

Abstract
Background
Gabapentin is the most commonly prescribed medication for the treatment of chronic musculoskeletal pain in cats. Despite this common and chronic usage, clinically relevant pharmacokinetic data is lacking.
Objectives
To evaluate the pharmacokinetics of clinically relevant dosing regimens of gabapentin in cats.
Animals
Eight research‐purpose mixed‐breed cats.
Methods
Cats were enrolled in a serial order, non‐randomized pharmacokinetic study. Gabapentin was administered as an IV bolus (5 mg/kg), orally (10 mg/kg) as a single dose or twice daily for 2 weeks, or as a transdermal gel (10 mg/kg) in serial order. Serial blood samples were collected up to 48 hours. Plasma concentrations were determined using Ultra Performance Liquid Chromatography‐Mass Spectrometry. Compartmental analysis was used to generate gabapentin time‐concentration models.
Results
After IV administration CL (median (range)) and terminal half‐life were 160.67 mL/kg*hr (119.63‐199.11) and 3.78 hours (3.12‐4.47), respectively. The oral terminal half‐life was 3.63 hours (2.96‐4.77), and 3.72 hours (3.12‐4.51) for single and repeated dosing. TMAX and CMAX, as predicted by the model were 1.05 hours (0.74‐2.11), and 12.42 μg/mL (8.31‐18.35) after single oral dosing, and 0.77 hours (0.58‐1.64), and 14.78 μg/mL (9.70‐18.41) after repeated oral dosing. Bioavailability after a single oral dose was 94.77% (82.46‐122.83).
Importance
Repeated oral dosing of gabapentin did not alter the drug's pharmacokinetics, making dose adjustments unnecessary with long‐term treatment. As prepared, the transdermal route is an inappropriate choice for drug administration. These relevant data are important for future studies evaluating potential efficacy of the medication for treating chronic pain states in cats.
Keywords: compounded, feline, pain, transdermal
ABBREVIATIONS
- API
active pharmaceutical ingredient
- AUC
area under the curve for the concentration versus time profile
- BCS
body condition score
- CL
systemic clearance
- CMAX
peak concentration
- EC50
half maximal effective concentration
- F
bioavailability, or fraction absorbed unchanged
- GABA
γ‐aminobutyric acid
- HPLC UV
high‐pressure liquid chromatography with ultraviolet detection
- hr
hours
- IACUC
institutional animal care and use committee
- IS
internal standard
- IV
intravenous
- K01 T1/2
absorption half‐life after oral administration
- K01
absorption rate constant to the central compartment
- K10 T1/2
elimination half‐life after oral administration
- K10
elimination rate constant from the central compartment
- K12
distribution rate constant from the central (1) to peripheral compartment (2)
- K21
distribution rate constant from the peripheral (2) to central compartment (1)
- LOD
limit of detection
- LOQ
limit of quantitation
- min
minutes
- NCSU CVM
North Carolina State University College of Veterinary Medicine
- r2
coefficient of determination
- SWFI
sterile water for injection
- T1/2
half‐life
- TLAG
lag time or delay for drug absorption following oral administration
- TMAX
time to peak concentration
- UPLC MS:MS
ultra performance liquid chromatography with tandem mass spectrometry
- USFDA
United States Food and Drug Administration
- USP
United States Pharmacopeia
- V1
apparent volume of the central compartment
- VAPs
venous access ports
- VSS
apparent volume of distribution at steady state
- α T1/2
plasma or distribution half‐life after IV administration
- β T1/2
elimination half‐life after IV administration
1. INTRODUCTION
There are no approved medications for the treatment of chronic musculoskeletal pain in cats, and only a limited number of analgesic therapeutics with data about efficacy.1, 2, 3, 4, 5 Before the studies reported here, we surveyed veterinarians regarding pharmaceuticals and dietary supplements used for the alleviation of chronic pain in the cat.6 The most frequently prescribed treatment was gabapentin.
Gabapentin, an analogue of the neurotransmitter γ‐aminobutyric acid (GABA), is a medication commonly used in human medicine for chronic (maladaptive) pain conditions, such as diabetic neuropathy.7, 8 The drug currently has United States Food and Drug Administration (USFDA) approval for postherpetic neuralgia, and as an adjunctive therapy for partial onset seizures in humans. It is proposed to alter trafficking of voltage‐gated calcium channel subunits9, 10 which have altered expression levels in rodent neuropathic pain models.11, 12 No clinical studies have been conducted and published assessing the efficacy of gabapentin for the treatment of chronic pain in cats.
The pharmacokinetics of single oral (10 mg/kg) and intravenous (IV; 4 mg/kg) doses of gabapentin in 6 adult spayed female cats has been described,13 but no other data are available on the pharmacokinetics of gabapentin in the cat. Due of this paucity of data relevant to the clinical use of the drug, the pharmacokinetics of gabapentin administered via intravenous, oral (both single and long‐term), and transdermal routes were investigated in the cat.
2. MATERIALS AND METHODS
2.1. Animals
Healthy purpose bred male castrated (5) and female spayed (3) domestic shorthaired cats were used in this study. All cats were 2 years old. Mean body weight was 4.8 kg, with a SD of ±0.8 kg, corresponding to a median body condition score (BCS) of 5 (4‐9). Cats were housed in climate‐controlled grouped housing (70°F with relative humidity between 30 and 70%) on a 12 : 12 light : dark cycle, with rotating enrichment toys and features. Cats were monitored at least twice daily for general health and wellbeing, with any abnormalities immediately reported to the staff veterinarian. Cats were fed according to their caloric needs, and allowed ad libitum access to water during the study. All study procedures were approved by the Institutional Animal Care and Use Committee at North Carolina State University (IACUC No. 16‐187).
2.2. Instrumentation and drug administration
Venous access ports (VAPs; Companion Port 5, Norfolk Access Technologies) were surgically implanted into the right jugular vein and threaded to the junction of the cranial vena cava and right atrium of all cats as previously described.14, 15 The port was placed at least 2 weeks before drug administration. This was to facilitate blood sample collection across the multiple phases of the study, and minimize stress to the cats.
On the respective day of drug administration, gabapentin was administered as an IV bolus (5 mg/kg; n = 8) in a cephalic vein, as an oral capsule (10 mg/kg; n = 7), or as a transdermal gel (10 mg/kg; n = 7) applied to the interior ear pinnae. Compounded gabapentin products were prepared according to United States Pharmacopeia (USP) standards <795> and <797>, as appropriate, and placed in a brown bag to protect them from light.16 Gabapentin for intravenous administration was formulated by adding 0.3 g of gabapentin active pharmaceutical ingredient (API; Letco Medical) to 30 mL of sterile water for injection (SWFI) for a final concentration of 10 mg/mL. The syringe‐to‐syringe method was used to create a homogenous mixture. After a 0.2 μm filter was used to sterilize the solution in the clean room, the filter's integrity was assessed via bubble point testing. The compound was then placed in a 30 mL glass vial, and refrigerated until injection, approximately 1 hour. Gabapentin for oral administration was formulated into 2 different strengths (50 and 75 mg) by triturating gabapentin API with lactose USP (Letco Medical) via geometric dilution and placing the respective triturates into #3 size, clear capsules (Letco Medical). Gabapentin for transdermal administration was compounded into a 200 mg/mL preparation, using ethoxydiglycol (solvent; Professional Compounding Centers of America), gabapentin API, and transdermal base (Lipoderm; Professional Compounding Centers of America). The mixture was made homogenous by using the syringe‐to‐syringe method. Strength of the compounded preparations was measured at an external lab using High‐Pressure Liquid Chromatography with Ultraviolet detection (HPLC UV; Campbell University Pharmaceutical Education & Research Center, Lillington, North Carolina), and in an internal lab following the USP standards (HPLC UV; 600 Controller/Pump, 717plus Autosampler, 2487 Dual Absorbance Detector, Waters).16
On the morning of the procedure, the area overlying the VAP was numbed with a topical 4% lidocaine cream (LMX4, Ferndale Laboratories). After approximately 1 hour, the area was clipped if necessary, and cleaned with a diluted iodine solution. A 22g right angle Huber needle infusion set (Norfolk Access Technologies) was then inserted through the skin into the port hub, and patency was assessed using heparinized saline solution (BD PosiFlush, Becton, Dickinson and Company). Before all samples, a 2‐3 mL “dump sample” was collected and set aside. The 2‐3 mL sample was then collected, and the “dump sample” returned to the cat. The catheter was then rinsed with either normal or heparinized saline solution. Blood samples were collected at 1, 3, 9, 15, 30, and 60 minutes, and then at 2, 4, 6, 8, 12, and 24 hours after IV administration. After administration of a single oral dose, samples were collected at 5, 15, 30, 45, 60, and 90 minutes, followed by collection at 2, 4, 8, 12, and 24 hours. After repeated oral administration (twice daily for 14 days) of the drug, additional samples were collected immediately before the second‐to‐last and last doses, as well as at 36 hours after the final dose. Samples were collected at 15, 30, 60, and 90 minutes, followed by collection at 2, 4, 8, 12, 24, 36, and 48 hours after transdermal application of the drug. Samples were immediately placed into K2EDTA tubes (BD Vacutainer, Becton, Dickinson and Company) for processing. Samples were centrifuged at 3000g for 15 minutes at 5°C, and plasma was aliquoted into 2 separate cryotubes and stored at −80°C until analyzed by Ultra Performance Liquid Chromatography with tandem Mass Spectrometry (UPLC‐MS:MS; Acquity UPLC, Xevo Triple Quadrupole Mass Spectrometer, Waters).
Cats received gabapentin by each route in a serial order of IV bolus, single oral, repeated oral (twice daily for 14 days around normal feeding times at 0:630‐07:00 and 15:30‐16:00), and then transdermal routes. Cats were fasted a minimum of 12 hours before dose administration on sampling days, and were allowed access to food after the 2 hours sample was collected. The single oral dose was administered without any food, and was immediately followed with a 5 mL oral bolus of water. The multiple oral doses were administered with a small amount of canned food (a/d Critical Care, c/d Urinary Care, Hill's Pet Nutrition), including on the sample collection day. The repeated oral phase was started immediately after the 24 hours sample was collected for the single oral administration, and was continued for 14 days. Otherwise, a washout period of at least 3 weeks was observed between study phases (IV and single oral, multiple oral and transdermal).
2.3. Gabapentin analysis
The concentration of gabapentin in feline plasma was quantified with UPLC‐MS:MS analysis of extracted samples using prepared calibration standards. An initial stock solution of 3000 μg/mL was prepared by dissolving 15 000 μg gabapentin reference standard in 5.0 mL of 50 : 50 acetonitrile : H2O (ACN : H2O) solvent. This was then serially diluted to create working solutions of 0.5, 2.5, 5.0, 25, 50, 250, 500, and 1500 μg/mL. The plasma calibration curve was prepared by diluting the working solutions with drug‐free feline plasma (extracted using EDTA, Equitech‐Bio, Inc) for a final calibration curve of 0.01, 0.05, 0.1, 0.5, 1.0, 5.0, 10.0, 30.0, and 60.0 μg/mL. The plasma calibration curve was prepared fresh each day of analysis. A pregabalin (Lyrica, Pfizer) internal standard was used as a control for variability in extraction, injection, and ionization.17 Pregabalin internal standard was prepared by emptying and dissolving a 150 mg capsule in 30 mL of methanol, for an initial concentration of 5 mg/mL. This solution/suspension was vortexed for approximately 5 minutes, and then centrifuged at 10 000 rct for 10 minutes. Supernatant was removed and serially diluted to achieve a final concentration of 0.05 ug/mL (B. Kukanich, personal communication, December 20, 2016).
Plasma concentrations of gabapentin API, m/z 172.1 → 154, and the internal standard (IS) pregabalin, m/z 160.2 → 142.1 were determined by UPLC MS MS. Pure analytical reference standard for pregabalin was not available at the time of assay validation, however, because previous reports have shown satisfactory results when using the finished product as an internal standard17, 18 we used Lyrica as the internal standard. A subsequent comparison between the pregabalin finished product and API (USP) was made to validate results. The standard curve was linear from 0.05 to 60 μg/mL (Ultra Performance Liquid Chromatography with tandem Mass Spectrometry = 0.997 ± 0.003; n = 3). A minimum of 4 replicates of 0.05, 0.5, 5, and 60 μg/mL were used to calculate intraday accuracies of 103.9%, 85.1%, 89.0%, and 101.6% respectively. Intraday precisions were 7.53%, 3.23%, 7.36%, and 7.71%, respectively. The pregabalin to gabapentin percent recoveries using either the API or the finished product were compared at spiked gabapentin concentrations of 0.05, 0.5, 5, and 60 μg/mL. With a minimum of 3 replicates at each concentration, the average ratios (API/finished product) were 1.08, 0.99, 1.02, and 0.99, respectively. The gabapentin limit of quantitation (LOQ) was determined to be 0.05 μg/mL, as it was the lowest concentration in the standard curve with acceptable accuracy (100 +/− 15%) and precision (<10%). The limit of detection (LOD) was 0.01 μg/mL, as the concentration was repeatedly measureable with a signal to noise ratio of at least 2, though variability precluded accurate quantification. Data points below the LOQ were excluded from analysis, because of the high coefficient of variability of concentrations below 0.05 μg/mL. Sample preparation involved combining 0.2 mL of plasma, 0.2 mL of pregabalin internal standard (0.05 μg/mL) and 0.8 mL methanol with 0.1% formic acid. The sample was then vortexed for 5 seconds, and centrifuged at 15 000g for 10 minutes. A volume of 1 mL of supernatant was removed and placed into a glass test tube to be evaporated for 50 minutes at 40°C. The precipitate was then reconstituted with 0.3 mL of 50 : 50 ACN : H2O, vortexed for 30 seconds, and then filtered using 0.2 μm injection vials. A volume of 5 μL was injected for each sample.
Separation was achieved at 40°C using a phenyl column (2.1 × 100 mm, 1.7 μm phenyl column, Waters), using mobile phase of A: water with 0.1% formic acid and B: acetonitrile with 0.1% formic acid at a flow rate of 0.4 mL/min.
2.4. Pharmacokinetic analysis
All pharmacokinetic analyses were performed using commercially available software (Phoenix WinNonlin, Certara). Nonlinear least squares regression was performed on plasma gabapentin concentrations after IV or oral administration, but not after transdermal administration. Data for all routes of administration were weighted by the reciprocal of the square of the observed plasma gabapentin concentration. 1‐, 2‐, and 3‐compartment models with elimination from the central compartment were fit to the data for IV administration. 1‐ and 2‐compartment models with both first‐order absorption in and elimination from the central compartment, with or without lag time were fit to the oral administration data. Models were selected based on observation of the residuals plots, visual inspection, and by use of the Akaike information criterion (AIC).19 Bioavailability values (F) for single oral dosing were calculated by use of the following equation:
where AUCoral and AUCIV were the Area Under the Curve after oral and IV administration, respectively; and doseIV and doseoral were the dose for IV and oral administration, respectively. The parameters V1 (central volume of distribution), K10, K12, K21, were estimated by use of the model for IV administration, and V (volume of distribution), K01, and K10 were estimated for both oral administration models. Other pharmacokinetic parameters were calculated by use of standard pharmacokinetic equations. The Wilcoxon signed‐rank test was used to compare pharmacokinetic parameters after single and repeated oral administration of gabapentin.
3. RESULTS
The strength of the compounded formulations were reported as (mean ± SD, when appropriate) 99% (n = 1), 99.7 ± 5.9% (n = 3), 98.0 ± 3.5% (n = 3), and 137% (n = 1) of the labeled strengths for the IV, 50 mg capsules, 75 mg capsules, and transdermal gel, respectively. Internal lab strength measurements indicated strengths of 92.6, 95, and 94.1% for the IV, 50 mg capsule, and 75 mg capsule formulations, respectively (n = 1).
The model that was the best fit for the plasma concentrations of gabapentin after IV administration was a 2‐compartment model (Figure 1). The actual dose administered ranged from 4.90 to 5.11 mg/kg. Pharmacokinetic parameters are available in Table 1.
Figure 1.
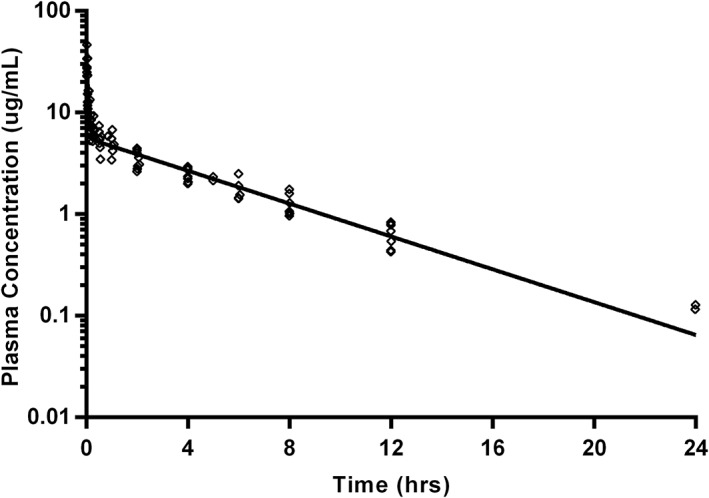
Gabapentin plasma concentrations after administration as an IV bolus (5 mg/kg) in 8 cats (open diamonds) with modeled time‐concentration curve (using geometric mean of derived parameters; solid line). Samples were collected at 1, 3, 9, 15, 30, and 60 minutes, and then at 2, 4, 6, 8, 12, and 24 hours after IV administration. Data below the limit of quantitation are excluded
Table 1.
Summary pharmacokinetics statistics after administration of gabapentin as a single IV bolus (5 mg/kg; n = 8), a single oral dose (10 mg/kg; n = 7), and repeated oral doses (10 mg/kg; n = 7)
| Parameter | IV | Single oral | Repeated oral |
|---|---|---|---|
| F (%) | * | 94.8 (82.5‐122.8) | * |
| K01 (1/hr) | * | 5.24 (1.77‐15.62) | 7.06 (1.32‐11.46) |
| K10 (1/hr) | 1.21 (0.87‐1.67) | 0.20 (0.14‐0.23) | 0.18 (0.15‐0.22) |
| K12 (1/hr) | 13.67 (8.35‐25.46) | * | * |
| K21 (1/hr) | 2.82 (1.88‐5.12) | * | * |
| V1 (mL/kg) | 129.09 (80.35‐171.50) | * | * |
| TLAG (hr) | * | 0.45a (0.24‐0.72) | 0.21a (0.01‐0.23) |
| A (μg/mL) | 32.77 (23.79‐55.89) | * | * |
| α (1/hr) | 16.97 (11.15‐31.74) | * | * |
| α T1/2 (hr) | 0.04 (0.02‐0.06) | * | * |
| B (μg/mL) | 5.61 (4.21‐6.80) | * | * |
| β (1/hr) | 0.18 (0.15‐0.22) | * | * |
| β T1/2 (hr) | 3.78 (3.12‐4.47) | * | * |
| AUC (hr*μg/mL) | 32.01 (24.61‐42.38) | 73.68 (55.71‐107.19) | 77.25 (66.26‐121.06) |
| CL (mL/kg/hr) | 160.67 (119.63‐199.11) | * | * |
| CMAX (μg/mL) | * | 12.42 (8.31‐18.35) | 14.78 (9.70‐18.41) |
| K01 T1/2 (hr) | * | 0.13 (0.04‐0.39) | 0.10 (0.06‐0.53) |
| K10 T1/2 (hr) | * | 3.53 (2.96‐4.78) | 3.90 (3.12‐4.51) |
| MRT (hr) | 5.17 (4.34‐6.05) | * | * |
| VSS (mL/kg) | 804.57 (643.71‐1049.90) | * | * |
| TMAX (hr) | * | 1.05 (0.74‐2.11) | 0.77 (0.58‐1.64) |
Results are presented as median (range). F, bioavailability, or fraction absorbed unchanged; K01, absorption rate constant to the central compartment; K10, elimination rate constant from the central compartment; K12, distribution rate constant from the central (1) to peripheral compartment (2); K21, distribution rate constant from the peripheral (2) to central compartment (1); V1, apparent volume of the central compartment; TLAG, lag time or delay for drug absorption following oral administration; A and B, and α and β = Coefficients and exponents, respectively, in the following equation used describe the drug disposition curve at time t: (A X e–αt) + (B X e–βt), where e is Euler's number (2.7183); T1/2, half‐life; α T1/2, plasma or distribution half‐life after IV administration; β T1/2, elimination half‐life after IV administration; AUC, area under the curve for the concentration versus time profile; CL, systemic clearance; CMAX, peak concentration; K01 T1/2, absorption half‐life after oral administration; K10 T1/2, elimination half‐life after oral administration; VSS, apparent volume of distribution at steady state, TMAX time to peak concentration.
Statistically significant difference (P = .0052).
A 1‐compartment model with lag time (Tlag) was the best fit for both single oral and repeated oral data (Figures 2 and 3, respectively). One cat was removed from study before oral or transdermal dosing because of complications secondary to the VAP. Data for 3 time points after single oral administration were excluded for 1 cat because they were clearly outliers, based on the extrapolation from the preceding plasma concentrations, and comparison to the other values obtained at those time points. A model could not be fit to the trough of pre‐dose time points during repeated oral dosing. The actual dose administered ranged from 9.04 to 12.5 mg/kg for both phases of oral administration. Pharmacokinetic parameters are summarized in Table 1. Wilcoxon signed‐rank tests reveal that only Tlag were significantly different (P = .016) between single and multiple oral dosing. The pharmacokinetic parameters calculated after single oral dosing were used to simulate the concentration time profile over the entirety of the multiple oral dosing regimen, with individual plasma values overlaid (Figure 4).
Figure 2.
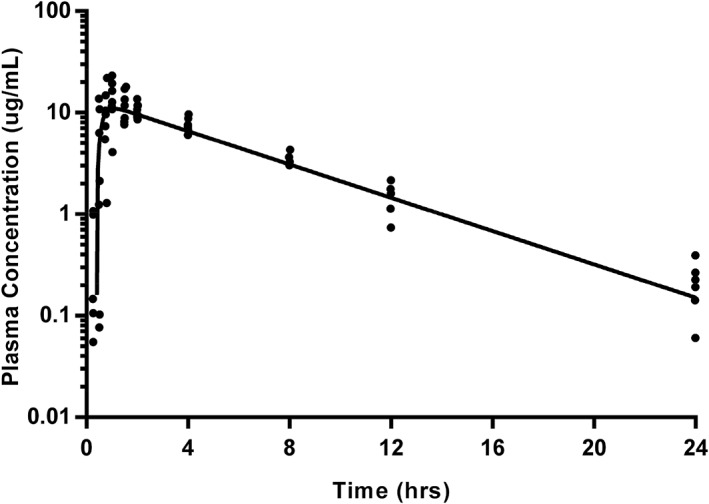
Gabapentin plasma concentrations after a single oral dose (10 mg/kg) in 7 cats (closed circles) with modeled time‐concentration curve (using geometric mean of derived parameters; solid line). Samples were collected at 5, 15, 30, 45, 60, and 90 minutes, followed by collection at 2, 4, 8, 12, and 24 hours after oral administration. Data below the limit of quantitation are excluded
Figure 3.
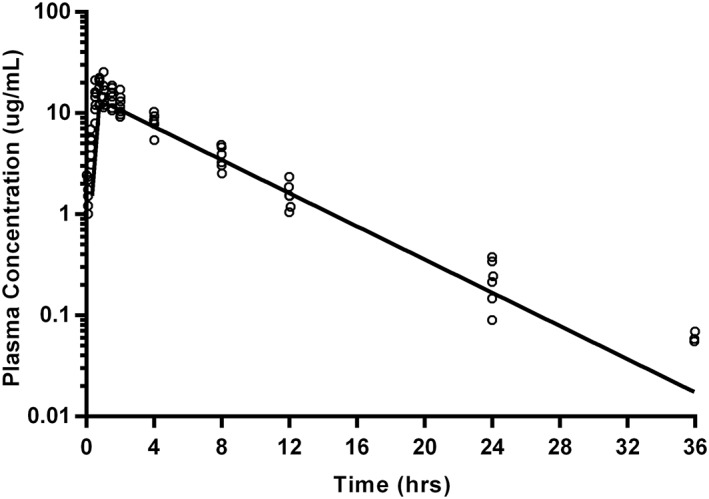
Gabapentin plasma concentrations after repeated oral dosing (10 mg/kg) in 7 cats (open circles) with modeled time‐concentration curve (using geometric mean of derived parameters; solid line). Samples were collected at 5, 15, 30, 45, 60, and 90 minutes, followed by collection at 2, 4, 8, 12, 24, and 36 hours after oral administration. Data below the limit of quantitation are excluded
Figure 4.
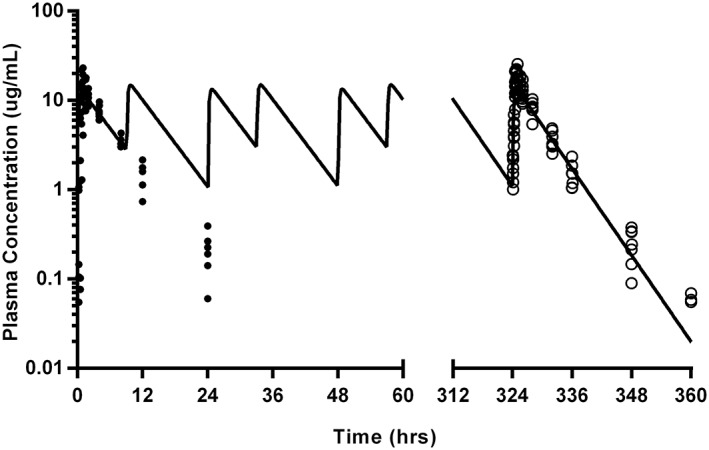
Gabapentin plasma concentrations after single (10 mg/kg; closed circles) and repeated oral dosing (10 mg/kg; open circles) in 7 cats with modeled time‐concentration curve (using geometric mean of derived repeated oral dosing parameters; solid line). Samples were collected at 5, 15, 30, 45, 60, and 90 minutes, followed by collection at 2, 4, 8, 12, and 24 hours after single oral dose administration. After the last repeated oral dose, samples were collected at 5, 15, 30, 45, 60, and 90 minutes, followed by collection at 2, 4, 8, 12, 24, and 36 hours. Data below the limit of quantitation are excluded
Because of erratic and poor transdermal absorption of gabapentin, the data was not modeled. Approximately 61% of collected samples had concentrations below the LOQ or LOD. The actual dose administered ranged from 9.68 to 10.05 mg/kg as labeled, or 13.26 to 13.77 mg/kg if corrected for the measured strength.
The only observed side effect during the study was mild‐moderate sedation, noted during the 2 hours after IV administration. Oral administration of the compounded capsules was well tolerated by all cats, with the majority of cats freely consuming the capsule during the multiple/long term administration phase. Soft Elizabethan collars were placed on all cats after transdermal application of the drug, in order to reduce the likelihood of transfer of the transdermal gel to oral ingestion. Three cats removed these collars at various time points, though they were not observed to have been actively grooming their ears and did not have correlating higher plasma drug concentrations than the other cats.
4. DISCUSSION
This report demonstrated that repeated oral dosing does not significantly impact gabapentin pharmacokinetics, and that the drug has poor bioavailability when administered as the transdermal gel compounded for this study.
While our values for clearance and terminal half‐lives are comparable to another report after a single IV bolus in cats, our results for the oral dose differ from data in that same report.13 Most notably, our maximum concentration (CMAX) was approximately 50% greater than previously reported (7.982 ± 1.053 μg/mL; 4.638‐10.550), with a slightly longer half‐life (2.95 ± 0.42 hours; 2.52‐3.52). These differences may be because of our reported higher bioavailability, higher dosages in some individuals, and differences in sampling sites and times. Ultimately, the reasons for the discrepancies between studies are undetermined.
There are no data for cats that describe the optimum plasma drug concentrations for gabapentin. While previously reported data and modeling suggests a half maximal effective concentration (EC50) ranging from 1.4 and 16.7 μg/mL for treatment of hyperalgesia in the rat20, 21, 22 and 5.4 μg/mL for the treatment of neuropathic pain in man,23 we cannot determine if these values will apply to cats. Calculation of the average plasma concentration after multiple doses results in a value of 6.11 μg/mL, with median trough concentrations of 2.55 and 1.49 μg/mL for the second‐to‐last and last dose, respectively. This would suggest that the current prescribing practices (10 mg/kg administered twice daily) would be insufficient to maintain plasma concentrations associated with efficacy in other species. Modeling to determine a potential dose and dosing interval, using a targeted minimum concentration of 5.4 μg/mL and CMAX of 16.7 μg/mL (based on the EC50 reported in man and rat, respectively) results in a suggested dose of approximately 8 mg/kg at an interval of 6 hours. Our data also suggest that gabapentin (as prepared in this study) has minimal transdermal absorption, and is not an appropriate route of administration. The transdermal vehicle was chosen because of its drug delivery properties, and its common use in practice.24, 25 These data are relevant given the increasing interest in compounding of transdermal preparations for cats.24, 25, 26
There were several sources of potential bias or error in our study. Treatments were administered in a serial order, without randomization. While clearance, volume of distribution, and other pharmacokinetic parameters are not expected to change significantly in healthy adult cats over period of 3 weeks (washout period), unpredictable or uncontrollable factors could have affected our results. Additionally, we wanted to compare the first and last doses in a contiguous dosing regimen in order to make the results more clinically relevant, and so the oral dosing (single and repeat) was set up serially. During the repeated oral dosing phase, gabapentin capsules were administered around the cats’ normal feeding times. This was done to emulate how owners are likely to time dosing. This resulted in varied inter‐dose intervals rather than rigid 12‐hour intervals, which could affect factors like accumulation. However, the accumulation ratio (AR) calculated using the median K10 after a single oral dose and a 12 hour dosing interval indicates minimal accumulation (AR = 1.09), which was similar to the value obtained when comparing the AUCs of the 2 dosing regimens (AR = 1.05). Any other factors potentially impacting the pharmacokinetics after repeated dosing (ie, induction or inhibition of metabolism) would still have occurred, if present. Data points below the LOQ were excluded from analysis because of the high variability observed during assay validation. This results in modeled time‐concentration curves that appear to poorly fit the data at later time points, and could affect parameters calculated by the terminal elimination phase. However, the models show good fit at preceding time points, and evaluation of residual plots, AIC values, and visual examination indicated that alternative models or weighing factors were unsatisfactory.
While the strengths of the IV solution and oral capsules were within the USP's acceptable range of ±10%, our transdermal gel was not. This resulted in higher than intended doses being administered, though absorption was still poor. It is possible that the transdermal preparations lacked adequate homogenization, which could have affected both drug kinetics during the study, and subsequent strength analysis. Cephalic catheters for IV bolus administration were placed either ipsi‐ or contralateral to the sampling ports, which could affect the detected plasma concentrations. This risk may be reduced by the placement of the catheter in the cranial vena cava/right atrium, but the risk remains. The use of a finished product (Lyrica) as our internal standard added a confounding factor to our results. We chose this internal standard because of its previous use in the veterinary literature, however, the use of an analytical grade standard would have eliminated this risk. Subsequent comparison of the assay using either the finished product or API revealed satisfactory agreement. Cats were fasted before drug administration, and compounded capsules were administered either with a water bolus (single oral dose) or with a small amount of food (multiple/long term dosing). It is known that gastrointestinal transit time (and therefore, absorption kinetics) are different between the fed and fasted state, as well as among different food particle sizes.27 The type of meal affects gabapentin absorption in people.28 There was also not an even distribution of sexes, and 1 cat would have been classified as obese. Finally, our research cats do not represent the “target” population of (typically older) cats with chronic or maladaptive pain.
CONFLICT OF INTEREST DECLARATION
Both Dr Baynes and Dr Papich are on the Scientific Review Committee for the Veterinary Pharmacology Research Foundation, who awarded the primary grant for the research performed. Dr Papich is the chair of the committee, and is also a member of the Board of Directors that awarded the funds. Dr Baynes’ and Papich's relationship to the study and its investigators was fully disclosed to the Scientific Review Committee and the Veterinary Pharmacology Research Foundation. They recused themselves from any discussions pertaining to the grant. The authors have no other conflicts of interest to disclose.
ACKNOWLEDGMENTS
The authors thank Mr Jim Yeatts for assistance conducting drug analysis. The authors also thank Dr Carrie Muller for her assistance during catheter implantation, drug administration, and sample collection. All work was performed at the North Carolina State University College of Veterinary Medicine (NCSU CVM) in Raleigh, North Carolina, USA, except for external determination of compounded product strength, which was performed at Campbell University Pharmaceutical Education & Research Center in Lillington, North Carolina, USA. The study was funded by the Veterinary Pharmacology Research Foundation Pharmacokinetics Grant (administered through the American Veterinary Medical Foundation), by the Comparative Pain Research Program, and by the Clinical Veterinary Pharmacy Residency Program at the NCSU CVM. This paper was presented as an oral presentation during the Annual CVM Research Forum at NCSU CVM on September 22, 2017.
OFF‐LABEL ANTIMICROBIAL DECLARATION
Cefazolin sodium was used in an off‐label manner during implantation of the jugular venous access ports.
INSTITUTIONAL ANIMAL CARE AND USE COMMITTEE (IACUC) OR OTHER APPROVAL DECLARATION
All procedures were performed according to a protocol (16‐187‐O) approved by the NCSU CVM IACUC.
Adrian D, Papich MG, Baynes R, Stafford E, Lascelles BDX. The pharmacokinetics of gabapentin in cats. J Vet Intern Med. 2018;32:1996–2002. 10.1111/jvim.15313
Funding information Clinical Veterinary Pharmacy Residency Program; Comparative Pain Research Program; Veterinary Pharmacology Research Foundation, Pharmacokinetics Grant (Administered by AVMF)
REFERENCES
- 1. Gruen ME, Thomson AE, Griffith EH, et al. A feline‐specific anti‐nerve growth factor antibody improves mobility in cats with degenerative joint disease‐associated pain: a pilot proof of concept study. J Vet Intern Med. 2016;32:1996‐2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gruen ME, Griffith E, Thomson A, et al. Detection of clinically relevant pain relief in cats with degenerative joint disease associated pain. J Vet Intern Med. 2014;28:346‐350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gunew MN, Menrath VH, Marshall RD. Long‐term safety, efficacy and palatability of oral meloxicam at 0.01‐0.03 mg/kg for treatment of osteoarthritic pain in cats. J Feline Med Surg. 2008;10:235‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lascelles BDX, DePuy V, Thomson A, et al. Evaluation of a therapeutic diet for feline degenerative joint disease. J Vet Intern Med. 2010;24:487‐495. [DOI] [PubMed] [Google Scholar]
- 5. Guedes AGP, Meadows JM, Pypendop BH, et al. Evaluation of tramadol for treatment of osteoarthritis in geriatric cats. J Am Vet Med Assoc 2018;252:565‐571. [DOI] [PubMed] [Google Scholar]
- 6. Adrian D, Rishniw M, Scherk M, et al. Prescribing practices of veterinarians in the treatment of chronic musculoskeletal pain in cats. J Feline Med Surg. In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Backonja M, Beydoun A, Edwards KR, et al. Gabapentin for the symptomatic treatment of painful neuropathy in patients with diabetes mellitus: a randomized controlled trial. J Am Med Assoc. 1998;280:1831‐1836. [DOI] [PubMed] [Google Scholar]
- 8. Moore RA, Wiffen PJ, Derry S, et al. Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2014:CD007938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kukkar A, Bali A, Singh N, et al. Implications and mechanism of action of gabapentin in neuropathic pain. Arch Pharm Res. 2013;36:237‐251. [DOI] [PubMed] [Google Scholar]
- 10. Patel R, Dickenson AH. Mechanisms of the gabapentinoids and α 2 δ‐1 calcium channel subunit in neuropathic pain. Pharmacol Res Perspect. 2016;4:e00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bauer CS, Nieto‐Rostro M, Rahman W, et al. The increased trafficking of the calcium channel subunit alpha2delta‐1 to presynaptic terminals in neuropathic pain is inhibited by the alpha2delta ligand pregabalin. J Neurosci. 2009;29:4076‐4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Luo ZD, Chaplan SR, Higuera ES, et al. Upregulation of dorsal root ganglion (alpha)2(delta) calcium channel subunit and its correlation with allodynia in spinal nerve‐injured rats. J Neurosci. 2001;21:1868‐1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Siao KT, Pypendop BH, Ilkiw JE. Pharmacokinetics of gabapentin in cats. Am J Vet Res. 2010;71:817‐821. [DOI] [PubMed] [Google Scholar]
- 14. Farrow HA, Rand JS, Burgess DM, et al. Jugular vascular access port implantation for frequent, long‐term blood sampling in cats: methodology, assessment, and comparison with jugular catheters. Res Vet Sci. 2013;95:681‐686. [DOI] [PubMed] [Google Scholar]
- 15. Aubert I, Abrams‐Ogg AC, Sylvestre AM, et al. The use of vascular access ports for blood collection in feline blood donors. Can J Vet Res. 2011;75:25‐34. [PMC free article] [PubMed] [Google Scholar]
- 16. United States Pharmacopeia and National Formulary (USP 37‐NF32) . Rockville, MD: United States Pharmacopeia Convention.
- 17. Kukanich B, Cohen RL. Pharmacokinetics of oral gabapentin in greyhound dogs. Vet J. 2011;187:133‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Malreddy PR, Coetzee JF, KuKanich B, et al. Pharmacokinetics and milk secretion of gabapentin and meloxicam co‐administered orally in Holstein‐Friesian cows. J Vet Pharmacol Ther. 2013;36:14‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yamaoka K, Nakagawa T, Uno T. Application of Akaike's information criterion (AIC) in the evaluation of linear pharmacokinetic equations. J Pharmacokinet Biopharm. 1978;6:165‐175. [DOI] [PubMed] [Google Scholar]
- 20. Taneja A, Troconiz IF, Danhof M, et al. Semi‐mechanistic modelling of the analgesic effect of gabapentin in the formalin‐induced rat model of experimental pain. Pharm Res. 2013;31:593‐606. [DOI] [PubMed] [Google Scholar]
- 21. Taneja A, Nyberg J, de Lange ECM, et al. Application of ED‐optimality to screening experiments for analgesic compounds in an experimental model of neuropathic pain. J Pharmacokinet Pharmacodyn. 2012;39:673‐681. [DOI] [PubMed] [Google Scholar]
- 22. Larsen MS, Keizer R, Munro G, et al. Pharmacokinetic/pharmacodynamic relationship of gabapentin in a CFA‐induced Inflammatory hyperalgesia rat model. Pharm Res. 2016;33:1133‐1143. [DOI] [PubMed] [Google Scholar]
- 23. Lockwood PA, Cook JA, Ewy WE, et al. The use of clinical trial simulation to support dose selection: application to development of a new treatment for chronic neuropathic pain. Pharm Res. 2003;20:1752‐1759. [DOI] [PubMed] [Google Scholar]
- 24. Delamaide Gasper JA, Barnes Heller HL, Robertson M, et al. Therapeutic serum phenobarbital concentrations obtained using chronic transdermal administration of phenobarbital in healthy cats. J Feline Med Surg. 2014;17:359‐363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benson KK, Zajic LB, Morgan PK, et al. Drug exposure and clinical effect of transdermal mirtazapine in healthy young cats: a pilot study. J Feline Med Surg. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hill KE, Mills PC, Jones BR, et al. Percutaneous absorption of methimazole: an in vitro study of the absorption pharmacokinetics for two different vehicles. J Vet Pharmacol Ther. 2015;38:581‐589. [DOI] [PubMed] [Google Scholar]
- 27. Sutton SC. Companion animal physiology and dosage form performance. Adv Drug Deliv Rev. 2004;56:1383‐1398. [DOI] [PubMed] [Google Scholar]
- 28. Gidal BE, Maly MM, Kowalski JW, et al. Gabapentin absorption: effect of mixing with foods of varying macronutrient composition. Ann Pharmacother. 1998;32:405‐409. [DOI] [PubMed] [Google Scholar]