

Abstract
A series of flavonoids 9a–f, 13b, 13d, 13e and 14a–f bearing diverse aliphatic amino moieties were designed, synthesized and evaluated for their cytotoxic activities against the ECA-109, A-549, HL-60, and PC-3 cancer cell lines. Most of the compounds exhibited moderate to good activities. The structure-activity relationships were studied, revealing that the chalcone skeleton is the most preferable for cytotoxic activities. Chalcone 9d was the most promising compound due to its high potency against the examined cancer cell lines (its IC50 values against ECA-109, A549, HL-60 and PC-3 cells were 1.0, 1.5, 0.96 and 3.9 μM, respectively).
Keywords: flavonoid, chalcone, synthesis, cytotoxic activity
1. Introduction
Flavonoids are a class of privileged structures that display a remarkable spectrum of biological activities [1]. Flavonoids are divided into six main categories: flavonols, flavones, flavanones, catechins, isoflavones and anthocyanidins, based on differences in their molecular backbone structures. A growing number of studies have indicated that flavonoids have important chemopreventive and chemotherapeutic effects on cancer [2,3]. Flavonoids, such as quercetin, catecholic xanthone and myricetin (Figure 1), show remarkable chemopreventive activities in a variety of bioassay systems and animal models [4,5,6]. More importantly, flavopiridol (Figure 1) that was identified as the first cyclin-dependent kinase (CDK) inhibitor has been tested in phase II clinical trials for the treatment of cancer [7,8].
Figure 1.

Structures of flavonoids with cytotoxic activity.
The structure-activity relationship (SAR) studies of flavopiridol revealed that the presence of the aliphatic amino on A ring is very important for CDK inhibitory activity [9]. On the other hand, the structural modifications of flavonoids with the introduction of nitrogen atoms have attracted considerable attention due to the observed improvements of the biological activities [10,11,12,13]. For example, 2'-aminochalcones demonstrated a significantly increased antitumor activity compared with chalcones that lack this function [11]. Another investigation showed that the introduction of aliphatic amino groups onto the flavonoids scaffold resulted in potent antitumor compounds [13].
Moreover, the functional aliphatic amino groups can enhance the aqueous solubility and drug-like character of candidate compounds [12,13]. Based on these observations, it is of interest to further evaluate the roles of basic amino functions in the antitumor activity of flavonoids. We speculated that introducing diverse flexible aliphatic amino groups to the flavonoid scaffold might be an effective way in discovering novel flavonoids with potential bioactivities. In this paper, we describe the detailed synthetic routes to the novel aliphatic amino substituted flavonoids 9, 13 and 14 (Figure 2) and their biological activities against ECA-109, A-549, HL-60, and PC-3 cancer cell lines. Moreover, we discuss the corresponding structure-activity relationships.
Figure 2.

Structures of synthesized compounds 9, 13 and 14.
2. Results and Discussion
2.1. Chemistry
2.1.1. Synthesis of Chalcone Derivatives 9a–f
The preparation of the target chalcones 9a–f is depicted in Scheme 1. Briefly, substrates 2 [14] and 3 [15] were prepared according to the described procedures. The dimethylated intermediate 3 was then treated with an equimolar amount o-chlorobenzaldehyde in a base-catalyzed Claisen-Schmidt reaction to afford chalcone 4 in good yield [16]. With the chalcone core established, all that remained was the introduction of the allyl group to position-3'. This involved O-allylation of chalcone 4 with allyl bromide, followed by Claisen rearrangement in N,N-dimethylaniline at 190 °C under N2. The position of the allyl function was confirmed by a nuclear Overhauser effect (NOE) experiment, which showed correlations between the OH-2' and H-1" signals. The synthesis of the target products 9a–f was achieved in moderate yields by the triethylamine-catalyzed reaction of the epoxychalcone 8 with suitable secondary amines in acetonitrile at room temperature. It is worth noting that direct epoxidation of the allylchalcone 6 with m-chloroperoxybenzoic acid yielded no product. As expected, after protection of the phenolic hydroxyl with an acetyl group, the epoxychalcone was formed after treatment of intermediate 7 with m-chloroperoxybenzoic acid, and the pure product epoxychalcone 8 was obtained.
Scheme 1.

Synthesis of chalcone derivatives 9a–f.
2.1.2. Synthesis of Flavone Derivatives 13b, 13d, 13e and 14a–f
The general method for the synthesis of the desired flavone derivatives 13b, 13d, 13e and 14a–f is outlined in Scheme 2. The allylchalcone 6 was cyclized in the presence of sodium acetate in 95% ethanol to produce flavanone 10 [17]. Subsequent oxidation of flavanone 10 with iodine through a modification of the procedure previously reported for the synthesis of analogous compounds [18] generated flavone 11. Preparation of epoxyflavone 12 was accomplished by the reaction of flavone 11 with m-chloroperoxybenzoic acid. Refluxing epoxyflavone 12 in ethanol with suitable secondary amines in the presence of triethylamine resulted in the formation of dimethoxyflavones 13a–f in high yields. The final steps required for completion of the synthesis target flavones were the selective demethylation of the methyl ether-protecting groups. The cleavage of aryl methyl ethers to liberate the corresponding phenolic compounds has been reviewed previously in the literature [19,20,21]. Our experiments showed that deprotection of dimethoxylflavone using a 2-fold excess of boron tribromide at room temperature successfully provided monomethoxyflavones 14a–f in good yields.
Scheme 2.

Synthesis of flavone derivatives 13b, 13d, 13e and 14a–f.
2.2. Biological Activities
The in vitro cytotoxic activities of the chalcone derivatives 9a–f and flavone derivatives 13b, 13d, 13e and 14a–f against four cancer cell lines, namely human esophageal carcinoma cell (ECA-109), human lung adenocarcinoma cell (A-549), human leukemia cell (HL-60) and human prostate carcinoma cell (PC-3), were assayed by the MTT method using taxol as the positive control. Table 1 summarizes the results expressed as IC50 values.
Table 1.
In vitro cytotoxic activity of compounds 9a–f, 13b, 13d, 13e and 14a–f a (IC50, µg/mL).
| Compounds | NR1R2 | ECA-109 b | A-549 c | HL-60 d | PC-3 e |
|---|---|---|---|---|---|
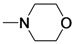
| 9a |
|
6.8 | 2.8 | 2.3 | 6.1 |
| 9b |

|
1.3 | 1.6 | 2.6 | 2.5 |
| 9c |

|
4.6 | 2.4 | 7.4 | 11.6 |
| 9d |

|
1.0 | 1.5 | 0.96 | 3.9 |
| 9e |
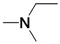
|
2.4 | 2.6 | 4.0 | 6.7 |
| 9f |
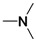
|
2.1 | 2.5 | 2.0 | 6.5 |
| 13b |
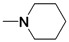
|
>50 | >50 | N.T. | N.T. |
| 13d |

|
>50 | >50 | N.T. | N.T. |
| 13e |
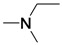
|
>50 | 34.1 | N.T. | N.T. |
| 14a |
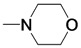
|
5.3 | 6.1 | 10.1 | 7.3 |
| 14b |

|
15.5 | 33.6 | 11.0 | 5.8 |
| 14c |
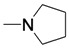
|
17.1 | 30.0 | 10.0 | 4.8 |
| 14d |
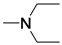
|
19.6 | 28.1 | 5.4 | 2.1 |
| 14e |
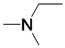
|
17.5 | 35.3 | 4.2 | 3.0 |
| 14f |
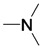
|
24.3 | 23.8 | 6.0 | 4.5 |
| Taxol | - | 12.8 × 10−3 | 13.6 × 10−3 | 2.2 × 10−3 | 5.3 × 10−3 |
a Data represent the mean values of three independent determinations; b Human esophageal carcinoma cell; c Human lung adenocarcinoma cell; d Human leukemia cell; e Human prostate carcinoma cell; N.T. not tested.
For the derivatives bearing the same substituents, the chalcones derivatives 9a–f had higher cytotoxic activity than the corresponding flavones 14a–f, with all IC50 values lower than 10 µg/mL against the human tumor cell lines tested (Table 1). Compound 9b with a piperidinyl substituent at position-3' displayed prominent cytotoxic activity, whereas introducing a morpholino or pyrrolidinyl substituent led to compounds 9a and 9c, respectively, which all demonstrated decreased cytotoxic activities. These results indicated that a smaller substituent and the introduction of additional heteroatoms were not suitable options for position-3'. Interestingly, further introducing open chain aliphatic amino groups afforded compounds 9d (diethylamino), 9e (ethylmethylamino) and 9f (dimethylamino), among which 9d exhibited excellent antitumor activity. This suggested that a bulkier substituent was favorable for the cytotoxic activity.
Because of the excellent activity of flavopiridol, the closed-ring derivatives 13 and 14 bearing the same substituents were synthesized and evaluated. Unfortunately, the formation of the B ring led to the loss of cytotoxic activity (compounds 13b, 13d and 13e). Meanwhile, the importance of an exposed hydroxyl and the significant difference between dimethoxyflavone and hydroxyflavone cores (e.g., 13b vs. 14b, 13d vs. 14d, 13e vs. 14e) was found.
Hydroxyflavones 14a–f displayed moderate cytotoxic activity, whereas dimethoxyflavones 13b, 13d and 13e had only marginal or no cytotoxic effects against the tested cell lines. Another interesting observation is that the flavone derivatives 14a–f with different substituent patterns at position-3'. displayed selective cytotoxic activity against PC-3, with IC50 values all lower than 10 µg/mL. Besides, the open chain aliphatic amino substituted flavones 14d, 14e and 14f showed significant cytotoxic activity against the HL-60 cell line.
3. Experimental
3.1. Chemistry
Melting points are not corrected and were recorded on a Buchi apparatus. IR spectra (KBr pellets, 400–4000 cm−1) were recorded on a Bruker VECTOR 22 FTIR spectrophotometer. 1H-NMR spectra and NOE spectra were recorded on a Bruker AM 400 instrument at 400 MHz (chemical shifts are expressed as δ values relative to TMS as internal standard). Mass spectra (MS), ESI (positive) were recorded on an Esquire-LC-00075 spectrometer.
3.1.1. 3-(2-Chlorophenyl)-1-(2-hydroxy-4,6-dimethoxyphenyl)prop-2-en-1-one (4)
To a stirred solution of 3 (0.39 g, 2 mmol) and 2-chlorobenzaldehyde (0.27 g, 3 mmol) in 80% EtOH (10 mL), KOH (1.0 g, 18 mmol) was added at 0 °C. The reaction mixture was stirred at room temperature overnight and then neutralized with 2N HCl and extracted with CH2Cl2 (2 × 30 mL). The combined organic fraction was washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was recrystallized from EtOH to give 0.57 g (89%) of the title compound 4 as a yellow solid. m.p. 135–137 °C (Ref. [22]: m.p. 136–137 °C); IR (KBr): 3454 (OH), 2953, 2870 (CH3), 1631 (C=O), 1557 (C=C), 1274 (Ar-O), 1064 (C-O), 750 cm−1; 1H-NMR (CDCl3) δ: 14.20 (s, 1H, OH-2'), 8.14 (d, 1H, J = 15.6 Hz, H-β), 7.87 (d, 1H, J = 15.6 Hz, H-α), 7.69 (m, 1H, H-6), 7.43 (m, 1H, H-3), 7.30 (m, 2H, H-4 and 5), 6.11 (s, 1H, H-5'), 5.96 (s, 1H, H-3'), 3.90 (s, 3H, OCH3), 3.84 (s, 3H, OCH3). ESI-MS: m/z 319 [M+1]+.
3.1.2. 1-(2-Allyloxy-4,6-dimethoxyphenyl)-3-(2-chlorophenyl)prop-2-en-1-one (5)
To a suspension of 4 (3.20 g, 10 mmol) and anhydrous K2CO3 in dry acetone, allyl bromide (9.1 mL, 10 mmol) was added with stirring. The resulting suspension was refluxed for 6 h, then the mixture was filtered, washed with acetone, and the filtrate was evaporated to dryness. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (6:1) as eluent to afford pure 5 (3.40 g, 95%) as a buff-colored liquid. IR (KBr): 3007 (=CH), 2944, 2849 (CH3, CH2), 1620 (C=O), 1535 (C=C), 1272 (Ar-O), 1081 (C-O) cm−1; 1H-NMR (CDCl3) δ: 7.79 (d, 1H, J = 16.0 Hz, H-β), 7.66 (m, 1H, H-6), 7.39 (m, 1H, H-3), 7.28 (m, 2H, H-4 and 5), 6.93 (d, 1H, J = 16.0 Hz, H-α), 6.16 (s, 1H, H-5'), 6.14 (s, 1H, H-3'), 5.95 (m, 1H, H-2"), 5.33 (dd, 1H, J1 = 16.8 Hz, J2 = 1.2 Hz, H-3"), 5.50 (m, 2H, H-1"), 3.84 (s, 3H, OCH3), 3.78 (s, 3H, OCH3). ESI-MS: m/z 359 [M+1]+.
3.1.3. 1-(3-Allyl-2-hydroxy-4,6-dimethoxyphenyl)-3-(2-chlorophenyl)prop-2-en-1-one (6)
A solution of 5 (4.00 g, 11.2 mmol) in N,N-dimethylaniline (60 mL) under N2 was heated to reflux for 4 h. The solvent was removed in vacuo, and the residue was recrystallized from EtOH and gave 3.05 g (75%) of title compound 6 as a yellow solid. m.p. 167–168 °C; IR (KBr): 3448 (OH), 2979, 2945 (CH3, CH2), 1630 (C=O), 1581 (C=C), 1274 (Ar-O), 1135 (C-O) cm−1; 1H-NMR (CDCl3) δ: 13.93 (s, 1H, OH-2'), 8.06 (d, 1H, J = 15.6 Hz, H-β), 7.79 (d, 1H, J = 15.6 Hz, H-α), 7.65 (m, 1H, H-6), 7.39 (m, 1H, H-3), 7.26 (m, 2H, H-4 and 5), 5.97 (s, 1H, H-5'), 5.94 (m, 1H, H-2"), 4.95 (m, 2H, H-3"), 3.91 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 3.31 (m, 2H, H-1"). ESI-MS: m/z 359 [M+1]+.
3.1.4. 2-Allyl-6-[3-(2-chlorophenyl)acryloyl]-3,5-dimethoxyphenyl acetate (7)
Acetic anhydride (1.27 mL, 13.5 mmol) was added to a solution of 6 (1.60 g, 5 mmol) in pyridine (20 mL). The reaction mixture was stirred at room temperature overnight, then neutralized with 2N HCl, and extracted with CH2Cl2 (3 × 40 mL). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (5:1) as eluent to give pure 7 (1.70 g, 85%) as a yellow solid. m.p. 106–109 °C; IR (KBr): 2979, 2948, 2846 (CH3, CH2), 1762, 1660 (C=O), 1612 (C=C), 1205 (Ar-O), 1091 (C-O) cm−1; 1H-NMR (CDCl3) δ: 7.89 (d, 1H, J = 15.6 Hz, H-β), 7.65 (m, 1H, H-6), 7.40 (m, 1H, H-3), 7.30 (m, 2H, H-4 and 5), 7.06 (d, 1H, J = 15.6 Hz, H-α), 5.97 (s, 1H, H-5'), 5.90 (m, 1H, H-2"), 4.96 (m, 2H, H-3"), 3.91 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 3.32 (m, 2H, H-1"), 2.32 (s, 3H, COCH3). ESI-MS: m/z 401 [M+1]+.
3.1.5. 2-[3-(2-Chlorophenyl)acryloyl]-3,5-dimethoxy-6-oxiranylmethylphenyl acetate (8)
To a stirred solution of 7 (0.60 g, 1.5 mmol) in CH2Cl2 (20 mL) was added a solution of m-chloroperoxybenzoic acid (0.42 g, 2.5 mmol) in CH2Cl2 (40 mL) dropwise during 0.5 h, with the temperature being maintained 0–5 °C. The mixture was stirred at room temperature for 24 h, and then the CH2Cl2 layer was washed successively with aqueous sodium bicarbonate and brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (6:1) as eluent to give pure 8 (0.42 g, 65%) as a yellow solid. m.p.106–109 °C; IR (KBr): 2943, 2842 (CH3, CH2), 1762, 1660 (C=O), 1612 (C=C), 1207 (Ar-O), 1090 (C-O), 768 cm−1; 1H-NMR (CDCl3) δ: 7.89 (d, 1H, J = 15.6 Hz, H-β), 7.65 (m, 1H, H-6), 7.41 (m, 1H, H-3), 7.33 (m, 2H, H-4 and 5), 7.01 (d, 1H, J = 15.6 Hz, H-α), 6.43 (s, 1H, H-5'), 3.92 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 3.07 (m, 1H, H-2"), 2.70 (dd, 1H, J1 = 16.0 Hz, J2 = 4.0 Hz, H-1"α), 2.67 (dd, 1H, J1 = 15.6 Hz, J2 = 4.4 Hz, H-1"β), 2.59 (t, 1H, J = 7.6 Hz, H-3"α), 2.51 (dd, 1H, J1 = 5.6 Hz, J2 = 2.4 Hz, H-3"β), 2.25 (s, 3H, COCH3). ESI-MS: m/z 417 [M+1]+.
3.1.6. General Procedure to Obtain Target Chalcones 9a–f
To a stirred solution of 8 (0.15 g, 0.36 mmol) and a suitable secondary amine (3.6 mmol) in CH3CN (2 mL), a catalytic amount of triethylamine (0.1 mL) was added. The resulting mixture was stirred at room temperature for 24 h. Then acetonitrile was removed in vacuo, extracted with CH2Cl2, the combined organic extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc/Et3N (100:100:2.5) as eluent to give pure target chalcones 9a–f. In this manner, the following target chalcones were obtained.
3-(2-Chlorophenyl)-1-{2-hydroxy-3-[2-hydroxy-3-(morpholino-4-yl)propyl]-4,6-dimethoxyphenyl}prop-2-en-1-one (9a). Yield: 72%, m.p. 141–143 °C; IR (KBr): 3458 (OH), 2920, 2851 (CH3, CH2), 1626 (C=O), 1566 (C=C), 1267 (Ar-O), 1172 (C-O), 758 cm−1; 1H-NMR (CDCl3) δ: 13.93 (s, 1H, OH-2'), 8.11 (d, 1H, J = 15.6 Hz, H-β), 7.81(d, 1H, J = 15.6 Hz, H-α), 7.69 (m, 1H, H-6), 7.43 (m, 1H, H-3), 7.30 (m, 2H, H-4 and 5), 6.01 (s, 1H, H-5'), 4.00 (s, 3H, OCH3), 3.95 (s, 3H, OCH3), 3.81 (m, 1H, H-2"), 3.70 (m, 4H, CH2O, CH2 × 2), 3.10 (dd, 1H, J1 = 13.6Hz, J2 = 7.2 Hz, H-1"α), 2.91 (dd, 1H, J1 = 13.2 Hz, J2 = 7.2 Hz, H-1"β), 2.49 (m, 2H, H-3"), 2.24 (m, 4H, NCH2, CH2 × 2). ESI-MS: m/z 462 [M+1]+.
3-(2-Chlorophenyl)-1-{2-hydroxy-3-[2-hydroxy-3-(piperidin-1-yl)propyl]-4,6-dimethoxyphenyl} prop-2-en-1-one (9b). Yield: 75%, m.p. 135–136 °C; IR (KBr): 3443 (OH), 2936, 2887 (CH3, CH2), 1628 (C=O), 1611, 1576 (C=C), 1272 (Ar-O), 1107 (C-O), 754 cm−1; 1H-NMR (CDCl3) δ: 8.10 (d, 1H, J = 15.6 Hz, H-β), 7.81 (d, 1H, J = 15.6 Hz, H-α), 7.69 (m, H-6), 7.43 (m, 1H, H-3), 7.30 (m, 2H, H-4 and 5), 6.01 (s, 1H, H-5'), 3.94 (s, 3H, OCH3), 3.91 (s, 3H, OCH3), 3.91 (m, 1H, H-2"), 2.87 (dd, 1H, J1 =13.6 Hz, J2 = 6.4 Hz, H-1"α), 2.73 (dd, 1H, J1 = 13.2 Hz, J2 =6.8 Hz, H-1"β), 2.55 (m, 2H, H-3"), 2.32 (m, 4H, NCH2, CH2 × 2), 1.53 (m, 4H, NCH2CH2, CH2 × 2), 1.41 (m, 2H, CH2CH2CH2); 13C-NMR (DMSO-d6) δ: 192.2, 167.0, 164.1, 162.0, 141.1, 134.6, 133.3, 131.6, 130.8, 129.1, 128.4, 127.3, 108.9, 108.1, 90.2, 71.1, 60.7, 56.8, 56.6, 53.3, 33.2, 23.8, 21.2; ESI-MS: m/z 460 [M+1]+.
3-(2-Chlorophenyl)-1-{2-hydroxy-3-[2-hydroxy-3-(pyrrolidin-1-yl)propyl]-4,6-dimethoxyphenyl} prop-2-en-1-one (9c). Yield: 55%, m.p. 132–135 °C; IR (KBr): 3453 (OH), 2937, 2879 (CH3, CH2), 1628 (C=O), 1601, 1576 (C=C), 1274 (Ar-O), 1110 (C-O), 754 cm−1; 1H-NMR (CDCl3) δ: 13.95 (s, 1H, OH-2'), 8.10 (d, 1H, J = 15.6 Hz, H-β), 7.79 (d, 1H, J = 15.6 Hz, H-α), 7.69 (m, 1H, H-6), 7.43 (m, 1H, H-3), 7.31 (m, 2H, H-4 and 5), 6.02 (s, 1H, H-5'), 3.94 (s, 3H, OCH3), 3.91 (s, 3H, OCH3), 3.91 (m, 1H, H-2"), 2.87 (dd, 1H, J1 =13.2 Hz, J2 = 5.6 Hz, H-1"α), 2.79 (dd, 1H, J1 = 13.2 Hz, J2 =5.8 Hz, H-1"β), 2.69 (m, 4H, NCH2, CH2 × 2), 2.52 (m, 2H, H-3"), 1.73 (s, 4H, NCH2CH2, CH2 × 2). ESI-MS: m/z 446 [M+1]+.
3-(2-Chlorophenyl)-1-[3-(3-diethylamino-2-hydroxypropyl)-2-hydroxy-4,6-dimethoxyphenyl]prop-2-en-1-one (9d). Yield: 63%, m.p. 114–116 °C; IR (KBr): 3450 (OH), 2936, 2859 (CH3, CH2), 1625 (C=O), 1579 (C=C), 1272 (Ar-O), 1123 (C-O), 759 cm−1; 1H-NMR (CDCl3) δ: 13.94 (s, 1H, OH-2'), 8.10 (d, 1H, J = 15.6 Hz, H-β), 7.84 (d, 1H, J = 15.6 Hz, H-α), 7.74 (m, 1H, H-6), 7.43 (m, 1H, H-3), 7.31 (m, 2H, H-4 and 5), 5.98 (s, 1H, H-5'), 3.97 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 3.92 (m, 1H, H-2"), 3.03 (dd, 1H, J1 =13.6 Hz, J2 =7.2 Hz, H-1"α), 2.84 (dd, 1H, J1 = 13.6 Hz, J2 = 6.8 Hz, H-1"β), 2.39 (m, 6H, H-3" and NCH2), 0.94 (t, 6H, NCH2CH3, CH3 × 2); 13C-NMR (DMSO-d6) δ: 192.5, 169.6, 164.9, 162.1, 140.9, 134.2, 132.3, 130.3, 129.8, 127.2, 127.1, 126.8, 108.0, 105.1, 90.5, 70.8, 61.0, 55.2, 54.6, 47.7, 34.2, 12.2; ESI-MS: m/z 448 [M+1]+.
3-(2-Chlorophenyl)-1-[3-(3-ethylmethylamino-2-hydroxypropyl)-2-hydroxy-4,6-dimethoxyphenyl] prop-2-en-1-one (9e). Yield: 76%, m.p. 115–118 °C; IR (KBr): 3448 (OH), 2924, 2856 (CH3, CH2), 1628 (C=O), 1563 (C=C), 1271 (Ar-O), 1120 (C-O), 749 cm−1; 1H-NMR (CDCl3) δ: 8.12 (d, 1H, J = 15.6 Hz, H-β), 7.82 (d, 1H, J = 15.6 Hz, H-α), 7.70 (m, 1H, H-6), 7.44 (m, 1H, H-3), 7.30 (m, 2H, H-4 and 5), 6.02 (s, 1H, H-5'), 3.95 (s, 3H, OCH3), 3.92 (s, 3H, OCH3), 3.92 (m, 1H, H-2"), 2.88 (dd, 1H, J1 =13.2Hz, J2 = 5.2 Hz, H-1"α), 2.74 (dd, 1H, J1 = 13.6 Hz, J2 = 4.8 Hz, H-1"β), 2.41 (m, 4H, H-3" and NCH2), 2.24 (s, 3H, NCH3), 1.04 (t, 3H, NCH2CH3). ESI-MS: m/z 434 [M+1]+.
3-(2-Chlorophenyl)-1-[3-(3-dimethylamino-2-hydroxypropyl)-2-hydroxy-4,6-dimethoxyphenyl] prop-2-en-1-one (9f). Yield: 75%, m.p. 127–128 °C; IR (KBr): 3458 (OH), 2914, 2866 (CH3, CH2), 1629 (C=O), 1611, 1566 (C=C), 1256 (Ar-O), 1137 (C-O), 769 cm−1; 1H-NMR (CDCl3) δ: 14.03 (s, 1H, OH-2'), 8.12 (d, 1H, J = 15.6 Hz, H-α), 7.82 (d, 1H, J = 15.6 Hz, H-β), 7.69 (m, 1H, H-6), 7.43 (m, 1H, H-3), 7.30 (m, 2H, H-4 and 5), 6.02 (s, 1H, H-5'), 3.95 (s, 3H, OCH3), 3.92 (s, 3H, OCH3), 3.92 (m, 1H, H-2"), 2.87 (dd, 1H, J1 = 13.6Hz, J2 = 7.2 Hz, H-1"α), 2.75 (dd, 1H, J1 = 13.6 Hz, J2 = 6.8 Hz, H-1"β), 2.44 (t, 1H, J = 12.6 Hz, H-3"α), 2.26 (s, 6H, NCH3, CH3 × 2), 2.21 (dd, 1H, J1 = 13.6 Hz, J2 = 6.8 Hz, H-3"β). ESI-MS: m/z 420 [M+1]+.
3.1.7. 8-Allyl-2-(2-chlorophenyl)-5,7-dimethoxy-2,3-dihydro-4H-1-benzopyran-4-one (10)
To a solution of 6 (4.52 g, 12.6 mmol) in 95% EtOH (136 mL) was added NaOAc (12.90, 157 mmol). The mixture was heated at reflux for 24 h, then EtOH was removed in vacuo, H2O was added and the mixture was extracted with CH2Cl2 (3 × 50 mL). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (5:1) as eluent to afford pure flavanone 10 (3.84 g, 85%) as a colorless solid, m.p. 131–133 °C. IR (KBr): 3006 (C=CH), 2942, 2879 (CH3, CH2), 1676 (C=O), 1602, 1567 (C=C), 1265 (Ar-O), 1126 (C-O) cm−1; 1H-NMR (CDCl3) δ: 7.70 (m, 1H, H-6'), 7.26 (m, 3H, H-3', 4' and 5'), 6.15 (s, 1H, H-6), 5.90 (m, 1H, H-2"), 5.73 (dd, 1H, J1 = 13.2 Hz, J2 = 3.2 Hz, H-2), 4.95 (m, 2H, H-2"), 3.96 (s, 3H, OCH3), 3.91 (s, 3H, OCH3), 3.35 (m, 2H, H-1"), 2.99 (dd, 1H, J1 = 16.8 Hz, J2 = 3.6 Hz, H-3'α), 2.75 (dd, 1H, J1 = 16.4 Hz, J2 = 4.0 Hz, H-3'β). ESI-MS: m/z 359 [M+1]+.
3.1.8. 8-Allyl-2-(2-chlorophenyl)-5,7-dimethoxy-4H-1-benzopyran-4-one (11)
To a stirred solution of 10 (2.1 g, 5.85 mmol) in pyridine (42 mL) was added I2 (1.49 g, 5.86 mmol). The mixture was heated to 90 °C for 6 h, and then pyridine was removed in vacuo, followed by addition of solid Na2SO3 until fading of the color. The mixture was extracted with CH2Cl2 (3 × 50 mL); the combined organic extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (2:1) as eluent to give pure flavone 11 (1.00 g, 49%) as a colorless solid, m.p. 181–183 °C. IR (KBr): 3015 (C=CH), 2943, 2879 (CH3, CH2), 1663 (C=O), 1602, 1573 (C=C), 1217 (Ar-O), 1063 (C-O) cm−1; 1H-NMR (CDCl3) δ: 7.63 (m, 1H, H-6'), 7.52 (m, 1H, H-3'), 7.39 (m, 2H, H-4' and 5'), 6.54 (s, 1H, H-3), 6.46 (s, 1H, H-6), 5.92 (m, 1H, H-2"), 4.95 (m, 2H, H-3"), 4.02 (s, 3H, OCH3), 3.97 (s, 3H, OCH3), 3.54 (m, 2H, H-1"). ESI-MS: m/z 357 [M+1]+.
3.1.9. 2-(2-Chlorophenyl)-5,7-dimethoxy-8-oxiranylmethyl-4H-1-benzopyran-4-one (12)
To a stirred solution of 11 (3.28 g, 10 mmol) in CH2Cl2 (40 mL) was added a solution of m-chloroperoxybenzoic acid (0.42 g, 2.5 mmol) in CH2Cl2 (80 mL) dropwise during 0.5 h, and the mixture was stirred at room temperature for 24 h, then the CH2Cl2 layer was washed successively with aqueous sodium bicarbonate and brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (2:1) as eluent to give pure 12 (1.78 g, 68%) as colorless solid. m.p. 106–109 °C. IR (KBr): 2999, 2886 (CH3, CH2), 1645 (C=O), 1603, 1579 (C=C), 1265 (Ar-O), 1116 (C-O) cm−1; 1H-NMR (CDCl3) δ: 7.70 (m, 1H, H-6'), 7.28 (m, 3H, H-3', 4' and 5'), 6.52 (s, 1H, H-3), 6.47 (s, 1H, H-6), 4.01 (s, 3H, OCH3), 3.96 (s, 3H, OCH3), 3.30 (dd, 1H, J1 = 13.6 Hz, J2 = 6.0 Hz, H-1"α), 3.16 (m, 1H, H-2"), 2.90 (dd, 1H, J1 = 13.6 Hz, J2 = 6.4 Hz, H-1"β), 2.69 (t, 1H, J = 5.0 Hz, H-3"α), 2.51 (m, 1H, J1 = 4.8 Hz, J2 = 2.0 Hz, H-3"β). ESI-MS: m/z 373 [M+1]+.
3.1.10. General Procedure to Obtain Dimethoxyflavones 13a–f
To a stirred solution of 12 (0.19 g, 0.50 mmol) and a suitable secondary amine (5.0 mmol) in ethanol (2 mL), a catalytic amount of triethylamine (0.1 mL) was added. The resulting mixture was refluxed for 24 h. Then ethanol was removed in vacuo, and the residue was then extracted with CH2Cl2. The CH2Cl2 extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using EtOAc/EtOH/Et3N (100:2:2.5) as eluent to afford pure target chalcones 13a–f. In this manner, the following target dimethoxyflavones were obtained:
2-(2-Chlorophenyl)-8-[2-hydroxy-3-(morpholino-4-yl)propy]-5,7-dimethoxy-4H-1-benzopyran-4-one (13a). Yield: 75%, m.p. 145–147 °C; IR (KBr): 3427 (OH), 2939, 2874 (CH3, CH2), 1645 (C=O), 1612, 1601 (C=C), 1285 (Ar-O), 1066 (C-O) cm−1; 1H-NMR (CDCl3) δ: 7.68 (m, 1H, H-6'), 7.54 (m, 1H, H-3'), 7.39 (m, 2H, H-4' and 5'), 6.52 (s, 1H, H-3), 6.49 (s, 1H, H-6), 5.00 (brs, 1H, OH-2''), 4.04 (s, 3H, OCH3), 3.98 (s, 3H, OCH3), 4.01 (m, 1H, H-2"), 3.69 (m, 4H, CH2O, CH2 × 2), 3.10 (dd, 1H, J1 =13.6 Hz, J2 =7.2 Hz, H-1"α), 2.91 (dd, 1H, J1 = 13.2 Hz, J2 = 7.2 Hz, H-1"β), 2.49 (m, 2H, H-3"), 2.24 (m, 4H, NCH2, CH2 × 2). ESI-MS: m/z 460 [M+1]+.
2-(2-Chlorophenyl)-8-[2-hydroxy-3-(piperidin-1-yl)propyl]-5,7-dimethoxy-4H-1-benzopyran-4-one (13b). Yield: 78%, m.p. 149–153 °C; IR (KBr): 3434 (OH), 2932, 2847 (CH3, CH2), 1655 (C=O), 1612, 1600 (C=C), 1284 (Ar-O), 1120 (C-O), 770 cm−1; 1H-NMR (CDCl3) δ: 7.66 (m, 1H, H-6'), 7.52 (m, 1H, H-3'), 7.39 (m, 2H, H-4' and 5'), 6.52 (s, 1H, H-3), 6.46 (s, 1H, H-6), 4.00 (s, 3H, OCH3), 3.96 (s, 3H, OCH3), 3.95 (m, 1H,H-2"), 3.07 (dd, 1H, J1 =13.6 Hz, J2 = 6.4 Hz, H-1"α), 2.86 (dd, 1H, J1 = 13.2 Hz, J2 =6.8 Hz, H-1"β), 2.50 (brs, 1H, OH-2''), 2.32 (m, 2H, H-3"), 2.25 (m, 4H, NCH2, CH2 × 2), 1.52 (m, 4H, NCH2CH2, CH2 × 2), 1.39 (m, 2H, CH2CH2CH2). ESI-MS: m/z 458 [M+1]+.
2-(2-Chlorophenyl)-8-[2-hydroxy-3-(pyrrolidin-1-yl)propyl]-5,7-dimethoxy-4H-1-benzopyran-4-one (13c). Yield: 74%, m.p. 149–153 °C; IR (KBr): 3428 (OH), 2963, 2933 (CH3, CH2), 1643 (C=O), 1602 (C=C), 1376 (Ar-O), 1127 (C-O) cm−1; 1H-NMR (CDCl3) δ: 7.67 (m, 1H, H-6'), 7.52(m, 1H, H-3'), 7.40 (m, 2H, H-4' and'), 6.52 (s, 1H, H-3), 6.46 (s, 1H, H-6), 4.04 (s, 3H, OCH3), 4.00 (s, 3H, OCH3), 4.02 (m, 1H, H-2"), 3.06 (dd, 1H, J1 =13.6 Hz, J2 = 6.8 Hz, H-1"α), 2.92 (dd, 1H, J1 = 13.6 Hz, J2 =6.8 Hz, H-1"β), 2.31 (m, 6H, NCH2 × 2 and H-3"), 1.73 (s, 4H, NCH2CH2, CH2 × 2). ESI-MS: m/z 444 [M+1]+.
2-(2-Chlorophenyl)-8-(3-diethylamino-2-hydroxypropyl)-5,7-dimethoxy-4H-1-benzopyran-4-one (13d). Yield: 80%, m.p. 149–153 °C; IR (KBr): 3435 (OH), 2964, 2931 (CH3, CH2), 1654 (C=O), 1621, 1602 (C=C), 1384 (Ar-O), 1118 (C-O), 772 cm−1; 1H-NMR (CDCl3) δ: 7.69 (m, 1H, H-6'), 7.52 (m, 1H, H-3'), 7.36 (m, 2H, H-4' and 5'), 6.53 (s, 1H, H-3), 6.42 (s, 1H, H-6), 4.01 (s, 3H, OCH3), 3.97 (s, 3H, OCH3), 3.90 (m, 1H, H-2"), 3.07 (dd, 1H, J1 = 13.2 Hz, J2 = 6.4 Hz, H-1"α), 2.84 (dd, 1H, J1 = 13.2 Hz, J2 = 6.4 Hz, H-1"β), 2.39 (m, 6H, H-3" and NCH2), 0.94 (t, 6H, NCH2CH3, CH3 × 2); 13C-NMR (DMSO-d6) δ: 179.3, 165.3, 164.2, 161.9, 157.0, 134.2, 132.9, 130.1, 129.8, 128.0, 126.5, 125.2, 112.5, 107.1, 95.8, 70.8, 61.0, 56.2, 55.9, 47.5, 35.8, 12.6; ESI-MS: m/z 446 [M+1]+.
2-(2-Chlorophenyl)-8-(3-ethylmethylamino-2-hydroxypropyl)-5,7-dimethoxy-4H-1-benzopyran-4-one (13e). Yield: 82%, m.p. 149–153 °C; IR (KBr): 3424 (OH), 2968, 2938 (CH3, CH2), 1653 (C=O), 1602, 1578 (C=C), 1328 (Ar-O), 1120 (C-O), 763 cm−1; 1H-NMR (CDCl3) δ: 7.67 (m, 1H, H-6'), 7.51 (m, 1H, H-3'), 7.36 (m, 2H, H-4' and 5'), 6.52 (s, 1H, H-3), 6.45 (s, 1H, H-6), 4.01 (s, 3H, OCH3), 3.97 (s, 3H, OCH3), 3.92 (m, 1H, H-2"), 3.07 (dd, 1H, J1 = 13.6 Hz, J2 = 6.8 Hz, H-1"α), 2.85 (dd, 1H, J1 = 13.2 Hz, J2 = 6.8 Hz, H-1"β), 2.41 (m, 4H, H-3" and NCH2), 2.17 (s, 3H, NCH3), 0.96 (t, 3H, NCH2CH3). ESI-MS: m/z 432 [M+1]+.
2-(2-Chlorophenyl)-8-(3-dimethylamino-2-hydroxypropyl)-5,7-dimethoxy-4H-1-benzopyran-4-one (13f). Yield: 87%, m.p. 149–153 °C; IR (KBr): 3441 (OH), 2939 (CH3, CH2), 1653 (C=O), 1623, 1602 (C=C), 1327 (Ar-O), 1120 (C-O), 762 cm−1; 1H-NMR (CDCl3) δ: 7.66 (m, 1H, H-6'), 7.51 (m, 1H, H-3'), 7.38 (m, 2H, H-4' and 5'), 6.51 (s, 1H, H-3), 6.46 (s, 1H, H-6), 4.00 (s, 3H, OCH3), 3.97 (s, 3H, OCH3), 3.92 (m, 1H, H-2"), 3.05 (dd, 1H, J1 = 13.6 Hz, J2 = 6.8 Hz, H-1"α), 2.86 (dd, 1H, J1 = 13.2 Hz, J2 = 3.8 Hz, H-1"β), 2.37 (t, 1H, J =11.8 Hz, H-3"α), 2.18 (s, 6H, NCH3, CH3 × 2), 2.11 (dd, 1H, J1 = 12.4 Hz, J2 = 3.2 Hz, H-3"β). ESI-MS: m/z 418 [M+1]+.
3.1.11. General Procedure to Obtain Monomethoxyflavones 14a–f
To a stirred solution of dimethoxyflavone (0.2 mmol) in 1,2-dichloroethane (2 mL) at room temperature was added a solution of BBr3 (1 mol/L, 0.4 mL, 0.4 mmol). The resulting mixture was stirred at room temperature for 3 h. It was cooled to 0 °C, and quenched by water (10 mL). It was made alkaline with Na2CO3 solution and extracted with CH2Cl2. The CH2Cl2 extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using EtOAc/petroleum ether/Et3N (100:50:2.5) as eluent to give pure monomethoxyflavones 14a–f. In this manner, the following targets were obtained:
2-(2-Chlorophenyl)-5-hydroxy-8-[2-hydroxy-3-(morpholin-4-yl)propyl]-7-methoxy-4H-1-benzopyran-4-one (14a). Yield: 88%, m.p. 139–151 °C; IR (KBr): 3424 (OH), 2934, 2870 (CH3, CH2), 1658 (C=O), 1620, 1601 (C=C), 1278 (Ar-O), 1166 (C-O) cm−1; 1H-NMR (CDCl3) δ: 12.80 (s, 1H, OH-5), 7.66 (m, 1H, H-6'), 7.54 (m, 1H, H-3'), 7.42 (m, 2H, H-4' and 5'), 6.54 (s, 1H, H-6), 6.45 (s, 1H, H-3), 3.98 (m, 1H, H-2"), 3.92 (s, 3H, OCH3), 3.64 (m, 4H, -CH2-O, CH2 × 2), 3.03 (dd, 1H, J1 = 13.6 Hz, J2 = 6.8 Hz, H-1"α), 2.85 (dd, 1H, J1 = 13.6 Hz, J2 = 5.6 Hz, H-1"β), 2.56 (m, 2H, H-3"), 2.30 (m, 4H, -N-CH2, CH2 × 2). ESI-MS: m/z 446 [M+1]+.
2-(2-Chlorophenyl)-5-hydroxy-8-[2-hydroxy-3-(piperidin-1-yl)propyl]-7-methoxy-4H-1-benzopyran-4-one (14b). Yield: 85%, m.p. 127–128 °C; IR (KBr): 3430 (OH), 2930, 2845 (CH3, CH2), 1659 (C=O), 1612, 1600 (C=C), 1280 (Ar-O), 1121(C-O), 771 cm−1; 1H-NMR (CDCl3) δ: 12.80 (s, 1H, OH-5), 7.68 (m, 1H, H-6'), 7.51(m, 1H, H-3'), 7.42 (m, 2H, H-4' and 5'), 6.53 (s, 1H, H-3), 6.42 (s, 1H, H-6), 3.89 (m, 4H, OCH3 and H-2"), 3.56 (brs, 1H, OH-2"), 3.00 (dd, 1H, J1 =13.6 Hz, J2 = 6.0 Hz, H-1"α), 2.79 (dd, 1H, J1 = 13.2 Hz, J2 =6.0 Hz, H-1"β), 2.47 (m, 2H, H-3"), 2.23 (m, 4H, NCH2, CH2 × 2), 1.48 (m, 4H, NCH2CH2, CH2 × 2), 1.37 (m, 2H, CH2CH2CH2). ESI-MS: m/z 444 [M+1]+.
2-(2-Chlorophenyl)-5-hydroxy-8-[2-hydroxy-3-(pyrrolidin-1-yl)propyl]-7-methoxy-4H-1-benzopyran-4-one (14c). Yield: 72%, m.p. 134–137 °C; IR (KBr): 3424 (OH), 2963, 2933 (CH3, CH2), 1659 (C=O), 1602 (C=C), 1384 (Ar-O), 1117 (C-O), 765 cm−1; 1H-NMR (CDCl3) δ: 12.79 (s, 1H, OH-5), 7.67 (m, 1H, H-6'), 7.51 (m, 1H, H-3'), 7.37 (m, 2H, H-4' and 5'), 6.53 (s, 1H, H-3), 6.42 (s, 1H, H-6), 3.88 (s, 3H, OCH3), 3.78 (m, 1H, H-2"), 3.54 (brs, 1H, OH-2"), 3.00 (dd, 1H, J1 = 13.6 Hz, J2 = 4.8 Hz, H-1"α), 2.78 (dd, 1H, J1 = 13.6 Hz, J2 = 4.8 Hz, H-1"β), 2.48 (m, 2H, H-3"), 2.22 (s, 4H, NCH2, CH2 × 2), 1.73 (s, 4H, NCH2CH2, CH2 × 2). ESI-MS: m/z 430 [M+1]+.
2-(2-Chlorophenyl)-8-(3-diethylamino-2-hydroxypropyl)-5-hydroxy-7-methoxy-4H-1-benzopyran-4-one (14d). Yield: 77%, m.p. 114–115 °C; IR (KBr): 3437 (OH), 2962, 2929 (CH3, CH2), 1662 (C=O), 1619, 1600 (C=C), 1294 (Ar-O), 1114 (C-O), 775 cm−1; 1H-NMR (CDCl3) δ: 12.79 (s, 1H, OH-5), 7.69 (m, 1H, H-6'), 7.53 (m, 1H, H-3'), 7.40 (m, 2H, H-4' and 5'), 6.53 (s, 1H, H-3), 6.42 (s, 1H, H-6), 4.01 (m, 1H, H-2"), 3.90 (s, 3H, OCH3), 3.06 (dd, 1H, J1 =13.4 Hz, J2 = 6.4 Hz, H-1"α), 2.84 (dd, 1H, J1 = 13.2 Hz, J2 =6.4 Hz, H-1"β), 2.39 (m, 6H, H-3" and NCH2), 0.94 (t, 6H, NCH2CH3, CH3 × 2); 13C-NMR (DMSO-d6) δ: 183.3, 166.3, 165.2, 162.9, 157.0, 134.2, 132.9, 131.1, 130.8, 129.0, 126.9, 125.2, 108.5, 106.1, 95.8, 70.4, 60.8, 56.2, 55.9, 47.6, 36.2, 12.4; ESI-MS: m/z 432 [M+1]+.
2-(2-Chlorophenyl)-8-(3-ethylmethylamino-2-hydroxypropyl)-5-hydroxy-7-methoxy-4H-1-benzopyran-4-one (14e). Yield: 70%, m.p. 119–123 °C; IR (KBr): 3425 (OH), 2965, 2945 (CH3, CH2), 1662 (C=O), 1617, 1583 (C=C), 1332 (Ar-O), 1114 (C-O) cm−1; 1H-NMR (CDCl3) δ: 12.80 (s, 1H, OH-5), 7.70 (m, 1H, H-6'), 7.54 (m, 1H, H-3'), 7.44 (m, 2H, H-4' and 5'), 6.56 (s, 1H, H-3), 6.45 (s, 1H, H-6), 3.92 (s, 3H, OCH3), 3.90 (m, 1H, H-2"), 3.02 (dd, 1H, J1 = 12.6 Hz, J2 = 6.4 Hz, H-1"α), 2.82 (dd, 1H, J1 = 13.0 Hz, J2 = 6.4 Hz, H-1"β), 2.42 (m, 4H, H-3" and NCH2), 2.17 (s, 3H, NCH3), 0.98 (t, 3H, NCH2CH3). ESI-MS: m/z 418 [M+1]+.
2-(2-Chlorophenyl)-8-(3-dimethylamino-2-hydroxypropyl)-5-hydroxy-7-methoxy-4H-1-benzopyran-4-one (14f). Yield: 80%, m.p. 149–153 °C; IR (KBr): 3435 (OH), 2937 (CH3, CH2), 1662 (C=O), 1617, 1583 (C=C), 1287 (Ar-O), 1120 (C-O) cm−1; 1H-NMR (CDCl3) δ: 12.82 (s, 1H, OH-5), 7.69 (m, 1H, H-6'), 7.54 (m, 1H, H-3'), 7.45 (m, 2H, H-4' and 5'), 6.55 (s, 1H, H-3), 6.45 (s, 1H, H-6), 3.92 (s, 3H, OCH3), 3.91 (m, 1H, H-2"), 3.02 (dd, 1H, J1 = 13.6 Hz, J2 = 3.8 Hz, H-1"α), 2.83 (dd, 1H, J1 = 13.2 Hz, J2 = 3.2 Hz, H-1"β), 2.36 (t, 1H, J = 12.0 Hz, H-3"α), 2.20 (s, 6H, NCH3, CH3 × 2), 2.12 (dd, 1H, J1 = 12.0 Hz, J2 = 3.2 Hz, H-3"β); 13C-NMR (DMSO-d6) δ: 183.5, 166.0, 165.0, 162.8, 157.1, 134.6, 133.2, 131.4, 130.8, 128.4, 126.9, 124.8, 107.5, 105.7, 95.6, 70.2, 63.8, 56.0, 44.6, 36.0; ESI-MS: m/z 404 [M+1]+.
3.2. Cytotoxicity Assays
In vitro cytotoxicity assays were carried out using 96-well microplate cultures and MTT staining based on the reported method [23]. Briefly, the cells were cultured in RPMI-1640 medium supplemented with fetal bovine serum in 5% CO2 incubated at 37 °C. For the experiment, the drugs were dissolved in DMSO. After seeding 4,000 cells per well in a 96-well microplate, the cells were treated with the sample at four concentrations per compound. The controls received just DMSO. Cells were then incubated at 37 °C for 3 days, after which time 5 μL MTT in citrate buffer was added. Cells were stored again for 4 h. After removing the medium and adding DMSO (100 μL per well) into the microplate with shaking for 10 min, the formazan crystals (the product of MTT reacting with dehydrogenase existing in the mitochondria) were redissolved and the absorbance was measured on a model MR 7000 microtiter plate reader (Dynatech International Corporation, Edgewood, NY, USA) at a wavelength of 570 nm. The result was expressed as the drug concentration which inhibited cell growth by 50% as compared to the control (IC50). The IC50 values were calculated from regression lines obtained from the probit of the percent cell growth inhibition plotted as a function of the logarithm of the dose. The tumor cell lines panel consisted of ECA-109, A-549, HL-60 and PC-3. In all of these experiments, three replicate wells were used to determine each point and taxol was used as the positive control.
4. Conclusions
We have designed and synthesized a novel series of flavonoid derivatives bearing diverse aliphatic amino moieties. Their antitumor activities were evaluated against the ECA-109, A-549, HL-60, and PC-3 cancer cell lines using the MTT method and a preliminary SAR was described. We identified two chlacone derivatives 9b and 9d as the most promising candidates which have high potency with IC50 values lower than 5 µg/mL against all human tumor cell lines examined. Further investigations will focus on the introduction of other chalcone templates and investigation of their effects on cytotoxic activities.
Acknowledgments
The authors are grateful to the support from National Natural Science Foundation of China (No. 81200428) and the Projects of Science Technology Department of Zhejiang Province (No. 2013C31119).
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 4, 6, 9b, 10, 13d, 14d and 14f are available from the authors.
References
- 1.Middleton E., Jr., Kandaswami C., Theoharides T.C. The effects of plant flavonoids on mammalian cells: Implications for inflammation, heart disease, and cancer. Pharmacol. Rev. 2000;52:673–751. [PubMed] [Google Scholar]
- 2.Ren W.Y., Qiao Z.H., Wang H.W., Zhu L., Zhang L. Flavonoids: Promising anticancer agents. Med. Chem. Res. 2003;23:519–534. doi: 10.1002/med.10033. [DOI] [PubMed] [Google Scholar]
- 3.Li Y., Fang H., Xu W. Recent advance in the research of flavonoids as anticancer agents. Mini Rev. Med. Chem. 2007;7:663–678. doi: 10.2174/138955707781024463. [DOI] [PubMed] [Google Scholar]
- 4.Min K., Ebeler S.E. Quercetin inhibits hydrogen peroxide-induced DNA damage and enhances DNA repair in Caco-2 cells. Food Chem. Toxicol. 2009;47:2716–2722. doi: 10.1016/j.fct.2009.07.033. [DOI] [PubMed] [Google Scholar]
- 5.Lee B.W., Lee J.H., Lee S.T., Lee W.S., Jeong T.S., Park K.H. Antioxidant and cytotoxic activities of xanthones from Cudraruatricuspidata. Bioorg. Med. Chem. Lett. 2005;15:5548–5552. doi: 10.1016/j.bmcl.2005.08.099. [DOI] [PubMed] [Google Scholar]
- 6.Lopez-Lazaro M., Willmore E., Austin C.A. The dietary flavonoids myricetin and fisetin act as dual inhibitors of DNA topoisomerases I and II in cells. Mutation. Res-Gen. Tox. Ens. 2010;696:41–47. doi: 10.1016/j.mrgentox.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 7.Kelland L.R. Flavopiridol, the first cyclin-dependent kinase inhibitor to enter the clinic: Current status. Expert Opin. Investig. Drugs. 2000;9:2903–2911. doi: 10.1517/13543784.9.12.2903. [DOI] [PubMed] [Google Scholar]
- 8.Senderowicz A.M., Sausville E.A. Preclinical and clinical development of cyclin-dependent kinase modulators. J. Natl. Cancer Inst. 2000;92:376–387. doi: 10.1093/jnci/92.5.376. [DOI] [PubMed] [Google Scholar]
- 9.Murthi K.K., Dubay M., McClure C., Brizuela L., Boisclair M.D., Worland P.J., Mansuri M.M., Pal K. Structure-activity relationship studies of flavopiridol analogues. Bioorg. Med. Chem. Lett. 2000;10:1037–1041. doi: 10.1016/S0960-894X(00)00156-6. [DOI] [PubMed] [Google Scholar]
- 10.Dauzonne D., Folleas B., Martinez L., Chabot G.G. Synthesis and in vitro cytotoxicity of a series of 3-aminoflavones. Eur. J. Med. Chem. 1997;32:71–82. doi: 10.1016/S0223-5234(97)84363-2. [DOI] [Google Scholar]
- 11.Xia Y., Yang Z.Y., Xia P., Bastow K.F., Nakanishi Y., Lee K.H. Antitumor agents. Part 202: Novel 2'-amino chalcones: Design, synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2000;10:699–701. doi: 10.1016/S0960-894X(00)00072-X. [DOI] [PubMed] [Google Scholar]
- 12.Nielsen S.F., Larsen M., Boesen T., Schonning K., Kromann H. Cationic chalcone antibiotics. Design, Synthesis, And mechanism of action. J. Med. Chem. 2005;48:2667–2677. doi: 10.1021/jm049424k. [DOI] [PubMed] [Google Scholar]
- 13.Liu X.L., Go M.L. Antiproliferative properties of piperidinylchalcones. Bioorg. Med. Chem. 2006;14:153–163. doi: 10.1016/j.bmc.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 14.Chen Z.W., Hu Y.Z., Wu H.H., Jiang H.D. Design and synthesis of 3-(2-pyridyl)pyrazolo[1,5-a]pyrimidines as potent CRF1 receptor antagonists. Bioorg. Med. Chem. Lett. 2004;14:3949–3952. doi: 10.1016/j.bmcl.2004.05.061. [DOI] [PubMed] [Google Scholar]
- 15.Ichino K., Tanaka H., Ito K., Tanaka T., Mizuno M. Synthesis of helilandin B, pashanone, and their isomers. J. Nat. Prod. 1988;51:906–914. doi: 10.1021/np50059a015. [DOI] [PubMed] [Google Scholar]
- 16.Sekizaki H. Synthesis of 2-Benzylidene-3(2H)-benzofuran-3-ones (Aurones) by oxidation of 2’-hydroxychalcones with mercury(II) acetate. Bull. Chem. Soc. Jpn. 1988;61:1407–1409. doi: 10.1246/bcsj.61.1407. [DOI] [Google Scholar]
- 17.Parmar V.S., Gupta S., Sharma R.K., Sharma V.K. Synthesis of 2,3-dihydro-8-(3-hydroxy-3-methylbut-1-enyl)-7-methoxy-2-phenyl-4H-1-benzopyran-4-one: A novel structure for falciformin. J. Org. Chem. 1990;55:1193–1197. doi: 10.1021/jo00291a017. [DOI] [Google Scholar]
- 18.Maitrejean M., Comte G., Barron D., Kirat K.E., Conseil G., Pietro A.D. The flavanolignan silybin and its hemisynthetic derivatives, a novel series of potential modulators of p-glycoprotein. Bioorg. Med. Chem. Lett. 2000;10:157–160. doi: 10.1016/S0960-894X(99)00636-8. [DOI] [PubMed] [Google Scholar]
- 19.Chu H.W., Wu H.T., Lee Y.J. Regioselective hydroxylation of 2-hydroxychalcones by dimethyldioxirane towards polymethoxylated flavonoids. Tetrahedron. 2004;60:2647–2655. doi: 10.1016/j.tet.2004.01.023. [DOI] [Google Scholar]
- 20.Dao T.T., Chi Y.S., Kim J., Kim H.P., Kim S., Park H. Synthesis and PGE2 inhibitory activity of 5,7-dihydroxyflavones and their o-methylated flavone analogs. Arch. Pharm. Res. 2003;26:345–350. doi: 10.1007/BF02976690. [DOI] [PubMed] [Google Scholar]
- 21.Tabaka A.C., Murthi K.K., Pal K., Teleha C.A. Application of modified flavone closure for the preparation of racemic L86–8275. Org. Process Res. Dev. 1999;3:256–259. doi: 10.1021/op980215h. [DOI] [Google Scholar]
- 22.Boeck P. Antifungal activity and studies on mode of action of novel xanthoxyline-derived chalcones. Arch. Pharm. 2005;338:87–95. doi: 10.1002/ardp.200400929. [DOI] [PubMed] [Google Scholar]
- 23.Cardenas M., Marder M., Blank V.C., Roguin L.P. Antitumor activity of some natural flavonoids and synthetic derivatives on various human and murine cancer cell lines. Bioorg. Med. Chem. 2006;14:2966–2971. doi: 10.1016/j.bmc.2005.12.021. [DOI] [PubMed] [Google Scholar]