

Abstract
2-[18F]Fluoroethyl azide ([18F]FEA) can readily be obtained by nucleophilic substitution of 2-azidoethyl-4-toluenesulfonate with [18F]fluoride (half-life 110 min), and has become widely used as a reagent for ‘click’ labeling of PET tracers. However, distillation of [18F]FEA is typically required, which is time-consuming and unpractical for routine applications. In addition, copper(I)-catalyzed cycloaddition of [18F]FEA with non-activated alkynes, and with substrates containing labile functional groups, can be challenging. Herein, we report a highly efficient and practical ligand-accelerated one-pot/two-step method for ‘click’ labeling of small molecule tracers with [18F]FEA. The method exploits the ability of the copper(I) ligand bathophenanthrolinedisulfonate to accelerate the rate of the cycloaddition reaction. As a result, alkynes can be added directly to the crude reaction mixture containing [18F]FEA, and as cyclisation occurs almost immediately at room temperature, the reaction is tolerant to labile functional groups. The method was demonstrated by reacting [18F]FEA with a series of alkyne-functionalized 6-halopurines to give the corresponding triazoles in 55–76% analytical radiochemical yield.
Keywords: fluorine-18; click chemistry; BPDS; 2-[18F]fluoroethyl azide; 6-halopurine; automated synthesis; 1,2,3-triazole; PET; radiotracer
1. Introduction
Labeling with 18F is challenging due to the time constraints imposed by the short half-life (110 min), the need to manipulate minute amounts of [18F]fluoride, and the need for remote automated synthesis of tracers for clinical use. [18F]Fluoride can readily be produced with high specific activity (SA), and is by far the most common source of 18F for tracer synthesis. Labeling is typically achieved by nucleophilic aliphatic substitution of suitable leaving groups, such as sulfonates, with [18F]fluoride. Yet, 18F-labeling of aliphatic groups is sensitive to the presence of hydrogen bond donors, as well as neighbouring group effects, and as a result tracer synthesis often requires the use of prosthetic labeling reagents. The copper(I)-catalyzed cycloaddition reaction of azides and alkynes to give 1,2,3-triazoles has recently emerged as a highly versatile method for conjugation of 18F-labeled ‘click’ reagents to small molecules and peptides [1,2,3,4]. Despite the broad substrate scope, the efficiency of the reaction is dependent on the nature of the ‘click’ partners, and conjugation to substrates with low reactivity or labile functional groups can be challenging [5]. In addition, purification of the intermediate labeling reagent is often required prior to the cycloaddition reaction in order to limit the formation of side products.
Among the array of ‘click’ reagents reported, 2-[18F]fluoroethyl azide ([18F]FEA) is attractive as its small size makes it particularly suited for labeling of small molecule tracers [2,5,6,7,8]. However, purification can only be achieved by distillation, which is delicate, poorly compatible with automated synthesis modules, and also potentially hazardous as it can result in release of gaseous [18F]FEA. Herein, we reported a convenient and highly efficient one-pot method for ‘click’ labeling with [18F]FEA. The method exploits the ability of the copper chelator bathophenanthrolinedisulfonate (BPDS, Figure 1) to accelerate the cycloaddition reaction, which overcomes the need to purify the intermediate [18F]FEA, and enables conjugation to precursors with labile functional groups. The method was demonstrated by reacting [18F]FEA in the form of a crude reaction mixture with a series of alkyne-functionalized 6-halopurines in the presence of copper(II)/ascorbate and BPDS to give the corresponding triazoles in 40–57% isolated radiochemical yield and with >99% radiochemical purity.
Figure 1.
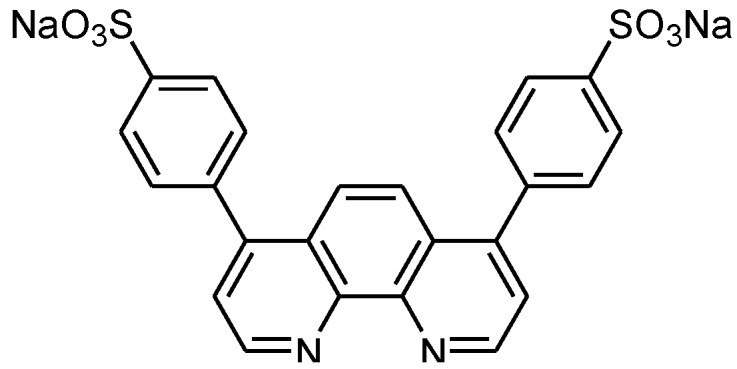
Chemical structure of BPDS.
2. Results and Discussion
2.1. Chemistry
The alkyne-functionalized 6-halopurines 2–5 (Scheme 1) and the corresponding triazole derivatives 6–9 were synthesized as labeling precursors and non-radioactive fluorinated reference compounds, respectively. The alkyne group was initially introduced into the purine scaffold by a base-promoted alkylation of 6-bromopurine and 6-chloropurine using propargyl bromide or p-tosylate-1-butyne (1) as previously reported [9]. The reaction provided both the N7-alkynyl (2a–5a, 10–20%) and the N9-alkynyl 6-halopurines (2b–5b, 40–60%), which were separated by flash chromatography. The N7-isomers were distinguished from the corresponding N9-isomers by down field shifts of H-2 and H-8 protons in 1H-NMR [10,11]. Compounds 2a, 3a, 4b and 5a were subsequently treated with FEA in the presence of catalytic amount of copper(II) sulfate and sodium ascorbate to provide the corresponding triazoles 6–9 in moderate to good yields.
Scheme 1.

Synthesis of the 6-halopurine alkynes 2–5 and the corresponding triazoles 6–9.
2.2. Radiochemistry
As formation of the N7-alkylated triazoles 6–8 under preparative conditions gave moderate yields (32–40%) with apparent decomposition of the corresponding precursors, we initially used non-radioactive FEA and the alkyne 2a to optimize the conditions for the cycloaddition reaction. Attempts to increase the reaction rate by the use of excess copper(II) sulfate and sodium ascorbate relative to the alkyne 2a failed to give the triazole 6 (15 min, rt), and instead resulted in formation of an unknown side product with a similar HPLC retention time to that of the target compound 6 [Figure 2(a)]. The use of ascorbic acid to allow formation of copper(I) under acidic conditions was also unsuccessful, and resulted in complete consumption of the precursor 2a with formation of another unidentified side product [Figure 2(b)]. To exclude the possibility that side product formation was caused by the in situ reduction of copper(II) sulfate, we investigated copper(I) chloride and triethylamine as an alternative catalytic system. However, we were unable to obtain the target triazole 6, and instead obtained product mixtures with similar HPLC profiles to that observed for the reaction of 2a and FEA in the presence of copper(II)/ascorbate [Figure 2(a)].
Figure 2.

Analytical HPLC profiles (UV at 265 nm) of the reaction mixtures resulting from treatment of 2a with non-radioactive FEA (1 equiv.) in the presence of excess copper(II) sulfate and (a) sodium ascorbate or (b) ascorbic acid. Unknown side products are labeled with *; (c) Reaction of 2a and FEA in the presence of the BPDS-copper(I) catalytic system; (d) Addition of 2a, copper(II) sulfate, BPDS and ascorbate to a crude reaction mixture containing [18F]FEA resulted in clean formation of the triazole [18F]6 (radioactivity profile shown as dotted line).
Purines are known to form complexes with copper ions [12,13], and it is plausible that the combined interactions of the alkyne group and nitrogens in 2a with copper(I) results in formation of a complex that impairs subsequent cyclisation with FEA through unfavorable steric or electronic interactions. Instead of increasing the rate of the cycloaddition reaction, addition of excess copper(I) relative to the alkyne 2a may therefore favor hydrolysis of the precursor 2a by activation of the chlorine in the 6-position. To explore this possibility, we investigated the use of BPDS as an auxiliary copper(I) chelator. The BPDS-copper(I) catalytic system was first identified from a fluorescent assay [14], and has since been used to accelerate triazole formation for reactions with peptides and substrates with high molecular weight for which the copper(II)/ascorbate system alone often provides unsatisfactory results [15,16]. Remarkably, in the presence of BPDS, copper(II) sulfate and sodium ascorbate, the cycloaddition of 2a with FEA occurred almost immediately at room temperature to give the target triazole 6 [Figure 2(c)].
Encouraged by the superb efficiency of the cycloaddition reaction in the presence of the BPDS-copper(I) catalytic system, we explored the possibility of preparing the triazoles [18F]6–[18F]9 in an one-pot reaction without distillation of the intermediate [18F]FEA. Indeed, sequential addition of alkyne 2a, copper(II) sulfate, BPDS and sodium ascorbate to a crude reaction mixture containing [18F]FEA resulted in formation of [18F]6 in 60% analytical radiochemical yield (RCY) within 1 min at room temperature. Provided that the alkyne 2a was used in excess relative to copper(II) sulfate and BPDS, no other radioactive product was observed, and the precursor 2a remained largely intact after the reaction [Figure 2(d)]. The apparent absence of non-radioactive triazoles in the product mixture was unexpected and suggests that the cycloaddition rate of FEA to alkynes far exceeds that for the corresponding 2-azidoethyl-4-toluenesulfonate precursor under these conditions. Comparable results were obtained for formation of triazoles [18F]7–9, which were obtained in 55–76% analytical and 40–57% isolated (decay-corrected) radiochemical yields (Table 1).
Table 1.
Radiochemical yields for formation of [18F]6–[18F]9.
| Entry | Product | RCY a (%) | |
|---|---|---|---|
| 1 | [18F]6 | 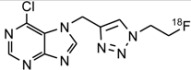 |
60 ± 1 (41) b |
| 2 | [18F]7 | 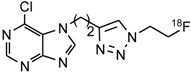 |
76 ± 1 (40) b,c |
| 3 | [18F]8 | 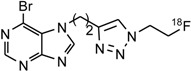 |
55 ± 2 (55) b |
| 4 | [18F]9 | 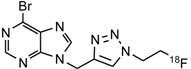 |
76 ± 1 (57) b |
a Average analytical decay-corrected radiochemical yields ± standard deviation (n = 3). b Isolated RCY in brackets (n = 1). c Isolated yield obtained from a fully automated radiosynthesis.
2.3. Automated radiosynthesis of purine [18F] 7
To evaluate the suitability of the one-pot method for routine tracer production we established an automated synthesis of the triazole-functionalized purine [18F]7 using a HBIII module (Scintomics). The configuration of the synthesis module is illustrated in Figure 3. [18F]Fluoride in water (~1.5 GBq) was trapped on an ion-exchange cartridge (QMA cartridge Sep-Pak® light, Waters) and released with a solution of Kryptofix 222 and potassium carbonate (0.5 mL of a solution 30 mM: 15 mM in acetonitrile:water = 85:15). Following azeotropic drying with anhydrous acetonitrile at 90 °C (2 × 0.5 mL), a solution of 2-azidoethyl-4-toluenesulfonate (8.4 µmol) in acetonitrile (0.4 mL) was added and the resulting mixture was allowed to react for 15 min at 80 °C, which resulted in formation of [18F]FEA in 80–90% analytical RCY. The reactor vial was cooled to room temperature with a stream of nitrogen, and a solution containing the alkyne 3 (10 µmol), CuSO4·5H2O (8 µmol) and BPDS (8 µmol) in a mixture of acetonitrile (0.15 mL) and phosphate buffer (0.2 mL, 0.1 M, pH = 7.4) was added, followed by a solution of sodium ascorbate (40 µmol in 0.1 mL H2O). After 1 min at room temperature the reaction was diluted with H2O (1 mL). HPLC analysis showed complete conversion of [18F]FEA to give [18F]7 in 75% analytical RCY. To enable purification on a semi-preparative column, we investigated the use of cartridge-based methods to clean up the product mixture prior to HPLC injection. However, the high polarity of [18F]7 made cartridge trapping challenging, and also complicated subsequent purification by HPLC.
Figure 3.

Schematic representation of the configuration of the HBIII synthesis module.
In order to maximize the trapping efficiency of [18F]7, we evaluated a number of solid phases, sizes of cartridges, and solvent systems. The highest RCY was obtained by dilution of the product mixture with water (30 mL) to give a total of 2% acetonitrile, followed by trapping of the crude product on a C-18 cartridge (Sep-Pak® Plus, Waters). The product was eluted with 25% acetonitrile in water (2 mL), the resulting solution was diluted with water (3 mL) and subsequently purified by HPLC (Chromolith® RP18-e using 9% acetonitrile with 0.1% formic acid as the eluent) to give [18F]7 in 40% decay-corrected RCY. While the radiochemical purity was > 99% [Figure 4(a)], the specific activity was still below 1 GBq/μmol. The use of a smaller cartridge (C-18 Sep-Pak® light, Waters) allowed release of the product in a lower solvent volume (20% ethanol in water, 1 mL), however the trapping was less efficient and led to loss of 15–20% of the product. The resulting solution was diluted with water (1 mL) and purified by HPLC as described above. The collected fraction was diluted with water to give a total concentration of 5% in acetonitrile, the product was trapped on C-18 Sep-Pak® light cartridge (Waters), the cartridge was washed with water (1 mL), and the product eluted with 20% ethanol in saline (0.8 mL). The resulting solution was diluted with saline for injection (2.4 mL) to give a total of 5% ethanol, and filtered on sterile filter (Millex-GV 0.22 µm, 13 mm, Millipore). When starting with 1–1.5 GBq [18F]fluoride, the formulated product [18F]7 was obtained in 9 ± 2% (n = 3) decay-corrected RCY after sterile filtration, with >99% radiochemical purity and a specific activity in the range of 5–7 GBq/μmol [Figure 4(b)]. The total synthesis time was 105 min.
Figure 4.

Analytical radio-HPLC profile (dotted line) and UV profile (265 nm, solid line) of [18F]7 obtained from the automated one-pot synthesis after using (a) Sep-Pak® Plus (Luna C18(2) column) and (b) and C-18 Sep-Pak® Light cartridge (Chromolith® performance column, RP18-e). Unidentified side product is labeled with *.
An inherent limitation of the one-pot method for cycloaddition of [18F]FEA with alkynes is that the total contents of the product mixture exceed the typical capacity of semi-preparative HPLC columns, and hence cartridge-based purification is required prior to sample loading. The high polarity of [18F]7 presented particular challenges in this respect as cartridge trapping and HPLC purification only was achievable when using aqueous solutions with very low concentrations of organic solvents. The poor retention of [18F]7 on reverse-phase stationary phases necessitated extensive dilution of product solutions, which added to the overall synthesis time, and also made it difficult to obtain the target compound with high specific activity. However, such complications should not affect tracers with more favorable physiochemical properties, in which case a shorter synthesis time with improved RCY and higher specific activity is likely to be achieved. While the outcome of the cycloaddition reaction will depend on the nature of the alkyne precursor, and the conditions used, in our hands we observed no competing reactions that are likely to complicate tracer purification.
It should be noted that one-pot ‘click’ labeling with [18F]FEA have been reported previously [17], and that BPDS has been used for one-pot labeling with other ‘click’ reagents [16]. However, in the absence of a chelator to accelerate the reaction rate, one-pot labeling with [18F]FEA is limited to highly reactive alkyne precursors. For ‘click’ reagents other than [18F]FEA, which readily can be purified with solid-phase extraction, one-pot 18F-labeling offer fewer benefits, which may explain the limited use of BPDS despite the astonishing improvement in reaction rates that can be achieved with this chelator.
3. Experimental
3.1. General
All reagents were purchased from Sigma-Aldrich (Dorset, UK) and used without further purification. NMR spectra were recorded on Bruker Avance (500 MHz or 600 MHz) spectrometers operating at the frequency of 500 or 600 MHz for 1H, and 125 or 150 MHz for 13C. Chemical shifts (δ) are reported in ppm downfield from the internal standard tetramethylsilane. High resolution mass spectra were recorded on a thermo Finnigan MAT900xp (CI/EI) or a Waters LCT Premier XE (ES) mass spectrometers. No-carrier-added aqueous [18F]fluoride was provided by St Thomas’ Hospital, King’s College London, UK. HPLC analyses were performed with an Agilent 1200 series system equipped with a diode array UV detector (results described are for UV at 265 nm), and a Raytest GABI star NaI detector. Analytical runs and HPLC purifications were carried out on a Chromolith® performance column, RP18-e, 100 × 4.6 mm and 100 × 10 mm (Merck), respectively. The solvent systems used were water and methanol, both containing 0.1% of formic acid. The flow rate was 3 mL/min for analytical and 5 mL/min for semi-preparative columns, respectively. Automated synthesis was carried out on HBIII module (Scintomics, Lindach, Germany). Isolated radiochemical yields (RCY) were measured using a Curiementor® 4 ion-chamber (PTW).
3.2. Chemistry
3.2.1. General Procedure for Preparation of the Alkyne-functionalized 6-Halopurines
To a mixture of the 6-halopurine (1.0 equiv.) and NaH (60% dispersed in oil, 1.1 equiv.) in anhydrous DMF was added the alkylating agent (1.3 equiv.) under inert atmosphere. After stirring for 24 h at room temperature, the reaction mixture was diluted with dichloromethane (DCM) and extracted three times with water. The organic layer was dried over MgSO4 and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of DCM and ethyl acetate (from 0% to 40% of ethyl acetate) to give the N7- and the N9-alkynyl analogues.
6-Chloro-7-propargylpurine (2a) and 6-chloro-9-propargylpurine (2b). A mixture of 6-chloropurine (500 mg, 3.24 mmol), and NaH (60% dispersed in oil, 142 mg, 3.56 mmol) in anhydrous DMF (15 mL) was treated with propargyl bromide (468 µL, 4.21 mmol) as described under the general alkylation procedure to give 2a (105 mg, 17%) and 2b (363 mg, 58%). Compound 2a: 1H-NMR (CDCl3, 600 MHz): δ 8.89 (s, 1H, H-2), 8.49 (s, 1H, H-8), 5.29 (d, J = 2.58 Hz, 2H, CH2C), 2.68 (t, J = 2.58 Hz, 1H, CCH). 13C-NMR (CDCl3): δ 162.2, 153.0, 148.3, 143.3, 122.3, 77.4, 75.1, 37.4. Accurate mass (EI-MS) m/z calcd for C8H5ClN4 (M)+ 192.0197, found 192.0204. Mp: 102 ± 1 °C. Compound 2b: 1H-NMR (CDCl3, 500 MHz): δ 8.74 (s, 1H, H-2), 8.33 (s, 1H, H-8), 5.06 (t, J = 2.46 Hz, 2H, CH2C), 2.58 (t, J = 2.55 Hz, 1H, CCH). 13C-NMR (CDCl3): δ 152.3, 151.3, 144.4, 131.6, 76.0, 75.1, 33.8. Accurate mass (EI-MS) m/z calcd for C8H5ClN4 (M)+ 192.0197, found 192.0190. Mp: 120 ± 1 °C.
6-Chloro-7-(1-butyne)purine (3a) and 6-chloro-9-(1-butyne)purine (3b). A mixture of 6-chloropurine (500 mg, 3.24 mmol), and NaH (60% dispersed in oil, 142 mg, 3.56 mmol) in anhydrous DMF (15 mL) was treated with 4-tosyloxy-1-butyne (1.56 g, 4.21 mmol) as described under the general alkylation procedure to give 3a (109 mg, 19%) and 3b (466 mg, 41%). Compound 3a: 1H-NMR (CDCl3, 600 MHz): δ 8.89 (s, 1H, H-2), 8.34 (s, 1H, H-8), 4.64 (t, J = 5.25 Hz, 2H, CH2CH2C), 2.84 (dt, J = 5.25 Hz, J = 2.15 Hz, 2H, CH2C), 2.01 (t, J = 2.15 Hz, 1H, CCH). 13C-NMR (CDCl3): δ 162.4, 152.7, 149.7, 142.8, 122.2, 78.7, 73.0, 45.9, 21.9. Accurate mass (EI-MS) m/z calcd for C9H7N4Cl (M)+ 206.0353, found 206.0359. Mp: 79 ± 1 °C. Compound 3b: 1H-NMR (CDCl3, 500 MHz): δ 8.74 (s, 1H, H-2), 8.25 (s, 1H, H-8), 4.46 (t, J = 5.30 Hz, 2H, CH2CH2C), 2.83 (m, 2H, CH2C), 2.06 (t, J = 2.15 Hz, 1H, CCH). 13C-NMR (CDCl3): δ 152.1, 151.7, 151.3, 145.6, 131.8, 79.4, 72.4, 43.2, 20.1. Accurate mass (EI-MS) m/z calcd for C9H7N4Cl (M)+ 206.0353, found 206.0353. Mp: 77 ± 2 °C.
6-Bromo-7-propargylpurine (4a) and 6-bromo-9-propargylpurine (4b). A mixture of 6-bromopurine (500 mg, 2.51 mmol), and NaH (60% dispersed in oil, 111 mg, 2.76 mmol) in anhydrous DMF (15 mL) was treated with propargyl bromide (365 µL, 5.03 mmol) as described under the general alkylation procedure to give 4a (115 mg, 19%) and 4b (361 mg, 61%). Compound 4a: 1H-NMR (CDCl3, 500 MHz): δ 8.81 (s, 1H, H-2), 8.51 (s, 1H, H-8), 5.33 (d, J = 2.56 Hz, 2H, CH2C), 2.67 (t, J = 2.56 Hz, 1H, CCH). 13C-NMR (CDCl3): δ 161.1, 152.8, 148.6, 133.3, 124.5, 77.4, 75.1, 37.2. Accurate mass (EI-MS) m/z calcd for C8H5BrN4 (M)+ 235.9697, found 235.9698. Mp: 103 ± 2 °C. Compound 4b: 1H-NMR (CDCl3, 500 MHz): δ 8.73 (s, 1H, H-2), 8.36 (s, 1H, H-8), 5.06 (d, J = 2.60 Hz, 2H, CH2C), 2.57 (t, J = 2.60 Hz, 1H, CCH). 13C-NMR (CDCl3): δ 152.3, 150.1, 144.2, 143.4, 134.3, 76.0, 75.0, 33.8. Accurate mass (CI-MS) m/z calcd for C8H6BrN4 (M+H)+ 236.9775, found 236.9779. Mp: 154 ± 1 °C.
6-Bromo-7-(1-butyne)purine (5a) and 6-bromo-9-(1-butyne)purine (5b). A mixture of 6-bromopurine (500 mg, 2.51 mmol), and NaH (60% dispersed in oil, 111 mg, 2.76 mmol) in anhydrous DMF (15 mL) was treated with 4-tosyloxy-1-butyne (732 mg, 3.27 mmol) as described under the general alkylation procedure to give 5a (58 mg, 9%) and 5b (276 mg, 44%). Compound 5a: 1H-NMR (CDCl3, 600 MHz): δ 8.85 (s, 1H, H-2), 8.41 (s, 1H, H-8), 4.67 (t, J = 6.24 Hz, 2H, CH2CH2C), 2.85 (dt, J = 6.24 Hz, J = 2.34 Hz, 2H, CH2C), 2.10 (t, J = 2.64 Hz, 1H, CCH). 13C-NMR (CDCl3): δ 161.3, 152.7, 150.0, 133.1, 124.5, 78.7, 73.1, 45.6, 21.9. Accurate mass (EI-MS) m/z calcd for C9H7N4Br (M)+ 249.9848, found 249.9856. Mp: 97 ± 1 °C. Compound 5b: 1H-NMR (CDCl3, 500 MHz): δ 8.67 (s, 1H, H-2), 8.25 (s, 1H, H-8), 4.44 (t, J = 5.0 Hz, 2H, CH2CH2C), 2.80 (m, 2H, CH2C), 2.05 (t, J = 2.59 Hz, 1H, CCH). 13C-NMR (CDCl3): δ 151.9, 150.4, 145.4, 143.3, 134.3, 79.4, 72.3, 43.2, 20.0. Accurate mass (EI-MS) m/z calcd for C9H7N4Br (M)+ 249.9848, found 249.9842. Mp: 94 ± 1 °C.
2-Fluoroethyl azide (FEA). To a solution of 2-fluoroethyl-4-toluenesulfonate [2] (1.1 g, 5.0 mmol) in dry DMF (15 mL) was added sodium azide (279 mg, 4.28 mmol). The resulting mixture was left to react for 24 h at ambient temperature, and used as a stock solution in subsequent reactions without any purification. WARNING: Attempts to isolate neat 2-fluoroethyl azide may result in an explosion.
3.2.2. General Procedure for Cycloaddition of 2-fluoroethyl azide (FEA) with Alkyne-functionalized Purines
A solution of the alkyne-functionalized 6-halopurine (1.0 equiv.) in DMF was subsequently reacted with FEA (1.3 equiv.) in the presence of sodium ascorbate (0.1 equiv.) and CuSO4·5H2O (0.05 equiv.) at room temperature for 4 h. The reaction mixture was diluted with DCM and the organic phase was washed with water. The organic layer was dried with MgSO4 and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica using a gradient of ethyl acetate and MeOH (from 0% to 10% of MeOH).
6-Chloro-7-[1-(2-fluoroethyl)-1H-[1,2,3]triazol-4-ylmethyl]-purine (6). To a solution of 2a (80 mg, 0.42 mmol) and FEA (48 mg, 0.54 mmol) in DMF (3 mL), was added sodium ascorbate (8.1 mg, 0.042 mmol) and CuSO4·5H2O (5.2 mg, 0.021 mmol) in H2O (0.5 mL) as described under the general procedure to give compound 6 in 40% yield (47 mg). 1H-NMR (CDCl3, 600 MHz): δ 8.87 (s, 1H, H-2), 8.49 (s, 1H, H-8), 7.75 (s, 1H, CH), 5.82 (s, 2H, CH2), 4.78 (dt, JF,H = 46.69 Hz, J1,2 = 4.44 Hz, 2H, CH2F), 4.68 (dt, JF,H = 27.13 Hz, J1,2 = 4.44 Hz, 2H, CH2CH2F). 13C-NMR (CDCl3): δ 162.1, 152.9, 149.4, 142.9, 142.2, 123.6, 122.2, 82.0, 80.9, 51.0, 50.9, 42.1. Accurate mass (CI-MS) m/z calcd for C10H10ClFN7 (M+H)+ 282.0670, found 282.0667. Mp: 143 ± 1 °C.
6-Chloro-7-{2-[1-(2-fluoroethyl)-1H-[1,2,3]triazol-4-yl]-ethyl}-purine (7). To a solution of 3a (30 mg, 0.15 mmol) and FEA (17 mg, 0.19 mmol) in DMF (2 mL) was added sodium ascorbate (2.9 mg, 0.015 mmol) and CuSO4·5H2O (1.8 mg, 0.007 mmol) in H2O (0.2 mL) as described under the general procedure to give compound 7 in 32% yield (14 mg). 1H-NMR (CDCl3, 600 MHz): δ 8.87 (s, 1H, H-2), 8.34 (s, 1H, H-8), 7.27 (s, 1H, CH), 4.94 (t, J1,2 = 6.6 Hz, 2H, CH2CH2), 4.74 (dt, JF,H = 46.8 Hz, J1,2 = 4.74 Hz, 2H, CH2F), 4.60 (dt, JF,H = 27.1 Hz, J1,2 = 4.5 Hz, 2H, CH2CH2F), 3.34 (t, J1,2 = 5.50 Hz, 2H, CH2CH2). 13C-NMR (CDCl3): δ 162.2, 152.7, 149.8, 142.9, 142.7, 123.2, 122.2, 82.1, 81.0, 50.8, 50.6, 46.4, 28.0. Accurate mass (ESI-MS) m/z calcd for C11H12ClFN7 (M+H)+ 296.0827, found 296.0835. Mp: 108 ± 2 °C.
6-Bromo-7-{2-[1-(2-fluoroethyl)-1H-[1,2,3]triazol-4-yl]-ethyl}-purine (8). To a solution of 5a (20 mg, 0.08 mmol) and FEA (9.2 mg, 0.10 mmol) in DMF (1.5 mL) was added sodium ascorbate (1.5 mg, 0.008 mmol) and CuSO4·5H2O (1.0 mg, 0.004 mmol) in H2O (0.1 mL) as described under the general procedure to give compound 8 in 37% yield (10 mg). 1H-NMR (CDCl3, 600 MHz): δ 8.82 (s, 1H, H-2), 8.08 (s, 1H, H-8), 7.27 (s, 1H, CH), 4.96 (t, J1,2 = 6.66 Hz, 2H, CH2CH2), 4.74 (dt, JF,H = 46.81 Hz, J1,2 = 4.74 Hz, 2H, CH2F), 4.61 (dt, JF,H = 27.13 Hz, J1,2 = 4.56 Hz, 2H, CH2CH2F), 3.35 (t, J1,2 = 6.60 Hz, 2H, CH2CH2). 13C-NMR (CDCl3): δ 161.3, 152.6, 150.0, 142.7, 133.1, 124.4, 123.3, 82.2, 81.0, 50.8, 50.6, 45.9, 28.1. Accurate mass (ESI-MS) m/z calcd for C11H12BrFN7 (M+H)+ 340.0322, found 340.0315. Mp: 120 ± 1 °C.
6-Bromo-9-[1-(2-fluoroethyl)-1H-[1,2,3]triazol-4-ylmethyl]-purine (9). To a solution of 4b (80 mg, 0.34 mmol) and FEA (39 mg, 0.44 mmol) in DMF (3.5 mL) was added sodium ascorbate (5.9 mg, 0.034 mmol) and CuSO4·5H2O (4.2 mg, 0.017 mmol) in H2O (0.4 mL) as described under the general procedure to give compound 9 in 60% yield (41 mg). 1H-NMR (CDCl3, 600 MHz): δ 8.73 (s, 1H, H-2), 8.36 (s, 1H, H-8), 7.81 (s, 1H, CH), 5.59 (s, 2H, CH2), 4.78 (dt, JF,H = 46.00 Hz, J1,2 = 4.20 Hz, 2H, CH2F), 4.67 (dt, JF,H = 27.00 Hz, J1,2 = 4.80 Hz, 2H, CH2CH2F). 13C-NMR (CDCl3): δ 152.2, 150.3, 145.0, 143.5, 141.8, 134.3, 124.1, 82.0, 80.8, 51.0, 50.8, 39.1. Accurate mass (ESI-MS) m/z calcd for C10H10BrFN7 (M + H)+ 326.0165, found 326.0160. Mp: 103 ± 1 °C.
3.3. Radiochemistry
3.3.1. One-pot Method for Cycloaddition of [18F]FEA to the Alkyne-Functionalized Purine Precursors
[18F]FEA was prepared as described by Glaser and Årstad. [2] Briefly, a mixture of Kryptofix 222 and potassium carbonate (30 mM : 15 mM dissolved in 0.5 mL acetonitrile-water = 4:1) was added to [18F]fluoride (~30 MBq) in water. The solvent was removed by heating at 90 °C under a stream of nitrogen. Acetonitrile (0.5 mL) was added, and the distillation continued. The procedure was repeated twice. After cooling to room temperature, a solution of 2-azidoethyl-4-toluenesulfonate (1.8 mg, 8.4 µmol in 0.4 mL anhydrous acetonitrile) was added. The reaction mixture was stirred at 80 °C. After 15 min, the crude mixture was allowed to reach ambient temperature prior to addition of the alkyne precursor (10 µmol in 0.15 mL acetonitrile), a solution of CuSO4·5H2O and bathophenanthrolinedisulfonic acid disodium salt hydrate (BPDS) (8 µmol each in 0.2 mL phosphate buffer 0.1 M, pH = 7.4), and sodium ascorbate (40 µmol in 0.1 mL H2O). After 1 min at room temperature the reaction was diluted with 1 mL H2O, and aliquots taken to determine the analytical and isolated RCYs.
[18F]6-Chloro-7-[1-(2-fluoroethyl)-1H-[1,2,3]triazol-4-ylmethyl]-purine ([18F]6). Hands-on reaction: analytical RCY: 60 ± 1% (n = 3); isolated RCY 41%. HPLC gradient system: the methanol content was increased from 5% to 12% over 7 min, then to 20% over 1 min and finally to 70% over 4 min. The flow rate was 3 mL/min and 5 mL/min for analytical and semi-preparative HPLC, respectively. Retention times of [18F]6 were 5.6 min and 8.6 min for analytical and semi-preparative runs, respectively.
[18F]6-Chloro-7-{2-[1-(2-fluoroethyl)-1H-[1,2,3]triazol-4-yl]-ethyl}-purine ([18F]7). Hands-on reaction: analytical RCY: 76 ± 1% (n = 3). Automated synthesis: analytical RCY: 75 ± 1% (n = 3); isolated RCY was 40% when using a C-18 Plus cartridge. When using a C-18 light cartridge the RCY was 9 ± 2% (n = 3) after sterile filtration, with a specific activity of 5-7 GBq/µmol. Analytical HPLC was carried out with a Luna C18(2) column (3µm, 50 × 4.6 mm, Phenomenex) with a flow rate of 1 mL/min using the following gradient: the methanol content was increased from 5% to 70% in 13 min. Semi-preparative HPLC with a Chromolith® performance column and a flow rate of 5 mL/min was carried out using an isocratic solvent system consisting of 9% acetonitrile in water with 0.1% formic acid. Retention time of [18F]7: 7.1 min and 17 min for analytical and semi-preparative runs, respectively. The specific activity was measured using a Chromolith® performance RP18-e (100 × 4.6 mm) column with a flow rate of 5 mL/min [Figure 4(b)]. The methanol content was increased from 5% to 20% over 5 min, and then to 50% over 3 min. A calibration curve was obtained by correlating the area under the peak (UV = 265 nm) with the injected mass of the non-radioactive reference compound 7 across a predetermined range (measurements in duplicate, linear fit with R2 = 99.4%). Following formulation of [18F]7, an aliquot was analyzed and the correlation curve was used to estimate the total mass in the sample (UV integration from 2–8 min). The specific activity was calculated as the activity in the aliquot divided on the estimated mass expressed as µmol of 7.
[18F]6-Bromo-7-{2-[1-(2-fluoroethyl)-1H-[1,2,3]triazol-4-yl]-ethyl}-purine ([18F]8). Hands-on reaction: analytical RCY: 55 ± 2% (n = 3); isolated RCY 55%. HPLC solvent gradient: the methanol gradient was increased from 5% to 20% in 5 min, kept constant at 20% for 2 min, and increased to 70% over 2 min. The flow rate was 3 mL/min and 5 mL/min for analytical and semi-preparative HPLC, respectively. Retention time of [18F]8: 5.6 min and 7.2 min for analytical and semi-preparative runs, respectively.
[18F]6-Bromo-9-[1-(2-fluoroethyl)-1H-[1,2,3]triazol-4-ylmethyl]-purine ([18F]9). Hands-on reaction: analytical RCY: 77 ± 1% (n = 3); isolated RCY 57%. Analytical HPLC was carried out with a Luna C18(2) column (3µm, 50 × 4.6 mm, Phenomenex) with a flow rate of 1 mL/min using the following gradient: the methanol content was increased from 20% to 45% in 6 min. Semi-preparative HPLC was carried out with a Chromolith® performance column using a flow rate of 5 mL/min. The methanol content was increased from 15% to 30% over 5 min. Retention time of [18F]9: 6.2 min and 4.1 min for analytical and semi-preparative runs, respectively.
4. Conclusions
Conditions for labeling of alkyne-functionalized 6-halopurines with [18F]fluoroethyl azide have been investigated. The use of the copper(II)/ascorbate catalytic system failed to yield the target triazoles in satisfactory yields, and addition of excess of copper(II) relative to the alkynes led to decomposition of the precursors. In contrast, when BPDS was used as an auxiliary copper(I) chelator the cycloaddition reaction proceeded almost immediately at room temperature to give the corresponding triazole-substituted purines. The high reaction efficiency in the presence of BPDS-copper(I) was exploited to develop a one-pot method for labeling that circumvents the need to distill [18F]fluoroethyl azide. The method enabled labeling of four 6-halopurines in 55–76% analytical radiochemical yield with short reaction time and under mild conditions (1 min at room temperature). The ability to carry out the reaction in one pot without the need for distillation makes this a highly convenient approach to prepare [18F]fluoroethyltriazoles.
Acknowledgments
This work was undertaken at UCLH/UCL which is funded in part by the Department of Health’s NIHR Biomedical Research Centres funding scheme (E.A.). The research leading to these results has received funding from the European Community’s Seventh Framework Programme (FP7/2007–2013) under grant agreement number 201380 (“Euripides”) (M.K. and E.G.).
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 6–9 are available from the authors.
References
- 1.Marik J., Sutcliffe J.L. Click for PET: rapid preparation of [18F]fluoropeptides using CuI catalyzed 1,3-dipolar cycloaddition. Tetrahedron Lett. 2006;47:6681–6684. doi: 10.1016/j.tetlet.2006.06.176. [DOI] [Google Scholar]
- 2.Glaser M, Årstad E. “Click labeling” with 2-[18F]fluoroethylazide for positron emission tomography. Bioconjug. Chem. 2007;18:989–993. doi: 10.1021/bc060301j. [DOI] [PubMed] [Google Scholar]
- 3.Glaser M, Robins E.G. ‘Click labeling’ in PET radiochemistry. J. Labelled Compd. Radiopharm. 2009;52:407–414. doi: 10.1002/jlcr.1656. [DOI] [Google Scholar]
- 4.Mamat C, Ramenda T, Wuest F.R. Recent applications of click chemistry for the synthesis of radiotracers for molecular imaging. Mini-Rev. Org. Chem. 2009;6:21–34. doi: 10.2174/157019309787316148. [DOI] [Google Scholar]
- 5.Smith G., Glaser M., Perumal M., Nguyen Q.-D., Shan B., Årstad E., Aboagye E.O. Design, Synthesis, and biological characterization of a Caspase 3/7 selective isatin labeled with 2-[18F]fluoroethylazide. J. Med. Chem. 2008;51:8057–8067. doi: 10.1021/jm801107u. [DOI] [PubMed] [Google Scholar]
- 6.Zhou D., Chu W., Dence C.S., Mach R.H., Welch M.J. Highly efficient click labeling using 2-[18F]fluoroethyl azide and synthesis of an 18FN-hydroxysuccinimide ester as conjugation agent. Nucl. Med. Biol. 2012;39:1175–1181. doi: 10.1016/j.nucmedbio.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laurens E., Dee Yeoh S., Rigopoulos A., Cao D., Cartwright G.A., O’Keefe G.J., Tochon-Danguy H.J., White J.M., Scott A.M., Ackermann U. Radiolabelling and evaluation of novel haloethylsulfoxides as PET imaging agents for tumor hypoxia. Nucl. Med. Biol. 2012;39:871–882. doi: 10.1016/j.nucmedbio.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 8.Smith G., Sala R., Carroll L., Behan K., Glaser M., Robins E., Nguyen Q-D., Aboagye E.O. Synthesis and evaluation of nucleoside radiotracers for imaging proliferation. Nucl. Med. Biol. 2012;39:652–665. doi: 10.1016/j.nucmedbio.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 9.Yan R., Sander K., Galante E., Rajkumar V., Badar A., Robson M., El-Emir E., Lythgoe M.F., Pedley R.B., Arstad E. A one-pot three-component radiochemical reaction for rapid assembly of 125I-labeled molecular probes. J. Am. Chem. Soc. 2013;135:703–709. doi: 10.1021/ja307926g. [DOI] [PubMed] [Google Scholar]
- 10.Kjellberg J., Johansson N.G. Charaterization of N7 and N9 alkylated purine by 1H and 13C NMR. Tetrahedron. 1986;42:6541–6544. doi: 10.1016/S0040-4020(01)88116-3. [DOI] [Google Scholar]
- 11.Manuprasad B.K., Murthy V.S., Shashikanth S., Rakshith D., Satish S., Raveesha K.A. Design, Synthesis and antibacterial activity of novel 1,3-thiazolidine purine nucleosides. Der Pharm. Chem. 2011;3:45–55. [Google Scholar]
- 12.Trávníček Z., Maloň M., Šindelář Z., Doležal K., Rolčík J., Kryštof V., Strnad M., Marek J. Preparation, Physicochemical properties and biological activity of copper(II) complexes with 6-(2-chlorobenzylamino)purine (HL1) or 6-(3-chlorobenzylamino)purine (HL2). The single-crystal X-ray structure of. [Cu(H+L2)2Cl3]Cl∙2H2O. J. Inorg. Biochem. 2001;84:23–32. doi: 10.1016/S0162-0134(00)00218-X. [DOI] [PubMed] [Google Scholar]
- 13.Colacio E., Cuesta R., Moreno J.M. Copper(I)-purine-phosphine complexes. Syntheses and molecular structures of [Cu2(µ-HL1)2(µ-dppm)(η1-dppm)2] and {[Cu3(µ3-Cl)2(µ-dppm)3][Cu(HL2)2]} (H2L1 = 8-Mercaptotheophylline and H2L2 = 8-Ethyl-3-methylxanthine) Inorg. Chem. 1997;36:1084–1087. doi: 10.1021/ic960928l. [DOI] [PubMed] [Google Scholar]
- 14.Lewis W.G., Magallon F.G., Fokin V.V., Finn M.G. Discovery and characterization of catalysts for azide-alkyne cycloaddition by fluorescence quenching. J. Am. Chem. Soc. 2004;126:9152–9153. doi: 10.1021/ja048425z. [DOI] [PubMed] [Google Scholar]
- 15.Hassane F.S., Frisch B., Schuber F. Targeted liposomes: convenient coupling of ligands to preformed vesicles using “click chemistry”. Bioconjug. Chem. 2006;17:849–854. doi: 10.1021/bc050308l. [DOI] [PubMed] [Google Scholar]
- 16.Gill H.S., Marik J. Preparation of 18F-labeled peptides using the copper(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition. Nat. Protoc. 2011;6:1718–1725. doi: 10.1038/nprot.2011.390. [DOI] [PubMed] [Google Scholar]
- 17.Glaser M., Solbakken M., Turton D.R., Pettitt R., Barnett J., Arukwe J., Karlsen H., Cuthbertson A., Luthra S.K., Årstad E. Methods for 18F-labeling of RGD peptides: Comparison of aminooxy [18F]fluorobenzaldehyde condensation with ‘click labeling’ using 2-[18F]fluoroethylazide, and S-alkylation with [18F]fluoropropanethiol. Amino Acids. 2009;37:717–724. doi: 10.1007/s00726-008-0200-0. [DOI] [PubMed] [Google Scholar]