

Abstract
Faced with the need to find new drugs for all kinds of diseases, science sees that Nature offers numerous classes of compounds showing an impressively high biological potential. Among those are the cyclodepsipeptides, hybrid structures composed of amino and hydroxy acids. In the past decades numerous cyclodepsipeptides have been isolated and their potential as drugs has been studied extensively. For several cyclodepsipeptides total syntheses both in solution and on solid-phase have been established, allowing the production of combinatorial libraries. In addition, the biosynthesis of specific cyclodepsipeptides has been elucidated and used for the chemoenzymatic preparation of nonnatural analogues. This review summarizes the recent literature on cyclic tetra- to decadepsipeptides, composed exclusively of α-amino- and α-hydroxy acids.
Keywords: natural product, cyclodepsipeptide, solid-phase synthesis, biological activity, total synthesis
Table of Contents
| 1. Introduction | 12369 |
| 2. Biosynthesis of Cyclodepsipeptides | 12370 |
| 3. Cyclotetradepsipeptides | 12371 |
| 3.1. AM-Toxins | 12371 |
| 4. Cyclopentadepsipeptides | 12374 |
| 4.1. Sansalvamide A, N-methylsansalvamide and neo-N-methylsansalvamide | 12374 |
| 4.2. Alternaramide | 12376 |
| 4.3. Zygosporamide | 12376 |
| 5. Cyclohexadepsipeptides | 12378 |
| 5.1. Enniatins and Beauvericin | 12378 |
| 5.1.1. Synthesis of Enniatins and Enniatin Derivatives | 12379 |
| 5.1.2. Biosynthesis of Enniatins and Beauvericin | 12381 |
| 5.2. Beauvenniatins | 12384 |
| 5.3. Hirsutellide A | 12385 |
| 5.4. Kutznerides | 12386 |
| 5.5. Monamycins | 12387 |
| 5.6. Himastatin | 12387 |
| 5.7. Paecilodepsipeptide A and Conoideocrellide A | 12390 |
| 5.8. Pullularins A-E | 12391 |
| 5.9. Hirsutatins A and B | 12392 |
| 6. Cycloheptadepsipeptides | 12392 |
| 6.1. HUN-7293 | 12392 |
| 7. Cyclooctadepsipeptides | 12395 |
| 7.1. Bassianolide | 12395 |
| 7.2. Verticlide | 12396 |
| 7.3. PF1022A and Emodepside | 12397 |
| 7.3.1. Syntheses of PF1022A | 12399 |
| 7.3.2. Synthesis of PF1022A-Analogues via Total Synthesis | 12401 |
| 7.3.3. PF1022A Analogues by Direct Derivatization of the Natural Product | 12404 |
| 7.3.4. Biosynthesis of PF1022A | 12406 |
| 7.3.5. Mode of Action of PF1022A and Emodepside | 12406 |
| 8. Cyclononadepsipeptides | 12408 |
| 8.1. BZR-cotoxins I-IV | 12408 |
| 8.2. Aureobasidins | 12409 |
| 9. Cyclodecadepsipeptides | 12410 |
| 9.1. Clavariopsin A and B | 12410 |
| 10. Summary | 12411 |
| Conflicts of Interest | 12411 |
| References | 12412 |
1. Introduction
Cyclodepsipeptides constitute a large family of peptide-related natural products consisting of hydroxy and amino acids linked by amide and ester bonds. The cyclodepsipeptides family can be subdivided into those with an irregular arrangement of ester groups (e.g., kutznerides) and those composed of regularly alternating amide and ester bonds (e.g., enniatins, PF1022A, valinomycine) [1]. Additional classes of cyclodepsipeptides contain β-hydroxy acids (e.g., theopapuamide, marformycins or neamphamide) or β-amino acids (e.g., destruxins) [2]. Cyclodepsipeptides show a broad spectrum of biological activities including antitumor, anthelmintic, insecticidal, antibiotic, antifungal, immunosuppressant, antiinflammatory and antimalarial activities. One plausible explanation for these manifold biological activities is, that the α-hydroxy acids mimic the corresponding α-amino acids. As a consequence depsipeptides are capable like natural peptides, consisting exclusively of ribosomal amino acids, to interact with numerous proteins. In addition, the cyclic nature and frequent N-methylation of amino acid residues confer resistance to hydrolyzing enzymes which results in enhanced oral bioavailability. Due to their unique structural and biological properties cyclodepsipeptides have emerged as promising lead structures for applications in crop protection (enniatins) as well as in human (aureobasidine) and veterinary medicine (PF1022). Emodepside®, a semisynthetic derivative of the regular cyclooctadepsipeptide PF1022A has been developed as a commercial anthelmintic.
The main source of cyclodepsipeptides are fermentations of various bacteria, actinomycetes and fungi which usually produce mixtures of structurally closely related structures [3,4]. Furthermore, numerous cyclodepsipeptides have been isolated from plants, algae, and cyanobacteria such as the cryptophycins, which are inhibitors of microtubule assembly with impressive in vivo activity against solid tumors [5]. In the recent past marine organisms came more and more into the focus of interest. For instance, solanamides A and B, isolated from the marine bacterium Photobacterium halotolerance, were found to inhibit virulence gene expression in the serious human pathogen, Staphylococcus aureus [6]. Halipeptins A-D, which show strong antimicrobial and antitumor activities, were isolated from the marine sponges Haliclona sp. and Leisosella cf. Arenifibrosa [7].
Solid-phase synthesis is the method of choice for the synthesis of peptides due to several remarkable advantages compared to solution synthesis as for instance (i) reagent and building block excesses to improve yields; (ii) simplified work-up procedures and (iii) parallel synthesis of peptides on fully automated synthesizers, to name only a few. In addition, the choice of a suitable carrier material such as the Kaiser oxime allows a cyclizative cleavage and reuse of the resin without the need for regeneration. The high-dilution conditions provided by the immense inner surface of solid-phase resins favor intramolecular cyclizations without the need for large solvent quantities [8]. However, the solid-phase synthesis of depsipeptides is by far not that developed as for peptides. The reasons are basically twofold. First, only a limited number of protecting groups for α-hydroxy acids and coupling reagents for ester formation is available. Second, the use of large excesses of hydroxy acids in the coupling reaction is hampered by their high prices and limited availability compared to ribosomal amino acids.
This review focuses on cyclic tetra- to decadepsipeptides, composed exclusively of α-amino- and α-hydroxycarboxlic acids. For other classes of depsipeptides the reader is referred to more specific literature [9,10,11,12].
2. Biosynthesis of Cyclodepsipeptides
In the living cell cyclodepsipeptides are synthesized by giant multi-domain nonribosomal peptide synthetases (NRPs), following the so-called thiol template mechanism which features the domain organization C1-A1-T1-C2-A2-MT-T2a-T2b (Figure 1). The biosynthesis comprises several steps: (1) activation of the α-amino acid and the α-hydroxy acid as adenylates (A = adenylation domain) using ATP as the co-substrate; (2) capture of those activated substrates as the corresponding thioesters by a phosphopantetheine linker (T = thiolation domain); (3) if necessary, N-methylation of amino acids (M domain); (4) condensation of the two substrates, covalently bound to the preceding T domain, forming an enzyme-bound intermediate (C = condensation domain) and (5) final release of the cyclodepsipeptide [13]. While bacterial NRPs use a thioesterase domain (TE) to perform the cyclization, NRPs of fungi terminate with a condensation-like domain (CT domain) [14]. A C-terminal reductase domain (R domain) catalyzes the reductive release of the depsipeptide from the synthetase [15]. Several modifications of the residues such as N-methylation, oxidation/reduction (Ox and KR domains), epimerization (E domains) and heterocycle formation (Cy cyclization domains) are performed on specialized domains of those peptide synthetases. In this way dipeptidol monomers are synthesized, which undergo head-to-tail oligmerization forming the desired regular, linear depsipeptide [16]. For instance, bassianolide, a regular cyclooctadepsipeptide, is synthesized as a cyclic tetramer of the dipeptide d-hydroxyisovalerate/N-methyl-l-leucine as a linear precursor which is finally cyclized on a C-domain [17].
Figure 1.

Biosynthesis of NRPs producing cyclodepsipeptides.
New, unnatural cyclodepsipeptide analogues can be made available by adding specific, unnatural amino and hydroxy acids either in vitro to the multi-domain peptide synthetases or in vivo to the producing strain of the microorganism [18].
3. Cyclotetradepsipeptides
3.1. AM-Toxins
AM-toxins (Figure 2), producued by Alternaria alternate, are host-specific phytotoxins which cause the spot disease on apple leaves of susceptible cultivars such as Indo and Starking Delicious [19]. AM-toxins consist of l-2-hydroxy-3-methylbutanoic acid (l-Hmb), l-alanine (l-Ala), a (para-substituted) l-2-amino-5-phenylpentanoic acid (l-App) and finally the highly reactive Michael-acceptor dehydroalanine which is supposed to be a major cause of the phytotoxic effects of AM-toxins.
Figure 2.
Structures of AM-toxins I-III (1–3).

l-Amp = l-2-Amino-5-(p-methoxyphenyl)pentanoic acid; l-App = l-2-amino-5-phenylpentanoic acid; l-Ahp = l-2-amino-5-(p-hydroxyphenyl)pentanoic acid.
To date, several total syntheses of AM-toxins I-III (1–3) were described [20,21]. In 1989 the Waki group presented a total synthesis of AM-toxin II (2) and [l-Phe3]AM-toxin II (8), an analogue of 2, in which l-2-amino-5-phenylpentanoic acid was replaced for a l-Phe (Scheme 1) [22]. The crucial step of this synthesis was the formation of the dehydroalanine residue which was established in the final step after deprotection using the Miller method (EDCI (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide) and CuCl) for the formal elimination of water.
Scheme 1.
Synthesis of 2 and its derivative 8 described by Waki et al.

Reagents and conditions: (a) Pd black, rt, 5 h; (b) 6 M HCl, dioxane, rt, 3 h; (c) Boc-l-Ser(Bzl)-ONSu, Et3N, DMF, 0 °C, 1 h; (d) HONSu (N-hydroxysuccinimide), EDCI*HCl (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide*hydrochloride), DMF, 4 °C, 24 h; (e) TFA, 0 °C, 30 min; (f) pyridine, DMF, rt, 2 d; (g) Pd black, HCOOH, rt, 2 h; (h) CuCl, EDC*HCl, reflux, 4 d.
In 1996 Shirahama et al., established another total synthesis of AM-toxin I (1, also called alternariolide) [23]. Here, d-β-phenylselenoalanine was used as precursor for dehydroalanine. Again, the double-bond was established in the final step but now by an oxidative elimination of the phenylselenyl group with anhydrous tert-butylhydroperoxide (TBHP) in CH2Cl2-TFE (5:1) in a yield of 87%. Miyashita and coworkers established the first solid-phase synthesis of AM-toxins on Wang resin [24]. The precursor d-Dap (d-2,3-diaminopropanoic acid) was converted into the dehydro amino acid by a Hofmann degradation with CH3I/KHCO3 in ethyl acetate. In 2001 Horikawa published an elegant solid-phase synthesis with selenocysteine as a central building block (Scheme 2). The role of selenocysteine was twofold: It was used as precursor for the dehydroalanine and as a linker to the resin. This strategy allowed the formation of the double bond in the final cleavage step [25].
Scheme 2.
Solid-phase synthesis of AM-toxin II (2) by Horikawa.

Reagents and conditions: (a) o-NO2-C6H4-SeCN, Bu3P/pyridine, rt, 2 h (83%); (b) Zn, AcOH/THF, rt, 2 h (82%); (c) succinic anhydride, DIPEA/CH2Cl2, rt, 3 h, (81%); (d) Gly-Wang resin, HATU, DIPEA/DMF, vortex, rt, overnight, (98% in Kaiser test); (e) Pd(PPh3)4, dimedone/THF, vortex, rt, 3 h; (f) NaNO2/AcOH aq., 30 min, (76%); (g) Cs2CO3, then allyl bromide, rt, overnight, (65%); (h) Boc-Ala, DCC, DMAP/DMF, rt, overnight (73%); (i) cat. Pd(PPh3)4, piperidine/THF, rt, 2 h, (93%); (j) l-App-OAll, HATU/DIPEA/DMF, rt, 6 h (88%); (k) 50% TFA/CH2Cl2, rt, 1 h (99%); (l) HATU, DIPEA/DMF, vortex, rt, overnight; (m) cat. Pd(PPh3)4, dimedone/THF, vortex, rt, 2 h; (n) 40% piperidine/DMF, vortex, rt, 1 h; (o) FDPP (pentafluorophenyl diphenylphosphinate), DIPEA/DMF, vortex, rt, 2 d; (p) TBHP (tert-butyl hydroperoxide)/CH2Cl2-TFE, vortex, rt, 7 h.
To evaluate the effect of substitution of an amide bond for an ester bond Konzone et al., synthesized a lactam analogue of AM-toxin I containing l-Hmb instead of l-Val [26]. Necrotic activity measurements on apple leaf however, indicated a considerable loss of phytotoxicity (0.1 µg/mL).
4. Cyclopentadepsipeptides
4.1. Sansalvamide A, N-methylsansalvamide and neo-N-methylsansalvamide
Sansalvamide (San A, 15) has been isolated from the organic extracts of the mycelium of a Fusarium species collected from the surface of the seagrass Halodule wrightii (Figure 3). Sansalvamide A consists of four hydrophobic α-amino acids (l-Phe, 2 l-Leu, l-Val) and one hydrophobic α-hydroxy acid (l-2-hydroxy-4-methylpentanoic acid) [27]. It exhibits cytotoxic activity against colon and melanoma cancer cell lines showing an in vitro IC50 value of 9.8 µg/mL towards HCT-116 colon carcinoma. Moreover IC50 values of 3.5 and 5.9 µg/mL were measured for the cancer cell-line COLO 205 and melanoma cell-line SK-MEL-2 [28].
Figure 3.

Structures of sansalvamides 15–17.
A N-methylated analogue of sansalvamide A, N-methylsansalvamide (16), was isolated by Cueto et al., from the extracts of a cultured marine fungus, strain CNL-619, collected in the US Virgin Islands. N-methylsansalvamide shows weak in vitro cytotoxicity in the NCI human tumor cell line screen (GI50 8.3 µM) [29].
Another analogue of sansalvamide A (15), neo-N-methylsansalvamide (17) was found in extracts from Fusarium solani KCCM90040, isolated from infected potatoes in Korea. Neo-N-methylsansalvamide (17) differs from cyclodepsipeptide 15 only by a different order of the identical residues. Neo-N-methylsansalvamide expressed in vitro cytotoxic effects on different human cancer cell lines. The EC50 values of neo-N-sansalvamide against the four human cancer cell lines A549, SK-OV-3, SK-MEL-2 and MES-SA were 10.7 ± 0.15, 11.2 ± 1.23, 10.02 ± 0.53, and 14.0 ± 0.74 µM, respectively [30].
A straightforward solid-phase synthesis was established by Silverman in 2000 [31]. The use of an arylsilane-based traceless linker allowed a synthesis in both N- and C-terminal direction (Scheme 3). This strategy was used to prepare sansalvamide analogue libraries. However, on solid-support only amide bonds were established by connecting a didepsipeptide building-block prepared in solution before. The final macrocyclization after removal of the protecting groups was accomplished with the coupling system HBTU (2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate), DIPEA (N,N-diisopropylethylamine) and NMP (N-methyl-2-pyrrolidone). Sansalvamide A (15) was obtained in a 10-step total synthesis with an overall yield of 67% and a purity of ˃95%. Remarkably, no high-dilution conditions were needed for the formation of the macrocycle. In addition, Silverman developed an alternative solid-phase strategy in which the ester-bond formation was performed directly on the support. This improved the efficiency of the synthesis considerably and allowed the automated synthesis of sansalvamide analogues. Protolytic cleavage of the product from the arylsilyl traceless linker was accomplished under acidic conditions with 50% TFA/CH2Cl2.
Scheme 3.
Solid-phase synthesis of 15 by Silverman and coworkers.

Reagents and conditions: (a) LiOH, THF/H2O (7:1), rt, 16 h; (b) HBTU, DIPEA, NMP, 16 h; (c) 2% thioanisole and 50% TFA in CH2Cl2, 15 min; (d) Fmoc-Leu-OH, HBTU, DIPEA, NMP, 6 h; (e) 20% piperidine in DMF 40 min; (f) Fmoc-Val-OH, HBTU, DIPEA, NMP, 6 h; (g) CHCl3/AcOH/NMM, Pd(PPh3)4, 3 h; (h) HBTU, DIPEA, NMP, 16 h; (i) 2% thioanisole and 50% TFA in CH2Cl2, 36 h; (j) allyl bromide, K2CO3, acetone, 48 h (95%); (k) Boc-Leu-OH, DMAP (cat.), DCC, CH2Cl2, 48 h (63%); (l) 50% TFA in CH2Cl2, 5 min; (m) O-allyl-Leu, HBTU, DIPEA, NMP, 16 h; (n) Fmoc-Leu-OH, HBTU, DIPEA, NMP, 6 h; (o) MSNT (1-(mesitylene-2-sulfonyl)-3-nitro-1,2,4-triazole), NMI (1-methylimidazole), CH2Cl2, 16 h.
It is worthwhile to note that the all-amide analogue of sansalvamide A (15), san A-amide, was shown to be 10-fold more active (IC50 0.98 µg/mL) in a cell-based cytotoxicity assay against HCT-116 human colon compared to the natural product 15 [32]. In the recent past, mainly the groups of Silverman and McAlpine prepared more than 100 sansalvamide A analogues, both on solid-phase and in solution. Some of those showed excellent chemotherapeutic activities [33,34,35,36,37,38,39].
4.2. Alternaramide
Alternaramide (25), composed of two d-Phe, two l-Pro and one l-Hiv (α-l-hydroxyisovaleric acid), was isolated from the fungus Alternaria sp. SF-5016 (Figure 4). Weak antibiotic activities against Bacillus subtilius and Staphylococcus aureus were reported [40]. A single total synthesis has been published to date [41].The residues involved in the macrocycle formation were found to be absolutely critical. A macrolactonization between the residues l-Pro and l-Hiv with excess of PyBOP and DIPEA gave a cyclization yield of only 5%. A macrolactamization between l-Hiv and l-Phe with the same reagent system afforded alternaramide (25) in 48% yield.
Figure 4.
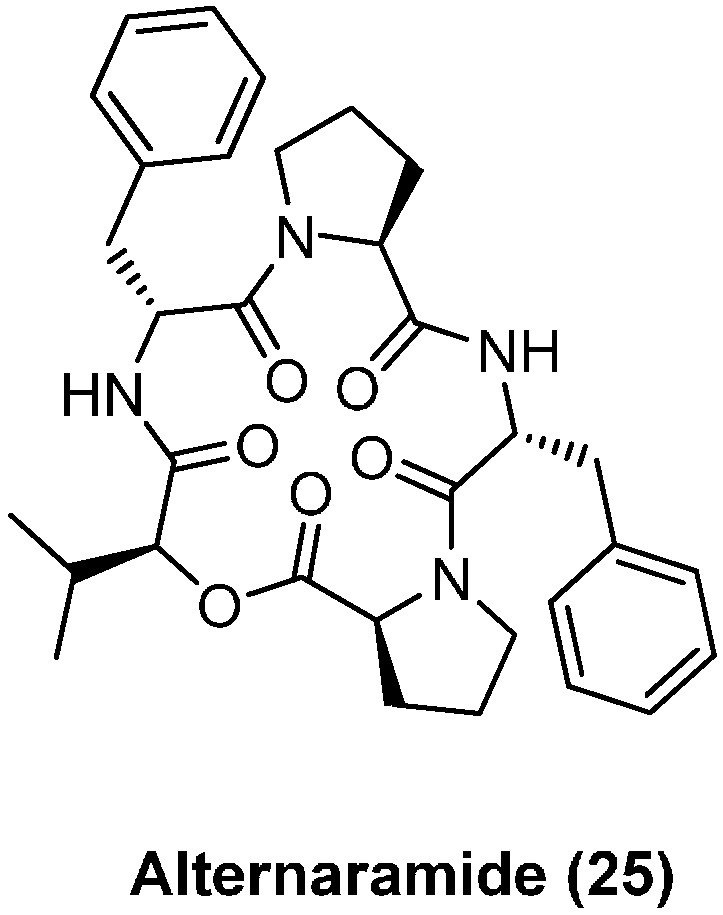
Structure of alternaramide (25).
4.3. Zygosporamide
Zygosporamide (26, Figure 5) is produced by the marine-derived fungus, Zygosporium masonii and is known as a potent and selective cytotoxic against SF-268 (GI50 6.5 nM) and RXF 393 (GI50 < 5.0 nM) cell lines [42].
Figure 5.
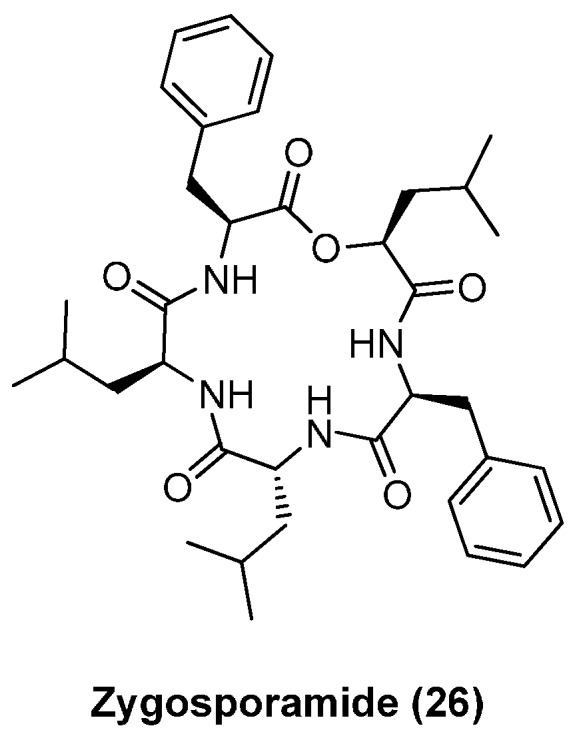
Structure of zygosporamide (26).
In order to establish structure-activity and structure-selectivity relationships, Ma developed a synthesis of zygosporamide (26) and prepared a small library of analogues (Scheme 4) [43]. The key step of that synthesis is the coupling of fragments 28 and 30 using the Yamaguchi esterification procedure. The HATU mediated macrocylization under high-dilution conditions gave zygosporamide in a yield of 50%.
Scheme 4.
Synthesis of zygosporamide.

Reagents and conditions: (a) l-Leu-OMe*HCl, EDC, HOBt, DIPEA, CH2Cl2 (92%); (b) aq. LiOH, THF, MeOH; (c) l-Phe-OMe*HCl, EDC, HOBt, DIPEA (95%); (d) l-Phe-OBn, BOP, DIPEA, MeCN (90%); (e) TCBC (2,4,6-trichlorobenzoyl chloride, Yamaguchi reagent), DIPEA, DMAP, toluene (80%); (f) Pd/C, H2, MeOH; (g) HATU, DIPEA, DMF/CH2Cl2, (1:4), 0.002 M (50%).
An alanine-scan and replacement of the hydroxy acid by the corresponding amino acid Leu revealed differing structure-activity relationships of cytotoxicity depending on the specific cancer cell line. (Figure 6, Table 1).
Figure 6.

Structures of zygosporamide analogues.
Table 1.
Biological activities of zygosporamide analogues.
| Compound | IC50 (µM) | ||||
|---|---|---|---|---|---|
| SF-268 | SF-295 | A549 | MDA-MB-231 | HCT-116 | |
| Zygosporamide (26) | 0.0065 | 15 | 7.4 | 8.5 | 11.5 |
| 31 | ~35 | 19.7 ± 8.8 | ~31 | 7.5 ± 6.3 | >50 |
| 32 | 10.4 ± 1.5 | 8.7 ± 0.9 | >50 | 5.0 ± 1.8 | 2.1 ± 0.4 |
| 33 | >50 | >50 | >50 | >50 | >50 |
| 34 | 8.8 ± 1.5 | ~6 | >50 | 2.8 ± 2.1 | 2.7 ± 0.6 |
| 35 | ~31 | ~32 | >50 | >50 | 7.8 ± 6.0 |
| 36 | 2.05 ± 2.0 | 14.4 | 21.0 ± 5.9 | >50 | 1.9 ± 1.3 |
5. Cyclohexadepsipeptides
5.1. Enniatins and Beauvericin
The enniatins and beauvericin (60) are known for a long time as fungal mycotoxins mainly from Fursarium spp. but also from Verticillium hemipterigenum and Halosarpheia sp. Additionally, beauvericin was isolated from the hypocrealean entomopathogens Beuveria bassiana, Peacilomyces fumoso-roseus and P. tenuipes.
Enniatins and beauvericin (60) form a family of regular cyclohexadepsipeptides consisting of three hydroxy acids and three N-methyl amino acids in an alternating order. Enniatins were first described over 60 years ago. Up to date 29 different enniatins were isolated and characterized, 18 of those as single compounds, the other ones as mixtures of structurally closely related homologs (Figure 7). N-Me-Val, N-Me-Leu and N-Me-Leu are the common amino acids, hydroxyisovaleric acid (Hiv) is the typical hydroxy acid found in most enniatins [44].
Figure 7.

Structures of enniatins 37–59 and beauvericin (60).
A wide range of biological activities has been reported for enniatins and the related beauvericin (60) such as inhibition of the mammalian cholesterol acyl transferase, binding to the γ-aminobutyric acid receptor, activating calcium-sensitive cell apoptose pathways and antiproliferative activity against different human cancer cell lines [45,46]. Recently it was shown that beauvericin also inhibits migration of the metastatic prostate cancer (PC-3M) and breast cancer (MDA-MB-231) cells [47]. In general, due to their ionophoric properties, enniatins and beauvericin (60) cause non-specific rat cortical membrane disruption in vitro [48].
5.1.1. Synthesis of Enniatins and Enniatin Derivatives
The first synthesis of an enniatin (enniatin B, 40) was described by Shemyakin in 1963 [49]. In order to establish a flow-synthesis for enniatin 40, a remarkably optimized synthesis was published by Ley in 2012 (Scheme 5) [50]. The coupling reagents EDCI for ester bond formation and Ghosez’s reagent (1-chloro-N,N,2-trimethylpropenylamine) for the formation of amide bonds gave excellent yields. In particular, in the critical macrocyclization step under high-dilution conditions Ghosez’s reagent proved superior. The Ley synthesis provided enniatin B in nine steps with an overall yield of 36%.
Scheme 5.
Total synthesis of enniatin B (40) by Ley et al.

Reagents and conditions: (a) EDCI, DMAP, CH2Cl2 (82%); (b) H2, Pd/C, EtOAc (100%); (c) 4 M HCl/1,4-dioxane (100%); (d) Ghosez's reagent (1-chloro-N,N,2-trimethyl-1-propenylamine), DIPEA, CH2Cl2 (91%); (e) 65, Ghosez's reagent, DIPEA, CH2Cl2, (86%); (f) H2, Pd/C, EtOAc (88%); (g) Ghosez's reagent, DIPEA, 0.005 M CH2Cl2 (56%).
In order to get more information on the potential of enniatins as lead structures for new drugs in the field of crop protection, medicine and animal health numerous analogues were synthesized during the past 20 years. In several cases improved or different biological activities were found. For instance, the replacement of one N-methyl-(l)-isoleucine (N-Me-Ile) in enniatin A by an N-methyl-(l)-alanine (N-Me-Ala) caused a 10-fold higher activity against the parasitic nematode H. contortus in sheep. Impressive anthelmintic activities were also found for the morpholino-phenyllactic acid containing enniatin derivatives 70a, 70b, and 71 (Table 2) which were available by nitration of the phenyl ring, reduction of the nitro-derivative and subsequent reductive amination (Scheme 6) [51].
Table 2.
Anthelmintic activity of enniatin derivatives 68–71.
| Enniatin Derivative | Anthelminctic Activity against H. Contortus |
|---|---|
| 68 | 0.25 a/0 b |
| 69c (para) | 0.10/1 |
| 70a (ortho) | 0.05/3 |
| 70b (meta) | 0.05/3 |
| 70c (para) | 0.10/1 |
| 71 | 0.10/3 |
(a) Dose in mg test substance kg−1 body weight (sheep); (b) 0 = < 50% egg reduction, 1 = 50%–75% egg reduction, 2 = 75%–95% egg reduction, 3 = ˃95% egg reduction.
Scheme 6.
Derivatization of enniatin analogue 68.

Reagents and conditions: (a) fumic nitric acid, 10 °C, 1 h: (b) 20% Pd(OH)2, H2; (c) 1. 2,5-dihydrofuran, ozone gas, MeOH, CH2Cl2, −60 °C, 2. NaBH3CN, −50 °C, 10 min.
Based on the observation that the exchange of N-Me-Ile by N-Me-Phe in enniatin A improves the anthelmintic activity, enniatin derivatives 73, 74, 77 and 78 were prepared by hydrogenation (PtO2, H2, 4–5 bar) of the phenylalanine residue. Even those cyclohexyl-enniatins showed a good to excellent anthelmintic in vivo activity in sheep (Scheme 7) [52].
Scheme 7.
Structures and activities of cyclohexyl-analogues of enniatins.

(a) Dose in mg test substance kg−1 body weight (sheep); (b) 0 = < 50% egg reduction; 1 = 50%–75% egg reduction; 2 = 75%–95% egg reduction; 3 = > 95% egg reduction.
5.1.2. Biosynthesis of Enniatins and Beauvericin
The first cloned and characterized cyclodepsipeptide synthesase was the enniatin synthetase ESyn, a 347-kDa multienzyme. Compared to other NRPSs, Esyn possesses only two substrate activation modules, EA and EB. While EA is responsible for the activation of α-d-hydroxy acids, EB activates amino acids [53]. The N-methylation of the amino acid residues is performed by the N-methyltransferase domain located in module EB. The methyl group is transferred from S-adenosylmethionine (AdoMet) to the amino acid. The occurrence of numerous natural enniatins is an evidence of a high tolerance of cyclodepsipeptide synthetases for different amino and hydroxy acids. Zocher and coworkers observed distinct amino acid specificities of two different enniatin producers, Fusarum lateritium (enniatin B and B1) and Fusarium sambucinum (enniatin A) [46]. Esyn from Fusarum lateritium shows a high affinity to l-Val, whereas Fusarium sambucinum prefers l-Ileu. Based on earlier studies, in which N-methyl-l-valine could be replaced successfully by l-α-aminobutyric acid, l-norvaline and l-leucine [54] additional enniatin analogues have been synthesized recently, following two different strategies, namely small scale in vitro syntheses using isolated cyclodepsipeptide synthetases and in vivo syntheses by feeding the enniatin producing fungi Fusarium scripi and Fursarium sambucinum [45]. In particular, the in vitro synthesis was intensively studied by Süssmuth in the recent past [55]. Albeit the amino acid substrates for Esyn are restricted to aliphatic, sterically not demanding side chains, several new enniatin analogues were made accessible via this elegant in vitro synthesis including N-methyl-l-alanine, N-methyl-l-serine, N-methyl-l-threonine and others. Remarkably, the substrate tolerance of Esyn for hydroxy acids is significantly broader. While the limitation to aliphatic residues still applies, bulky alkyl, halogen or hydroxy functionalized side-chains and even propargyl substituents are accepted. Using this chemoenzymatic approach, Süssmuth succeeded in preparing an enniatin B (40) library (Figure 8) containing a set of different α-d-hydroxy acid derivatives 79–97. However, the minute amounts of enniatin derivatives available by in vitro synthesis prevented thorough studies of biological activities up to now. Additional enniatin derivatives were obtained by simple adding of unusual substrates such as d-2-hydroxybuturic acid, d-lactic acid, l-alanine, l-Abu (l-α-aminobutyric acid) l-serine or l-threonine to the producing Fusarium strains [45].
Figure 8.

In vitro synthesis of hydroxy acid derivatives of 40.
Beauvericin synthetase (BbBEAS) consists of a single polypeptide chain with a molecular mass of about 351 kD. Analogous to enniatin biosynthesis, beauvericin (60) is also produced by a thiol template mechanism [56,57]. However, the two depsipeptide synthetases differ in their substrate selectivities. Beauvericin synthetase preferably accepts N-methyl-l-phenylalanine and some other aliphatic hydrophobic amino acids. The efficiency of incorporation into the cyclodepsipeptide framework decreases with the length of the side chain: N-methyl-l-phenylalanine was easily replaced by ortho-, meta- and para-fluoro substituted phenylalanine derivatives, as well as by N-methyl-l-leucine, N-methyl-l-norleucine and N-methyl-l-isoleucine residues.
In contrast, N-methyl-l-valine a constituent of enniatin B is not accepted. Molnar published six new beauvericin (104–109) analogues prepared via a precursor-directed biosynthesis with the Beauveria bassiana ATCC 7159 strain (Figure 9) [58]. With dl-2-hexabutyric acid (Hbu) and dl-3-fluorophenylalanine as substrates six further beauvericin derivatives 98–103 were obtained by Nilanonta et al., from the fungal strain Peacilomyces tenuipes BCC 1614 [59].
Figure 9.

Derivatives 98–109 of beauvericin via biosynthesis.
In 2012 Süssmuth, for the first time, used recombinant beauvericin synthetase BbBEAS isolated from E. coli to generate new beauvericin analogues. In a subsequent whole cell in vivo approach the production of beauvericins by mutational biosynthesis using a KIVR knockout strain of B. bassiana was studied [60]. Similar to Esyn also BbBEAS shows a broader substrate tolerance for hydroxy acids as exemplified by cyclodepsipeptides 109–119 (Figure 10).
Figure 10.

In vitro and in vivo synthesis of beauvericin analogues 109–119.
5.2. Beauvenniatins
A few publications describe hybrid structures between the aliphatic, enniatin-type and aromatic, beauvericin-type cyclohexadepsipeptides, the so-called beauvenniatins (Figure 11, Table 3). Beauvenniatins A–E (127–131) were isolated from Acremonium sp., strain BCC 28424 and beauvenniatins F, G1, G2, G3, H1, H2, and H3 (120–126) from strain BCC 2629 [61,62].
Figure 11.

Structures of beauvenniatins 120–131.
Table 3.
Biological activity of beauvenniatins 120–131.
| Beauvenniatin | Cytotoxicity (IC50) | Antimalaria (IC50) | Antituberculosis (IC50) | ||
|---|---|---|---|---|---|
| NCI-H187 g | KB h | Vero i | |||
| 127 | 1.0 a | 1.3 a | 2.2 a | 3.0 c | 3.13 e |
| 130 | 0.92 a | 1.6 a | 1.3 a | 3.0 c | 3.13 e |
| 131 | 6.6 a | 14 a | 7.0 a | 3.4 c | ˃50 e |
| 128 | ˃50 a | ˃50 a | ˃50 a | ˃10 c | ˃50 e |
| 129 | 7.6 a | 9.0 a | 8.3 a | 2.9 c | 25 e |
| 120 | 2.29 ± 1.26 b | 1.05 ± 0.05 b | 5.5 b | 3.8 ± 0.1 d | 1.07 f |
| 121–123 | 1.23 ± 0.49 b | 1.06 ± 0.06 b | 4.1 b | 3.9 ± 0.4 d | 2.18 f |
| 124–126 | 1.45 ± 0.38 b | 1.15 ± 0.11 b | 1.9 b | 3.6 ± 0.9 d | 4.45 f |
(a) IC50 (µg/mL); (b) IC50 (µM); (c) antimalarial activity against P. falciparium K1, showed in IC50 (µg/mL); (d) antimalarial activity against P. falciparium K1, showed in IC50 (µM); (e) antituberculosis activity against M. tuberculosis H37Ra, showed in IC50 (µg/mL); (f) antituberculosis activity against M. tuberculosis H37Ra, showed in IC50 (µg/mL); (g) human small-cell lung cancer; (h) oral human epidermoid carcinoma; (i) African green monkey kidney fibroblasts.
5.3. Hirsutellide A
Hirsutellide A (132, Figure 12), another 18-membered cyclodepsipeptide, was isolated from the cell extracts of Hirsutella kobayasii BCC 1660 [63]. It exhibits antimycobacterial activity (MIC 6–12 µg/mL; activity against Mycobacterium tuberculosis) and also weak in vitro antimalarial activity (IC50 2.8 µg/mL; activity against Plasmodium falciparum). However no cytotoxic effects were found against Vero cells at 50 µg/mL . A conventional solution synthesis, using standard reagents for ester (DCC, (N,N'-dicyclohexylcarbodiimide) and amide bond (HATU) formation was developed by Xu [64,65]. In fact, the macrocylization reaction turned out problematic. BOP-Cl (bis-(2-oxo-3-oxazolidinyl)-phosphinic chloride) afforded only a low yield of 15% of the cyclic depsipeptide, FDDP a slightly improved yield of 22%.
Figure 12.

Structure of hirsutellide A and its stereoisomer.
A drastically improved cyclization yield (61%) was obtained for the stereoisomer 133 of hirustellide A with AgBF4 as cyclization reagent [66]. The authors suggest that the silver ion acts as a template, to which the carbonyl and the oxygen or amino groups coordinate. This arrangement in close spatial neighborhood facilitates then the difficult cyclization.
5.4. Kutznerides
In 2006 Pohanka and Vasiliauskas isolated a novel class of cyclohexadepsipeptides from the actinomycete Kutzneria sp. 744. The kutznerides 134–142 inhibit the growth of the pathogenic fungi Pythium undalatum, Ceratobasidium bicorne and Fusarium avenaceum and thus illustrate attractive lead structures for novel antifungals (Figure 13) [67,68]. These compounds 134–142 represent remarkable structures as they do not contain a single ribosomal amino acid. With respect to the tricyclic tryptophane analogue, the kutznerides pose a formidable challenge for a total synthesis.
Figure 13.
Structure of kutznerides 134–142.

MecPGly = 2-(1-methylcyclopropyl)-d-glycine, OHdiMeBu = (S)-2-hydroxy-3,3-dimethylbutanoic acid, MeSer = O-methyl-l-serine, OHGlu = 3-hydroxy-d-glutamic acid, diCPlC = (2S,3aR,8aS)-6,7-dichloro-3a-hydroxypyrrolidino[2,3-b]indole-2-carboxylic acid.
The kutznerides were tested against diverse pathogens shown in Table 4. The most potent compounds were the trichloro kutzernerides 2 (135) and 8 (141), with promising MIC values against Erwina carotovora and Staphylococcus aureus. In 2007 Walsh identified the biosynthesis gene cluster comprising six nonribosomal peptide synthetase (NRPS) modules distributed over three proteins and a variety of tailoring enzymes including halogenases [69]. The study provides an interesting insight into the biochemical transformations involved in the synthesis of the unique residues of the kutznerides.
Table 4.
MIC values of kutznerides against various pathogens.
| Kutzneride | MIC values (µM) of Pathogens | ||
|---|---|---|---|
| Drechslera sorokiniana (agricultural fungus) | Erwina carotovora (agricultural bacterium) | Staphylococcus aureus (human bacterium) | |
| 1 (134) | 230 | 60 | 12 |
| 2 (135) | 110 | 6 | 6 |
| 3 (136) | 260 | 12 | 9 |
| 4 (137) | ˃590 a | 120 | 140 |
| 5 (138) | ˃240 a | 120 | 120 |
| 6 (139) | ˃400 a | ˃230 a | ˃230 a |
| 7 (140) | ˃420 a | 180 | 120 |
| 8 (141) | 230 | 6 | 6 |
| 9 (142) | ˃230 | 230 | 60 |
(a) No inhibition at the indicated concentration.
5.5. Monamycins
The monamycins 143–155 were isolated from Streptomyces jamaicensis more than fifty years ago (Figure 14) [70]. In 1970 Hall described potassium, caesium and rubidium complexes of the monamycins 143–155 and attributed their antibacterial effects against Staphylococcus aureus (IC50 0.5–2.0 µg/mL) to their ionophoric properties causing lysis of the bacterial cell [71]. This class of cyclodepsipeptides attracted much interest when pronounced antibiotic effects against resistant strains of Gram-positive bacteria were found [72]. Bromomonamycin analogues of chloromonamycins G1, G2, G3, H1, H2, and I were made available by cultivating the fungus in a chloride-free medium to which NaBr was added as halogen source [73]. Unfortunately, the bromomonamycins showed only a reduced antibacterial activity. The monamycins contain the unique building block (3S,5S)-5-hydroxypiperazic acid which was first synthesized by Hale via a chiral-pool approach starting from d-mannitol [72].
Figure 14.

Structures of different monamycins 143–155.
5.6. Himastatin
Himastatin (156) is a highly interesting cyclodepsipeptide due to its strong antimicrobial activity in particular against Gram-positive bacteria and its unique symmetric structure (Figure 15, Table 5) [74]. Himastatin has been found in a strain of Strepthomyces hygroscopicus (ATCC 53653) collected in the state of Himachal Pradesh in India. Himastatin represents a dimeric cyclohexadepsipeptide consisting of two tricyclic tryptophan derivatives that are connected by a biphenyl bridge. With (3R,5R)-5-hydroxypiperazic acid a second unusual building is incorporated in the cyclodepsipeptide framework [75,76,77].
Figure 15.

Structure of himastatin (156).
Table 5.
Antimicrobial activities of 156 [71].
| Organism | MIC (µg/mL) | Organism | MIC (µg/mL) |
|---|---|---|---|
| E. faecalis A20688 | 0.5 | E. coli A20697 | ˃500 |
| E. faecalis A25707 | 0.5 | E. coli A9751 | 2 |
| E. faecalis A25707 | 0.25 | Klebsiella pneumoniae A9664 | ˃500 |
| S.aureus A9537 | 0.5 | K. pneumonia A20468 | ˃500 |
| S. aures A20698 | 1 | Proteus vulgaris A21559 | ˃500 |
| S. aureus A24407 | 1 | Pseudonomas aeruginosa A9843 | ˃500 |
| Bacillus subtilis A9506-A | 2 | P. aeruginosa A20235) | ˃500 |
| Escherichia coli A15119 | ˃500 | P. aeruginosa A20235 | ˃500 |
The structure of himastatin (156) was mainly deduced by degradation experiments. Based on NMR studies Bristol Myers scientists attributed a trans-arrangement of the carboxamide and the OH-group in the tryptophan-biphenyl cleavage product (Figure 16) [78]. However, in a seminal total synthesis of himastatin Danishefsky proved the trans-arrangement to be wrong [79].
Figure 16.

Proposed (157) and proven (158) structure of the central himastatin degradation product.
In a second total synthesis Danishefsky obtained the correct himastatin stereoisomer which was identical with natural himastatin in all spectroscopic properties [80]. This elegant synthesis was based on two separate fragments, a pentadepsipeptide acid (165, Scheme 8) and a pyrroloindoline dimer (171, Scheme 9) which were coupled only in the final stage, establishing the dimeric structure of himastatin in a one-pot reaction.
Scheme 8.
Total synthesis of himastatin: fragment 165.

Reagents and conditions: (a) Fmoc-l-Leu, EDCl, DMAP, CH2Cl2; (b) piperidine, CH3CN, 76% (over 2 steps; (c) 161, HATU, HOAt, collidine, CH2Cl2, 95%; (d) 163, collidine, CH2Cl2; (e) piperidine, CH3CN, 96% (over 2 steps); (f) Troc-d-valine, IPCC, Et3N, DMAP, CH2Cl2, (g) ZnCl2, CH3NO2; (h) TBSOTf, 2,6-lutidine, CH2Cl2; (i) [Pd(PPh)3]4, PhSiH3, THF, 72% (over 3 steps).
Scheme 9.
Total synthesis of himastatin 156: final steps.

Reagents and conditions: (a) TrCl (trityl chloride), TEA, THF; (b) tert-butyl isourea, CH2Cl2, rt, 76% (over 2 steps); (c) DMDO, CH2Cl2, −78 °C; (d) HOAc, MeOH, CH2Cl2, rt, 76% (over 2 steps); (e) CbzCl, py, CH2Cl2; (f) TBSCl, DBU, DMF, rt (61%); (g) ICl, 2,6-di-tert-butylpyridine, CH2Cl2 (73%); (h) Me6Sn2, [Pd(PPh3)4], THF, 60 °C (87 °C); (i) 170, [Pd2dba3], Ph3As, DMF, 45 °C (83%); (j) TBAF, THF; (k) TMESCl, DBU, DMF (95%); (l) Pd/C, H2, EtOAc; (m)Fmoc-HOSu, py, CH2Cl2 (96%); (n) TESOTf, lutidine, CH2Cl2, 0 °C to rt; (o) allyl alcohol, DBAD, PH3P, THF (81%), (p) piperidine, CH3CN (74%); (q) HATU, HOAt, collidine, CH2Cl2, −10 °C to rt (60%); (r) 1. [Pd(PPh3)4], PhSiH3, THF; 2. Pb/Cd, NH4OAc, THF (56%); (s) HOAt, HATU, DIPEA, DMF; (t) TBAF, THF, HOAc (35%).
Isohimastatin (173), originally proposed to be himastatin, the monomers of himastatin and isohimastatins (174 and 175), obtained in the course of the total synthesis (Figure 17), were also tested against various Gram-positive bacteria but were found all inactive.
Figure 17.

Analogues of himastatin.
Inspired by its unique structure, Ju explored the biosynthesis of himastatin (156) [81]. Himastatin is biosynthesized by a nonribosomal peptide synthetase (NRP) assembly line, that requires prior- and post-oxidative modifications for the formation of the 3-hydroxy-d-Pip residue, the oxygenation of l-Trp and the subsequent symmetrical biaryl coupling. Ju found three cytochrome P450 enzymes responsible for the complete biosynthesis of himastatin: (i) HmtT transforms l-Trp to the tricyclic pyrroloindole unit; (ii) HmtN regio- and stereoselectively hydroxylates d-Pip; and (iii) HmtS catalyses the biaryl coupling.
5.7. Paecilodepsipeptide A and Conoideocrellide A
Very recently, the cyclohexadepsipeptide conoideocrellide A (177) was isolated from Conoideocrella tenuis BCC 18627, an insect pathogenic fungus together with its linear precursors conoideocrellides B-D (Figure 18) [82]. A structurally closely related cyclodepsipeptide, paecilodepsipeptide A (176), was isolated from Paecilomyces cinnamomeus BCC 9616, another insect pathogenic fungus. Paecilodepsipeptide A has activity against the malarial parasite Plasmodium falciparum K1 with an IC50 of 4.9 µM, and, furthermore, it shows cytotoxicity against two cancer cell lines, KB (IC50 = 5.9 µM) and BC (IC50 = 6.6 µM). Interestingly, only the cyclic depsipeptide showed biological activity, but not the linear precursors [83].
Figure 18.

Structure of paecilodepsipeptide A and conoideocrellide A.
In 2008 Yang and coworkers detailed a total synthesis of paecilodepsipeptide A (176) in solution (Scheme 10). The highest yield (72%) in the critical macrocylization reaction was obtained with HATU as coupling reagent under high-dilution conditions [84].
Scheme 10.
Total synthesis of paecilodepsipeptide A 176.

Reagents and conditions: (a) EDCl/HOBt/Et3N/CH2Cl2, 89%; (b) H2, Pd/C; (c) 179 EDCl/HOBt/Et3N/CH2Cl2, 86%; (d) EDCl/HOBt/Et3N/CH2Cl2, 90%; (e) H2, Pd/C MeOH; (f) EDCl/HOBt/Et3N/CH2Cl2, 76%; (g) 184, EDCl/HOBt/Et3N/CH2Cl2, 75%; (h) TBAF/THF; (i) HATU/DIPEA/DM, 72%.
5.8. Pullularins A-E
From the fungus Pullularia sp. BCC 8613 four cyclohexadepsipeptides, pullularins A–D (186–189) were isolated and characterized by Isaka in 2007 [85]. Pullularin A (186) (Figure 19) shows antimalarial, antiviral (herpes) and antitubercular activity. All pullularines share an unusual O-prenyl-l-tyrosine residue and a single ester bond in the cycle. In 2012 an additional cyclodepsipeptide, pullularin E (190) was published [86]. Pullularin E (190) is characterized by an O-isopentenyl substituted tyrosine instead of a O-dimethylallyl residue as found in the pullularins A–D (186–189).
Figure 19.

Structures of pullularins A-E (186–190).
5.9. Hirsutatins A and B
Hirsutatins A (191) and B (192, Figure 20) as well as the hirsutellones A-E were isolated from the insect pathogenic fungus Hirsutella nivea BCC 2594 [87,88]. While hirsutatin B 192 shows antimalarial activity against Plasmodium falciparum K1 (IC50 5.8 µg/mL) 191 was found inactive even at a concentration of 20 µg/mL. In contrast to the enniatins, in which the amino acids usually have the l- and the hydroxy acids the d-configuration, the hirsutatine building blocks all have the l-configuration.
Figure 20.

Structure of hirsutatin A (191) and B (192).
6. Cycloheptadepsipeptides
6.1. HUN-7293
Isolated first in 1992 from a fungal broth heptadepsipeptide HUN-7293 (193, Figure 21) has emerged as a potent lead structure for the treatment of autoimmune diseases and inflammatory disorders [89]. It acts as an inhibitor of inducible cell adhesion molecule expression (VCAM-1: IC50 1.0 nM, ICAM-1: IC50 24.0 nM, E-selectin: IC50 24.0 nM). Structurally HUN-7293 consists of six l-amino acids, four of which nonribosomal, and one d-α-hydroxy acid.
Figure 21.

Structure of HUN-7293 (193).
The first total synthesis of HUN-7293 (193) was described by Boger in 1999 [90]. Two strategies were envisaged (Scheme 11) for the critical macrocyclization. In the first route, a linear heptadepsipeptide was prepared, using a modified Mitsonobu reaction to form the ester bond in the final step. After simultaneous removal of the protecting groups under acidic conditions a macrolactamization with EDCI-HOAt afforded the desired product in a yield of 71%. The second route, a macrolactonization, however did not give any cyclodepsipeptide HUN-7293 neither with a Mitsunobu reaction nor with the Steglich esterification method.
Scheme 11.

Synthesis of HUN-7293 (193).
With an efficient total synthesis at hand, an aza-analogue of HUN-7293 (aza HUN-7293, 209) was prepared [91]. In contrast to the macrocyclization conditions used for the total synthesis of the natural product, the coupling system BOP ((benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexa-fluoro-phosphate)/DMAP was shown to be superior because epimerization was significantly reduced. An alternative method for preparing aza-HUN-7293 by a straightforward modification of the natural product was published by Schreiner and coworkers [92]. The authors successfully described the synthesis of aza-HUN-7293 (209) by a ring opening reaction and subsequent substitution of the d-hydroxy acid by the corresponding d-amino acid (Scheme 12).
Scheme 12.

Synthesis of aza-Hun-7293 (209) by modification of the natural product.
In order to establish structure-activity relationships Boger used solution-phase parallel synthesis to prepare HUN-7293 analogues, including an alanine and a N-methyl deletion scan [90]. However, to varying degrees all analogues showed reduced activities against VCAM-1 and ICAM-1 (Figure 22).
Figure 22.

HUN-7293 analogues and their biological activities.
7. Cyclooctadepsipeptides
7.1. Bassianolide
Bassianolide (222, Figure 23) was isolated from Beauveria bassiana, Lecanicilium sp. (formerly Verticillium lecanii) and from the wood-decaying fungus Xylaria sp. BCC1067 [93]. Bassianolide belongs to the regular 24-membered cyclooctadepsipeptides and consists of four N-methyl-l-leucines and four d-hydroxyisovaleric acids. Manifold biological activities have been reported for bassianolide, among those insecticidal activity and cytotoxicity against different cancer cell lines. An impressive anthelmintic activity was observed with a worm (Ascaridia galli in chicken) reduction of ˃90% at a dosage of ≥10 mg/kg [94,95].
Figure 23.
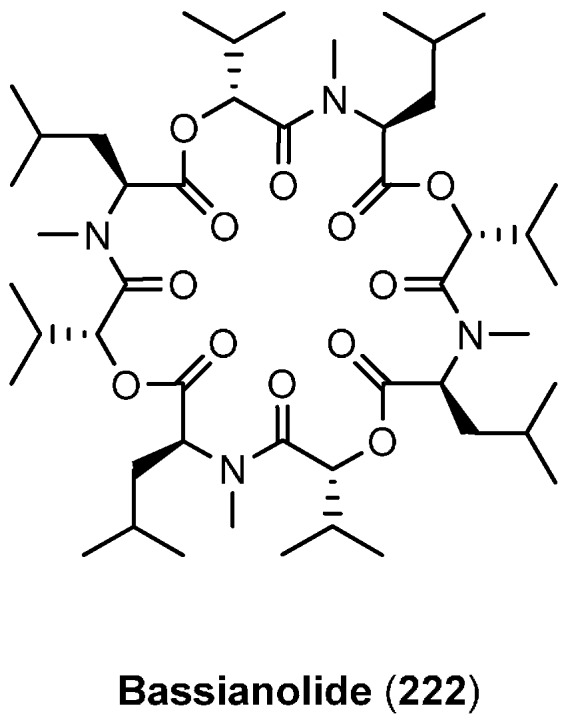
Structure of bassianolide (222).
Bassianolide (222) has been synthesized in solution with standard methodology by successive couplings of Cbz-l-N-methylleucine with t-butyl-d-hydroxyisovalerate, protecting group removal and subsequent macrocyclization under high-dilution conditions [96].
7.2. Verticlide
Verticilide (A1, 223), another cyclooctadepsipeptide is produced by Verticillium sp. FKI-1033 (Figure 24). Similar to bassianolide verticilide is highly symmetrical and composed of four N-methyl-l-alanines and four d-hydroxyheptanoic acids. Verticilide (223) has been shown to bind selectively to the insect ryanodine receptor, a major target for modern insecticides, in the low micromolar range (IC50 4.2 µM) [97]. Ryanodine receptors belong to a group of calcium channels found in skeletal muscle, smooth muscle and heart muscle cells. In contrast to mammalians, which have three types of ryanodine receptors (RyR), insects have only a single RyR. Several state of the art synthetic insecticides such as the anthranilic diamides bind to insect ryanodine receptors with nanomolar affinities. In addition to verticilide A1, three related cyclodepsipeptides, verticilides A2 (224), A3 (225) and the cyclohexadepsipeptide B1 (226) (Figure 25), were isolated from the fungal strain FKI-2679 by Oshiro in 2012 [98]. In addition to their ryanodine-receptor binding all verticilides inhibit efficiently cholesterol acetyltransferases ACAT1 and ACAT2 with IC50 values from 0.23 to 11 µM (Table 6).
Figure 24.

Structure of verticilide (223).
Figure 25.

Structures of verticilide analogues 224–226.
Table 6.
Inhibition of cholesterol acetyltransferases ACAT1 and ACAT2.
| Verticilde | IC50 (µM) | |
|---|---|---|
| ACAT1 | ACAT2 | |
| A1 (223) | 2.5 | 0.23 |
| A2 (224) | 4.8 | 0.55 |
| A3 (225) | 3.5 | 0.36 |
| B1 (226) | 11 | 1.3 |
A solution synthesis of verticilide (223) based on a Boc/benzyl ester protecting group strategy was published in 2006 by Omura. Including the precedent deprotection of the N- and C-termini, the macrocyclization step under high dilution conditions afforded a most remarkable 94% yield of verticilide. Overall, cyclodepsipeptide 223 was obtained in 13 steps with 66% yield (Scheme 13) [99].
Scheme 13.
Total synthesis of verticilides (223).

Reagents and conditions: (a) EDCl, DMAP; (b) 4 M HCl; (c) H2 Pd/C; (d) PyBrop, DIPEA; (e) PyBrop, DIPEA, CH2Cl2 (0.005 M).
7.3. PF1022A and Emodepside
PF1022A (235, Figure 26), the most prominent cyclooctadepsipeptide, was isolated in 1992 by Sasaki, who also described the potent anthelmintic activity against Ascaridia galli in chicken with no adverse toxic effects on the host animals [100]. PF1022A (235) is a metabolite of Mycelia sterilia (Rosselinia sp.) originally isolated from the leaves of Camellia japonica. PF1022A consists of four l-N-methylleucines, two d-phenyllactic acids and two d-lactic acids, which are linked together in a regular pattern giving the molecule a two-fold axis of symmetry. Due to its high and broad in vitro and in vivo anthelmintic activity against a multitude of nematodes, infecting humans, pets, sheep, cattle and horses, PF1022A became the most relevant lead structure in the search for novel anthelmintic drugs during the past two decades. Its unique resistance breaking mode of action, renders PF1022A superior to all standard commercial anthelmintics to which resistances have been emerged in different degrees. A semisynthetic derivative of PF1022A, emodepside (236, bis-para morpholino-PF1022A) has been introduced into the market (Profender® and Procox®) in 2008 as a broad spectrum anthelmintic.
Figure 26.
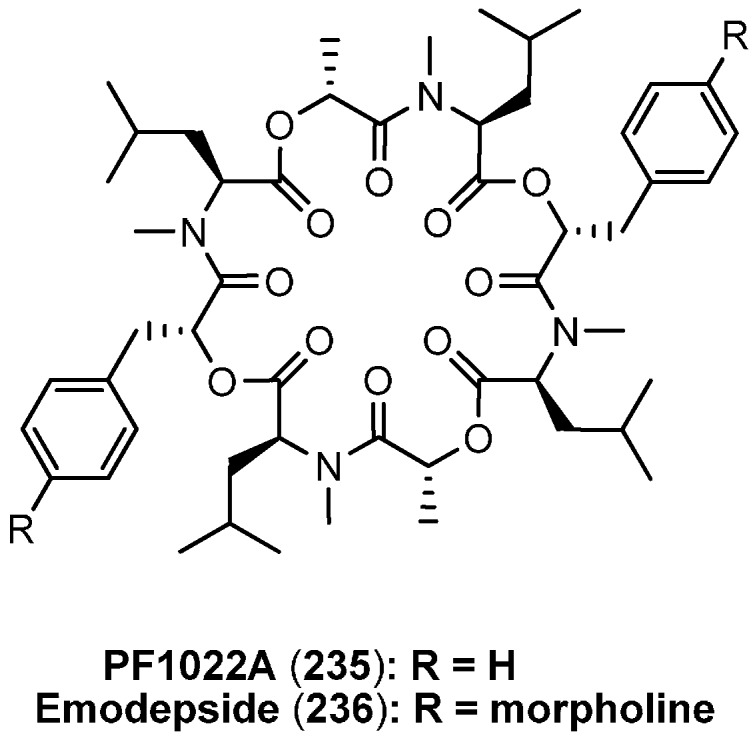
Structures of PF1022A (235) and emodepside (236).
In the fermentation broth of PF1022A (235), several structurally similar cyclooctadepsipeptides were found, isolated and tested for their anthelmintic activities (Figure 27) [101]. However, all homologues showed a reduced anthelmintic activity against A. galli in chicken.
Figure 27.
PF1022A and analogues 237–243 isolated from the fermentation broth.

(a) >90% worm reduction against Ascaridia galli in chicken.
7.3.1. Syntheses of PF1022A
In the past 20 years several total syntheses, both in solution and on solid-phase have been reported for PF1022A and analogues [102,103,104,105]. The C2-axis of symmetry reduces the effort to the synthesis of an appropriate substituted linear tetradepsipeptide which is subsequently dimerized and cyclized (Scheme 14).
Scheme 14.

Retrosynthetic study of PF1022A (235).
One of the first total syntheses was described by Nelson in 1994 employing the N-Boc/benzylester protecting group strategy. Both amide and ester bonds of the linear depsipeptides were formed with DCC. The macrocylization reaction with BOP as coupling reagent BOP/NMM under high dilution conditions (1 mM) yielded PF1022A in a yield of 50%.
A more efficient total synthesis was described by Scherkenbeck in 1995 favoring a N-benzyl/tert-butylester protecting group strategy (Scheme 15). The didepsipeptides were established with concomitant inversion of the configuration of the l-hydroxy acids introduced in the synthesis. As a consequence, only the inexpensive natural l-amino acids were needed as starting material for this total synthesis strategy. All amide bonds were established with BOP-Cl, which is known to be highly efficient in the coupling of N-methyl amino acids. In particular, the macrocyclization with this reagent succeeded in amazingly high yields of 85%–90% under high-dilution conditions. This remarkably high yield of an usually critical macrocyclization can be attributed to a conformer with a cis-amide bond between a Lac and a N-Me-Leu reside, found in the linear PF1022A precursor in solution. That cis-arrangement orients the N- and C-termini close together and thus facilitates the ring-closure.
Scheme 15.
Total synthesis of PF1022A (235) in solution by Scherkenbeck et al.

Reagents and conditions: (a) NaNO2, 6 N HCl, from 0 °C to rt; (b) isobutene, conc. H2SO4, CH2Cl2, rt, autoclave, 48 h; (c) Cs2CO3, ethanol, rt, 5 h, then NBn-l-Leu-OtBu, DMSO, rt, 20 h; (d) Pd(OH)2, H2, ethanol, rt, 6 h; (e) HCl (gas), CH2Cl2, 0 °C, 2 h; (f) DIPEA, BOP-Cl, CH2Cl2, from 0 °C to rt, 2 h; (g) DIPEA, BOP-Cl, CH2Cl2, rt, 24 h (2 cycles).
In 1997 Lee published the first solid-phase synthesis of PF1022A (235) analogues, using the Kaiser-oxime resin and a Boc protecting group strategy [106]. Unfortunately, the yields of this synthesis were low and only two chain-elongation steps were performed on the resin, establishing exclusively amide-bonds. PyBrop, used for the coupling steps works especially well, like BOP-Cl, for bulky and N-alkylated amino acids. The starting tetradepsipeptide was prepared in solution as well as the didepsipeptides. As a consequence, all ester bonds were formed in solution. The Kaiser oxime is one of the rare solid-supports which allows a cyclizative cleavage under almost neutral conditions [107,108]. Thus, PF1022 analogues were obtained in about 55% yield after refluxing the deprotected linear precursor in ethyl acetate for 2 days. In the same manner Lee prepared ɛ-lactam and related analogues of PF1022A (Figure 29) [109,110].
Figure 29.

Conformationally restricted analogues of PF1022A (235).
In 2012 Scherkenbeck published the first real solid-phase synthesis of PF1022A based on the Wang resin and a Fmoc/THP-ether protecting group strategy [111]. That strategy allowed a stepwise coupling of N-methyl amino acids and hydroxy acids in an alternating sequence on solid support (Scheme 16). The Kaiser-oxime turned out unsuitable for this 17 step synthesis due to the formation of inseparable side-products which became dominant particular in the last steps. Much better results were obtained on Wang-resin with HATU as coupling reagent for amide bonds and a combination of DIC/DMAP/HOBt for the ester bonds. The overall yield obtained for the linear cyclooctadepsipeptide 265 was in the range of 13%–16%, correlating with 89% average yield for each step. The macrocyclization with BOP-Cl was accomplished in solution after removal of the protecting groups in a yield of 81%.
Scheme 16.
Stepwise solid-phase synthesis of PF1022A by Scherkenbeck et al.

Reagents and conditions: (a) DIC, HOBt, THF, rt, 16 h; (b) p-TsOH, DCM/MeOH 97:3, rt, 2x1 h; (c) 266, DIC, HOBt, DMAP, THF, rt, 16 h; (d) 25% piperidine/THF, rt, 30 min; (e) 267, HATU, DIPEA, THF, rt, 16 h.
A segment total synthesis based on couplings of the didepsipeptides prepared in solution developed by the same group worked well on both Wang- and Kaiser-resin (Scheme 17) [112]. The advantage of this procedure is, that in the coupling steps solely amide bonds are formed which succeeds in higher yields as ester formation. Expectedly, a cyclizative cleavage strategy in a solution of DIPEA and HOAc in DCM was used to remove the final product from Kaiser oxime. The overall yield of PF1022A prepared on Kaiser-oxime was 30% and that of emodepside remarkable 45% compared to Wang resin where PF1022 (235) was obtained only in 25% yield including the separate solution cyclization.
Scheme 17.
Segment solid-phase synthesis of PF1022A (235) and emodepside (236).

Reagents and conditions: (a) Kaiser oxime: i. 25% TFA/DCM, rt, 30 min; ii. Boc-MeLeu-Lac-OH, HATU, DIPEA, DMF, rt, 16 h; iii. 25% TFA/DCM, rt, 30 min; Wang resin: i. 25% piperidine/THF, rt, 30 min; ii. Fmoc-MeLeu-Lac-OH, HATU, DIPEA, THF, rt, 16 h; iii. 25% piperidine/THF, rt, 30 min; (b) Kaiser oxime: i. Boc-MeLeu-PhLac-OH or Boc-MeLeu-morphPhLac-OH, HATU, DIPEA, DMF, rt, 16 h; ii. 25% TFA/DCM, rt, 30 min; Wang resin: i. Fmoc-MeLeu-PhLac-OH, HATU, DIPEA, THF, rt, 16 h; ii. 25% piperidine/THF rt, 30 min; (c) Kaiser oxime: i. Boc-MeLeu-Lac-OH, HATU, DIPEA, DMF, rt, 16 h; ii. 25% TFA/DCM, rt, 30 min; Wang resin: i. Fmoc-MeLeu-Lac-OH, HATU, DIPEA, THF, rt, 16 h; ii. 25% piperidine/THF, rt, 30 min; (d) Kaiser oxime: DIPEA, AcOH, DCM, rt, 16 h; Wang resin: i. 50% TFA/DCM, rt, 1 h: ii. BOP-Cl, DIEA, rt, 48 h.
7.3.2. Synthesis of PF1022A-Analogues via Total Synthesis
In order to establish structure-activity relationships, numerous PF1022A analogues were prepared either by total synthesis or by semisynthetic modification of the natural product. The significance of the N-methyl groups was tested by replacing them with N-ethyl, N-propyl and N-isopropyl groups. While the N-ethyl PF1022 analogue had almost the same anthelmintic activity as the natural product, the N-propyl and N-isopropyl derivatives were clearly less active. A PF1022A analogue lacking all N-methyl groups was found almost inactive, too. The exchange of N-Me-Leu for N-Me-Ile, N-Me-Val, N-Me-Nva (N-methyl-norvaline), N-Me-Ala and N-Me-Phe also resulted in reduced anthelmintic activities (Figure 28).
Figure 28.

Biological activity of various PF1022A-derivatives.
Based on the hypothesis, that only one half of the C2-symmetric PF1022A cycle is necessary for biological activity, unsymmetric PF1022A derivatives were synthesized by replacing the N-methyl-leucines in one half of the molecule by N-methyl-alanine and N-methyl-norvaline residues [113]. However, also those derivatives were found considerably less anthelmintically active. The crucial role of the hydroxy acids becomes already evident from a comparison of PF1022A (235) with PF1022B (237) and PF1022C (238) which show a reduced anthelmintic activity against H. contortus at least by a factor of 10 (Figure 27). Anyway, the position of choice for structural modifications turned out to be the phenyllactic acid residues as exemplified by emodepside (236).
Another line of research besides total syntheses of PF1022A analogues, derivatization of the natural product and backbone modifications was addressed to the question of conformational flexibility of the cyclooctadepsipeptide macrocycle. In solution PF1022A exists as a mixture of two conformers, one of which contains a single cis-amide bond between a N-Me-Leu and a Lac residue as part of a β-turn [114]. In order to elucidate the role of the β-turn, Scherkenbeck replaced a N-Me-Leu-Lac moiety by the more rigid d-Pro-l-Pro dipeptide and the bicyclic “Nagai-Sato” (BTD) bicyclic β-turn mimic (Figure 29) [115]. Analogous, conformationally restricted analogues 285–287 and 289–291 were reported by Dutton [116]. Cyclodepsipeptides 282 and 284 gave full control of H. contortus at a dose of 0.1 mg/kg in sheep while the d-Pro-l-Pro analogue 283 was found almost inactive (Figure 29). These results indicate that by an appropriate choice of the β-turn mimetic at least partially non-peptidic PF1022A analogues may be feasible with improved metabolic stability.
Research published by Scherkenbeck on the novel class of azadepsipeptides goes in the opposite direction and aims at imposing more flexibility on the cyclooctadepsipeptide framework [117]. In a lengthy synthesis (Scheme 18) the azadepsipeptide analogue 305 was prepared and tested for its anthelmintic activity. Again, the anthelmintic activity was reduced considerably. However, the reason for that remains speculative because two parameters have been changed at the same time. The α-carbon of leucine was replaced by a nitrogen atom and at the same time the chirality at that position was destroyed.
Scheme 18.

Synthesis of aza-PF1022A (305).
7.3.3. PF1022A Analogues by Direct Derivatization of the Natural Product
PF1022A has a remarkable stability against strongly acidic conditions. Therefore, classical aromatics chemistry such as nitrations, sulfonylations, halogenations and Friedel-Crafts acylations have been used to prepare a multitude of PF1022A analogues by modification of the natural product (Scheme 19) [118]. On the contrary, Palladium catalyzed coupling reactions do not work at all, probably due to complexation of the Palladium in the PF1022A macrocycle.
Scheme 19.

Derivatization of PF1022A (235).
A catalytic oxidation of the benzene rings of PF1022A (235) with RuCl3 provides the mono- or bis-carboxylic acids 308 and 309, which represent valuable starting materials for the synthesis of diverse heteroaromatic systems such as benzothiazoles and benzimidazoles [119]. The tert-butyl and related alkyl ethers of PF1022H, a side-product from fermentation, showed very promising anthelmintic profiles. An alternative to the minute amounts of PF1022H accessible from the fermentation broth is based on the bis-anilino PF1022A (313) which is available by nitration of PF1022A with fumic nitric acid and subsequent hydrogenation. Formation of the diazonium salt and aqueous work-up immediately provided semisynthetic PF1022H (243) in excellent yields (80%). In addition, the bis-anilino PF1022A (313) has been used for the preparation of urethanes, carbamates and heterocylic analogues. Emodepside (236) was prepared from the bis-anilino derivative 313 by alkylation with the bis-tosylates in high yields.
Friedel-Crafts acylation of PF1022A with acetyl chloride followed by bromination provides the α-bromo ketones 318 and 319 as starting materials for Hantzsch syntheses of pyrroles, thiazoles and additional heteroaromatics. By systematic modification of the phenyl rings or replacement by other heteroaromatics several highly anthelmintic PF1022A derivatives were identified over the past decade.
Further modifications were directed to the PF1022A backbone including partial reductions of the amide groups and chemoselective transformations of the carbonyl groups to oximes and thioamides [120]. With increasing amounts of BH3 THF and reaction times it is possible to reduce two, three or all amide bonds. Due to a lower steric hindrance the MeLeu-Lac amide bonds are reduced first, followed by the MeLeu-PheLac bonds (Figure 30). An anthelmintic screening against H. contortutus in sheep demonstrated the mono- and bis-reduction products 327 and 328 highly active, while the tri- and tetra-reduced products 329 and 330 retained only weak anthelmintic activity.
Figure 30.

Backbone modification of PF1022A by reduction using BH3·THF.
Additional backbone-modified PF1022A derivatives were prepared by chemoselective thionation of the amide carbonyl groups [121]. With Belleau’s reagent (334, 0.5 molar equiv. in tetrahydrofuran) a mono-thionated product was obtained, with an excess of Lawesson’s reagent (333) all amide carbonyl groups were transformed into thioamides, respectively (Scheme 20). Subsequent reaction of the thioamides with substituted hydroxylamines in the presence of Hg(OAc)2 afforded the corresponding amidoximes some of which showing better anthelmintic activity than PF1022A [122].
Scheme 20.

Thionation of PF1022A and conversion to amidoximes.
7.3.4. Biosynthesis of PF1022A
PF1022A (235) is biosynthesized by the nonribosomal peptide synthetase PFSYN (350 kDa) following the usual thiotemplate mechanism [123]. The substrate tolerance of PFSYN for hydroxy acids was explored comprehensively by Süssmuth (Figure 31) [124]. PFSYN was shown to accept a broad variety of aromatic, heteroaromatic and aliphatic residues which were successfully incorporated into the cyclooctadepsipeptide framework allowing the synthesis of a broad spectrum of PF1022A derivatives. In contrast to the α-hydroxy activating domain of enniatin synthetase (ESYN), PFSYN is capable of activating both d-lactates and d-phenyllactates. In particular mentionable with respect to more advanced analogues is the acceptance of hydroxy acids with para-halogenated phenyl rings and propargyl side-chains.
Figure 31.

PF1022A derivatives obtained by in vitro synthesis.
7.3.5. Mode of Action of PF1022A and Emodepside
PF1022A (235) and its semi-synthetic commercial derivative emodepside (236) show high activity against several parasitic gastrointestinal nematodes in combination with low toxicity to mammals. Most important, 235 and 236 are fully effective against levamisol-, benzimidazol- and ivermectin-resistant nematodes in sheep and cattle. This fact strongly supports the hypothesis of a novel and unique mode of action for PF1022A and related cyclooctadepsipeptides [125]. The application of PF1022A or emodepside induces the release of inhibitoric, postsynaptic acting neurotransmitters, resulting in the paralysis of pharyngeal muscles and the somatic musculature which finally causes the death of the nematode. The elucidation of the molecular mode of action of PF1022A was an odyssey for almost 20 years. Beginning with the ionophoric properties as the toxic principle of PF1022A and related cyclodepsipeptides, followed by the suggestion of a direct interaction with the GABA receptor, mode of action studies then postulated the HC110-R receptor, related to the mammalian G-protein coupled receptor latrophilin, to be the molecular target of PF1022A. Latrophilin is the known receptor of latrotoxin, the poison of the black widow spider. Most recent studies propose the SLO-1 potassium channel as the real target eventually together with HC110-R [126].
8. Cyclononadepsipeptides
8.1. BZR-cotoxins I-IV
BZR-cotoxins (Figure 32) were isolated from the plant pathogenic fungus Bipolaris zeicola. The BZR-cotoxin complex consists of three cyclononadepsipeptides (BRZ-cotoxin I-III, (368–370) and the cyclooctadepsipeptide BZR-cotoxin IV (371). Among those four cyclodepsipeptides BZR-cotoxin IV was considered the most important one [127,128,129,130].
Figure 32.

Structures of BZR-cotoxin I-IV.
8.2. Aureobasidins
Aureobasidins, isolated from the black yeast Aureobasidium pullulans R106, show high antifungal activities (Figure 33). To date over 30 aureobasidin analogues were characterized, all of which are composed of one hydroxy and eight amino acids [131,132]. A central step in the total synthesis of aureobasidin A (372) by Kuromoe is the PyBrop mediated macrocycle formation between l-allo-isoleucine (l-aIle) and l-Pro [133]. Another total synthesis, described by Cooper and coworkers uses fragment couplings and a cyclization between the residues l-Phe and l-Val [134].
Figure 33.

Structure and activity of aureobasidin A (372).
Recently Maharani et al., described a total synthesis of an synthetic analogue of (372), [2S,3S]-aureobasidin L (376), on chlorotrityl resin [135]. The critical macrocyclization was accomplished by formation of an amide bond between Val (N-terminus) and Pro (C-terminus) (Scheme 21).
Scheme 21.
Total synthesis of aureobasidin derivative 376 on solid-phase.

Reagents and conditions: (a) 1. Fmoc-l-Pro, CH2Cl2, DIPEA; 2. MeOH; 3. piperidine in DMF; (b) 1. Fmoc-l-N-MePhe, HBTU/HOBt, HATU/HOAt, or BTC (triphosgene)/collidine, DIPEA, CH2Cl2/DMF; 2. piperidine in DMF; (c) 1. Fmoc-l-Phe, HATU/HOAt, DIPEA, CH2Cl2/DMF; 2. piperidine in DMF; (d) Fmoc-l-N-MeVal, HBTU/HOBt or HATU/HOAt, DIPEA, CH2Cl2/DMF; 2. piperidine in DMF; (e) 1. Fmoc-l-N-MeVal-l-Hmp-OH, BTC/collidine, DIPEA, THF; 2. 20% piperidine in DMF (two cycles) or piperidine/DBU/DMF (two cycles); (f) 1. Fmoc-l-Leu, BTC/collidine, DIPEA, THF then HATU/HOAt, DIPEA, CH2Cl2/DMF; 2. 20% piperidine in DMF (two cycles) or piperidine/DBU/DMF, 20 min; (g) 1. Fmoc-l-N-MeVal, BTC/collidine, DIPEA, THF; 2. piperidine/DBU/DMF, 20 min; (h) 1. Fmoc-l-Val, BTC/collidine, DIPEA, THF then HATU/HOAt, DIPEA, CH2Cl2/DMF; 2. piperidine/DBU/DMF, 20 min; (i) 1. TFA/CH2Cl2, 15 min; 2. HATU, DIPEA, CH2Cl2.
9. Cyclodecadepsipeptides
9.1. Clavariopsin A and B
The antifungal active Clavariopsins A (377) and B (378) (Figure 34) were isolated from the culture broth of the aquatic hyphomycetes Clavariopsis aquatic. These decacyclodepsipeptides induce a swelling of fungal hyphae by inhibition fungal cell wall biosynthesis [136,137].
Figure 34.

Structures of clavariopsins A and B and their antifungal activity.
10. Summary
This review gives an overview of natural depsipeptides with ring-sizes between 12 and 30 atoms and composed exclusively of α-amino acids and at least one α-hydroxy acid. Most of these macrocyclic depsipeptides show impressive biological activities in the field of crop protection, animal health or medicine. The high incidence of biological activity in this family of natural products can be attributed to the cyclic nature which improves the metabolic stability and the occurrence of α-hydroxy acids which mimic α-amino acids. The biosynthesis of cyclodepsipeptides is accomplished nonribosomally by cyclodepsipeptide synthetases, which comprise large multifunctional proteins. The cyclodepsipeptide synthases show a relatively broad tolerance for α-hydroxy acids and have been applied for in vitro syntheses of new cyclodepsipeptides, simply by adding synthetic α-hydroxy acids to those enzymes. Numerous cyclodepsipeptides such as the AM-toxins, the kutznerides and the monamycins contain unusual nonribosomal residues which present formidable challenges also for the total synthesis of these compounds. Highly efficient syntheses both in solution and on solid-phase have been developed for enniatins and in particular for the cyclooctadepsipeptide PF1022A. The most critical step in almost all syntheses is the final macrocyclization which due to entropic reasons needs high-dilution conditions and often provides only moderate yields of the cyclization product. Chemical syntheses, in vitro syntheses with the cyclodepsipeptide synthetases and feeding experiments using the cyclodepsipeptide producing strains have been employed for the efficient production of analogues of the natural cyclodepsipeptides.
Presently, the most successful story is the development of the semi-synthetic PF1022A derivative emodepside as a commercial anthelmintic. Intelligent combinations of classical total syntheses, chemical derivatizations and (bio)combinatorial approaches will provide additional commercial cyclodepsipeptides in the fields of medicine and plant protection in the near future, underlining the importance of this particular class of natural compounds.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Dory Y.L., Mellor J.M. Improved methods of synthesis of valinomycins. Tetrahedron Lett. 1989;30:1695–1698. doi: 10.1016/S0040-4039(00)99557-1. [DOI] [Google Scholar]
- 2.Hinaje M., Ford M., Banting L., Arkle S., Khambay B. An investigation of the ionophoric characteristics of destruxin A. Arch. Biochem. 2002;405:73–77. doi: 10.1016/S0003-9861(02)00275-8. [DOI] [PubMed] [Google Scholar]
- 3.Davies J.S. The cyclization of peptides and depsipeptides. J. Pept. Sci. 2003;9:471–501. doi: 10.1002/psc.491. [DOI] [PubMed] [Google Scholar]
- 4.Davidson B.S. Ascidians: procedure of amino acid-derived metabolites. Chem. Rev. 1993;93:1771–1791. doi: 10.1021/cr00021a006. [DOI] [Google Scholar]
- 5.Moore R.E. Cyclic peptides and depsipeptides from cyanobacteria: A review. J. Ind. Microbiol. 1996;16:134–143. doi: 10.1007/BF01570074. [DOI] [PubMed] [Google Scholar]
- 6.Kitir B., Baldry M., Ingmer H., Olsen C.A. Total synthesis and structural validation of cyclodepsipeptides solonamide A and B. Tetrahedron. 2014 in press. [Google Scholar]
- 7.Li W., Schlecker A., Ma D. Total synthesis of antimicrobial and antitumor cyclic depsipeptides. Chem. Commun. 2010;46:5403–5420. doi: 10.1039/c0cc00629g. [DOI] [PubMed] [Google Scholar]
- 8.Lüttenberg S., Sondermann F., Scherkenbeck J. A practical method for the generation of Kaiser-oxime resin. Tetrahedron Lett. 2013;54:907–908. [Google Scholar]
- 9.Stolze S.C., Kaiser M. Case studies of the synthesis of bioactive cyclodepsipeptide natural products. Molecules. 2013;18:1337–1367. doi: 10.3390/molecules18021337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Humano K., Kinoshita M., Furuya K., Miyamoto M., Takamatsu Y., Hemmi A., Tanzawa K. Leualacin, a novel calcium blocker from Hapsidospora irregularis. J. Antibiot. 1991;45:899–905. doi: 10.7164/antibiotics.45.899. [DOI] [PubMed] [Google Scholar]
- 11.Kikuchi M., Nosaka K., Akaji K., Konno H. Solid phase total synthesis of callipeltin E isolated from marine sponge Latrunculia sp. Tetrahedron Lett. 2011;52:3872–3875. doi: 10.1016/j.tetlet.2011.05.062. [DOI] [Google Scholar]
- 12.Zhou X., Huang H., Li J., Song Y., Jiang R., Liu J., Zhang S., Hua Y., Ju J. New anti-infective cycloheptadepsipeptide congeners and absolute stereochemistry from the deep sea-derived Streptomyces drozdowiczii SCIO 10141. Tetrahedron. 2014 in press. [Google Scholar]
- 13.Finking R., Marahiel M.A. Biosynthesis of nonribosomal peptides. Annu. Rev. Microbiol. 2004;58:453–488. doi: 10.1146/annurev.micro.58.030603.123615. [DOI] [PubMed] [Google Scholar]
- 14.Gao X., Haynes S.W., Ames B.D., Wang P., Vien L.P., Walsh C.T., Tang Y. Cyclization of fungal nonribosomal peptides by a terminal condensation-like domain. Nat. Chem. Biol. 2012;8:823–830. doi: 10.1038/nchembio.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kopp F., Mahlert C., Grünewald J., Marahiel M.A. Peptide macrocyclization: the reductase of the nostocyclopeptide synthetase triggers the self-assembly of a macrocyclic imine. J. Am. Chem. Soc. 2006;128:16478–16479. doi: 10.1021/ja0667458. [DOI] [PubMed] [Google Scholar]
- 16.Xu Y., Orozo R., Wijeratne E.M.K., Gunatilaka A.A.L., Stock S.P., Molnar I. Biosynthesis of the cyclooligomer depsipeptide beauvericin, a virulence factor of the entomopathogenic fungus Beauveria bassiana. Chem. Biol. 2008;15:898–907. doi: 10.1016/j.chembiol.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 17.Yu D., Xu F., Zi J., Wang S., Gage D., Zeng J., Zhan J. Engineered production of fungal anticancer cyclooligomer depsipeptides in Saccharomyces cerevisae. Metab. Eng. 2013;18:60–68. doi: 10.1016/j.ymben.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 18.Süssmuth R., Müller J., von Döhren H., Molnar I. Fungal cyclooligomer depsipeptides: From classical biochemistry to combinatorial biosynthesis. Nat. Prod. Rep. 2011;28:99–124. doi: 10.1039/c001463j. [DOI] [PubMed] [Google Scholar]
- 19.Miyashita M., Nakamori T., Miyagawa H., Akamatsu M., Ueno T. Inhibitor activity of analogs of AM-toxin, a host-specific phytotoxin from the Alternaria alternate apple pathotype, on photosynthetic O2 evolution in apple leaves. Biosci. Biotechnol. Biochem. 2003;67:635–638. doi: 10.1271/bbb.67.635. [DOI] [PubMed] [Google Scholar]
- 20.Lee S., Aoyagi H., Shimohigashi Y., Izumiya N. Synthesis of cyclotetradepsipeptides, AM- toxin I and its analogs. Tetrahedon Lett. 1976;11:843–846. doi: 10.1016/S0040-4039(00)92899-5. [DOI] [Google Scholar]
- 21.Shimohigashi Y., Lee S., Kato T., Izumiya N., Ueno T., Fukami H. Synthesis and necrotic activity of dihydro-AM-toxin I. Agric. Biol. Chem. 1977;8:1533–1534. doi: 10.1271/bbb1961.41.1533. [DOI] [Google Scholar]
- 22.Ueda K., Waki M., Teruya Y., Izumiya N. Facile synthesis of AM-toxin II. Bull. Chem. Soc. Jpn. 1989;62:1635–1638. doi: 10.1246/bcsj.62.1635. [DOI] [Google Scholar]
- 23.Hashimoto K., Sakai M., Okuno T., Shirahama H. β-Phenylselenoalanine as a dehydroalanine precursor-efficient synthesis of alternariolide (AM-toxin I) Chem. Commun. 1996:1139–1140. doi: 10.1039/cc9960001139. [DOI] [Google Scholar]
- 24.Miyashita M., Nakamori T., Murai T., Miyagawa H., Akamatsu M., Ueno T. Facile synthesis of AM-toxins and analogs as cyclic depsipeptides by the solid-phase method. Biosci. Biotechnol. Biochem. 1998;9:1799–1801. doi: 10.1271/bbb.62.1799. [DOI] [PubMed] [Google Scholar]
- 25.Horikawa E., Kodaka M., Nakahara Y., Okuno H., Nakamura K. Solid-phase synthesis of dehydropeptide, AM-toxin II, using a novel selenyl linker by side-chain tethered strategy. Tetrahedon Lett. 2001;42:8337–8339. doi: 10.1016/S0040-4039(01)01790-7. [DOI] [Google Scholar]
- 26.Konzono T., Kanmera T., Kato T., Ueno T., Izumiya N. Synthesis of lactam analog of AM-toxin I. Agric. Biol. Chem. 1983;47:2631–2632. doi: 10.1271/bbb1961.47.2631. [DOI] [Google Scholar]
- 27.Kunicki J.B., Peterson M.N., Alexander L.D., Ardi V.C., McConnell J.R., McAlpine S.R. Synthesis and evaluation of biotinylated sansalvamide A analogs and their modulation of Hsp90. Bioorg. Med. Chem. Lett. 2011;21:4716–4719. doi: 10.1016/j.bmcl.2011.06.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belofsky G.N., Jensen P.R., Fenical W. Sansalvamide: A new cytotoxic cyclic depsipeptide produced by a marine fungus of the genus Fusarium. Tetrahedron Lett. 1999;40:2913–2916. doi: 10.1016/S0040-4039(99)00393-7. [DOI] [Google Scholar]
- 29.Cueto M., Jensen P.R., Fenical W. N-methylsansalvamide, a cytotoxic cyclic depsipeptide from a marine fungus of the genus Fusarium. Phytochemistry. 2000;55:223–226. doi: 10.1016/S0031-9422(00)00280-6. [DOI] [PubMed] [Google Scholar]
- 30.Song H., Lee H., Lee C. A new cytotoxic cyclic pentadepsipeptide, neo-N-methylsansalvamide produced by Fusarium solani KCCM90040, isolated from potato. Food Chem. 2011;126:472–478. doi: 10.1016/j.foodchem.2010.11.023. [DOI] [Google Scholar]
- 31.Lee Y., Silverman R.B. Rapid, high-yield, solid-phase synthesis of the antitumor antibiotic sansalvamide A using a side-chain-tethered phenylalanine building block. Org. Lett. 2000;2:3743–3746. doi: 10.1021/ol0002830. [DOI] [PubMed] [Google Scholar]
- 32.Gu W., Liu S., Silverman R.B. Solid-phase, Pd-catalyzed silicon-aryl carbon bond formation. Synthesis of sansalvamide A peptide. Org. Lett. 2002;4:4171–4174. doi: 10.1021/ol0269392. [DOI] [PubMed] [Google Scholar]
- 33.Carroll C.L., Johnston J.V.C., Kekec A., Brown J.D., Parry E.P., Cajica J., Medina I., Cook K.M., Corral R., Pan P., et al. Synthesis and cytotoxicity of novel sansalvamide A derivatives. Org. Lett. 2005;7:3481–3484. doi: 10.1021/ol051161g. [DOI] [PubMed] [Google Scholar]
- 34.Liu S., Gu W., Lo D., Ding X.Z., Ujiki M., Adrian T.E., Stoff G.A., Silverman R.B. N-Methylsansalvamide A peptide analogs. Potent antitumor agents. J. Med. Chem. 2005;48:3630–3638. doi: 10.1021/jm048952t. [DOI] [PubMed] [Google Scholar]
- 35.Davis M.R., Styers T.J., Rodriguez R.A., Pan P., Vasko R.C., McAlpine S.R. Synthesis and cytotoxicity of a new class of potent decapeptide macrocycles. Org. Lett. 2008;10:177–180. doi: 10.1021/ol702403r. [DOI] [PubMed] [Google Scholar]
- 36.Alexander L.D., Sellers R.P., Davis M.R., Ardi V.C., Johnson V.A., Vasko R.C., MaAlpine S.R. Evaluation of di-sansalvamide A derivatives: Synthesis, structure-activity relationship, and mechanism of action. J. Med. Chem. 2009;52:7927–7930. doi: 10.1021/jm901566c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davis M.R., Singh E.K., Wahyudi H., Alexander L.D., Kunicki J.B., Nazarova L.A., Fairweather K.A., Giltrap A.M., Jolliffe K.A., Mc Alpine S.R. Synthesis of sansalvamide A peptidomimetics: Triazole, oxazole, thiazole and pseudoproline containing compounds. Tetrahedron. 2012;68:1029–1051. doi: 10.1016/j.tet.2011.11.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramsey D.M., McConnel J.R., Alexander L.D., Tanaka K.W., Vera C.M., McAlpine S.R. An Hsp90 modulator that exhibits a unique mechanistic profile. Bioorg. Med. Chem. Lett. 2012;22:3287–3290. doi: 10.1016/j.bmcl.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu S., Yang Y., Zhao C., Huang J., Han C., Han J. Effect of the 4'-substituted phenylalanine moiety of sansalvamide A peptide on antitumor activity. Med. Chem. Commun. 2014;5:463–467. doi: 10.1039/c3md00294b. [DOI] [Google Scholar]
- 40.Kim M., Sohn J., Ahn J., Oh H. Alternaramide, a cyclic depsipeptide from the marine-derived fungus Alternaria sp. SF-5016. J. Nat. Prod. 2009;72:2065–2068. doi: 10.1021/np900464p. [DOI] [PubMed] [Google Scholar]
- 41.Horton A.E., May O.S., Elsegood M.R.J., Kimber M.C. Total synthesis of the marine-derived cyclic depsipeptide alternaramide. Synlett. 2011;22:797–800. [Google Scholar]
- 42.Oh D., Jensen P.R., Fenical W. Zygosporamide, a cytotoxic cyclic depsipeptide from the marine-derived fungus Zygosporium masonii. Tetrahedron Lett. 2006;47:8625–8628. doi: 10.1016/j.tetlet.2006.08.113. [DOI] [Google Scholar]
- 43.Wang Y., Zhang F., Zhang Y., Liu J.O., Ma D. Synthesis and antitumor activity of cyclodepsipeptide zygosporamide and its analogues. Bioorg. Med. Chem. Lett. 2008;18:4385–4387. doi: 10.1016/j.bmcl.2008.06.066. [DOI] [PubMed] [Google Scholar]
- 44.Sy-Cordero A.A., Pearce C.J., Oberlies N.H. Revisting the enniatins: A review of their isolation, biosynthesis, structure determination, and biological activities. J. Antibiot. 2012;65:541–549. doi: 10.1038/ja.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krause M., Lindemann A., Glinski M., Hornbogen T., Bonse G., Jeschke P., Thielking G., Gau W., Kleinkauf H., Zocher R. Directed biosynthesis of new enniatins. J. Antibiot. 2001;54:797–804. doi: 10.7164/antibiotics.54.797. [DOI] [PubMed] [Google Scholar]
- 46.Pieper R., Kleinkauf H., Zocher R. Enniatin synthetases from different fusaria exhibiting distinct amino acid specificities. J. Antibiot. 1992;45:1273–1277. doi: 10.7164/antibiotics.45.1273. [DOI] [PubMed] [Google Scholar]
- 47.Zhan J., Burns A.M., Liu M.X., Faeth S.H., Gunatilaka A.A.L. Search for cell motility and angiogenesis inhibitors with potential anticancer activity: Beauvericin and other constitutions of two endophytic strains of Fusarium oxysporum. J. Nat. Prod. 2007;70:227–232. doi: 10.1021/np060394t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu X., Xu R., Ouyang Z., Qian C., Shen Y., Wu X., Gu Y., Xu Q., Sun Y. Beauvericin ameliorates experimental colitis by inhibiting activated T cells via downregulation of the PI3K/Akt signaling pathway. PLoS One. 2013;8:1–11. doi: 10.1371/journal.pone.0083013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shemyakin M.M., Ovchinnikov Y.A., Kiryushkin A.A., Ivanov V.T. The structure and total synthesis of enniatin B. Tetrahedron Lett. 1963;14:885–890. [Google Scholar]
- 50.Hu D.X., Bielitza M., Koos P., Ley V.S. A total synthesis of the ammonium ionophore, (−)-enniatin B. Tetrahedron Lett. 2012;53:4077–4079. doi: 10.1016/j.tetlet.2012.05.110. [DOI] [Google Scholar]
- 51.Jeschke P., Benet-Buchholz J., Harder A., Etzel W., Schindler M., Gau W., Weiss H. Synthesis and anthelmintic activity of substituted (R)-phenyllactic acid containing cyclohexadepsipeptides. Bioorg. Med. Chem. Lett. 2006;16:4410–4415. doi: 10.1016/j.bmcl.2006.05.040. [DOI] [PubMed] [Google Scholar]
- 52.Jeschke P., Harder A., Etzel W., Schindler M., Thielking G. Synthesis and anthelmintic activity of cyclohexadepsipeptides with cyclohexylmethyl side chains. Bioorg. Med. Chem. Lett. 2007;17:3690–3695. doi: 10.1016/j.bmcl.2007.04.037. [DOI] [PubMed] [Google Scholar]
- 53.Glinski M., Urbanke C., Hornbogen T., Zocher R. Enniatin synthetase is a monomer with extended structure: evidence for an intermolecular mechanism. Arch. Microbiol. 2002;178:267–273. doi: 10.1007/s00203-002-0451-1. [DOI] [PubMed] [Google Scholar]
- 54.Madry N., Zocher R., Grodzki K., Kleinkauf H. Selective synthesis of depsipeptides by the immobilized multienzyme enniatin synthetase. Appl. Microbiol. Biotechnol. 1984;20:83–86. [Google Scholar]
- 55.Feifel S.C., Schmiederer T., Hornbogen T., Berg H., Süssmuth R.D., Zocher R. In vitro synthesis of new enniatins: Probing the α-hydroxy carboxylic acid binding pocket of the multienzyme enniatin synthetase. ChemBioChem. 2007;8:1767–1770. doi: 10.1002/cbic.200700377. [DOI] [PubMed] [Google Scholar]
- 56.Peeters H., Zocher R., Kleinkauf H. Synthesis of beauvericin by a multifunctional enzyme. J. Antibiot. 1988;41:352–359. doi: 10.7164/antibiotics.41.352. [DOI] [PubMed] [Google Scholar]
- 57.Peeters H., Zocher R., Madry N., Oelrichs P.B., Kleinkauf H., Kraepelin G. Cell-free synthesis of the depsipeptide beauvericin. J. Antibiot. 1983;36:1762–1766. doi: 10.7164/antibiotics.36.1762. [DOI] [PubMed] [Google Scholar]
- 58.Xu Y., Zhan J., Wijeratne E.M.K., Burns A.M., Gunatilaka A.A.L., Molnar I. Cytotoxic and antihaptotactic beauvericin analogues from precursor-directed biosynthesis with the insect pathogen Beauveria bassiana ATCC 7159. J. Nat. Prod. 2007;70:1467–1471. doi: 10.1021/np070262f. [DOI] [PubMed] [Google Scholar]
- 59.Nilanonta C., Isaka M., Kittakoop P., Trakulnaleamsai S., Tanticharoen M., Thebtaranonth Y. Precursor-directed biosynthesis of beauvericin analogs by the insect pathogenic fungus Paecilomyces tenuipes BCC 1614. Tetrahedron. 2002;58:3355–3360. doi: 10.1016/S0040-4020(02)00294-6. [DOI] [Google Scholar]
- 60.Matthes D., Richter L., Müller J., Denisiuk A., Feifel S.C., Xu Y., Espinosa-Artiles P., Süssmuth R.D., Molnar I. In vitro chemoenzymatic and in vivo biocatalytic synthesis of new beauvericin analogues. Chem. Commun. 2012;48:5674–5676. doi: 10.1039/c2cc31669b. [DOI] [PubMed] [Google Scholar]
- 61.Isaka M., Yangchum A., Sappan M., Suvannakad R., Srikitikulchai P. Cyclohexadepsipeptides from Acremonium sp. BCC 28424. Tetrahedron. 2011;67:7929–7935. doi: 10.1016/j.tet.2011.08.041. [DOI] [Google Scholar]
- 62.Bunyapaiboonsri T., Vongvilai P., Auncharoen P., Isaka M. Cyclohexadepsipeptides from the filamentous fungus Acremonium sp. BCC 2629. Helv. Chim. Acta. 2012;95:963–972. doi: 10.1002/hlca.201100482. [DOI] [Google Scholar]
- 63.Vongvanich N., Kittakoop P., Isaka M., Trakulnaleamsai S., Vimuttipong S., Tanticharoen M., Thebtaranonth Y., Hirsutellide A. a new antimycobacterial cyclohexadepsipeptide from the entomopathogenic fungus Hirsutella kobayasii. J. Nat. Prod. 2002;65:1346–1348. doi: 10.1021/np020055+. [DOI] [PubMed] [Google Scholar]
- 64.Xu Y., Chen L., Duan X., Meng Y., Jiang L., Li M., Zhao G., Li Y. Total synthesis of hirsutellide A. Tetrahedron Lett. 2005;46:4377–4379. doi: 10.1016/j.tetlet.2005.04.087. [DOI] [Google Scholar]
- 65.Xu Y., Duan X., Li M., Jian L., Zhao G., Meng Y., Chen L. Synthesis of the key precursor of hirsutellide A. Molecules. 2005;10:259–264. doi: 10.3390/10010259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu Y., Chen L., Ma Y., Li J., Cao X. Silver-ion mediated macrocyclization to form cyclohexadepsipeptide. Synlett. 2007;12:1901–1904. [Google Scholar]
- 67.Pohanka A., Menkis A., Levenfors J., Broberg A. Low-abundance kutznerides from Kutznerica sp. 744. J. Nat. Prod. 2006;69:1776–1781. doi: 10.1021/np0604331. [DOI] [PubMed] [Google Scholar]
- 68.Broberg A., Menkis A., Vasiliauskas R. Kutznerides 1–4, depsipeptides from the actinomycete Kutzeria sp. 744 inhabiting mycorrhizal roots of Picea abies seedlings. J. Nat. Prod. 2006;69:97–102. doi: 10.1021/np050378g. [DOI] [PubMed] [Google Scholar]
- 69.Fujimori D.G., Hrvatin S., Neumann C.S., Strieker M., Marahiel M.A., Walsh C.T. Cloning and characterization of the biosynthetic gene cluster for kutnerides. PNAS. 2007;104:16498–16503. doi: 10.1073/pnas.0708242104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bevan K., Davies J.S., Hall M.J., Hassal C.H., Morton R.B., Phillips D.A.S., Ogihira Y., Thomas W.A. The monamycins, a new family of cyclodepsipeptide antibiotics. Experientia. 1970;26:122–123. doi: 10.1007/BF01895528. [DOI] [PubMed] [Google Scholar]
- 71.Hall M.J. Mode of action of monamycin. Evidence for the formation of a complex between the monamycin cyclodepsipeptide antibiotics and some cations in solution. Biochem. Biophys. Res. Commun. 1970;38:590–598. doi: 10.1016/0006-291x(70)90622-4. [DOI] [PubMed] [Google Scholar]
- 72.Hale K.J., Jogiya N., Manaviazar S. Monamycin synthetic studies. Pt 1. An enantiospecific total synthesis of (3S,5S)-5-hydroxypiperazic acid from d-Mannitol. Tetrahedron Lett. 1998;39:7163–7166. [Google Scholar]
- 73.Hall M.J., Handford B.O., Hassyl C.H., Phillips D.A.S., Rees A.V. Bromomonamycins, unnatural analogues of the monamycin cyclodepsipeptide antibiotics: Production, isolation, and biological activity. Antimircob. Agents Chemother. 1973;3:380–383. doi: 10.1128/AAC.3.3.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lam K.S., Heslter G.A., Mattel J.M., Mamber S.W., Forenza S. Himastatin, a new antitumor antibiotic from Streptomyces hygroscopius. I. Taxonomy of producing organism, fermentation and biological activity. J. Antibiot. 1990;8:956–960. doi: 10.7164/antibiotics.43.956. [DOI] [PubMed] [Google Scholar]
- 75.Left J.E., Schröder D.R., Krishan B.S., Matron J.A. Himastatin, a new antitumor antibiotic from Streptomyces hygroscopicus. II. Isolation and characterization. J. Antibiot. 1990;8:961–966. doi: 10.7164/antibiotics.43.961. [DOI] [PubMed] [Google Scholar]
- 76.Left J.E., Schroeder D.R., Golik J., Matson J.A., Doyle T.W., Lam K.S., Hill S.E., Lee M.S., Whitney J.L., Krishnan B.S. Himastatin, a new antitumor antibiotic from Streptomyces hygroscopicus. III. Structural elucidation. J. Antibiot. 1996;49:299–311. doi: 10.7164/antibiotics.49.299. [DOI] [PubMed] [Google Scholar]
- 77.Ruiz-Sanchis R., Savina S.A., Albericio F., Alvarez M. Structure, bioactivity and synthesis of natural products with hexahydropyrrolo[2,3-b]indole. Chem. Eur. J. 2011;17:1388–1408. doi: 10.1002/chem.201001451. [DOI] [PubMed] [Google Scholar]
- 78.Kamenecka T.M., Danishefsky S.J. Discovery through total synthesis: A retrospective on the himastatin problem. Chem. Eur. J. 2001;7:41–63. doi: 10.1002/1521-3765(20010105)7:1<41::AID-CHEM41>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 79.Kamenecka T.M., Danishefsky S.J. Studies in the total synthesis of himastatin: A revision of the stereochemical assignment. Angew. Chem. Int. Ed. 1998;37:2993–2995. doi: 10.1002/(SICI)1521-3773(19981116)37:21<2993::AID-ANIE2993>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 80.Kamenecka T.M., Danishefsky S.J. Total synthesis of himastatin: Confirmation of the revised stereostructure. Angew. Chem. Int. Ed. 1998;37:2995–2998. doi: 10.1002/(SICI)1521-3773(19981116)37:21<2995::AID-ANIE2995>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 81.Ma J., Wang Z., Huang H., Luo M., Zuo D., Wang B., Sun A., Cheng Yi., Zhang C., Ju J. Biosynthesis of himastatin: assembly line and characterization of three cytrochrome P450 enzymes involved in the post-tailoring oxidative steps. Angew. Chem. Ind. Ed. 2011;50:7797–7802. doi: 10.1002/anie.201102305. [DOI] [PubMed] [Google Scholar]
- 82.Isaka M., Palasarn S., Supothina S., Komwijit S., Luangsa-ard J. Bioactive compounds from the scale insect pathogenic fungus Conoideocrella tenius BCC 18627. J. Nat. Prod. 2011;74:782–789. doi: 10.1021/np100849x. [DOI] [PubMed] [Google Scholar]
- 83.Isaka M., Palasarn S., Lapanun S., Striklung K. Peacilodepsipeptide A, an antimalarial and antitumor cyclohexadepsipeptide from the insect pathogenic fungus Peacilomyces cinnamomeus BCC 9616. J. Nat. Prod. 2007;70:675–678. doi: 10.1021/np060602h. [DOI] [PubMed] [Google Scholar]
- 84.Yang M.J., Wu J., Yang Z.D., Zhang Y.M. First total synthesis of peacilodepsipeptide A. Chin. Chem. Lett. 2009;20:527–530. doi: 10.1016/j.cclet.2009.01.032. [DOI] [Google Scholar]
- 85.Isaka M., Berkaew P., Intereya K., Komwijit S., Sathitkunanon T. Antiplasmodial and antiviral cyclohexadepsipeptides from the endophytic fungus Pullularia sp. BCC 8613. Tetrahedron. 2007;63:6855–6860. [Google Scholar]
- 86.Ebrahim W., Kjer J., El Amrani M., Wray V., Lin W., Ebel R., Lai D., Proksch P. Pullularins E and F, two new peptides from the endophytic fungus Bionecteria ochroleuca isolated from the mangrove plant Sonneratia caseolaris. Mar. Drugs. 2012;10:1081–1091. doi: 10.3390/md10051081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Isaka M., Palasarn S., Sriklung K., Kocharin K. Cyclohexadepsipeptides from the insect pathogenic fungus Hirsutella nivea BB 2594. J. Nat. Prod. 2005;68:1680–1682. doi: 10.1021/np050246n. [DOI] [PubMed] [Google Scholar]
- 88.Isaka M., Rugseree N., Maithip P., Kongsaeree P., Prabpai S., Thebtaranonth Y. Hirsutellones A-E, antimycobacterial alkaloids from the insect pathogenic fungus Hirsutella nivea BCC 2594. Tetrahedron. 2005;61:5577–5583. doi: 10.1016/j.tet.2005.03.099. [DOI] [Google Scholar]
- 89.Chen Y., Bilban M., Foster C.A., Boger D.L. Solution-phase parallel synthesis of a pharmacophore library of HUN-7293 analogues: A general chemical mutagenesis approach to defining structure-function properties of naturally occurring cyclic (depsi)peptides. J. Am. Chem. Soc. 2002;124:5431–5440. doi: 10.1021/ja020166v. [DOI] [PubMed] [Google Scholar]
- 90.Boger D.L., Keim H., Oberhauser B., Schreiner E.P., Foster C.A. Total synthesis of HUN-7293. J. Am. Chem. Soc. 1999;121:6197–6205. doi: 10.1021/ja990918u. [DOI] [Google Scholar]
- 91.Boger D.L., Chen Y., Foster C.A. Synthesis and evaluation of aza HUN-7293. Bioorg. Med. Chem. Lett. 2000;10:1741–1744. doi: 10.1016/S0960-894X(00)00336-X. [DOI] [PubMed] [Google Scholar]
- 92.Schreiner E.P., Kern M., Steck A. A convenient protocol for selective cleavage of 2-hydroxy acid amides. Application to semisynthesis of the cyclic heptapeptide aza HUN-7293. J. Org. Chem. 2002;67:8299–8304. doi: 10.1021/jo025979g. [DOI] [PubMed] [Google Scholar]
- 93.Kanaoka M., Isogai A., Murakoshi S., Ichinoe M., Suzuki A., Tamura S. Bassianolide, a new insecticidal cyclodepsipeptide from Beauveria bassiana and Verticillium lecanii. Agric. Biol. Chem. 1978;42:629–635. doi: 10.1271/bbb1961.42.629. [DOI] [Google Scholar]
- 94.Nakajyo S., Shimizu K., Kometani A., Suzuki A., Ozaki H., Urakawa N. On the inhibitory mechanism of bassianolide, a cyclodepsipeptide, in acetylcholine-induced contraction in guinea-pig taenia coli. Jpn. J. Pharmacol. 1983;33:573–582. doi: 10.1254/jjp.33.573. [DOI] [PubMed] [Google Scholar]
- 95.Jirakkakul J., Punya J., Pongpattanakitshote S., Paungmoung P., Vorapreeda N., Tachaleat A., Klomnara C., Tanticharoen M., Cheevadhanarak S. Identification of the nonribosomal peptide synthetase gene responsible for bassianolide synthesis in wood-decaying fungus Xylaria sp. BCC1067. Microbiology. 2008;154:995–1006. doi: 10.1099/mic.0.2007/013995-0. [DOI] [PubMed] [Google Scholar]
- 96.Kanaoka M., Isogai A., Suzuki A. Synthesis of bassianolide. Tetrahedron Lett. 1977;46:4049–4050. doi: 10.1016/S0040-4039(01)83423-7. [DOI] [Google Scholar]
- 97.Shiomi K., Matsui R., Kakei A., Yamaguchi Y., Masuma R., Hatano H., Arai N., Isozaki M., Tanaka H., Kobayashi S., et al. Verticilide, a new ryanodine-binding inhibitor, produced by Verticilium sp. FKI-1033. J. Antibiot. 2010;63:77–82. doi: 10.1038/ja.2009.126. [DOI] [PubMed] [Google Scholar]
- 98.Ohshiro T., Matsudo D., Kazuhiro T., Uchida R., Nonaka K., Masuma R., Tomada H. New verticilides, inhibitors of acyl-CoA: Cholesterol acyltransferase, produced by Verticillium sp. FKI-2679. J. Antibiot. 2012;65:255–262. doi: 10.1038/ja.2012.12. [DOI] [PubMed] [Google Scholar]
- 99.Monma S., Sunazuka T., Nagai K., Arai T., Shiomi K., Matsui R., Omura S. Verticilide: Elucidation of absolute configuration and total synthesis. Org. Lett. 2006;8:5601–5604. doi: 10.1021/ol0623365. [DOI] [PubMed] [Google Scholar]
- 100.Sasaki T., Takagi M., Yaguchi T., Miyadoh S., Okada T., Koyama M. A new anthelmintic cyclodepsipeptide, PF1022A. J. Antibiot. 1992;45:692–697. doi: 10.7164/antibiotics.45.692. [DOI] [PubMed] [Google Scholar]
- 101.Sakanaka O., Okada Y., Ohyama M., Matsumoto M., Takahashi M., Murai Y., Iinuma K., Harder A., Mencke N. Novel Cyclic Depsipeptide PF1022A Derivatives. WO97/11064. Patent. 1997
- 102.Dutton F.E., Nelson S.J. Synthesis of PF1022A, an anthelmintic cyclodepsipeptide. J. Antibiot. 1994;47:1322–1327. doi: 10.7164/antibiotics.47.1322. [DOI] [PubMed] [Google Scholar]
- 103.Ohyama M., Iinuma K., Isogai A., Suzuki A. Total synthesis of the anthelmintic cyclodepsipeptide, PF1022A. Biosci. Biotechnol. Biochem. 1994;58:1193–1194. doi: 10.1271/bbb.58.1193. [DOI] [Google Scholar]
- 104.Scherkenbeck J., Plant A., Harder A., Mencke N. A highly efficient synthesis of the anthelmintic cyclooctadepsipeptide PF1022A. Tetrahedron. 1995;51:8459–8470. doi: 10.1016/0040-4020(95)00461-G. [DOI] [Google Scholar]
- 105.Kobayashi M., Nanba T., Toyama T., Saito A. Synthesis and anthelmintic activity of the cyclodepsipeptide PF1022A. Annu. Rep. Sankyo Res. Lab. 1994;46:67–75. [Google Scholar]
- 106.Lee B.H. Solid-phase synthesis of cyclooctadepsipeptide PF1022A analogs using a cyclization-cleavage method with oxime-resin. Tetrahedron Lett. 1997;38:757–760. doi: 10.1016/S0040-4039(96)02454-9. [DOI] [Google Scholar]
- 107.Nishino N., Xu M., Mihara H., Fujimoto T. Sequence dependent cyclization-cleavage of dipeptides from the oxime resin and its prevention. Bull. Chem. Soc. Jpn. 1991;65:991–994. doi: 10.1246/bcsj.65.991. [DOI] [Google Scholar]
- 108.Kumagai H. Efficient preparation of cyclic peptide mixtures by solid phase synthesis and cyclization cleavage with oxime resin. Tetrahedron Lett. 1995;36:4837–4840. doi: 10.1016/0040-4039(95)00906-S. [DOI] [Google Scholar]
- 109.Dutton F.E., Lee B.H. Epsilon-lactam analogs of the anthelmintic cyclodepsipeptide PF1022A. Tetrahedron Lett. 1998;39:5313–5316. doi: 10.1016/S0040-4039(98)01075-2. [DOI] [Google Scholar]
- 110.Lee B.H., Dutton F.E., Thompson D.P., Thomas E.M. Generation of a small library of cyclodepsipeptides PF1022A analogues using a cyclization-cleavage method with oxime resin. Bioorg. Med. Chem. Lett. 2002;12:353–356. doi: 10.1016/S0960-894X(01)00748-X. [DOI] [PubMed] [Google Scholar]
- 111.Lüttenberg S., Sondermann F., Scherkenbeck J. Anthelmintic PF1022A: Stepwise solid-phase synthesis of a cyclodepsipeptide containing N-methyl amino acids. Tetrahedron. 2012;68:2068–2073. doi: 10.1016/j.tet.2011.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Scherkenbeck J., Lüttenberg S., Ludwig M., Brüchner K., Kotthaus A. Segment solid-phase total synthesis of the anthelmintic cyclooctadepsipeptides PF1022A and emodepside. Eur. J. Org. Chem. 2012:1546–1553. doi: 10.1002/ejoc.201101421. [DOI] [Google Scholar]
- 113.Scherkenbeck J., Jeschke P., Harder A. PF1022A and related cyclodepsipeptides—A novel class of anthelmintics. Curr. Top. Med. Chem. 2002;2:759–777. doi: 10.2174/1568026023393624. [DOI] [PubMed] [Google Scholar]
- 114.Scherkenbeck J., Dyker H., Gondol D., Harder A., Plant A., Reichel F. Synthesis, conformational studies and anthelmintic activity of a constrained PF1022A analogue. Pest. Sci. 1999;55:457–461. doi: 10.1002/(SICI)1096-9063(199904)55:4<457::AID-PS927>3.0.CO;2-U. [DOI] [Google Scholar]
- 115.Dyker H., Harder A., Scherkenbeck J. Chimeric cyclodepsipeptides as mimetics for the anthelmintic PF1022A. Bioorg. Med. Chem. Lett. 2004;14:6129–6132. doi: 10.1016/j.bmcl.2004.07.097. [DOI] [PubMed] [Google Scholar]
- 116.Dutton F.E., Lee B.H., Johnson S.S., Coscarelli E.M., Lee P.H. Restricted conformation analogues of an anthelmintic cyclodepsipeptide. J. Med. Chem. 2003;46:2057–2073. doi: 10.1021/jm020482k. [DOI] [PubMed] [Google Scholar]
- 117.Dyker H., Scherkenbeck J., Gondol D., Goehrt A., Harder A. Azadepsipeptides: Synthesis and evaluation of a novel class of peptidomimetics. J. Org. Chem. 2001;66:3760–3766. doi: 10.1021/jo001749v. [DOI] [PubMed] [Google Scholar]
- 118.Ohyama M., Okada Y., Takahashi M., Sakanaka O., Matsumoto M., Atsumi K. Structure-activity relationship of anthelmintic cyclooctadepsipeptides. Biosci. Biotechnol. Biochem. 2011;75:1354–1363. doi: 10.1271/bbb.110129. [DOI] [PubMed] [Google Scholar]
- 119.Ohyama M. Chemistry of anthelmintic cyclodepsipeptides. Sci. Report of Meiji Seika Kaisha. 2006;45:8–34. [Google Scholar]
- 120.Dyker H., Harder A., Mencke N., Scherkenbeck J., Georg von S. Desoxycyclodepsipeptides and Their Use for Combatting Endoparasites. WO19980554469A1. Patent. 1998
- 121.Jeschke P., Harder A., Etzel W., Gau W., Thielking G., Bonse G., Iinuma K. Synthesis and anthelmintic activity of thioamide analogues of cyclic octadepsipeptides such as PF1022A. Pest. Manag. Sci. 2001;57:1000–1006. doi: 10.1002/ps.382. [DOI] [PubMed] [Google Scholar]
- 122.Jeschke P., Harder A., Samson-Himmelstjerna G.V., Etzel W., Gau W., Thielking G., Bonse G. Synthesis of anthelmintically active N-methylated amidoxime analogues of the cyclic octadepsipeptide PF1022A. Pest. Manag. Sci. 2002;58:1205–1215. doi: 10.1002/ps.590. [DOI] [PubMed] [Google Scholar]
- 123.Weckwerth W., Miyamoto K., Iinuma K., Krause M., Glinski M., Storm T., Bonse G., Kleinkauf H., Zocher R. Biosynthesis of PF1022A and related cyclooctadepsipeptides. J. Biol. Chem. 2000;275:17909–17915. doi: 10.1074/jbc.M001084200. [DOI] [PubMed] [Google Scholar]
- 124.Müller J., Feifel S.C., Schmiederer T., Zocher R., Süssmuth R.D. In vitro synthesis of new cyclodepsipeptides of the PF1022A-type: Probing the α-d-hydroxy acid tolerance of PF1022 synthetase. ChemBioChem. 2009;10:323–328. doi: 10.1002/cbic.200800539. [DOI] [PubMed] [Google Scholar]
- 125.Harder A., Schmitt-Wrede H., Krücken J., Marinovski P., Wunderlich F., Willson J., Amliwala K., Holden-Dye L., Walker R. Cyclooctadepsipeptides—An anthelmintically active class of compounds exhibiting a novel mode of action. Int. J. Antimicrob. Ag. 2003;22:318–331. doi: 10.1016/S0924-8579(03)00219-X. [DOI] [PubMed] [Google Scholar]
- 126.Guest M., Bull K., Walker R.J., Amliwala K., O’Connor V., Harder A., Holden-Dye L., Hopper A.N. The calcium-activated potassium channel, SLO-1, is required for the action of the novel cyclo-octadepsipeptide anthelmintic, emodepside, in Caenorhabditis elegans. Int. J. Parasitol. 2007;37:1577–1588. doi: 10.1016/j.ijpara.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 127.Ueda K., Xiao J., Doke N., Nakatsuka S. Isolation and structure of BZR-cotoxin IV produced by Bipolaris zeicola race 3, the cause of leaf spot disease in corn. Tetrahedron Lett. 1995;36:741–744. doi: 10.1016/0040-4039(94)02331-5. [DOI] [Google Scholar]
- 128.Ueda K., Xiao J., Doke N., Nakatsuka S. Structure of BZR-cotoxin I produced by Bipolaris zeicola race 3, the cause of leaf spot disease in corn. Tetrahedron Lett. 1994;35:7033–7036. doi: 10.1016/0040-4039(94)88218-5. [DOI] [Google Scholar]
- 129.Ueda K., Xiao J., Doke N., Nakatsuka S. Structure of BZR-cotoxin II produced by Bipolaris zeicola race 3, the cause of leaf spot disease in corn. Tetrahedron Lett. 1994;33:5377–5380. doi: 10.1016/S0040-4039(00)79098-8. [DOI] [Google Scholar]
- 130.Ueda K., Xiao J., Doke N., Nakatsuka S. Structure of BZR-cotoxin III produced by Bipolaris zeicola race 3, the cause of leaf spot desease in corn. Nat. Prod. Lett. 1994;6:43–48. doi: 10.1080/10575639508044086. [DOI] [Google Scholar]
- 131.Ikai K., Shiomi K., Takesako K., Mizutani S., Yamamoto J., Ogawa Y., Ueno M., Kato I. Structures of aureobasidins B to R. J. Antibiot. 1991;44:1187–1198. doi: 10.7164/antibiotics.44.1187. [DOI] [PubMed] [Google Scholar]
- 132.Yoshikawa Y., Ikai K., Umeda Y., Ogawa A., Takesako K., Kato I. Isolation, structures, and antifungal activities of new aureobasidins. J. Antibiot. 1993;46:1347–1354. doi: 10.7164/antibiotics.46.1347. [DOI] [PubMed] [Google Scholar]
- 133.Kurome T., Inami K., Inoue T., Ikai K., Takesako K., Kato I., Shiba T. Total synthesis of an antifungal cyclic depsipeptide aureobasidin A. Tetrahedron. 1996;52:4327–4346. doi: 10.1016/0040-4020(96)00132-9. [DOI] [Google Scholar]
- 134.Jao E., Cooper A.B., Rane D.F., Saksena A.K., Desai J., Wang J., Girijavallabhan V.M., Ganguly A.K. Total synthesis of the antifungal cyclic depsipeptides sch 57697 and aureobasidin A. Tetrahedron Lett. 1996;37:5661–5664. doi: 10.1016/0040-4039(96)01199-9. [DOI] [Google Scholar]
- 135.Maharani R., Brownlee R.T.C., Hughes A.B., Abbott B.M. A total synthesis of a highly N-methylated cyclodepsipeptide [2S,3S]-aureobasidin L using solid-phase methods. Tetrahedron. 2014;70:2351–2358. doi: 10.1016/j.tet.2014.02.036. [DOI] [Google Scholar]
- 136.Suzuki Y., Ojika M., Sakagami Y., Kajda K., Fudou R., Kameyama T. New cyclic depsipeptide antibiotics, clavariopsin A and B, produced by an aquatic hyphomycetes, Clavariopsis aquatica. 2. Structure analysis. J. Antibiot. 2001;54:22–28. doi: 10.7164/antibiotics.54.22. [DOI] [PubMed] [Google Scholar]
- 137.Kaika K., Fudou R., Kameyama T., Tubaki K., Suzuki Y., Ojika M., Sakagami Y. New cyclic depsipeptide antibiotics, clavariopsins A and B, produced by an aquatic hyphomycetes, Clavariopsis aquatica. 1. Taxonomy, fermentation, isolation, and biological properties. J. Antibiot. 2001;54:17–21. doi: 10.7164/antibiotics.54.17. [DOI] [PubMed] [Google Scholar]