

Abstract
Hispidin oligomers are styrylpyrone pigments isolated from the medicinal fungi Inonotus xeranticus and Phellinus linteus. They exhibit diverse biological activities and strong free radical scavenging activity. To rationalize the antioxidant activity of a series of four hispidin oligomers and determine the favored mechanism involved in free radical scavenging, DFT calculations were carried out at the B3P86/6-31+G (d, p) level of theory in gas and solvent. The results showed that bond dissociation enthalpies of OH groups of hispidin oligomers (ArOH) and spin density delocalization of related radicals (ArO•) are the appropriate parameters to clarify the differences between the observed antioxidant activities for the four oligomers. The effect of the number of hydroxyl groups and presence of a catechol moiety conjugated to a double bond on the antioxidant activity were determined. Thermodynamic and kinetic studies showed that the PC-ET mechanism is the main mechanism involved in free radical scavenging. The spin density distribution over phenoxyl radicals allows a better understanding of the hispidin oligomers formation.
Keywords: hispidin, antioxidant activity, DFT, BDE, PC-ET
1. Introduction
Styrylpyrones form a small group of natural phenolics of which goniothalamin (1, Figure 1), a cytotoxic plant derivative, is the best known representative [1]. A fascinating series of styrylpyrone oligomers and adducts was isolated from mushrooms belonging to the Inonotus and Phellinus genera (Hymenochaetaceae family), with hispidin (2, Figure 1) as a prototype [2]. The key structural difference with goniothalamin-type compounds lies in the presence of a highly oxidisable 4-hydroxy group, which can be held responsible for the reactivity of hispidin and its derivatives. These lignicolous mushrooms are traditionally used in Russia and western Siberia as folk medicines for the treatment of various serious diseases including cancer, stomach, liver or heart diseases [3,4,5]. Most of these diseases are related to oxidative stress, which results from the overproduction of free radicals and specifically reactive oxygen species [6].
Figure 1.

Molecular structures of compounds 1–6.
Experimental and theoretical studies of the structure-antioxidant activity relationships of natural polyphenols proved that this activity mainly depends on phenolic hydroxyl groups, and their capacity to donate a hydrogen atom to quench free radicals. In addition to the experimental studies on the quantitative structure-antioxidant activity relationship (QSAR), quantum chemical calculations showed to be a potent tool to rationalize structure-antioxidant activity of polyphenolic compounds [7,8,9,10,11,12,13,14,15,16]. Density functional theory (DFT) has become a very popular tool to rationalize QSAR, since it successfully describes the hydrogen atom transfer of OH phenolic groups to quench free radicals. Several structural features are known to enhance the antioxidant activity. Among them are (i) the presence of a catechol moiety, which strongly participates in the free radical scavenging capacity of flavonoids [17], oligomers of guaiacol [11], chalcones [10], hydroxybenzohydroxamic acid derivatives [18], as well as hispidin oligomers [19]; and (ii) 3-OH and C2=C3 double bond in flavonoids [12,17].
In the present paper, we report the study of the structure-antioxidant activity relationships of a series of hispidin oligomers using DFT calculations. A series of five hispidin oligomers were isolated from culture broths of I. xeranticus and P. linteus [19]. They include hispidin (2), isohispidin (3), 3,14'-bihispidinyl (4), hypholomin B (5) and 1,1-distyrylpyrylethan (6) (Figure 1). Compounds 3–5 are believed to be biosynthesized by dimerization of hispidin. Their antioxidant activity or, more specifically, ability to scavenge DPPH (1,1-diphenyl-2-picrylhydrazyl) free radical was determined by Jung et al. [19] through Blois’ method [20].
2. Results and Discussion
2.1. Hispidin Dimerization
Various styrylpyrone metabolites can be biogenerated by dimerization or oligomerization process [21]. For instance, hispidin oligomers 4 and 5 are likely to derive from dimerization of hispidin. The dimerization of hispidin is believed to proceed through a free-radical chain reaction. In the initiation step, the oxidation of hispidin leads to the formation of an unstable ArOH+• radical cation. This is followed by a proton loss (heterolytic rupture of the OH bond of ArOH+•), thus forming a hispidinyl ArO• radical. The hispidinyl and isohispidinyl radicals display a strong electronic spin density on oxygen and few carbon atoms (Figure 2). During the propagation and termination steps, the coupling between hispidinyl and isohispidinyl radicals generates various dimers. For instance, the coupling between the C3 of 4-O• free radical and C13 of the 11-O• hispidinyl radicals leads to the formation of dimer 4.
Figure 2.

Electronic spin density distribution of i-O• hispidinyl and isohispidinyl radicals (a) 4-O•, 11-O• and 12-O• hispidinyl radicals; (b) 2-O•, 11-O• and 12-O• isohispidinyl radicals.
2.2. Structure-Antioxidant Activity Relationships
2.2.1. Tautomerism Effect
The tautomerization of hispidin (2) leads to the formation of stable isohispidin (3, Figure 3a). The equilibrium is a two-step reaction, with 4-oxohispidin (7) as intermediate state. Two transition states were determined in the gas phase and in PCM at B3P86/6-31+G (d, p) levels. As can be seen in the coordinate reaction diagrams (Figure 3b,c), the initial compound hispidin is more stable than the final isohispidin. The equilibrium shifts toward the lower Gibbs energy and the formation of hispidin should be thus favored. However, the presence of two transition states on both sides of intermediate state 7 indicates that the intermediate must overcome an activation energy barrier before reverting to 2 or 3. The activation energies calculated in gas between hispidin and TS1, and between the intermediate and TS2 are 52 and 61 kcal/mol, respectively. PCM has negligible effects on activation energies with a variation of less than 2 kcal/mol. The high activation barriers can be related to the one-step direct intramolecular hydrogen transfer mechanisms that take place through highly strained four-membered transition states. An alternative mechanism that would explicitly involve a solvent molecule would allow considering an indirect hydrogen transfer through a six-membered cycle transition state that would be far less strained and thus significantly decrease the energy barriers. Such an approach has been employed in several studies. Emamian et al. studied the solvent effect on the tautomerization of pyridazinone into pyridazole by combining explicit and solvation models and highlighted the significance of explicit solvent molecules involved in six or higher membered TSs in reducing the activation barriers [22]. Chahkandi et al. investigated the effect of 1-3 water assisted molecules on the activation energies of the tautomerism reaction of 1,3,4-oxadiazole derivatives and found that the energy barriers were strongly reduced in presence of water molecules [23]. We thus used an hybrid model i.e., PCM + one solvent molecule (Figure 3c). Taking into account an assisting solvent molecule results in a significant decrease of the energy barriers to 27 and 31 kcal/mol for TS1 and TS2, respectively. This shows that an intramolecular mechanism is much less likely than a solvent assisted mechanism.
Figure 3.

(a) Tautomerism of hispidin to isohispidin; (b) tautomerism chemical profiles in gas and solvent; and (c) tautomerism chemical profile in hybrid model (PCM + one assisted solvent molecule).
From these results, it appears that hispidin is more stable compared to isohispidin and that interconversion is unlikely at room temperature, thus explaining the description of hispidin and isohispidin as two distinct compounds.
BDEs were calculated for both tautomers in order to study possible differences in antioxidant activity. The results showed similar BDEs for both tautomers (Table 1). For instance, the BDE difference between the active OH groups in hispidin and isohispidin is less than 0.2 kcal/mol and 0.3 kcal/mol in the gas phase and PCM, respectively.
Table 1.
BDEs and IPs of hispidin and isohispidin calculated using the B3P86 hybrid functionals and 6-31+G(d,p) and 6-311+G(d,p) basis sets.
| Compounds | Gas | PCM | ||
|---|---|---|---|---|
| B3P86/6-31+G(d,p) | B3P86/6-311+G(d,p) | B3P86/6-31+G(d,p) | B3P86/6-311+G(d,p) | |
| BDE (kcal/mol) | ||||
| Hispidin | ||||
| 4-OH | 90.0 | 88.2 | 90.6 | 89.5 |
| 11-OH | 78.9 | 78.7 | 80.3 | 80.0 |
| 12-OH | 75.0 | 74.6 | 76.2 | 75.9 |
| Isohispidin | ||||
| 2-OH | 79.2 | 79.0 | 80.9 | 80.6 |
| 11-OH | 79.0 | 78.9 | 80.3 | 80.0 |
| 12-OH | 75.2 | 74.9 | 76.5 | 76.1 |
| IP(eV) | ||||
| Hispidin | 7.75 | 7.82 | 6.24 | 6.30 |
| Isohispidin | 7.96 | 8.10 | 6.39 | 6.39 |
2.2.2. Isomerism Effect
Styrylpyrone oligomers often exist in isomeric and conformer forms. We had previously tested the conformation effect on the antioxidant activity of a series of guaiacol oligomers and showed that the stable conformers have similar BDEs, IPs, HOMO and LUMO molecular orbitals parameters [11]. For hispidin oligomers, due to the restricted rotation of C7=C8, only cis and trans isomers need to be considered. We calculated BDEs, IPs and HOMO and LUMO distribution for cis and trans hispidin isomers (Table 2 and Figure 4a). Similar BDEs values were obtained for both isomers (differences less than 0.6 kcal/mol in solvent) and IP (7.3 eV). The HOMO and LUMO molecular orbitals were also found very similar for both cis and trans isomers (Figure 4). The trans isomer is more stable than cis isomer by 9 kcal/mol. In trans isomer, the structure is totally planar, while the structure of the cis isomer slightly deviates out of plane due to the steric hindrance between the aromatic and lactone rings. However, conjugation over the entire molecule is observed for both cis and trans isomers (Figure 4a).
Table 2.
BDEs and IPs of hispidin oligomers calculated at B3P86/6-31+G(d,p) level.
| Compounds | BDEs (kcal/mol) | IP (eV) | IC50 (µmol/L) | |||||
|---|---|---|---|---|---|---|---|---|
| 4-OH | 11-OH | 12-OH | 4'-OH | 11'-OH | 12'-OH | |||
| (a) Gas phase | ||||||||
| Hispidin (trans) | 90.0 | 78.9 | 75.0 | - | - | - | 7.75 | 1.31 ± 0.81 |
| Hispidin (cis) | 89.1 | 78.2 | 76.0 | - | - | - | 7.95 | 1.31 ± 0.81 |
| 3,14'-Bihispidinyl | 83.6 | 78.9 | 74.8 | 89.2 | 77.1 | 74.6 | 7.08 | 0.90 ± 0.61 |
| Hypholomine B (syn) | - | 77.4 | 73.9 | 89.9 | 77.5 | 77.5 | 7.7 | 0.31 ± 0.22 |
| Hypholomine B (anti) | - | 88.6 | 75.4 | 91.2 | 77.8 | 77.7 | 7.7 | 0.31 ± 0.22 |
| 1,1-Distyrylpyrylethan | 101.2 | 78.8 | 74.6 | 99.2 | 88.6 | 74.7 | 7.2 | 0.37 ± 0.15 |
| (b) PCM | ||||||||
| Hispidin (trans) | 90.6 | 80.3 | 76.2 | - | - | - | 6.24 | 1.31 ± 0.81 |
| Hispidin (cis) | 90.9 | 79.3 | 77.0 | - | - | - | 6.30 | 1.31 ± 0.81 |
| 3,14'-Bihispidinyl | 84.8 | 80.4 | 76.3 | 90.2 | 79.3 | 76.6 | 6.10 | 0.90 ± 0.61 |
| Hypholomine B (syn) | - | 79.1 | 75.7 | 92.8 | 79.4 | 79.2 | 6.3 | 0.31 ± 0.22 |
| Hypholomine B (anti) | - | 84.7 | 76.3 | 92.7 | 79.9 | 79.7 | 6.3 | 0.31 ± 0.22 |
| 1,1-Distyrylpyrylethan | 97.2 | 79.7 | 76.1 | 94.4 | 85.4 | 76.2 | 6.2 | 0.37 ± 0.15 |
Figure 4.

HOMO and LUMO distributions for styrylpyrones (a) cis and trans hispidin (2) isomers; (b) syn and anti hypholomine B (5) isomers.
In case of dimer 5, the orientation of the lactone and aromatic rings with respect to the covalent bond (C7'-C8') in the tetrahydrofuran ring leads to syn- and anti-isomers. Recently, we showed that syn and anti terrein isomers displayed similar antioxidant activities [24]. Urbaniak et al. showed that E and Z ferulic acid have similar free radical scavenging capacity [25]. In accordance with hispidin isomers results, both syn- and anti-dimer 5 isomers showed similar theoretical results (Table 2 and Figure 4b). For the other hispidin oligomers we only considered the anti-isomers, as they were identified experimentally as the stable isomers. For dimers 4 and 6, the free rotation of the C-C covalent bonds (linking hispidin moieties) gives rise to diverse conformers. Two conformers were obtained for 4 with torsion angles of 60 and 120 degrees. For these two conformers, the hydrogen bonds between (i) OH groups and (ii) OH groups and carbonyl group were taken into consideration. Calculations showed that the conformation had little influence on theoretical parameters. Therefore, only the most stable conformer with the lowest energy and intramolecular hydrogen bonds was considered.
2.2.3. BDEs Analysis
On the basis of IC50 (the capacity of hispidin oligomer to scavenge 50% of DPPH free radical) values (Table 2), hispidin oligomers were capable of scavenging DPPH free radical in the following order: hypholomine B > 1,1-distyrylpyrylethane > 3,14'-bihispidinyl > hispidin [19]. For each hispidin oligomer, the lowest BDEs were obtained for 12-OH and 12'-OH groups of catechol rings (Table 2). The BDEs values are relatively close to each other, with differences less than 0.6 kcal/mol in PCM. Such differences do not explain the observed results. The lowest BDEs being associated with 12-OH groups come from the fact that the electronic spin density distribution of 12-OH phenoxyl radical is delocalized over the radical, which is not the case for other i-OH radicals (Figure 5). The high antioxidant activity of dimers 4–6 with regard to hispidin is probably related to the number of catechol and OH groups (Figure 1). This result was consistent with the good antioxidant role of the catechol moiety generally observed for polyphenols. The BDEs values of hispidin oligomers are relatively similar, thus the correlation to experiment results is comparatively weak. To improve such correlation, we calculated BDEd parameter (BDEs calculated after a second hydrogen atom transfer), which showed a good correlation to the antioxidant activity of guaiacol oligomers [11].
Figure 5.

Spin density distribution for i-O● styrylpyrone radicals (a) hispidin; (b) 3,14'-bihispidinyl; (c) hypholomine B.
2.2.4. Double BDEs Analysis
In Table 3, we report BDEd for OH groups (after a second hydrogen atom transfer) and IPd for hispidin oligomers in the gas phase and PCM. For each hispidin oligomers, the lowest BDEd is obtained for the 11-OH group. The lowest of all BDEd is obtained for the 11-OH group of 5 with a value of 77.4 kcal/mol. This BDEd is in a good agreement with the experimental results, which showed that oligomer 5 is the most potent antioxidant among tested hispidin oligomers with the lowest IC50 value (Table 3). The BDEd of 11-OH groups of 4 and 5 are relatively similar (Table 2). The high antioxidant activity of 5 with respect to 4 could be explained by the low BDE of a third hydrogen atom transfer (BDEt). These results are in agreement with recent results reported by Amić et al. who uncovered the significant role of BDEd and IPd for a double PCET mechanism of free radical scavenging potency in flavonoids [26].
Table 3.
BDEd and IPd of hispidin oligomers calculated at B3P86/6-31+G(d,p) level.
| Compounds | BDEd (kcal/mol) | IPd (eV) | IC50 (µmol/L) | ||||
|---|---|---|---|---|---|---|---|
| 4-OH | 11-OH | 4'-OH | 11'-OH | 12'-OH | |||
| (a) Gas phase | |||||||
| Hispidin | 99.3 | 81.8 | - | - | - | 7.9 | 1.31 ± 0.81 |
| 3,14'-Bihispidinyl | 89.4 | 82.2 | 106.9 | 82.1 | 87.6 | 7.4 | 0.90 ± 0.61 |
| Hypholomine B ( anti) | - | 81.2 | 111.0 | 96.2 | 95.7 | 7.9 | 0.31 ± 0.22 |
| 1,1-Distyrylpyrylethan | 108.4 | 81.7 | 100.8 | 97.2 | 90.6 | 7.4 | 0.37 ± 0.15 |
| (b) PCM | |||||||
| Hispidin | 93.9 | 78.2 | - | - | - | 6.3 | 1.31 ± 0.81 |
| 3,14'-Bihispidinyl | 89.4 | 78.9 | 99.0 | 84.5 | 89.0 | 6.2 | 0.90 ± 0.61 |
| Hypholomine B ( anti) | - | 77.4 | 101.1 | 97.6 | 97.0 | 6.3 | 0.31 ± 0.22 |
| 1,1-Distyrylpyrylethan | 101.3 | 78.8 | 98.5 | 97.5 | 91.2 | 6.2 | 0.37 ± 0.15 |
2.2.5. Effect of the Number of OH Groups and Conjugation of the Catechol Moiety
QSAR studies of antioxidant activity of polyphenols confirmed the significant role of the number of hydroxyl groups and catechol moieties conjugated to double bond in increasing the antioxidant activity [17,27]. These effects are well illustrated on the present series of hispidin derivatives. For instance, hispidin 2, with three hydroxyl groups, is less active than its dimers 4 and 5 with five and six hydroxyl groups respectively. Hypholomine B (5) possesses two catechol moieties; on one side the catechol moiety is conjugated to a double bond (C7=C8) and on the other side the catechol is attached to a simple covalent bond (C7'-C8'). The difference between BDEs values of 12'-OH and 12-OH is 2.3 kcal/mol. This difference demonstrates the importance of a double bond conjugated to a catechol moiety, which extends the electronic spin delocalization over the radical (Figure 5c). The improvement of the antioxidant activity of dimer 4 compared to hispidin 2 can be related to the elongation of the conjugated chain. This result is in good agreement with a recent study by Lu et al. who showed that the elongation of resveratrol chain significantly increases the antioxidant activity [28].
2.3. Thermodynamical and Kinetic Study
2.3.1. Thermodynamic of the PC-ET Mechanism
To correlate theoretical parameters and experimental results for DPPH free radical scavenging activity, free Gibbs energies (ΔG) were evaluated for the reaction between hispidin oligomers and DPPH or CH3OO• free radicals. For both free radicals, the PC-ET and ET-PT mechanisms were studied (Figure 6 and Table 4). ΔGET values of the first step of the ET-PT mechanism are strongly influenced by the PCM [10,11], where the solvent induces a decrease of 55–70 and 90–116 kcal/mol for DPPH and CH3OO•, respectively. Even in PCM, the ET mechanism is endergonic. It must be stressed that the ET physical process is just a first step that is followed by heterolytic bond dissociation. ΔGET−PT results from these two steps. The reactions between hispidin oligomers and DPPH radical are exergonic with ΔGPC−ET close to −4 kcal/mol (Table 5a).
Figure 6.

Chemical profiles for PC-ET and ET-PT mechanisms.
Table 4.
Thermodynamics (ΔGPC−ET and ΔGET in kcal/mol) of the DPPH and CH3OO• free radical scavenging by styrylpyrone derivatives.
| ΔGPC−ET | ΔGET | |||||||
|---|---|---|---|---|---|---|---|---|
| DPPH | CH3OO• | DPPH | CH3OO• | |||||
| Gas | PCM | Gas | PCM | Gas | PCM | Gas | PCM | |
| (a) B3P86//First PC-ET | ||||||||
| Hispidin | −4.27 | −0.12 | −8.76 | −8.13 | 88.50 | 22.20 | 143.63 | 41.55 |
| 3,14'−Bihispidinyl | −4.10 | 0.09 | −8.59 | −7.91 | 74.25 | 19.45 | 129.38 | 38.80 |
| Hypholomine B | −3.29 | −0.28 | −7.78 | −8.28 | 87.50 | 22.63 | 142.63 | 41.98 |
| Pinilidin | −4.54 | −0.15 | −9.02 | −8.16 | 75.65 | 20.21 | 130.78 | 39.56 |
| (b) B3P86//Second PC-ET | ||||||||
| Hispidin | 4.04 | 3.42 | −0.45 | −4.58 | 93.84 | 25.88 | 148.97 | 45.22 |
| 3,14'-Bihispidinyl | 2.41 | 2.06 | −2.07 | −5.94 | 81.36 | 22.98 | 136.49 | 42.32 |
| Hypholomine B | 1.67 | 1.13 | −2.82 | −6.88 | 92.45 | 25.64 | 147.59 | 44.99 |
| Pinilidin | 4.09 | 2.04 | −0.40 | −5.97 | 82.16 | 23.00 | 137.29 | 42.34 |
| (c) MPWB1K//First PC-ET | ||||||||
| Hispidin | −2.26 | 1.34 | −5.78 | −5.07 | 43.94 | 26.24 | 163.06 | 46.97 |
| 3,14'-Bihispidinyl | −2.00 | 1.51 | −5.33 | −4.90 | 81.13 | 24.85 | 146.25 | 45.58 |
| Hypholomine B | −1.18 | 1.22 | −4.71 | −5.19 | 97.51 | 27.24 | 162.63 | 47.96 |
| Pinilidin | −2.36 | 1.39 | −5.89 | −4.41 | 85.23 | 25.53 | 150.35 | 123.55 |
| (d) MPWB1K//Second PC-ET | ||||||||
| Hispidin | 18.25 | 17.15 | 0.13 | −3.66 | 109.41 | 40.95 | 163.91 | 50.20 |
| 3,14'-Bihispidinyl | 16.92 | 15.79 | 13.10 | 9.38 | 96.93 | 38.05 | 162.05 | 50.87 |
| Hypholomine B | 15.88 | 14.85 | 12.35 | 8.44 | 108.2 | 40.72 | 173.14 | 61.44 |
| Pinilidin | 18.30 | 15.76 | 14.77 | 9.35 | 97.73 | 38.07 | 162.85 | 58.79 |
Table 5.
Calculated thermodynamics (ΔG in kcal/mol) and kinetics (ΔG# in kcal/mol and K in M−1·s−1) parameters of PC-ET mechanism of the CH3OO• (R•) free radical scavenging by styrylpyrones.
| ΔG | ΔG# | KTST | KTST/W | KTST/ST | |
|---|---|---|---|---|---|
| (a) B3P86//Gas phase | |||||
| Hispidin + R• → [Hispidin-H]• + RH | −8.76 | 4.89 | 1.61*109 | 4.29*109 | 1.59*1010 |
| 3,14'-Bihispidinyl + R• → [3,14'-Bihispidinyl -H]• + RH | −8.59 | 5.15 | 1.04*109 | 2.73*109 | 3.63*109 |
| Hypholomine B + R• → [Hypholomine B -H]• + RH | −7.78 | 6.00 | 2.49*109 | 6.93*108 | 2.74*109 |
| Pinilidin + R• → [Pinilidin -H]• + RH | −9.02 | 4.89 | 1.61*109 | 4.08*109 | 1.28*1010 |
| (b) B3P86//PCM | |||||
| Hispidin + R• → [Hispidin-H]• + RH | −8.13 | 8.55 | 3.32*106 | 1.52*107 | 1.20*107 |
| 3,14'-Bihispidinyl + R• → [3,14'-Bihispidinyl -H]• + RH | −7.91 | 11.30 | 3.20*104 | 1.62*105 | 1.02*105 |
| Hypholomine B + R• → [Hypholomine B -H]• + RH | −8.28 | 8.22 | 5.87*106 | 2.66*107 | 1.84*107 |
| Pinilidin + R• → [Pinilidin -H]• + RH | −8.16 | 5.66 | 1.62*109 | 1.28*109 | 1.80*1010 |
| (c) MPWB1K//Gas phase | |||||
| Hispidin + R• → [Hispidin-H]• + RH | −5.78 | 16.96 | 2.36 | 6.30 | 2.68*101 |
| 3,14'-Bihispidinyl + R• → [3,14'-Bihispidinyl -H]• + RH | −5.33 | 17.20 | 1.58 | 4.12 | 7.35 |
| Hypholomine B + R• → [Hypholomine B -H]• + RH | −4.71 | 18.02 | 3.96*10−1 | 1.10 | 5.66 |
| Pinilidin + R• → [Pinilidin -H]• + RH | −5.89 | 17.24 | 1.47 | 3.73 | 13.31 |
| (d) MPWB1K//PCM | |||||
| Hispidin + R• → [Hispidin-H]• + RH | −5.07 | 17.64 | 7.40*10−1 | 3.38 | 1.47*102 |
| 3,14'-Bihispidinyl + R• → [3,14'-Bihispidinyl -H]• + RH | −4.9 | 20.40 | 7.03*10−3 | 3.56*10−2 | 2.41 |
| Hypholomine B + R• → [Hypholomine B -H]• + RH | −5.19 | 17.82 | 5.46*10−1 | 2.48 | 1.49*102 |
| Pinilidin + R• → [Pinilidin -H]• + RH | −4.41 | 16.02 | 1.14*101 | 5.10*101 | 3.51*103 |
ΔGPC−ET values for a second PC-ET are shown in Table 4b. In case of peroxyl radical scavenging by hispidin oligomers, the second PC-ET is thermodynamically favored with a ΔGPC−ET close to −6 kcal/mol.
2.3.2. Kinetics of the Reaction Styrylpyrone + CH3OO• → [styrylpyrone-H]• + CH3OOH
According to the thermodynamic data of ET-PT, the first step was clearly the limiting step (e.g., for the hispidin reactivity with CH3OO•, ΔGET = +41.55 kcal/mol and ΔGPT = −49.68 kcal/mol in PCM. The ET transition states cannot be calculated exactly, but the activation free energy is obviously higher than ΔGET (i.e., higher than 41.55 kcal/mol). The transition state is close to the energy required for a vertical transition, which was calculated to be at least 3–4 kcal/mol higher than ΔGET. Thus, the energy of activation would be significantly higher for ET than for the first PC-ET mechanism, which makes the ET rates higher than those of PC-ET. This indicates that PC-ET was faster than ET-PT. PC-ET appears as a major mechanism, while ET-PT would be minor for this series of compounds. The relatively small barriers (or high rate constants) obtained for the major mechanism (PC-ET) seem to indicate that the reaction is mainly thermodynamically governed (Table 5). It is known that the hybrid functional B3P86 underestimates activation barriers. Indeed, our results appear as too low. Better results were obtained with hybrid functional MBWB1K (Table 5).
These results highlight the importance of thermodynamic parameters (BDE, BDED, and ΔGPC−ET) to rationalize the antioxidant activity of hispidin oligomers. In addition to BDE and BDEd parameters, the abstraction of the hydrogen atom depends on steric hindrance and electrostatic interaction between the ArOH and •OOCH3 in the transition states. The activation energies decrease with the increase of electrostatic interactions. In the current study, the activation energy was found inversely proportional to dH…OOCH3 (R2 = 72%) and proportional to dArO…H (R2 = 69%) in the transition states.
3. Methodology
It is well known that polyphenols ArOH (e.g., styrylpyrones) scavenge free radicals (R•) by their ability to donate hydrogen atoms from hydroxyl groups via the following probable mechanisms:
-
(i)
Proton Coupled-Electron Transfer (PC-ET) versus Hydrogen atom transfer (HAT)
ArOH + R• → ArO• + RH In the above reaction, the electron and proton are transferred from the active phenolic hydroxyl group to the free radical in a single step. This type of reactions can be subdivided into two distinct subclasses, hydrogen atom transfer (HAT) and proton coupled electron transfer (PC-ET) [14,29,30]. In HAT, the proton and electron are transferred together, as a hydrogen atom. In PC-ET mechanism, the proton and electron are transferred between different sets of orbitals [14]. Depending on the structure of reactants and the environment (e.g., solvent, enzyme), this reaction can proceed via HAT or PC-ET [31]. Various criteria can be used to distinguish between HAT and PC-ET. Tishchenko et al. used electronic adiabacity and nonadiabactity criterion to determine the mechanism involved in the reactivity of phenol with •NH2 and •OOCH3 radicals [31]. With regard to this criterion, HAT is electronically adiabatic, while PC-ET involves electronic nonadiabatic effects [31]. By using orbital-based criterion, Mayer et al. found that the degenerate hydrogen atom exchange reaction between phenoxyl radical (PhO•) and phenol (PhOH) involves a transfer of a proton and an electron between two different sets of molecular orbitals (PC-ET mechanism), while the reaction between benzyl radical and toluene involves the transfer of proton and electron between the same sets of molecular orbitals (HAT mechanism) [14]. As shown by Mayer et al. and Oksana et al. the reactivity of phenols with free radicals such CH3OO• and PhO• involve a PC-ET mechanism [14,31]. Therefore, one can consider that the reactivity of the current series of hispidin oligomers (ArOH) with CH3OO• proceeds through a PC-ET mechanism. The natural bond orbitals (NBO) charges were calculated for the reactants (hispidin oligomers and peroxy radical) and TS structures using both hybrid functionals B3P86 and MPWB1K in gas and solvent. The results showed a slight variation of NBO charges of the abstracted hydrogen atom between the TS and the reactants. For instance, a slight increase of 0.02 was obtained in gas phase with MPWB1K hybrid functional. However, the sum of the oxygen atoms (bold atoms) in the reactants [Ph-O-H + •O-OCH3] and transition state [Ph-O…H…O-OCH3]• are −0.90 and −1.06, respectively. Thus, −0.16 more negative charge resides on the oxygen at the TS than in the reactants. In the PC-ET mechanism of phenoxyl/phenol, Mayer et al. found −0.17 more negative charge residing on oxygen atoms at the TS than in the reactants [14]. From this results, the reactivity of the hispidin oligomers with CH3OO• can be considered as proceeding through a PC-ET mechanism.
The O-H bond dissociation is homolytic. Therefore, the bond dissociation enthalpy (BDE) of OH groups is considered as the main parameter that governs this mechanism. BDEs are calculated for each OH group of hispidin oligomers by the following formula:
where H is the enthalpy that takes into account temperature-dependent corrections [zero point energy (ZPE), translational, rotational and vibrational energies at 298K]; H (ArOH, 298K) and H (ArO•, 298K) are the enthalpies of the hispidin oligomer and its corresponding radical (obtained after homolytic OH bond dissociation), respectively; and H (H•, 298K) is the enthalpy of hydrogen radical. BDE is an intrinsic parameter that helps to estimate the capacity of a compound to lose an hydrogen atom. The lower is the BDE, the easier is the OH bond dissociation and the more important is its role in the antioxidant reactivity. The PC-ET mechanism [Eq. (1)] depends as well on the reactivity of the antioxidant with the free radical R• (i.e., DPPH, CH3OO•). The reaction is thermodynamically favorable (exergonic) if its Gibbs free energy (ΔG) is negative. The PC-ET mechanism can be followed by a second hydrogen atom transfer from the phenoxyl radical (i.e., radical obtained from the first hydrogen atom transfer). Double bond dissociation enthalpy (BDEd) becomes an important parameter when calculated BDEs are similar. BDEd are calculated by the following formula:BDE = H(ArO•, 298K) + H(H•, 298K) − H(ArOH, 298K), (1)
where H ([ArO•− H], 298K) and H (ArO•, 298K) are the enthalpies of phenoxyl radical obtained after the first and second hydrogen atom transfer, respectively. As for BDE, the lower is the BDEd, the easier is the O-H bond rupture of phenoxyl radical, and the more important is its role in the antioxidant reactivity. We have demonstrated in a previous study the significant role of BDEd parameter to rationalize the antioxidant activity of a series of guaiacol oligomers [11].BDEd = H([ArO•− H], 298K) + H(H•, 298K) − H(ArO•, 298K), (2) -
(ii)
Electron Transfer-Proton Transfer (ET-PT)
ArOH + R• → ArOH+•+ R− → ArO• + RH The ET-PT mechanism consists of two steps. In the first step, an electron transfer (ET) from the styrylpyrone to the free radical leads to the formation of radical cation ArOH+•. In the second step, a heterolytic O-H bond dissociation of the radical cation (i.e., proton loss) leads to the formation of a phenoxyl radical. This mechanism is governed by the ionization potential (IP) of the radical cation and the Gibbs free energy reaction of the first step. The lower is the IP value, the easier is the electron transfer and the higher is the antioxidant activity.
-
(iii)
Sequential Proton Loss Electron Transfer (SPLET)
ArOH → ArO− + H+ ArO− + R• → ArO• + R− R− + H+ → RH The SPLET mechanism consists of three steps. In the first step, a heterolytic bond dissociation of a phenolic hydroxyl group leads to the formation of a phenoxyl anion and the release of a proton. In the second step, an electron transfer from the phenoxyl anion to the free radical leads to the formation of a phenoxyl radical and an anion (R−). In the last step, the protonation of R− leads to the formation of RH. This mechanism is strongly favored under alkaline conditions (e.g., high pH), which may help in the proton of the first step [32,33].
-
(iv)
Adduct formation (AF)
ArOH + R• → [ArOH-R]• → stable adducts The AF mechanism is more specific and is observed between (a) carbon centered radicals and double bonds; or (b) hydroxyl radicals and aromatic rings. Numerous side reactions may occur that lead to stable adducts from [ArOH-R]•. In the present study, we focused on the first two mechanisms PC-ET and ET-PT. The identification of the major mechanism was carried out in two stages, which were (i) determination of the most active OH group (i.e., those with low BDEs); and (ii) thermodynamic and kinetic studies of the reactivity of the active OH groups with free radicals (e.g., CH3OO• and DPPH). Both mechanisms lead to the formation of the same final products and thus possess the same free Gibbs reaction energy. Consequently, they are a similar from a thermodynamic point of view (ΔGPC−ET = ΔGET−PT). The favored mechanism was determined kinetically by calculating the activation free energies of PC-ET and ET-PT mechanisms (ΔG#).
All theoretical calculations (conformational studies, optimization and frequency calculations) were carried out using DFT methods as implemented in Gaussian09 [34]. The hybrid functional B3B86 proved to be the most suitable for structure-antioxidant activity of polyphenols and is deemed particularly adapted for phenol BDEs by giving a high accuracy compared to the experimental values [12]. Based on these studies, we extended the use of B3P86 hybrid functionals to hispidin derivatives 2–6. To test the basis set effect, we calculated BDEs for hispidin (2) and its tautomer isohispidin (3) using double and triple basis sets 6-31+G(d,p) and 6-311+G(d,p), respectively. The obtained BDEs exhibited a difference lower than 0.4 kcal/Mol for the active OH group (12-OH) in gas or PCM (Table 1). Such a difference is low enough to be considered as a part of the global error. Consequently, we used double basis set 6-31+G(d, p) for hispidin oligomers 4–6. The geometries, enthalpies (H), and Gibbs energies (G) of reactants, intermediates, and products were determined at B3P86/6-31+G(d, p) level.
All ground states were confirmed by vibrational frequency analysis (i.e., no imaginary frequency). The transition states (TSs) were confirmed by the presence of one imaginary frequency, which was assigned to the vibration mode corresponding to the reaction studied (e.g., bond formation, bond breaking). The TS Gibbs free energy allowed us to evaluate the free ΔG#. The activation free energies are known to be underestimated by B3P86, whereas the hybrid functional MPWB1K appeared adequate to reproduce ΔG# [35], especially for hydrogen atom transfer reactions [36]. All calculations related to the kinetic study (i.e., reactant, product, and TS energies) were performed using B3P86 and MPWB1K hybrid functionals. The solvent effects (MeOH) were taken into account implicitly using the polarizable continuum model (PCM). In PCM, the substrate is embedded into a cavity surrounded by a dielectric continuum characterized by its dielectric constant (εMeOH = 32.613). Different PCM models are available. The calculations were performed using the integral equation formalism variant (IEFPCM) as implemented in the Gaussian09 program [34]. The use of an explicit solvent-surrounded OH phenol group had been investigated and PCM was confirmed to give a better description of BDEs [37]. Trouillas et al. tested a hybrid model (i.e., one or two water molecules surrounding OH groups + PCM) for quercetin flavonoid. They showed that resulting BDEs were slightly different from those obtained with PCM only [10]. Regarding the kinetic study of the PC-ET mechanism, the determination of rate constants is more reliable than calculation of activation energies (ΔG#) only, since the former would take into account the tunneling in hydrogen shift [38,39]. These rate constants were calculated using transition state theory (TST) according to the following Equation (3):
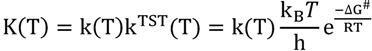 |
(3) |
where k(T) is the transmission coefficient. It represents the quantum tunneling through the reaction barrier. The commonly and easiest used method for evaluating the tunneling is the Wigner method as in Equation (4) below [40]:
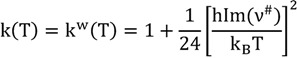 |
(4) |
where ν# is the transition state imaginary frequency corresponding to the reaction coordinate, T the temperature, and h and kB are Planck’s and Boltzmann’s constants, respectively. The imaginary frequency is used to estimate the width of the barrier. However, no term accounts for the barrier height (activation energy) in Winger’s method.
Skodje and Truhlar proposed an improvement over Wigner’s method [41]. The Skodje and Truhlar method accounts for both the barrier width through the imaginary frequency, and the barrier height. The transmission coefficients are obtained by using the following expressions [Equations (5) and (6)]:
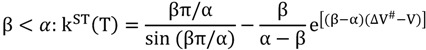 |
(5) |
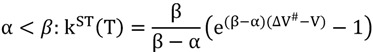 |
(6) |
where:
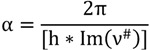 |
(7) |
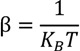 |
(8) |
ΔV# is the zero-point-including potential energy difference between the transition state and the reactants.
4. Conclusions
In the present study, we highlighted the role of different descriptors BDEs, BDEd, number of OH groups, and conjugated double bond to a catechol group on the antioxidant activity of a series of hispidin oligomers. Theoretical results showed that BDEd is most significant parameter to understand and rationalize the experimental antioxidant activity values of hispidin derivatives. Thermodynamic and kinetic studies showed that the PC-ET mechanism is the favored mechanism for the title compounds to scavenge free radicals.
Acknowledgments
The authors acknowledge the Ministry of Higher Education (MOHE) for financial support under the Fundamental Research Grant Scheme (FRGS) with reference numbers 600-RMI/FRGS 5/3 (12/2012) and 600-RMI/ST/FRGS 5/3/Fst (4/2011), Universiti Teknologi MARA for financial support under the Cumulative Impact Factor Initiative (CIFI) Grant Scheme with reference number UiTM 600-RMI/DANA 5/3/CIFI (117/2013). The authors thank Patrick Trouillas, University of Limoges, France, for providing access to supercomputer CALI (CAlcul en LImousin).
Author Contributions
El Hassane Anouar and Jean-Frédéric F. Weber designed the research; Syed Adnan Ali Shah, Normahanim Binti Hassan and Najoua El Moussaoui performed research and analyzed the data; Rohaya Ahmad, Mohd Zulkefeli and Jean-Frédéric F. Weber analyzed the data and El Hassane Anouar wrote the paper. All authors read and approved the final manuscript.
Conflictts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Not available.
References
- 1.Jewers K., Davis J.B., Dougan J., Manchanda A.H., Blunden G., Kyi A., Wetchapinan S. Goniothalamin and its distribution in four Goniothalamus species. Phytochemistry. 1972;11:2025–2030. doi: 10.1016/S0031-9422(00)90168-7. [DOI] [Google Scholar]
- 2.Fiasson J.-L. Distribution of styrylpyrones in the basidiocarps of various Hymenochaetaceae. Biochem. Syst. Ecol. 1982;10:289–296. doi: 10.1016/0305-1978(82)90002-3. [DOI] [Google Scholar]
- 3.Zhu T., Guo J., Collins L., Kelly J., Xiao Z.J., Kim S.H., Chen C.Y. Phellinus linteus activates different pathways to induce apoptosis in prostate cancer cells. Br. J. Cancer. 2007;96:583–590. doi: 10.1038/sj.bjc.6603595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saar M. Fungi in khanty folk medicine. J. Ethnopharmacol. 1991;31:175–179. doi: 10.1016/0378-8741(91)90003-V. [DOI] [PubMed] [Google Scholar]
- 5.Huang N.L. A mysterious medicinal fungus in Russia: Inonotus obliquus. Edible Fungi China. 2002;21:7–8. [Google Scholar]
- 6.Margaill I., Plotkine M., Lerouet D. Antioxidant strategies in the treatment of stroke. Free Radic. Biol. Med. 2005;39:429–443. doi: 10.1016/j.freeradbiomed.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 7.Lemanska K., Szymusiak H., Tyrakowska B., Zielinski R., Soffers A.E., Rietjens I.M. The influence of pH on antioxidant properties and the mechanism of antioxidant action of hydroxyflavones. Free Radic. Biol. Med. 2001;31:869–881. doi: 10.1016/S0891-5849(01)00638-4. [DOI] [PubMed] [Google Scholar]
- 8.Priyadarsini K.I., Maity D.K., Naik G.H., Kumar M.S., Unnikrishnan M.K., Satav J.G., Mohan H. Role of phenolic O-H and methylene hydrogen on the free radical reactions and antioxidant activity of curcumin. Free Radic. Biol. Med. 2003;35:475–484. doi: 10.1016/S0891-5849(03)00325-3. [DOI] [PubMed] [Google Scholar]
- 9.Kozlowski D., Marsal P., Steel M., Mokrini R., Duroux J.-L., Lazzaroni R., Trouillas P. Theoretical investigation of the formation of a new series of antioxidant depsides from the radiolysis of flavonoid compounds. Radiat. Res. 2007;168:243–252. doi: 10.1667/RR0824.1. [DOI] [PubMed] [Google Scholar]
- 10.Kozlowski D., Trouillas P., Calliste C., Marsal P., Lazzaroni R., Duroux J.-L. Density functional theory study of the conformational, electronic, and antioxidant properties of natural chalcones. J. Phys. Chem. A. 2007;111:1138–1145. doi: 10.1021/jp066496+. [DOI] [PubMed] [Google Scholar]
- 11.Anouar E., Calliste C.A., Kosinova P., di Meo F., Duroux J.L., Champavier Y., Marakchi K., Trouillas P. Free radical scavenging properties of guaiacol oligomers: A combined experimental and quantum study of the guaiacyl-moiety role. J. Phys. Chem. A. 2009;113:13881–13891. doi: 10.1021/jp906285b. [DOI] [PubMed] [Google Scholar]
- 12.Trouillas P., Marsal P., Siri D., Lazzaroni R., Duroux J.-L. A DFT study of the reactivity of OH groups in quercetin and taxifolin antioxidants: The specificity of the 3-OH site. Food Chem. 2006;97:679–688. doi: 10.1016/j.foodchem.2005.05.042. [DOI] [Google Scholar]
- 13.Trouillas P., Marsal P., Svobodova A., Vostalova J., Gazak R., Hrbac J., Sedmera P., Kren V., Lazzaroni R., Duroux J.-L., et al. Mechanism of the Antioxidant Action of Silybin and 2,3-Dehydrosilybin Flavonolignans: A Joint Experimental and Theoretical Study. J. Phys. Chem. A. 2008;112:1054–1063. doi: 10.1021/jp075814h. [DOI] [PubMed] [Google Scholar]
- 14.Mayer J.M., Hrovat D.A., Thomas J.L., Borden W.T. Proton-coupled electron transfer versus hydrogen atom transfer in benzyl/toluene, methoxyl/methanol, and phenoxyl/phenol self-exchange reactions. J. Am. Chem. Soc. 2002;124:11142–11147. doi: 10.1021/ja012732c. [DOI] [PubMed] [Google Scholar]
- 15.Wang L.-F., Zhang H.-Y. Unexpected role of 5-OH in DPPH radical-scavenging activity of 4-thiaflavans. Revealed by theoretical calculations. Bioorg. Med. Chem. Lett. 2004;14:2609–2611. doi: 10.1016/j.bmcl.2004.02.066. [DOI] [PubMed] [Google Scholar]
- 16.Luzhkov V.B. Mechanisms of antioxidant activity: The DFT study of hydrogen abstraction from phenol and toluene by the hydroperoxyl radical. Chem. Phys. 2005;314:211–217. doi: 10.1016/j.chemphys.2005.03.001. [DOI] [Google Scholar]
- 17.Rice-Evans C., Miller N.J., Paganga G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic. Biol. Med. 1996;20:933–956. doi: 10.1016/0891-5849(95)02227-9. [DOI] [PubMed] [Google Scholar]
- 18.Bayat A., Fattahi A. Investigation of the scavenging mechanism of tyrosyl radical by hydroxybenzohydroxamic acid derivatives: A DFT study. Comp. Theor. Chem. 2013;1018:35–44. [Google Scholar]
- 19.Jung J.Y., Lee I.K., Seok S.J., Lee H.J., Kim Y.H., Yun B.S. Antioxidant polyphenols from the mycelial culture of the medicinal fungi Inonotus xeranticus and Phellinus linteus. J. Appl. Microbiol. 2008;104:1824–1832. doi: 10.1111/j.1365-2672.2008.03737.x. [DOI] [PubMed] [Google Scholar]
- 20.Blois M.S. Antioxidant determinations by the use of a stable free radical. Nature. 1958;181:1199–1200. doi: 10.1038/1811199a0. [DOI] [Google Scholar]
- 21.Lee I.-K., Yun B.-S. Styrylpyrone-class compounds from medicinal fungi Phellinus and Inonotus spp., and their medicinal importance. J. Antibiot. 2011;64:349–359. doi: 10.1038/ja.2011.2. [DOI] [PubMed] [Google Scholar]
- 22.Emamian S.R., Domingo L.R., Tayyari S.F. Tautomerism in pyridazin-3 (2H)-one: A theoretical study using implicit/explicit solvation models. J. Mol. Graph. Model. 2014;49:47–54. doi: 10.1016/j.jmgm.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 23.Chahkandi B., Tayyari S.F., Bakhshaei M., Chahkandi M. Investigation of simple and water assisted tautomerism in a derivative of 1, 3, 4-oxadiazole: A DFT study. J. Mol. Graph. Model. 2013;44:120–128. doi: 10.1016/j.jmgm.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 24.Trabolsy Z.B.K.A., Anouar E.H., Zakaria N.S.S., Zulkeflee M., Hasan M.H., Zin M.M., Ahmad R., Sultan S., Weber J.-F.F. Antioxidant activity, NMR, X-ray, ECD and UV/visible spectra of (+)-terrein: Experimental and theoretical approaches. J. Mol. Struct. 2014;1060:102–110. [Google Scholar]
- 25.Urbaniak A., Szeląg M., Molski M. Theoretical investigation of stereochemistry and solvent influence on antioxidant activity of ferulic acid. Comp. Theor. Chem. 2013;1012:33–40. doi: 10.1016/j.comptc.2013.02.018. [DOI] [Google Scholar]
- 26.Amić A., Marković Z., Dimitrić Marković J.M., Stepanić V., Lučić B., Amić D. Towards an improved prediction of the free radical scavenging potency of flavonoids: The significance of double PCET mechanisms. Food Chem. 2014;152:578–585. doi: 10.1016/j.foodchem.2013.12.025. [DOI] [PubMed] [Google Scholar]
- 27.Rastija V., Medić-Šarić M. QSAR study of antioxidant activity of wine polyphenols. Eur. J. Med. Chem. 2009;44:400–408. doi: 10.1016/j.ejmech.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 28.Lu L., Zhu S., Zhang H., Zhang S. Improvement of antioxidative activity of resveratrol by elongating conjugated chain: A DFT theoretical study. Comp. Theor. Chem. 2013;1019:39–47. doi: 10.1016/j.comptc.2013.06.019. [DOI] [Google Scholar]
- 29.Hammes-Schiffer S. Proton-coupled electron transfer: Classification scheme and guide to theoretical methods. Energ. Environ. Sci. 2012;5:7696–7703. doi: 10.1039/c2ee03361e. [DOI] [Google Scholar]
- 30.Hammes-Schiffer S. Theoretical perspectives on proton-coupled electron transfer reactions. Acc. Chem. Res. 2001;34:273–281. doi: 10.1021/ar9901117. [DOI] [PubMed] [Google Scholar]
- 31.Tishchenko O., Truhlar D.G., Ceulemans A., Nguyen M.T. A unified perspective on the hydrogen atom transfer and proton-coupled electron transfer mechanisms in terms of topographic features of the ground and excited potential energy surfaces as exemplified by the reaction between phenol and radicals. J. Am. Chem. Soc. 2008;130:7000–7010. doi: 10.1021/ja7102907. [DOI] [PubMed] [Google Scholar]
- 32.Di Meo F., Lemaur V., Cornil J., Lazzaroni R., Duroux J.-L., Olivier Y., Trouillas P. Free Radical Scavenging by Natural Polyphenols: Atom versus Electron Transfer. J. Phys. Chem. A. 2013;117:2082–2092. doi: 10.1021/jp3116319. [DOI] [PubMed] [Google Scholar]
- 33.Musialik M., Litwinienko G. Scavenging of dpph radicals by vitamin E is accelerated by its partial ionization: The role of sequential proton loss electron transfer. Org. Lett. 2005;7:4951–4954. doi: 10.1021/ol051962j. [DOI] [PubMed] [Google Scholar]
- 34.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., et al. Gaussian 09. Revision A.02. Gaussian, Inc. ; Wallingford, CT, USA: 2009. [Google Scholar]
- 35.Zhao Y., Truhlar D.G. Hybrid meta density functional theory methods for thermochemistry, thermochemical kinetics, and noncovalent interactions: The MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and van der Waals Interactions. J. Phys. Chem. A. 2004;108:6908–6918. doi: 10.1021/jp048147q. [DOI] [Google Scholar]
- 36.Tejero I., González-García N., González-Lafont À., Lluch J.M. Tunneling in green tea: Understanding the antioxidant activity of catechol-containing compounds. a variational transition-state theory study. J. Am. Chem. Soc. 2007;129:5846–5854. doi: 10.1021/ja063766t. [DOI] [PubMed] [Google Scholar]
- 37.Guerra M., Amorati R., Pedulli G.F. Water effect on the oh dissociation enthalpy of para-substituted phenols: A DFT study. J. Org. Chem. 2004;69:5460–5467. doi: 10.1021/jo0495236. [DOI] [PubMed] [Google Scholar]
- 38.McMahon R.J. Chemical reactions involving quantum tunneling. Science. 2003;299:833–834. doi: 10.1126/science.1080715. [DOI] [PubMed] [Google Scholar]
- 39.Sirjean B., Dames E., Wang H., Tsang W. Tunneling in hydrogen-transfer isomerization of n-alkyl radicals. J. Phys. Chem. A. 2011;116:319–332. doi: 10.1021/jp209360u. [DOI] [PubMed] [Google Scholar]
- 40.Wigner E. Calculation of the rate of elementary association reactions. J. Chem. Phys. 1937;5:720–725. doi: 10.1063/1.1750107. [DOI] [Google Scholar]
- 41.Skodje R.T., Truhlar D.G. Parabolic tunneling calculations. J. Phys. Chem. 1981;85:624–628. [Google Scholar]