trifluoromethyl)benzoic acid exhibited only a very mild intrinsic activity towards M. kansasii (MICs ≥ 250 μmol/L), whereas other mycobacteria were completely insusceptible at 1,000 µmol/L.
Table 1.
Antimycobacterial activity of salicylanilide 4-(trifluoromethyl)benzoates 1.
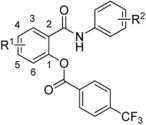 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC [μmol/L] | ||||||||||||||||||
| R1 | R2 |
Mtb.
331/88 |
M. avium
330/88 |
M. kansasii
235/80 |
M. kansasii
6509/96 |
|||||||||||||
| 14 d | 21 d | 14 d | 21 d | 7 d | 14 d | 21 d | 7 d | 14 d | 21 d | |||||||||
| 1a | 4-Cl | 3-Cl | 2 | 4 | 8 | 16 | 0.5 | 2 | 4 | 2 | 8 | 8 | ||||||
| 1b | 5-Cl | 3-Cl | 4 | 4 | 8 | 8 | 1 | 4 | 4 | 2 | 8 | 8 | ||||||
| 1c | 4-Cl | 4-Cl | 2 | 2 | 4 | 8 | 1 | 2 | 2 | 1 | 4 | 4 | ||||||
| 1d | 5-Cl | 4-Cl | 4 | 4 | 4 | 8 | 1 | 4 | 4 | 1 | 4 | 4 | ||||||
| 1e | 4-Cl | 3,4-diCl | 1 | 1 | 4 | 4 | 1 | 2 | 4 | 1 | 2 | 4 | ||||||
| 1f | 5-Cl | 3,4-diCl | 1 | 2 | 2 | 8 | 1 | 2 | 4 | 1 | 2 | 4 | ||||||
| 1g | 4-Cl | 3-Br | 1 | 2 | 2 | 8 | 2 | 4 | 4 | 1 | 4 | 4 | ||||||
| 1h | 5-Cl | 3-Br | 4 | 8 | 8 | 16 | 4 | 8 | 8 | 2 | 8 | 8 | ||||||
| 1i | 4-Cl | 4-Br | 2 | 2 | 8 | 8 | 1 | 4 | 8 | 2 | 8 | 8 | ||||||
| 1j | 5-Cl | 4-Br | 2 | 4 | 8 | 8 | 1 | 4 | 4 | 2 | 4 | 4 | ||||||
| 1k | 4-Cl | 3-F | 8 | 8 | 16 | 32 | 8 | 16 | 16 | 8 | 16 | 16 | ||||||
| 1l | 5-Cl | 3-F | 4 | 4 | 8 | 16 | 4 | 8 | 8 | 8 | 8 | 16 | ||||||
| 1m | 4-Cl | 4-F | 8 | 8 | 16 | 32 | 4 | 16 | 16 | 8 | 16 | 16 | ||||||
| 1n | 5-Cl | 4-F | 4 | 8 | 4 | 4 | 2 | 8 | 8 | 4 | 8 | 8 | ||||||
| 1o | 4-Cl | 4-CF3 | 1 | 1 | 1 | 4 | 1 | 2 | 2 | 1 | 2 | 2 | ||||||
| 1p | 5-Cl | 4-CF3 | 2 | 2 | 2 | 8 | 2 | 2 | 4 | 1 | 4 | 4 | ||||||
| 1q | 4-Cl | 3-CF3 | 2 | 2 | 4 | 16 | 2 | 4 | 8 | 1 | 4 | 8 | ||||||
| 1r | 4-Br | 4-CF3 | 1 | 1 | 2 | 4 | 1 | 2 | 4 | 1 | 2 | 4 | ||||||
| INH | 0.5–1 | 0.5–1 | ≥250 | ≥250 | >250 | >250 | >250 | 2–4 | 4–8 | 8–16 | ||||||||
| PAS | 62.5 | 62.5 | 32 | 125 | 125 | 1000 | >1000 | 32 | 125 | 500 | ||||||||
| CF3-BA | >1,000 | >1,000 | >1,000 | >1,000 | 1,000 | >1,000 | >1,000 | 250 | 1,000 | 1,000 | ||||||||
INH = isoniazid; PAS = p-aminosalicylic acid; CF3-BA = 4-(trifluoromethyl)benzoic acid; Two best MICs for each strain are given in bold.
In general, the most active derivatives share a trifluoromethyl moiety (compounds 1o–r) or two chlorines (compounds 1e, 1f) on the aniline ring; on the other side, fluorine atom provided only a minimal benefit. Esters of 5-chlorosalicylanilides showed a higher antimycobacterial activity than those derived from 4-chlorosalicylanilides, with fluorinated molecules 1k–n being an exception.
Importantly, salicylanilide 4-(trifluoromethyl)benzoates 1 were assayed being predominantly more active than parent salicylanilides [23] – e.g., up to eight times for 1f, 1n and 1o against M. avium or 1e for M. tuberculosis. In some cases, esters and parent phenolic molecules share the same MIC values, e.g., 1b, 1k or 1d for M. tuberculosis. Sporadically, the esterification led to the decreased potency; namely 1i, 1k, 1m and 1o against M. kansasii, 1m and 1r for M. avium. 5-Bromo-2-hydroxy-N-[4-(trifluoromethyl)phenyl]benzamide (SAL-7) represents only one salicylanilide, which esterification did not improve activity against any mycobacterial strain. Additionally, the most active compounds (i.e., with at least one MIC against any strain ≤ 1 μmol/L) were evaluated against five MDR-TB strains and one XDR-TB strain with different resistance patterns (Table 2).
Table 2.
MICs of salicylanilide 4-(trifluoromethyl)benzoates 1 towards MDR- and XDR-TB.
| MIC [μmol/L] | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 |
Mtb. 7357/1988 |
Mtb. 9449/2006 |
Mtb. 53/2009 |
Mtb. 234/2005 |
Mtb. Praha 1 |
Mtb. Praha 131 (XDR-TB) | |||||||
| 14 d | 21 d | 14 d | 21 d | 14 d | 21 d | 14 d | 21 d | 14 d | 21 d | 14 d | 21 d | |||
| 1a | 4-Cl | 3-Cl | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| 1b | 5-Cl | 3-Cl | 8 | 8 | 4 | 4 | 8 | 8 | 8 | 8 | 4 | 4 | 8 | 8 |
| 1c | 4-Cl | 4-Cl | 2 | 2 | 2 | 2 | 4 | 4 | 4 | 4 | 2 | 2 | 2 | 2 |
| 1d | 5-Cl | 4-Cl | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 2 | 4 | 2 | 4 |
| 1e | 4-Cl | 3,4-diCl | 1 | 2 | 2 | 2 | 2 | 4 | 1 | 2 | 1 | 2 | 2 | 2 |
| 1f | 5-Cl | 3,4-diCl | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 1 | 2 | 1 | 2 |
| 1g | 4-Cl | 3-Br | 2 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 1 | 2 | 1 | 2 |
| 1i | 4-Cl | 4-Br | 2 | 4 | 2 | 4 | 4 | 4 | 2 | 2 | 2 | 2 | 2 | 2 |
| 1j | 5-Cl | 4-Br | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 |
| 1o | 4-Cl | 4-CF3 | 0.5 | 1 | 1 | 1 | 1 | 1 | 0.5 | 1 | 0.5 | 0.5 | 0.5 | 1 |
| 1r | 4-Br | 4-CF3 | 1 | 1 | 1 | 1 | 1 | 2 | 0.5 | 1 | 0.5 | 1 | 0.5 | 1 |
MIC values of 1 μmol/L and lower are given in bold.
Importantly, all evaluated esters showed a significant activity towards MDR- and XDR-TB strains independently of the particular resistances. Interestingly, not only esters with the best activity towards M. tuberculosis (MICs ≤ 1 μmol/L) were assayed, as in previous studies [9,13,24], as even additional derivatives with MICs in the range of 2–4 μmol/L, were also powerful. All of these derivatives affected the growth of drug-resistant M. tuberculosis at equal or almost equal concentrations required for the inhibition of drug-sensitive one; drug-resistant strains were more susceptible to esters 1j, 1o and 1r than M. tuberculosis H37Rv. These findings indicate that salicylanilide 4-(trifluoromethyl)benzoates do not share any cross-resistance with conventionally used drugs (INH, rifamycines, EMB, STM, OFX, clofazimine, aminoglycosides) and they may be prospective and perspective agents for combating drug-resistant TB.
4-(Trifluoromethyl)benzoates 1 possess potent in vitro antimycobacterial properties; although sharing a high lipophilicity, their efficacy did not surpassed esters with N-acetyl-L-phenylalanine [9] with MDR- and XDR-TB being an exception, but esters 1 exhibited lower MICs than, e.g., salicylanilide benzenesulfonates [10] or esters with N-benzyloxycarbonyl amino acids [25]. Carbamates showed better in vitro activity against M. tuberculosis including drug-resistant strains, while atypical mycobacteria were inhibited at similar concentrations [24]. When compared to closely related salicylanilide benzoates [13], the introduction of trifluoromethyl moiety into a benzoic acid ring did not improved activity, although the presence of this substituent in parent salicylanilides resulted unambiguously in the excellent antimycobacterial properties.
2.3. Isocitrate Lyase Inhibition Assay
Salicylanilide 4-(trifluoromethyl)benzoates were evaluated for their inhibition of mycobacterial ICL to elucidate their impact on one of the known targets for latent TB infection (Table 3). Compounds with dual effect on both the actively growing and latent mycobacterial populations may bring an appreciable benefit.
Table 3.
ICL inhibition activity of salicylanilide 4-(trifluoromethyl)benzoates 1.
| R1 | R2 | % ICL inhibition at 10 μmol/L (± standard deviation) | |
|---|---|---|---|
| 1a | 4-Cl | 3-Cl | 18 ± 1.9 |
| 1b | 5-Cl | 3-Cl | 13 ± 1.7 |
| 1c | 4-Cl | 4-Cl | 12 ± 2.7 |
| 1d | 5-Cl | 4-Cl | 12 ± 2.3 |
| 1e | 4-Cl | 3,4-diCl | 18 ± 1.5 |
| 1f | 5-Cl | 3,4-diCl | 17 ± 1.6 |
| 1g | 4-Cl | 3-Br | 27 ± 0.2 |
| 1h | 5-Cl | 3-Br | 21 ± 1.8 |
| 1i | 4-Cl | 4-Br | 18 ± 1.2 |
| 1j | 5-Cl | 4-Br | 14 ± 4.0 |
| 1k | 4-Cl | 3-F | 25 ± 0.8 |
| 1l | 5-Cl | 3-F | 20 ± 0.5 |
| 1m | 4-Cl | 4-F | 23 ± 1.4 |
| 1n | 5-Cl | 4-F | 17 ± 1.3 |
| 1o | 4-Cl | 4-CF3 | 13 ± 1.2 |
| 1p | 5-Cl | 4-CF3 | 17 ± 2.3 |
| 1q | 4-Cl | 3-CF3 | 19 ± 0.5 |
| 1r | 4-Br | 4-CF3 | 23 ± 0.1 |
| 3-NP | 25 ± 4.1 | ||
| INH | 0 | ||
INH = isoniazid; 3-NP = 3-nitropropionic acid; Inhibition rate values comparable to 3-NP or higher are given in bold.
Salicylanilide 4-(trifluoromethyl)benzoates 1 showed 12%–27% inhibition of isocitrate lyase at 10 µmol/L concentration. Six esters demonstrated ≥ 20% enzyme inhibition rate (1g, 1h, 1k, 1l, 1m, 1r) with superiority of the 3-bromoaniline derivative 1g; this molecule also possessed higher potency than 3-NP, a well-known standard. Derivatives of 5-chlorosalicylic acid mostly exhibited higher potency than those related to 4-chlorosalicylic acid (1a vs. 1b, 1g vs. 1h, 1k vs. 1l, etc.). The substitution of aniline ring at the position 3 is linked with enhanced enzyme inhibition when compared to 4-substituted anilines (e.g., 1a vs. 1c, 1g vs. 1i, 1o vs. 1q).
Salicylanilide 4-(trifluoromethyl)benzoates 1 produced significantly and consistently higher ICL-1 inhibition than previously reported salicylanilide derivatives – salicylanilides, their pyrazinoates, benzoates, benzenesulfonates or esters with N-acetyl-L-phenylalanine [4].
The additional ICL inhibition assay was performed with the derivatives at the concentration of 100 µmol/L; nevertheless, some of the compounds precipitated in the medium after a very short period during the investigation. This effect at the higher concentration may be render due to a high lipophilicity.
Obviously, even though salicylanilide 4-(trifluoromethyl)benzoates 1 exhibited a significant ICL-1 inhibition, their concentration values are higher (IC50 > 10 µmol/L) in comparison with MICs necessary for the mycobacterial growth arrest. Importantly, the inhibition of isocitrate lyase does not facilitate the elimination of mycobacteria during the acute phase of infection – in contrast to the chronic phase [3]. Therefore there is not a direct relationship of in vitro MICs of salicylanilides towards growing mycobacteria and the ICL inhibition, especially when the suppression of ICL may affect the slowly- or non-growing mycobacterial subpopulations existing on the nutrition poor substrates. Thus, the salicylanilide derivatives share a complex and still not fully elucidated mechanism of action for actively growing mycobacteria, since more cellular targets have been identified for this chemical group (e.g., [4,5,15]). Further experiments dealing with the mechanism(s) of action are appropriate.
2.4. Antibacterial Evaluation
Table 4 overviews the antibacterial activity. For the remaining ten esters (1a, 1c, 1d, 1f, 1g, 1i, 1l, 1n, 1q, 1r), whose MICs are not reported, it was not possible to determine them. Similarly as in previously reported antifungal assay [22], they could not be dissolved in the testing medium well or they precipitated rapidly in them due to an escalated lipophilicity. 4-(Trifluoromethyl)benzoic acid showed no antibacterial action under our conditions (MIC > 500 µmol/L).
Table 4.
MICs of salicylanilide 4-(trifluoromethyl)benzoates 1 towards selected bacteria.
| MIC/IC90 [μmol/L] | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | S. aureus CCM 4516/08 | MRSA H 5996/08 | S. epidermidis H 6966/08 | Enterococcus sp. J 14365/08 | E. coli CCM 4517 | ||||||
| 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | |||
| 1b | 5-Cl | 3-Cl | 0.98 | 1.95 | 0.98 | 1.95 | 0.98 | 1.95 | 7.81 | 15.62 | >250 | >250 |
| 1e | 4-Cl | 3,4-diCl | 0.49 | 0.98 | 0.49 | 0.98 | 0.49 | 0.98 | 0.49 | 3.9 | >500 | >500 |
| 1h | 5-Cl | 3-Br | 0.98 | 0.98 | 0.49 | 0.98 | 0.49 | 0.98 | 31.25 | 250 | >500 | >500 |
| 1j | 5-Cl | 4-Br | 0.49 | 0.98 | 0.49 | 0.98 | 0.49 | 0.98 | 1.95 | 7.81 | 250 | >250 |
| 1k | 4-Cl | 3-F | 3.9 | 3.9 | 0.98 | 1.95 | 3.9 | 3.9 | 31.25 | >250 | >250 | >250 |
| 1m | 4-Cl | 4-F | 7.81 | 7.81 | 15.62 | 15.62 | 31.25 | >500 | 500 | >500 | >500 | >500 |
| 1o | 4-Cl | 4-CF3 | 3.9 | 7.81 | 3.9 | 7.81 | 3.9 | 7.81 | 7.81 | 62.5 | >125 | >125 |
| 1p | 5-Cl | 4-CF3 | 0.49 | 0.98 | 0.49 | 0.98 | 0.49 | 0.98 | 0.49 | 3.9 | 31.25 | 250 |
| PNC | 0.98 | 0.98 | 62.5 | 125 | 250 | 250 | 7.81 | 15.62 | >500 | >500 | ||
MRSA: methicillin-resistant Staphylococcus aureus; PNC: benzylpenicillin; One or two best MIC(s) for each strain are given in bold.
All soluble derivatives showed a very good activity against Gram-positive cocci, in some cases (1e, 1j, 1p, particularly 1b, 1h and 1k) even excellent MICs lower than 1 μmol/L, despite the methicillin-resistance. Based on MIC values, these esters act likely mainly as bactericidal agents. MICs against S. aureus were comparable to benzylpenicillin and, moreover, 4-(trifluoromethyl)benzoates 1 exceeded its activity for MRSA, S. epidermidis and partly for Enterococcus (1e, 1j, 1p). Among Gram-negative bacteria, only E. coli possessed a partial susceptibility from 31.25 μmol/L (1p), while the growth of P. aeruginosa and both strains of K. pneumoniae was not reduced up to the concentration of 500 μmol/L.
Due to a limited number of assayed esters, it is difficult to discuss the structure-activity relationship. In the pair of 1o and 1p, the 5-chloro derivative (1p) showed lower MICs and when compared 1k vs. 1m, 3'-F moiety resulted in a higher activity.
Based on limited data, 4-(trifluoromethyl)benzoates displayed more significant activity against Gram-positive bacteria than salicylanilide benzoates [11], pyrazinoates [12] or esters with N-acetyl-L-phenylalanine [9].
2.5. Cytotoxicity Evaluation
Since salicylanilide derivatives have displayed cytotoxicity for human cells (e.g., [4,9,20,24]), we examined in vitro cytotoxicity of six 4-(trifluoromethyl)benzoates, which shared the highest rate of MDR-TB growth suppression (1e, 1f, 1g, 1j, 1o, 1r), as well as esters with the lowest MICs for Staphylococcus sp. (1e, 1j, 1p). For comparison, seven parent salicylanilides (SAL-1 – SAL-7) and 4-(trifluoromethyl)benzoic acid were also investigated in the liver HepG2 cells model (Table 5). The parameter IC50 was used as a measure of toxicity, which allowed the quantitative comparison of the toxicity among the tested compounds.
Table 5.
IC50 and SI for selected salicylanilide 4-(trifluoromethyl)benzoates 1.
| R1 | R2 | IC50 [µmol/L] HepG2 | SI for Mtb. H37Rv 331/88 | SI for MDR-TB strains | SI for XDR-TB strain | SI for Staphylococcus sp. | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 24 h | 14 d | 21 d | 14 d | 21 d | 14 d | 14 d | 24 h | 48 h | |||
| 1e | 4-Cl | 3,4-diCl | 3.13 | 3.13 | 3.13 | 1.57–3.13 | 0.78–1.57 | 1.57 | 1.57 | 6.39 | 3.19 |
| 1f | 5-Cl | 3,4-diCl | 4.46 | 4.46 | 2.23 | 2.23–4.46 | 2.23 | 4.46 | 2.23 | - | - |
| 1g | 4-Cl | 3-Br | 2.63 | 2.63 | 1.32 | 1.32–2.63 | 1.32 | 2.63 | 1.32 | - | - |
| 1j | 5-Cl | 4-Br | 4.82 | 2.41 | 1.21 | 2.24 | 2.24 | 4.82 | 2.41 | 9.84 | 4.92 |
| 1o | 4-Cl | 4-CF3 | 1.33 | 1.33 | 1.33 | 1.33–2.66 | 1.33–2.66 | 2.66 | 1.33 | 0.34 | 0.17 |
| 1p | 5-Cl | 4-CF3 | 1.77 | 0.89 | 0.89 | - | - | - | - | 3.61 | 1.81 |
| 1r | 4-Br | 4-CF3 | 2.03 | 2.03 | 2.03 | 2.03–4.06 | 1.02–2.03 | 4.06 | 2.03 | - | - |
| SAL-1 | 5-Cl | 3,4-diCl | 0.84 | 0.21 | 0.10 | - | - | - | - | - | - |
| SAL-2 | 4-Cl | 3,4-diCl | 1.80 | 0.45 | 0.45 | - | - | - | - | - | - |
| SAL-3 | 5-Cl | 3-Br | 1.54 | NT | NT | - | - | - | - | - | - |
| SAL-4 | 4-Cl | 4-Br | 1.98 | 0.5 | 0.5 | - | - | - | - | - | - |
| SAL-5 | 5-Cl | 4-CF3 | 0.36 | 0.18 | 0.18 | - | - | - | - | - | - |
| SAL-6 | 4-Cl | 4-CF3 | 0.69 | 0.17 | 0.17 | - | - | - | - | - | - |
| SAL-7 | 5-Br | 4-CF3 | 1.67 | 1.67 | 1.67 | - | - | - | - | - | - |
| CF3-BA | 563.70 | - | - | - | - | - | - | - | - | ||
CF3-BA = 4-(trifluoromethyl)benzoic acid. SI = IC50/MIC100; MIC values of salicylanilides against M. tuberculosis were taken from reference [23].
IC50 of 4-(trifluoromethyl)benzoates 1 were within the range 1.33–4.82 μmol/L and IC50 of parent salicylanilides of 0.36–1.98 μmol/L. For all pairs salicylanilide and its 4-(trifluoromethyl)benzoate, the esterification alleviated mildly the strong cytotoxicity of the parent salicylanilides containing free phenolic group, which is probably mainly responsible for the cytotoxicity. Its temporary masking leads to the derivatives with somewhat higher IC50 values – from 1.2-fold (1r) up to 3.7-fold for 1e and 1p, but still in similar concentration range. 4-(Trifluoromethyl)benzoic acid was many time less toxic with IC50 of 563.7 µmol/L.
Selectivity indexes (SI) ranges from 0.1 up to 1.67 for M. tuberculosis for parent salicylanilides, while esters with a significant activity against MDR-TB showed SI of 0.89–4.46 for drug-sensitive M. tuberculosis and of 0.78–4.46 for drug-resistant strains. SI values for S. aureus were within 0.17–9.84; SI of 1j almost reached the value of 10, which is generally considered being borderline. With respect to the purposed cytotoxicity reduction of the parent salicylanilides, the results are not satisfactory.
When compared to other salicylanilide esters, 4-(trifluoromethyl)benzoates showed about ten times higher cytotoxicity than salicylanilide carbamates [24], esters with N-acetyl-L-phenylalanine [9] or even more than esters with N-benzyloxycarbonyl amino acids [25], while it was approximately comparable with salicylanilide benzenesulfonates and benzoates [4].
However, salicylanilide esterification may represent one successful way to reduce their cytotoxicity for human cells as it was demonstrated especially for esters with N-protected α-amino acids [9,25] and carbamates [24]; it is necessary to search new type(s) of acid(s), because 4-(trifluoromethyl)benzoic acid brought a very mild and still insufficient benefit.
From other point of view, salicylanilides and their 4-(trifluoromethyl)benzoates may be considered as potential anticancer agents. One study [20] reported some salicylanilides being proposed and synthesized as potential cytotoxic agents against hepatocellular carcinoma. All of our salicylanilide derivatives showed IC50 values similar to the most active salicylanilides reported by Zhu et al. [20] (1.3 and 1.7 μmol/L); even three salicylanilides (SAL-1, SAL-5, SAL-6) were more active with 5-chloro-2-hydroxy-N-[4-(trifluoromethyl)phenyl]benzamide (SAL-5) being superior with IC50 of 0.36 μmol/L. Especially salicylanilide 4-(trifluoromethyl)benzoates 1 could be more convenient because previously it was demonstrated that some 3- and 4-(trifluoromethyl)benzoate esters of scaffolds with an intrinsic activity showed superiority among other esters [26,27].
The mechanism of cytotoxicity seems to be multiple – besides non-specific damage of cellular structures and function by the in vivo released phenolic group [8], it has been described a mild inhibition of human methionine aminopeptidase [4], inhibition of EGFR protein tyrosine kinases [17,19,20,28,29] or other human enzymes [18,19].
3. Experimental
3.1. Chemistry
The synthesis and characterization (m.p., IR and NMR spectra, elemental analyses) of the presented salicylanilide 4-(trifluoromethyl)benzoates [2-(phenylcarbamoyl)phenyl 4-(trifluoromethyl)benzoates; Scheme 1] 1a–r was published recently by Krátký et al. [22].
3.2. Antimycobacterial Evaluation
All compounds were tested for their in vitro against Mycobacterium tuberculosis 331/88 (H37Rv; dilution of the strain was 10−3), nontuberculous mycobacteria: Mycobacterium avium 330/88 [resistant to INH, RIF, ofloxacin (OFX), and ethambutol (EMB); dilution 10−5], one strain of Mycobacterium kansasii 235/80 from the Czech National Collection of Type Cultures (CNCTC) (dilution 10−4) and a clinically isolated strain of M. kansasii 6509/96 (dilution 10−5). The micromethod for the determination of MICs was used. Antimycobacterial activities were determined in the Šula´s semisynthetic medium (SEVAC, Prague, Czech Republic). The tested compounds were added to the medium as solutions in dimethyl sulfoxide (DMSO). The following concentrations were used: 1000, 500, 250, 125, 62.5, 32, 16, 8, 4, 2, 1, 0.5, 0.25, and 0.125 μmol/L. The MICs were determined after incubation at 37 °C for 14 and 21 days, for M. kansasii additionally for 7 days. MIC (in μmol/L) was the lowest concentration at which the complete inhibition of mycobacterial growth occurred. The first-line anti-tuberculosis drug isoniazid (INH) and structurally similar para-aminosalicylic acid (PAS), a second-line oral agent, were chosen as the reference drugs.
The most active esters were evaluated at similar conditions and concentrations against six MDR-TB strains (dilution 10−3) with different resistance patterns. All strains are resistant to INH, RIF, rifabutine, and streptomycin (STM); an additional resistance was present in some cases: 7357/1998 and 234/2005 both resistant to INH, rifamycines, STM, EMB, and OFX; 9449/2006 resistant to INH, rifamycines, and STM; 53/2009 resistant to INH, rifamycines, STM, and EMB; Praha 1 resistant to INH, rifamycines, STM, EMB, and clofazimine; and Praha 131 resistant to INH, rifamycines, STM, EMB, OFX, gentamicin and amikacin (i.e., XDR-TB strain). The following concentrations were used: 1000, 500, 250, 125, 62.5, 32, 16, 8, 4, 2, 1, 0.5, 0.25, and 0.125 μmol/L.
3.3. Isocitrate Lyase Inhibition Assay (ICL1)
The Mycobacterium tubeculosis H37Rv genomic DNA was used as a template. The icl gene (Rv0467) of 1.28 kb was amplified using PCR. The amplified DNA was cloned into the pET-28b(+) plasmid vector Novagen (Merck KGaA, Darmstadt, Germany) using NdeI and HindIII restriction sites. The recombinant plasmid was transferred into E. coli HB101. DNA sequencing was employed to confirm that the inserted coding sequence had no mutations. For bacterial expression, 25 mL culture volumes were inoculated with BL21(DE3) cells containing the recombinant plasmid and allowed to grow until an optical density of OD595 = 0.6 was achieved. Then, the culture was induced with 1 mmol/L isopropyl-β-D-thiogalactopyranoside solution and incubated at 30 °C for additional 4 h. The cells were harvested at 4 °C by centrifugation at 6,000 g for 10 min. The resulting pellet was resuspended in BugBuster Protein Extraction Reagent Novagen (Merck KGaA, Darmstadt, Germany). The cell debris was removed by centrifugation, and the histidine-tagged protein was purified using an Äkta purifier (Amersham Biosciences, Valley Stream, NY, USA). The purity of the protein was confirmed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie blue staining of the gel. The protein concentration was determined by the Bradford method [30].
The isocitrate lyase activity was assayed according to the protocol reported by Dixon and Kornberg [31]. The enzyme assay was optimised in the final volume of 100 μL using 96-well plates (NUNC, Schoeller, Prague, Czech Republic) and the final concentration of tested compound in the reaction mixture 10 μmol/L. The reaction buffer contained 50 mmol/L of KH2PO4, 4 mmol/L of MgCl2·6H2O, 4 mmol/L of phenylhydrazine hydrochloride, 12 mmol/L of L-cysteine, H2O and KOH to pH 7.0. The isocitrate cleavage was measured by the change in the absorbance at 324 nm, which is associated with the formation of glyoxylate phenyl hydrazone. Each tested compound was dissolved in DMSO to prepare a 1 mmol/L solution, and 1 μL of this solution was added to 93.9 μL of reaction buffer. Then, it was added 0.1124 μL of the recombinant enzyme in phosphate buffer and glycerol solution with concentration of 0.58 mg/mL (Bradford). Finally the reaction was started by the addition of 0.2 μmol of (+)-potassium Ds-threo-isocitrate in solution.
Isoniazid was employed as a negative control (inhibition 0%), and 3-nitropropionic acid (3-NP) served as the positive control. All of the control compounds were added to the reaction mixture the same way like tested compounds and also their concentration in the reaction mixture was 10 μmol/L. The inhibitory activity of DMSO alone (1 μL) was subtracted from the activities of the evaluated compounds.
3.4. Antibacterial Evaluation
The in vitro antibacterial activity was assayed against eight Gram-positive and Gram-negative strains: Staphylococcus aureus CCM 4516/08, methicillin-resistant Staphylococcus aureus H 5996/08 (MRSA), Staphylococcus epidermidis H 6966/08, Enterococcus sp. J 14365/08; Escherichia coli CCM 4517, Klebsiella pneumoniae D 11750/08, ESBL-positive Klebsiella pneumoniae J 14368/08, and Pseudomonas aeruginosa CCM 1961.
The microdilution broth method modified according to standard M07-A07 in Mueller-Hinton broth (HiMedia Laboratories, Mumbai, India) adjusted to pH 7.4 (±0.2) was used. The tested compounds were dissolved in DMSO to the final concentrations ranging from 500 to 0.49 mmol/L. Benzylpenicillin (penicillin G) was used as a comparative drug. Bacterial inoculum in sterile water was prepared to match 0.5 McFarland scale (1.5 ± 108 CFU/mL). The minimum inhibitory concentrations were assayed as 90% (IC90) or higher reduction of growth when compared to the control. The determination of results was performed visually and spectrophotometrically (at 540 nm). The values of MICs were determined after 24 and 48 h of incubation in the darkness at 35 °C (±0.1) in a humid atmosphere.
3.5. Cytotoxicity Evaluation (HepG2 Cells)
Seven esters and their parent salicylanilides with the most robust antimicrobial activity were tested for cytotoxicity in the human hepatocellular liver carcinoma cell line HepG2 (passages 21–24; ECACC, Salisbury, UK) using a standard colorimetric method measuring a tetrazolium salt reduction (CellTiter(R) 96 AQueous One Solution Assay, Promega G3580, Madison, WI, USA).
The cells were routinely cultured in Minimum Essentials Eagle Medium (Sigma-Aldrich, Darmstadt, Germany) supplemented with 10% foetal bovine serum (PAA; Biotech, Prague, Czech Republic), 1% of L-glutamine solution (Sigma-Aldrich), and non-essential amino acids solution (Sigma-Aldrich) in a humidified atmosphere containing 5% CO2 at 37 °C. For subculturing, the cells were harvested after trypsin/EDTA (Sigma-Aldrich) treatment at 37 °C. The cells treated with the tested substances were used as experimental groups. Untreated HepG2 cells were used as control groups.
The cells were seeded in density 1 × 104 cells per well in a 96-well plate with microscopic control. Next day, the cells were treated with each of the tested substances. Because of the low solubility of some investigated compounds in water they were dissolved in a very small amount of DMSO and in a small volume added to cell culture; a final solution contained less than 1% DMSO. The most of the tested compounds were prepared in incubation concentrations of 0.01–20 µM in triplicates. 4-(Trifluoromethyl)benzoic acid was applied in the concentrations of 10–4,000 µM in triplicates. The following types of controls were included: determination of 100% viability and 0% viability (the cells treated by 10% DMSO), no cell control and vehiculum controls. These checking samples were prepared in triplicates.
The treated cells were incubated together with controls at 37 °C for 24 h in 5% CO2 atmosphere. After this time reagent from the kit CellTiter 96 AQueous One Solution Cell Proliferation Assay was added. The CellTiter 96 assay is based on the reduction of tetrazolium salt MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] to water-soluble formazan dye by metabolically active cells. The reduction of the reagent is attributed to availability of NADH or NADPH. The decline in levels of these metabolically important compounds in the cell causes that the production of formazan is reduced. The tested plate was incubated for 2 h at 37 °C and 5% CO2 and after this time, the absorbance was recorded at 490 nm using a 96-well plate reader (TECAN, Infinite M200, Grödig, Austria).
The results were expressed as inhibitory concentration which is necessary to inhibit cell viability to 50% from the maximal (control) viability (IC50). A standard toxicological parameter IC50 was calculated in each of the tested substances using GraphPad Prism software (version 5.02; GraphPad Software Inc., San Diego, CA, USA).
4. Conclusions
Eighteen salicylanilide 4-(trifluoromethyl)benzoates with a known antifungal activity underwent additional biological screening for the antimycobacterial and antibacterial activity, cytotoxicity and inhibition of mycobacterial isocitrate lyase. All compounds showed a significant antimycobacterial activity including against drug-resistant strains; in some cases comparable or even better than isoniazid. 3,4-Dichloro- and trifluoromethyl moieties represent the optimal substitution of aniline ring. Esters caused consistently a mild inhibition of isocitrate lyase; moreover, some derivatives are comparable to 3-nitropropionic acid. The substitution of aniline ring at the position 3 enhances this action. Although salicylanilide 4-(trifluoromethyl)benzoates failed with respect to our intention of finding the most antimycobacterial active salicylanilide esters, they offer very low MICs against Gram-positive strains and IC50 values for HepG2 cells.
In contrast to the aniline ring, the introduction of a 4-trifluoromethyl moiety into an acyl part of the salicylanilide esters did not provide sharply improved antimycobacterial activity, when compared to salicylanilide benzoates. The most potent esters are still considerably cytotoxic in micromolar values, although less than parent salicylanilides. Their cytotoxicity was mostly a slightly higher than for corresponding benzoates. In summary, salicylanilides and their 4-(trifluoromethyl)benzoates may be promising initial step compounds for anticancer research with advantageous broad-spectrum antimicrobial properties.
Acknowledgments
This work was financially supported by IGA NT 13346 (2012). This publication is a result of the project implementation: Support of establishment, development, and mobility of quality research teams at the Charles University, project number CZ.1.07/2.3.00/30.0022, supported by The Education for Competitiveness Operational Programme (ECOP) and co-financed by the European Social Fund and the state budget of the Czech Republic.
We thank Ida Dufková for the excellent performance of antibiotic susceptibility tests (under the supervision of Vladimír Buchta), Vladimír Wsól for the supervision of enzyme inhibition assay and Jaroslava Urbanová, M.A., for the language help.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 1a–r and SAL-1 – SAL-7 are available from the authors.
References
- 1.WHO. 2011/2012 Tuberculosis Global Facts. [(accessed on 12 December 2012)]. Available online: http://www.who.int/tb/publications/2011/factsheet_tb_2011.pdf.
- 2.Caminero J.A. Extensively drug-resistant tuberculosis: Is its definition correct? Eur. Respir. J. 2008;32:1413–1415. doi: 10.1183/09031936.00094708. [DOI] [PubMed] [Google Scholar]
- 3.McKinney J.D., Bentrup K.H., Munoz-Elias E.J., Miczak A., Chen B., Chan W.T., Swenson D., Sacchettini J.C., Jacobs W.R., Russell D.G. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. 2000;406:735–738. doi: 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- 4.Krátký M., Vinšová J., Novotná E., Mandíková J., Wsól V., Trejtnar F., Ulmann V., Stolaříková J., Fernandes S., Bhat S., Liu J.O. Salicylanilide derivatives block Mycobacterium tuberculosis through inhibition of isocitrate lyase and methionine aminopeptidase. Tuberculosis. 2012;92:434–439. doi: 10.1016/j.tube.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 5.Krátký M., Vinšová J. Advances in mycobacterial isocitrate lyase targeting and inhibitors. Curr. Med. Chem. 2012;19:6126–6137. doi: 10.2174/092986712804485782. [DOI] [PubMed] [Google Scholar]
- 6.Cabrera C.E., Gómez R.F., Zuñiga A.E., Corral R.H., López B., Chávez M. Epidemiology of nosocomial bacteria resistant to antimicrobials. Colomb. Med. 2011;42:117–125. [Google Scholar]
- 7.Scientific Blueprint for Tuberculosis Drug Development. [(accessed on 12 December 2012)]. Available online: http://www.tballiance.org/downloads/publications/TBA_Scientific_Blueprint.pdf.
- 8.Krátký M., Vinšová J. Salicylanilide ester prodrugs as potential antimicrobial agents—A review. Curr. Pharm. Des. 2011;17:3494–3505. doi: 10.2174/138161211798194521. [DOI] [PubMed] [Google Scholar]
- 9.Krátký M., Vinšová J., Buchta V., Horvati K., Bösze S., Stolaříková J. New amino acid esters of salicylanilides active against MDR-TB and other microbes. Eur. J. Med. Chem. 2010;45:6106–6113. doi: 10.1016/j.ejmech.2010.09.040. [DOI] [PubMed] [Google Scholar]
- 10.Krátký M., Vinšová J., Rodriguez N.G., Stolaříková J. Antimycobacterial activity of salicylanilide benzenesulfonates. Molecules. 2012;17:492–503. doi: 10.3390/molecules17010492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krátký M., Vinšová J., Buchta V. In vitro antibacterial and antifungal activity of salicylanilide benzoates. ScientificWorldJournal. 2012;12 doi: 10.1100/2012/290628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krátký M., Vinšová J., Buchta V. In vitro antibacterial and antifungal activity of salicylanilide pyrazine-2-carboxylates. Med. Chem. 2012;8:732–741. doi: 10.2174/157340612801216346. [DOI] [PubMed] [Google Scholar]
- 13.Krátký M., Vinšová J., Stolaříková J. Antimycobacterial assessment of salicylanilide benzoates including multidrug-resistant tuberculosis strains. Molecules. 2012;17:12812–12820. doi: 10.3390/molecules171112812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng T.J.R., Wu Y.T., Yang S.T., Lo K.H., Chen S.K., Chen Y.H., Huang W.I., Yuan C.H., Guo C.W., Huang L.Y., et al. High-throughput identification of antibacterials against methicillin-resistant Staphylococcus aureus (MRSA) and the transglycosylase. Bioorg. Med. Chem. 2010;18:8512–8529. doi: 10.1016/j.bmc.2010.10.036. [DOI] [PubMed] [Google Scholar]
- 15.Garner A.L., Gloeckner C., Tricoche N., Zakhari J.S., Samje M., Cho-Ngwa F., Lustigman S., Janda K.D. Design, synthesis, and biological activities of closantel analogues: Structural promiscuity and its impact on Onchocerca volvulu. J. Med. Chem. 2011;54:3963–3972. doi: 10.1021/jm200364n. [DOI] [PubMed] [Google Scholar]
- 16.Fomovska A., Wood R.D., Mui E., Dubey J.P., Ferreira L.R., Hickman M.R., Lee P.J., Leed S.E., Auschwitz J.M., Welsh W.J., et al. Salicylanilide inhibitors of Toxoplasma gondii. J. Med. Chem. 2012;55:8375–8391. doi: 10.1021/jm3007596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding N., Zhang W., Xiao H.L., Wang P., Li Y.X. Synthesis and biological evaluation of a series of novel salicylanilides as inhibitors of EGFR protein tyrosine kinases. Chin. Chem. Lett. 2012;23:529–532. doi: 10.1016/j.cclet.2012.03.016. [DOI] [Google Scholar]
- 18.Steffen J.D., Coyle D.L., Damodaran K., Beroza P., Jacobson M.K. Discovery and structure-activity relationships of modified salicylanilides as cell permeable inhibitors of poly(ADP-ribose) glycohydrolase (PARG) J. Med. Chem. 2011;54:5403–5413. doi: 10.1021/jm200325s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zuo M., Zheng Y.W., Lu S.M., Li Y., Zhang S.Q. Synthesis and biological evaluation of N-aryl salicylamides with a hydroxamic acid moiety at 5-position as novel HDAC–EGFR dual inhibitors. Bioorg. Med. Chem. 2012;20:4405–4412. doi: 10.1016/j.bmc.2012.05.034. [DOI] [PubMed] [Google Scholar]
- 20.Zhu Z.W., Shi L., Ruan X.M., Yang Y., Li H.Q., Xu S.P., Zhu H.L. Synthesis and antiproliferative activities against Hep-G2 of salicylanide derivatives: potent inhibitors of the epidermal growth factor receptor (EGFR) tyrosine kinase. J. Enzym. Inhib. Med. Chem. 2011;26:37–45. doi: 10.3109/14756361003671060. [DOI] [PubMed] [Google Scholar]
- 21.Liu X.H., Lv P.C., Li B., Zhu H.L., Song B.A. Synthesis, structure, and antibacterial activity of novel 5-arylpyrazole derivatives. Aust. J. Chem. 2008;61:223–230. doi: 10.1071/CH07253. [DOI] [Google Scholar]
- 22.Krátký M., Vinšová J. Antifungal activity of salicylanilides and their esters with 4-(trifluoromethyl)benzoic acid. Molecules. 2012;17:9426–9442. doi: 10.3390/molecules17089426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waisser K., Bureš O., Holý P., Kuneš J., Oswald R., Jirásková L., Pour M., Klimešová V., Kubicová L., Kaustová J. Relationship between the structure and antimycobacterial activity of substituted salicylanilides. Arch. Pharm. Pharm. Med. Chem. 2003;336:53–71. doi: 10.1002/ardp.200390004. [DOI] [PubMed] [Google Scholar]
- 24.Imramovský A., Vinšová J., Férriz J.M., Doležal R., Jampílek J., Kaustová J., Kunc F. New antituberculotics originated from salicylanilides with promising in vitro activity against atypical mycobacterial strains. Bioorg. Med. Chem. 2009;17:3572–3579. doi: 10.1016/j.bmc.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 25.Férriz J.M., Vávrová K., Kunc F., Imramovský A., Stolaříková J., Vavříková E., Vinšová J. Salicylanilide carbamates: Antitubercular agents active against multidrug-resistant Mycobacterium tuberculosis strains. Bioorg. Med. Chem. 2010;18:1054–1061. doi: 10.1016/j.bmc.2009.12.055. [DOI] [PubMed] [Google Scholar]
- 26.Liu X.H., Song B.A., Bhadury P.S., Zhu H.L., Cui P., Hou K.K., Xu H.L. Novel 5-(3-(substituted)-4,5-dihydroisoxazol-5-yl)-2-methoxyphenyl derivatives: Synthesis and anticancer activity. Aust. J. Chem. 2008;61:864–869. doi: 10.1071/CH07395. [DOI] [Google Scholar]
- 27.Wada K., Ohkoshi E., Morris-Natschke S.L., Bastow K.F., Lee K.H. Cytotoxic esterified diterpenoid alkaloid derivatives with increased selectivity against a drug-resistant cancer cell line. Bioorg. Med. Chem. Lett. 2012;22:249–252. doi: 10.1016/j.bmcl.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deng W., Guo Z., Guo Y., Feng Z., Jiang Y., Chu F. Acryloylamino-salicylanilides as EGFR PTK inhibitors. Bioorg. Med. Chem. Lett. 2006;16:469–472. doi: 10.1016/j.bmcl.2005.06.088. [DOI] [PubMed] [Google Scholar]
- 29.Liechti C., Séquin U., Bold G., Furet P., Meyer T., Traxler P. Salicylanilides as inhibitors of the protein tyrosine kinase epidermal growth factor receptor. Eur. J. Med. Chem. 2004;39:11–26. doi: 10.1016/j.ejmech.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 30.Bradford M.M. Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 31.Dixon G.H., Kornberg H.L. Assay methods for key enzymes of the glyoxylate cycle. Biochem. J. 1959;72:P3. [Google Scholar]