

Abstract
An efficient method for the synthesis of a variety of pyrimidine derivatives 3a–t by reaction of barbituric acids 1a,b as Michael donor with nitroalkenes 2a–k as Michael acceptor using an aqueous medium and diethylamine is described. This 1,4-addition strategy offers several advantages, such as using an economic and environmentally benign reaction media, high yields, versatility, and shorter reaction times. The synthesized compounds were identified by 1H-NMR, 13C-NMR, CHN, IR, and MS. The structure of compound 3a was further confirmed by single crystal X-ray structure determination.
Keywords: Michael reactions, barbituric acid, aqueous media, green chemistry
1. Introduction
The Michael reaction is one of the most powerful tools for the formation of carbon–carbon bonds in organic synthesis [1,2,3,4,5,6]. The addition of various active methines compounds to nitroalkenes has received increased attention since the conjugated addition products are aliphatic nitro compounds. These are recognized as versatile synthetic building blocks which can be either transformed into biologically active compounds such as tetrahydropyrans, amino acids, pyrrolidines and lactones [7,8,9], or readily converted into other functionalities such as ketones, amines, carboxylic acids, nitrile oxides, etc. [10,11,12,13]. Extensive studies have been devoted to the development of catalytic systems for Michael additions of various active methines such as pronucleophiles to nitroalkenes including cinchona organocatalysts [14], enzymes [15], various chiral amines [16,17], transition metal-free organocatalysts [18,19,20] and chiral metal complexes [21,22,23,24,25].
Recently, the utilization of water as a solvent has emerged as an extensively investigated topic in organic transformations for its environmental friendly character, low cost and properties conferring unique selectivity and reactivity [26]. For these reasons, the development of synthetically useful reactions that take place in water is of considerable topical interest.
Barbituric acids constitute an interesting family of pyrimidinetrione heterocycles [27,28,29]. They are well-known in medicinal chemistry as sedatives, hypnotics, anticonvulsants and anxiolytic agents [30,31,32]. Barbituric acids are also particularly utilized as nucleophiles. In continuation of our research program [33,34,35,36,37], we have investigated the reaction of barbituric acids, as nucleophiles with nitroalkenes as Michael acceptors in water using diethylamine to afford multifunctional pyrimidine systems for biological and pharmacological evaluation. To the best of our knowledge, this is the first successful method of this type using aqueous diethylamine as reaction medium.
2. Results and Discussion
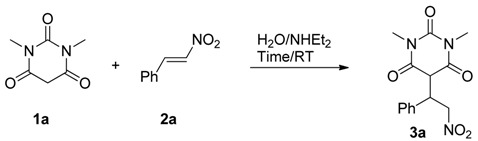
We initiated our investigations by using 1,3-dimethylbarbituric acid (1a) as C-based nucleophile and nitroalkene (2a) as a Michael acceptor for the 1,4-addition strategy in the presence of aqueous diethylamine at ambient temperature, as shown in Table 1. These are the optimal reaction conditions for the construction of Michael product by this strategy [38].
Table 1.
Screening of conditions for the Michael addition reaction of the model substrate a.
| Entry | Condition | Time | Yield (%) b |
|---|---|---|---|
| 1 | Et2NH/H2O | 1 | 99 |
| 2 | iPr2NH/H2O | 4 | 85 |
| 3 | (Cyclohexyl)2NH/H2O | 4 | 82 |
| 4 | Morpholine/H2O | 3 | 78 |
| 5 | NaOH/H2O | 6 | 65 |
| 6 | Et2NH | 10 | 10 |
| 7 | H2O | 10 | 0 |
a All reactions were carried out with 1,3-dimethylbarbituric acid 1a (1.5 mmol), nitroalkene 2a (1.5 mmol) and amine (1.5 mmol) in water (1.5 mL) for the specified time. b Yield of isolated product.
As expected, the Michael product 3a was obtained in quantitative yield after 1 h (Table 1, entry 1). In addition, other secondary amines were examined. It was found that iPr2NH generated the Michael product in a significant yield of 85% (Table 1, entry 2). Additionally, (cyclohexyl)2NH and morpholine afforded the product in substantial yields of 82% and 78%, respectively (Table 1, entries 3, 4). NaOH was also examined and was found to be less efficient in the reaction. Only a moderate yield was obtained (Table 1, entry 5). In the absence of amine (entry 6) or water (entry 7), the reaction either could not be performed or proceeded very slowly. The best results, with respect to yield, were obtained by performing the reaction with the combined promoting effects of both H2O and Et2NH.
It is well known that the rate for organic reactions conducted in aqueous media require either hydrogen bonding activation of functional groups by water or the repulsive hydrophobic interactions of the reactants [39,40,41]. We thus envisioned that hydrogen bonding activation in the presence of amine as a base generates an enolate, the catalyst of choice for the Michael strategy (Scheme 1).
Scheme 1.

A possible mechanistic pathway.
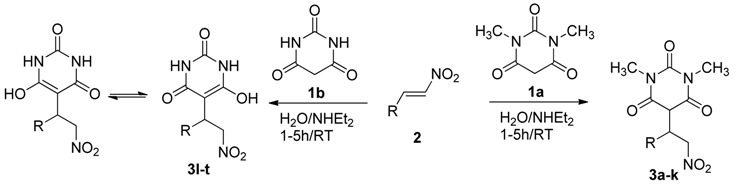
With the optimal reaction conditions in hand, the substrate scope was then investigated. First, 1,3-dimethylbarbituric acid (1a) as C-based nucleophile was reacted with eight different phenyl-type substituted nitroalkenes as Michael acceptors (Table 2, entries 1–8). The reactions proved to work well with a range of nitroalkenes bearing either electron-withdrawing or electron-donating groups that produced the desired products with excellent yields (88%–99%).
Table 2.
Michael addition reaction of barbituric acid derivatives 1 to nitroolefin 2 catalyzed by Et2NH in water at room temperature a.
| Entry | 3 | R | Yield (%) b |
|---|---|---|---|
| 1 | 3a | Ph | 99 |
| 2 | 3b | p-CH3Ph | 96 |
| 3 | 3c | p-BrPh | 92 |
| 4 | 3d | p-ClPh | 91 |
| 5 | 3e | 2,4-Cl2Ph | 90 |
| 6 | 3f | 2,6-Cl2Ph | 91 |
| 7 | 3g | p-CH3OPh | 89 |
| 8 | 3h | p-NO2Ph | 88 |
| 9 | 3i | Ferrocene | 93 |
| 10 | 3j | CH3 | 96 |
| 11 | 3k | Thiophene | 95 |
| 12 | 3l | Ph | 97 |
| 13 | 3m | p-CH3Ph | 94 |
| 14 | 3n | p-BrPh | 88 |
| 15 | 3o | p-ClPh | 89 |
| 16 | 3p | 2,4-Cl2Ph | 85 |
| 17 | 3q | 2,6-Cl2Ph | 86 |
| 18 | 3r | p-CH3OPh | 88 |
| 19 | 3s | Ferrocene | 92 |
| 20 | 3t | p-NO2Ph | 87 |
a All reactions were carried out with barbituric acid 1 (1.5 mmol), nitroalkene 2 (1.5 mmol) and amine (1.5 mmol) in water (1.5 mL) for the specified time. b Yield of isolated product.
Next, we applied the conditions to reactions of barbituric acid (1b) with eight different phenyl-type substituted nitroalkenes. Interestingly, the Michael product was separated in enol form and not in keto form in very good to excellent yields (Table 2, entries 13–20). Finally, to show the generality of the procedure, we tested ferrocene nitroalkene as Michael acceptor and the products were obtained in very good yields (Table 2, entries 9, 19). A further development has also been achieved by our group. Thus, Michael addition of barbiturate to either aliphatic nitroalkenes or heteroaromatic nitroalkenes affords the pyrimidine adduct (Table 2, entries 10, 11) with excellent yields (96% and 95%, respectively). Thus, the methodology not only suitable for aromatic nitroalkenes, but also aliphatic and heteroaromatic nitroalkenes.
The X-ray structure of 3a (Figure 1) was obtained by single crystal structure determination from a single crystal grown from CHCl3/Et2O as a solvent. The structure shows interesting characteristics and also the hydrogen-bonding interactions are listed (see Supporting Information). The packing of the molecules in the crystal structure is stabilized by C–H⋅ ⋅ ⋅O hydrogen bonds into a three-dimensional framework structure.
Figure 1.

ORTEP representation of the structure of 3a.
3. Experimental
3.1. General Information
All the chemicals were purchased from Sigma-Aldrich and Fluka (Seelze, Germany), and were used without further purification unless otherwise stated. All melting points were measured on a Gallenkamp melting point apparatus in open glass capillaries and are uncorrected. IR Spectra were measured as KBr pellets on a Nicolet 6700 FT-IR spectrophotometer (Madison, WI, USA). The NMR spectra were recorded on a Varian Mercury Jeol-400 NMR spectrometer (Tokyo, Japan).1H-NMR (400 MHz), and 13C-NMR (100 MHz) were run in either deuterated dimethylsulphoxide (DMSO-d6) or deuterated chloroform (CDCl3). Chemical shifts (δ) are referred in terms of ppm and J-coupling constants are given in Hz. Mass spectra were recorded on a Jeol JMS-600 H instrument (Tokyo, Japan). Elemental analysis was carried out on a Perkin Elmer 2400 Elemental Analyzer (Vernon Hills, Illinois, IL, USA); CHN mode.
3.2. General Procedure for Synthesis of Nitroalkenes 2a–k
Equimolar amounts of aryl aldehyde and nitromethane (1 equiv.) were dissolved in 95% ethyl alcohol (100 mL) at room temperature and then a solution of sodium hydroxide (1 equiv.) in ethyl alcohol (30 mL) was added from a dropping funnel at a rate of 5 mL per minute. The solution of the nitromethane and aldehyde in alcohol was then vigorously stirred at room temperature for 1 h. As the reaction proceeded, the insoluble sodium salt of the condensation product precipitated. After all of the alkali had been added and with the temperature kept below 20 °C, ice water (about 30 mL) was slowly added until the precipitate just dissolved. This clear, cold solution was slowly added to a solution of 15% HCl (50 mL) and more cold water (300 mL) was added. A fine, light yellow precipitate was immediately formed and filtered with suction after standing for half an hour. The product thus formed was quite pure and was found to be satisfactory as a starting product for subsequent preparations without further purification.
3.3. General Procedure for Michael Addition for the Synthesis of 3a–t
A mixture of nitroalkene 2a–k (1.5 mmol), barbituric acid derivatives 1a,b (1.5 mmol), and Et2NH (1.5 mmol) in degassed H2O (1.5 mL) was stirred at room temperature for 1 h until TLC showed complete disappearance of the reactants. The product either precipitated or was in an oily form. The reaction mixture was acidified using 1M HCl extract with DCM/10% EtOH then washed with brine and dried over MgSO4 to afford 3a–t.
1,3-Dimethyl-5-(2-nitro-1-phenylethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (3a) According to the general procedure, 3a was prepared from 1,3-dimethylbarbituric acid (1a) and (E)-(2-nitrovinyl)benzene (2a) as white crystals (453 mg, 1.48 mmol, 99%); m.p. 85 °C; IR (KBr) ν/cm−1 1738, 1671, 1552, 1372, 1228; 1H-NMR (CDCl3) δ 7.29 (m, 3H, Ph), 7.03 (m, 2H, Ph), 5.28 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 5.02 (dd, 1H, J 13.9, 5.8, CH2NO2), 4.50 (m, 1H, CHPh), 3.84 (d, 1H, J = 3.6 Hz, COCHCO), 3.12(s, 3H, CH3), 3.07(s, 3H, CH3); 13C-NMR (CDCl3) δ 166.8, 150.5, 133.7, 129.4, 127.4, 76.5, 51.5, 45.5, 28.5, 28.3; LC/MS (ESI) m/z 305 [M]+; Anal. for C14H15N3O5; calcd: C, 55.08; H, 4.95; N, 13.76; Found: C, 55.10; H, 4.95; N, 13.75.
The structure of 3a was confirmed by X-ray crystal structure analysis (Bruker AXS GmbH, Karlsruhe, Germany). CCDC-933479 contains the supplementary crystallographic data for this compound. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. A colorless cubic crystal of the compound suitable for X-ray analysis was formed in CHCl3/Et2O at room temperature after 2 days.
1,3-Dimethyl-5-(2-nitro-1-(p-tolyl)ethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (3b) According to the general procedure, 3b was prepared from 1,3-dimethylbarbituric acid (1a) and (E)-1-methyl-4-(2-nitrovinyl)benzene (2b) as an oily product (460 mg, 1.44 mmol, 96%). IR (KBr) ν/cm−1 1738, 1671, 1552, 1372, 1228; 1H-NMR (CDCl3) δ 7.08 (d, 2H, J = 8.0 Hz, Ph), 6.91 (d, 2H, J = 8.0 Hz, Ph), 5.25 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 4.99 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 4.45 (m, 1H, CHPh), 3.82 (d, 1H, J = 3.6 Hz, COCHCO), 3.13 (s, 3H, CH3), 3.08 (s, 3H, CH3), 2.28 (s, 3H, CH3); 13C-NMR (CDCl3) δ 166.9, 166.9, 150.6, 139.3, 130.5, 129.9, 127.3, 51.5, 45.3, 28.5, 28.3, 21.1; LC/MS (ESI): m/z 319 [M]+; Anal. for C15H17N3O5; calcd: C, 56.42; H, 5.37; N, 13.16; Found: C, 56.41; H, 5.36; N, 13.17.
5-(1-(4-Bromophenyl)-2-nitroethyl)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (3c) According to the general procedure, 3c was prepared from 1,3-dimethylbarbituric acid (1a) and (E)-1-bromo-4-(2-nitrovinyl) benzene (2c) as a yellow powder (530 mg, 1.38 mmol, 92%); m.p.: 99 °C; IR (KBr) ν/cm−11738, 1668, 1552, 1375; 1H-NMR (DMSO-d6) δ 7.51 (d, 2H, J = 7.3 Hz, Ph), 7.07 (d, 2H, J = 7.3 Hz, Ph), 5.40 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 5.23 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 4.29 (bs, 1H, CHPh), 4.17 (bs, 1H, COCHCO), 3.06 (s, 3H, CH3), 2.96 (s, 3H, CH3); 13C-NMR (DMSO-d6) δ 167.8, 167.5, 166.4, 151.3, 135.0, 132.2, 132.1, 131.8, 130.3, 122.1, 76.6, 52.1, 44.0, 28.5, 28.3; LC/MS (ESI) m/z 384 [M]+; Anal. for C14H14BrN3O5; calcd: C, 43.77; H, 3.67; Br, 20.80; N, 10.94;Found: C, 43.79; H, 3.65; Br, 20.81; N, 10.94.
5-(1-(4-Chlorophenyl)-2-nitroethyl)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (3d) According to the general procedure, 3d was prepared from 1,3-dimethylbarbituric acid (1a) and (E)-1-chloro-4-(2-nitrovinyl)benzene (2d) as an oily product (462 mg, 1.36 mmol, 91%). IR (KBr) ν/cm−1 1738, 1669, 1552, 1423, 1238; 1H-NMR (DMSO-d6) δ 7.44 (d, 2H, J = 8.0 Hz, Ph), 7.13 (d, 2H, J = 8.0 Hz, Ph), 5.41 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 5.23 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 4.30 (bs, 1H, CHPh), 4.16 (bs, 1H, COCHCO), 3.06 (s, 3H, CH3), 2.95 (s, 3H, CH3); 13C-NMR (DMSO-d6) δ 167.8, 167.5, 166.4, 151.3, 135.0, 132.2, 132.1, 131.8, 130.3, 122.1, 76.6, 52.1, 44.0, 28.5, 28.3; LC/MS (ESI) m/z 339 [M]+; Anal. for C14H14ClN3O5; calcd: C, 49.49; H, 4.15; Cl, 10.44; N, 12.37;Found: C, 49.50; H, 4.14; Cl, 10.43; N, 12.36.
5-(1-(2,4-Dichlorophenyl)-2-nitroethyl)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (3e) According to the general procedure, 3e was prepared from 1,3-dimethylbarbituric acid (1a) and (E)-2,4-dichloro-1-(2-nitrovinyl) benzene (2e) as a yellow powder (505 mg, 1.35 mmol, 90%); m.p.: 119 °C; IR (KBr) ν/cm−1 1738, 1667, 1551, 1423, 1221;1H-NMR (DMSO-d6) δ 7.65 (s, 1H, Ph), 7.52 (d, 1H, J = 8.0 Hz, Ph), 7.45(d, 1H, J = 8.0 Hz, Ph), 5.31–5.22 (m, 3H, CH2NO2 and COCHCO), 4.83 (bs, 1H, CHPh); 13C-NMR (DMSO-d6): δ 167.3, 166.4, 151.7, 135.0, 130.9, 129.6, 127.9, 75.7, 51.6, 28.8, 28.7; LC/MS (ESI) m/z 374 [M]+; Anal. for C14H13Cl2N3O5; calcd: C, 44.94; H, 3.50; Cl, 18.95; N, 11.23; Found: C, 44.92; H, 3.51; Cl, 18.90; N, 11.25.
5-(1-(2,6-Dichlorophenyl)-2-nitroethyl)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (3f) According to the general procedure, 3f was prepared from 1,3-dimethylbarbituric acid (1a) and (E)-2,6-dichloro-1-(2-nitrovinyl)benzene (2f) as a white powder (510 mg, 1.36 mmol, 91%); m.p.: 140 °C; IR (KBr) ν/cm−1 1738, 1670, 1551, 1423, 1228; 1H-NMR (DMSO-d6) δ 7.50 (d, 2H, J = 8.0 Hz, Ph), 7.36 (t, 1H, J = 8.0 Hz, Ph), 5.32 (d, 2H, J = 6.6 Hz, CH2NO2), 5.00 (m, 1H, CHPh), 4.11 (d, 1H, J = 11.0 Hz, COCHCO), 3.08 (s, 3H, CH3), 3.03 (s, 3H, CH3); 13C-NMR (DMSO-d6) δ 167.7, 166.8, 152.0, 134.7, 132.5, 131.2, 130.6, 129.3, 75.6, 50.47, 28.8, 28.7; LC/MS (ESI) m/z 374[M]+; Anal. for C14H13Cl2N3O5; calcd: C, 44.94; H, 3.50; Cl, 18.95; N, 11.23;Found: C, 44.92; H, 3.51; Cl, 18.90; N, 11.25.
5-(1-(4-Methoxyphenyl)-2-nitroethyl)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (3g) According to the general procedure, 3g was prepared from 1,3-dimethylbarbituric acid (1a) and (E)-1-methoxy-4-(2-nitrovinyl) benzene (2g) as an oily product (447 mg, 1.33 mmol, 89%). IR (KBr) ν/cm−1 1738, 1670, 1551, 1423, 1228; 1H-NMR (DMSO-d6) δ 6.96 (d, 2H, J = 8.0 Hz, Ph), 6.85 (d, 2H, J = 8.0 Hz, Ph), 5.41 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 5.16 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 4.21 (m, 1H, CHPh), 4.04 (d, 1H, J = 2.9 Hz, COCHCO), 3.69 (s, 3H, CH3), 2.99 (s, 3H, CH3), 2.93 (s, 3H, CH3); 13C-NMR (DMSO-d6) δ 168.2, 167.7, 159.6, 151.3, 129.1, 126.6, 114.6, 77.0, 55.6,52.4, 44.7,28.4, 28.3 ; LC/MS (ESI) m/z 335[M]+; Anal. for C15H17N3O6; calcd: C, 53.73; H, 5.11; N, 12.53; Found: C, 53.74; H, 5.10; N, 12.52.
1,3-Dimethyl-5-(2-nitro-1-(4-nitrophenyl)ethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (3h) According to the general procedure, 3h was prepared from 1,3-dimethylbarbituric acid (1a) and (E)-1-nitro-4-(2-nitrovinyl)benzene (2h) as a dark brown powder (462 mg, 1.32 mmol, 88%); m.p.: 170 °C; IR (KBr) ν/cm−1 1738, 1646, 1551, 1366, 1217; 1H-NMR (DMSO-d6) δ 7.50 (d, 2H, J = 8.0 Hz, Ph), 7.38 (d, 2H, J = 8.0 Hz, Ph), 5.32 (d, 2H, J = 6.6 Hz, CH2NO2), 5.00 (m, 1H, CHPh), 4.11 (d, 1H, J = 11 Hz, COCHCO), 3.08 (s, 3H, CH3), 3.03 (s, 3H, CH3); 13C-NMR (DMSO-d6) δ 167.7, 166.8, 152.0, 136.7, 134.7, 132.5, 131.2, 130.6, 129.3, 75.6, 50.4, 28.8, 28.7; LC/MS (ESI) m/z 350[M]+; Anal. for C14H14N4O7; calcd: C, 48.00; H, 4.03; N, 15.99; Found: C, 48.01; H, 4.01; N, 16.01.
1,3-Dimethyl-5-(2-ferrocenyl)ethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (3i) According to the general procedure, 3i was prepared from 1,3-dimethylbarbituric acid (1a) and (E)-1-ferrocenyl-2-nitroethene (2i) as a dark purple powder (595 mg, 1.39 mmol, 93%); m.p.: 139 °C; IR (KBr) ν/cm−1 1738, 1671, 1551, 1423, 1367, 1228; 1H-NMR (DMSO-d6) δ 5.17 (d, 2H, J = 7.3 Hz, CH2NO2), 4.21 (bs, 5H, ferrocene), 4.00 (m, 1H, CH-ferrocene), 3.92 (d, 1H, J = 2.9 Hz, COCHCO), 3.38 (bs, 4H, ferrocene), 3.03 (s, 3H, CH3), 2.88 (s, 3H, CH3), 2.93 (s, 3H, CH3); 13C-NMR (DMSO-d6) δ 168.4, 167.6, 151.4, 83.2, 76.7, 69.4, 69.3, 68.7, 68.3, 67.7, 66.0, 52.2, 28.5, 28.2 ; LC/MS (ESI) m/z 427[M]+; Anal. for C19H21FeN3O5; calcd: C, 53.41; H, 4.95; N, 9.84; Found: C, 53.42; H, 4.94; N, 9.85.
1,3-Dimethyl-5-(1-nitropropan-2-yl)pyrimidine-2,4,6(1H,3H,5H)-trione (3j) According to the general procedure, 3j was prepared from 1,3-dimethylbarbituric acid (1a) and (E)-1-nitroprop-1-ene (2j) as a yellowish oily product (350 mg, 1.44 mmol, 96%). IR (KBr) ν/cm−1 1745, 1665, 1325, 1225; 1H-NMR (DMSO-d6) δ 4.93 (dd, H, J = 11.2, 5.2 Hz, CH2NO2), 4.70 (dd, H, J = 11.2, 5.2 Hz, CH2NO2), 3.98 (d, 1H, J = 2.4, COCHCO), 3.23 (m, 1H, CH), 3.20 (s, 3H, CH3), 3.02 (s, 3H, CH3), 0.95 (d, 3H, J = 5.6, CH3); 13C-NMR (DMSO-d6) δ 167.4, 167.3, 165.8, 151.4, 78.4, 50.3, 28.2, 27.7, 13.6; LC/MS (ESI) m/z 244[M]+; Anal. for C9H13N3O5; calcd: C, 44.44; H, 5.39; N, 17.28; Found: C, 44.46; H, 5.40; N, 17.31.
1,3-Dimethyl-5-(2-nitro-1-(thiophen-2-yl)ethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (3k) According to the general procedure, 3k was prepared from 1,3-dimethylbarbituric acid (1a) and (E)-2-(2-nitrovinyl)thiophene (2k) as a brown semisolid product (443 mg, 1.42 mmol, 95%). IR (KBr) ν/cm−11767, 1680, 1558, 1343, 1225;1H-NMR (DMSO-d6) δ 7.45 (brs, 1H, thiophene), 6.95 (brs, 1H, thiophene), 6.89 (brs, 1H, thiophene), 5.45 (d, 1H, J = 10.8 Hz, CH2NO2), 5.20 (dd, 1H, J = 16.8, 8.8 Hz, CH2NO2), 4.66 (brs, 1H, COCHCO), 4.26 (brs, 1H, CHCH2NO2), 3.12 (s, 3H, CH3), 3.05 (s, 3H, CH3); 13C-NMR (DMSO-d6) δ 169.5, 168.7, 137.9, 135.9, 134.8, 129.8, 79.5, 29.1, 28.5, 27.5; LC/MS (ESI) m/z 312[M]+; Anal. for C12H13N3O5S; calcd: C, 46.30; H, 4.21; N, 13.50; Found: C, 46.32; H, 4.22; N, 13.48.
6-Hydroxy-5-(2-nitro-1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (3l) According to the general procedure, 3l was prepared from barbituric acid (1b) and (E)-(2-nitrovinyl)benzene (2a) as a white powder (401 mg, 1.45 mmol, 97%); m.p.: 165 °C; IR (KBr) ν/cm−1 3204, 3015, 2920, 1740, 1550, 1363, 1217;1H-NMR (DMSO-d6) δ 11.31 (s, 2H, NH), 7.40 (m, 3H, Ph), 7.13 (m, 2H, Ph), 5.44 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 5.28 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 4.33 (m, 1H, CHPh); 13C-NMR (DMSO-d6) δ 169.7, 169.5, 150.7, 135.8, 129.3, 128.8, 128.3, 79.7, 77.4, 51.3, 43.6; LC/MS (ESI) m/z 277 [M]+; Anal. for C12H11N3O5; calcd: C, 51.99; H, 4.00; N, 15.16; Found: C, 52.01; H, 4.02; N, 15.15.
6-Hydroxy-5-(2-nitro-1-(p-tolyl)ethyl)pyrimidine-2,4(1H,3H)-dione compound with diethylamine (1:1) (3m) According to the general procedure, 3m was prepared from barbituric acid (1b) and (E)-1-methyl-4-(2-nitrovinyl)benzene (2b) as a white powder (513 mg, 1.41 mmol, 94%); m.p.: 210 °C; IR (KBr) ν/cm−1 3210, 3015, 1738, 1686, 1574, 1374; 1H-NMR (DMSO-d6) δ 9.22 (s, 2H, NH), 7.27 (d, 2H, J = 8.0 Hz, Ph), 6.99 (d, 2H, J = 8.0 Hz, Ph), 5.35 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 5.09 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 4.74(m, 1H, CHPh), 2.92 (m, 1H and 4H, COCHCO and CH3CH2NHCH2CH3), 2.21 (s, 3H, CH3), 1.14 (t, 6H, J = 7.3 Hz, CH3CH2NHCH2CH3); 13C-NMR (DMSO-d6) δ 164.7, 152.5, 141.2, 135.0, 128.7, 128.1, 85.0, 79.5, 41.9, 21.1, 11.5; LC/MS (ESI) m/z 364[M]+; Anal. for C15H17N3O5; calcd: C, 56.03; H, 6.64; N, 15.38; Found: C, 56.05; H, 6.65; N, 15.39.
5-(1-(4-Bromophenyl)-2-nitroethyl)-6-hydroxypyrimidine-2,4(1H,3H)-dione (3n) According to the general procedure, 3n was prepared from barbituric acid (1b) and (E)-1-bromo-4-(2-nitrovinyl) benzene (2c) as a yellow powder (566 mg, 1.32 mmol, 88%); m.p.: 130 °C; IR (KBr) ν/cm−1: 3204, 3015, 2920, 1740, 1550, 1363, 1217; 1H-NMR (DMSO-d6) δ 9.22 (s, 2H, NH), 7.42 (d, 2H, J = 8.0 Hz, Ph), 7.18 (d, 2H, J = 8.0 Hz, Ph), 5.42 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 5.27 (m, 2H, CH2NO2 and CHPh); 13C-NMR (DMSO-d6) δ 169.5, 169.3, 150.7, 135.1, 133.4, 130.3, 129.3, 79.7, 77.1, 51.3, 33.2; LC/MS (ESI) m/z 356 [M]+; Anal. for C12H10BrN3O5; calcd: C, 40.47; H, 2.83; Br, 22.44; N, 11.80; Found: C, 40.50; H, 2.85; Br, 22.45; N, 11.83.
5-(1-(4-Chlorophenyl)-2-nitroethyl)-6-hydroxypyrimidine-2,4(1H,3H)-dione (3o) According to the general procedure, 3o was prepared from barbituric acid (1b) and (E)-1-chloro-4-(2-nitrovinyl) benzene (2d) as a white powder (512 mg, 1.33 mmol, 89%); m.p.: 80 °C; IR (KBr) ν/cm−1 3208, 3018, 2980, 1740, 1699, 1555, 1363, 1217; 1H-NMR (DMSO-d6) δ 9.22 (s, 2H, NH), 7.55 (m, 4Ph), 5.37 (dd, 1H, J = 13.9 Hz, CH2NO2), 5.07 (dd, 1H, J = 13.9 Hz, CH2NO2), 4.73 (m, 1H, CHPh); 13C-NMR (DMSO-d6) δ 164.4, 152.3, 143.5, 132.2, 131.8, 131.4, 130.5, 119.2, 84.8, 78.9, 29.4; LC/MS (ESI) m/z 311 [M]+; Anal. for C12H10ClN3O5; calcd:C, 46.24; H, 3.23; Cl, 11.37; N, 13.48; Found: C, 46.21; H, 3.22; Cl, 11.40; N, 13.45.
5-(1-(2,4-Dichlorophenyl)-2-nitroethyl)-6-hydroxypyrimidine-2,4(1H,3H)-dione compound with diethylamine (1:1) (3p) According to the general procedure, 3p was prepared from barbituric acid (1b) and (E)-2,4-dichloro-1-(2-nitrovinyl)benzene (2e) as a beige powder (534 mg, 1.27 mmol, 85%); m.p.: 190 °C; IR (KBr) ν/cm−1 3151, 2986, 1697, 1590, 1376; 1H-NMR (DMSO-d6) δ 9.18 (s, 2H, NH), 8.30 (bs, 1H, OH), 7.83 (d, 2H, J = 8.8 Hz, Ph), 7.44(s, 1H, Ph), 7.30 (d, 2H, J = 8.8 Hz, Ph), 5.35 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 5.14 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 4.90 (m, 1H, CHPh), 2.92 (m, 1H and 4H, COCHCO and CH3CH2NHCH2CH3), 2.21 (s, 3H, CH3), 1.14 (t, 6H, J = 7.3 Hz, CH3CH2NHCH2CH3); 13C-NMR (DMSO-d6) δ 165.0, 152.5, 140.2, 134.1, 132.7, 131.7, 128.5, 127.3, 82.5, 77.3, 41.9, 21.1, 11.5; LC/MS (ESI) m/z 419 [M]+; Anal. for C16H20Cl2N4O5; calcd: C, 45.84; H, 4.81; Cl, 16.91; N, 13.36; Found: C, 45.87; H, 4.83; Cl, 16.90; N, 13.35.
5-(1-(2,6-Dichlorophenyl)-2-nitroethyl)-6-hydroxypyrimidine-2,4(1H,3H)-dione (3q) According to the general procedure, 3q was prepared from barbituric acid (1b) and (E)-2,6-dichloro-1-(2-nitrovinyl) benzene (2f) as a yellow powder (540 mg, 1.29 mmol, 86%); m.p.: 130 °C; IR (KBr) ν/cm−1 3155, 2986, 1740, 1670, 1551, 1423, 1228;1H-NMR (DMSO-d6) δ 11.38 (s, 2H, NH), 7.63 (d, 2H, J = 8.8 Hz, Ph), 7.46 (s, 1H, Ph), 7.36 (d, 2H, J = 8.8 Hz, Ph), 5.37 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 5.25 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 5.04 (m, 1H, CHPh); 13C-NMR (DMSO-d6) δ 169.6, 168.4, 151.3, 133.1, 130.9, 129.8, 129.2, 75.8, 49.2; LC/MS (ESI) m/z 419 [M]+; Anal. for C12H9Cl2N3O5; calcd: C, 41.64; H, 2.62; Cl, 20.49; N, 12.14; Found: C, 41.65; H, 2.61; Cl, 20.50; N, 12.13.
6-Hydroxy-5-(1-(4-methoxyphenyl)-2-nitroethyl)pyrimidine-2,4(1H,3H)-dione compound with diethylamine (1:1) (3r) According to the general procedure, 3r was prepared from barbituric acid (1b) and (E)-4-methoxy-1-(2-nitrovinyl)benzene (2g) as a yellow powder (501 mg, 1.32 mmol, 88%); m.p.: 152 °C; IR (KBr) ν/cm−1 3459, 3016, 2970, 1740, 1571, 1365; 1H-NMR (DMSO-d6) δ 9.07 (s, 2H, NH), 7.32 (d, 2H, J = 8.8 Hz, Ph), 6.75(s, 1H, Ph), 7.30 (d, 2H, J = 8.8 Hz, Ph), 5.31 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 5.06 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 4.68 (m, 1H, CHPh), 3.68 (s, 3H, OCH3), 2.93 (m, 1H and 4H, COCHCO and CH3CH2NHCH2CH3), 1.15 (t, 6H, J = 7.3 Hz, CH3CH2NHCH2CH3); 13C-NMR (DMSO-d6) δ 164.6, 157.9, 152.5, 136.4, 129.3, 113.5, 85.0, 79.8, 55.4, 41.9, 11.6; LC/MS (ESI) m/z 380[M]+; Anal. for C17H24N4O6; calcd: C, 53.68; H, 6.36; N, 14.73; Found: C, 53.68; H, 6.36; N, 14.73.
5-(2-Ferrocenyl)ethyl)6-Hydroxypyrimidine-2,4(1H,3H)-dione compound with diethylamine(1:1) (3s) According to the general procedure, 3s was prepared from barbituric acid (1b) and (E)-1-ferrocenyl-2-nitroethene (2i) as a brown powder (550 mg, 1.38 mmol, 92%); m.p.: 190 °C; IR (KBr) ν/cm−1 3449, 3016, 2970, 1738, 1546, 1365, 1217;1H-NMR (DMSO-d6) δ 8.98 (s, 2H, NH), 5.16 (t, 1H, J = 9.5 Hz, CH2NO2), 4.89 (dd, 1H, J = 13.9, 5.8 Hz, CH2NO2), 4.53(m, 1H, CHPh), 4.25-3.93 (m, 10H, ferrocene and COCHCO), 2.92 (bs, 4H, CH3CH2NHCH2CH3), 1.15 (bs, 6H, CH3CH2NHCH2CH3); 13C-NMR (DMSO-d6) δ 164.5, 152.5, 92.3, 84.7, 79.6, 69.3, 68.8, 67.8, 66.7, 66.4, 41.8, 34.7, 11.6; LC/MS (ESI) m/z 399 [M]+; Anal. for C17H17FeN3O5; calcd: C, 51.15; H, 4.29; N, 10.53; Found: C, 51.17; H, 4.30; N, 10.54.
6-Hydroxy-5-(2-nitro-1-(4-nitrophenyl)ethyl)pyrimidine-2,4(1H,3H)-dione compound with diethylamine (1:1) (3t). According to the general procedure, 3t was prepared from barbituric acid (1b) and (E)-4-nitro-1-(2-nitrovinyl)benzene (2h) as a yellow powder (515 mg, 1.3 mmol, 87%); m.p.: 185 °C; IR (KBr) ν/cm−1 3445, 3015, 2970, 1738, 1575, 1373, 1216;1H-NMR (DMSO-d6) δ 9.18 (s, 2H, NH), 8.13(d, 1H, J = 7.3 Hz, Ph), 7.70(d, 1H, J = 8.0 Hz, Ph), 7.56(t, 1H, J = 8.0 Hz, Ph), 7.36(t, 1H, J = 7.3 Hz, Ph), 5.57(dd, 1H, J = 11.7, 8.0 Hz, CH2NO2), 5.11 (t, 1H, J = 8.0 Hz, COCHCO), 5.06 (dd, 1H, J = 11.7, 8.0 Hz, CH2NO2), 3.37 (bs, 1H, CHPh), 2.90 (q, J = 7.3 Hz, 4H, CH3CH2NHCH2CH3), 1.13 (t, 6H, J = 7.3 Hz, CH3CH2NHCH2CH3); 13C-NMR (DMSO-d6) δ 164.9, 152.5, 149.5, 138.6, 133.0, 131.4, 127.6, 123.6, 84.5, 78.0, 41.9, 35.1, 11.5; LC/MS (ESI) m/z 395[M]+; Anal. for C16H21N5O7; calcd: C, 48.61; H, 5.35; N, 17.71; Found: C, 48.59; H, 5.34; N, 17.68.
4. Conclusions
A very convenient procedure for the syntheses of pyrimidine derivatives by Michael addition of cyclic 1,3-dicarbonyl compounds to a range of nitroalkenes using a simple NHEt2/H2O medium has been developed. The reaction scope is substantial and a number of substituted barbituric acids and nitroalkenes could be successfully applied to give multifunctional pyrimidine derivatives. These reactions gave high yields of products in short periods of time. The study of the full scope of this asymmetric transformation and its application in the synthesis of biologically active molecules are currently underway in our laboratory.
Acknowledgments
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding the work through the research group project Number RGP-VPP-257.
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/19/1/1150/s1.
Author Contributions
A.B. proposed the subject, designed the study. H.J.A.-N. and M.W. carried out the synthesis of all the products. A.M.A.-M. and Y.N.M. monitor progress of the ongoing research of the proposed project, helped in the results and discussion. H.G. and H.-K.F. carried out the X-ray crystallography part. A.B. prepared draft the manuscript. All the authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 3a–t are available from the authors.
References
- 1.Sibi M.P., Manyem S. Enantioselective conjugate additions. Tetrahedron. 2000;56:8033–8061. doi: 10.1016/S0040-4020(00)00618-9. [DOI] [Google Scholar]
- 2.Berner O.M., Tedeschi L., Enders D. Asymmetric Michael additions to nitroalkenes. Eur. J. Org.Chem. 2002;2002:1877–1894. doi: 10.1002/1099-0690(200206)2002:12<1877::AID-EJOC1877>3.0.CO;2-U. [DOI] [Google Scholar]
- 3.Christoffers J., Baro A. Construction of quaternary stereocenters: New perspectives through enantioselective Michael reactions. Angew. Chem. Int. Ed. 2003;42:1688–1690. doi: 10.1002/anie.200201614. [DOI] [PubMed] [Google Scholar]
- 4.Notz W., Tanaka F., Barbas C.F., III. Enamine-based organocatalysis with proline and diamines: The development of direct catalytic asymmetric Aldol, Mannich, Michael, and Diels-alder reactions. Acc. Chem. Res. 2004;37:580–591. doi: 10.1021/ar0300468. [DOI] [PubMed] [Google Scholar]
- 5.Ballini R., Bosica G., Fiorini D., Palmieri A., Petrini M. Conjugate additions of nitroalkanes to electron-poor alkenes: Recent results. Chem. Rev. 2005;105:933–971. doi: 10.1021/cr040602r. [DOI] [PubMed] [Google Scholar]
- 6.Sulzer-Moss S., Alexakis A. Chiral amines as organocatalysts for asymmetric conjugate addition to nitroolefins and vinyl sulfones via enamine activation. Chem. Commun. 2007;30:3123–3135. doi: 10.1039/b701216k. [DOI] [PubMed] [Google Scholar]
- 7.Krause N., Hoffmann-Roder A. Recent advances in catalytic enantioselective Michael additions. Synthesis. 2001;2:171–196. doi: 10.1055/s-2001-10803. [DOI] [Google Scholar]
- 8.Ono N. The Nitro Group in Organic Synthesis. Wiley-VCH; NewYork, NY, USA: 2001. [Google Scholar]
- 9.Czekelius C., Carreira E.M. Convenient transformation of optically active into chiral aldoximes and nitriles. Angew. Chem. Int. Ed. 2005;44:612–615. doi: 10.1002/anie.200461879. [DOI] [PubMed] [Google Scholar]
- 10.Calderari G., Seebach D. Asymmetrische Michael-additionen. Stereoselektive alkylierung chiraler, nicht racemischer enolate durch nitroolefine. Herstellung enantiomerenreiner γ-aminobuttersäure- und bernsteinsäure-derivate. Helv. Chim. Acta. 1985;68:1592–1604. doi: 10.1002/hlca.19850680611. [DOI] [Google Scholar]
- 11.Rosini G., Ballini R. Functionalized nitroalkanes as useful reagents for alkyl anion synthons. Synthesis. 1988;1988:833–847. doi: 10.1055/s-1988-27726. [DOI] [Google Scholar]
- 12.Barrett A.G.M., Graboski G. Conjugated nitroalkenes: Versatile intermediates in organic synthesis. Chem. Rev. 1986;86:751–762. doi: 10.1021/cr00075a002. [DOI] [Google Scholar]
- 13.Ballini R., Petrini M. Recent synthetic developments in the nitro to carbonyl conversion (Nef reaction) Tetrahedron. 2004;60:1017–1047. doi: 10.1016/j.tet.2003.11.016. [DOI] [Google Scholar]
- 14.Shi M., Lei Z.Y., Zhao M.X., Shi J.W. A highly efficient asymmetric Michael addition of anthrone to nitroalkenes with cinchona organocatalysts. Tetrahedron Lett. 2007;48:5743–5746. doi: 10.1016/j.tetlet.2007.06.107. [DOI] [Google Scholar]
- 15.Cai J.F., Guan Z., He Y.H. The lipase-catalyzed asymmetric C–C Michael addition. J. Mol. Catal. B Enzym. 2011;68:240–244. doi: 10.1016/j.molcatb.2010.11.011. [DOI] [Google Scholar]
- 16.Nigmatov A.G., Kuchurov I.V., Siyutkin D.E., Zlotin S.G. Enantioselective addition of carbon acids to α-nitroalkenes: The first asymmetric aminocatalytic reaction in liquefied carbon dioxide. Tetrahedron Lett. 2012;53:3502–3505. doi: 10.1016/j.tetlet.2012.04.123. [DOI] [Google Scholar]
- 17.Chen F.X., Shao C., Wang Q., Gong P., Zhang D.Y., Zhang B.Z., Wang R. An enantioselective Michael addition of malonate to nitroalkenes catalyzed by low loading demethylquinine salts in water. Tetrahedron Lett. 2007;48:8456–8459. doi: 10.1016/j.tetlet.2007.09.168. [DOI] [Google Scholar]
- 18.Albrecht B., Harald G. Asymmetric Organocatalysis—From Biomimetic Concepts to Applications in Asymmetric Synthesis. Wiley-VCH; New York, NY, USA: 2005. [Google Scholar]
- 19.Hayashi Y., Gotoh T., Hayasji T., Shoji M. Diphenylprolinol silyl ethers as efficient organocatalysts for the asymmetric Michael reaction of aldehydes and nitroalkenes. Angew. Chem. Int. Ed. 2005;44:4212–4215. doi: 10.1002/anie.200500599. [DOI] [PubMed] [Google Scholar]
- 20.Huang H.B., Jacobsen E.N. Highly enantioselective direct conjugate addition of ketones to nitroalkenes promoted by a chiral primary amine-thiourea catalyst. J. Am. Chem. Soc. 2006;128:7170–7171. doi: 10.1021/ja0620890. [DOI] [PubMed] [Google Scholar]
- 21.Watanabe M., Ikagawa A., Wang H., Murata K., Ikariya T. Catalytic enantioselective Michael addition of 1,3-dicarbonyl compounds to nitroalkenes catalyzed by well-defined chiral ru amido complexes. J. Am. Chem. Soc. 2004;126:11148–11149. doi: 10.1021/ja046296g. [DOI] [PubMed] [Google Scholar]
- 22.Evans D.A., Seidel D. Ni(II)-Bis[(R,R)-N,N′-dibenzylcyclohexane-1,2-diamine]Br2 catalyzed enantioselective Michael additions of 1,3-dicarbonyl compounds to conjugated nitroalkenes. J. Am. Chem. Soc. 2005;127:9958–9959. doi: 10.1021/ja052935r. [DOI] [PubMed] [Google Scholar]
- 23.Lu S.F., Du D.M., Xu J., Zhang S.W. Asymmetric Michael addition of nitroalkanes to nitroalkenes catalyzed by C2-symmetric tridentate bis(oxazoline) and bis(thiazoline) zinc complexes. J. Am. Chem. Soc. 2006;128:7418–7419. doi: 10.1021/ja0604008. [DOI] [PubMed] [Google Scholar]
- 24.Dijk E.W., Boersma A.J., Feringa B.L., Roelfes G. On the role of DNA in DNA-based catalytic enantioselective conjugate addition reactions. Org. Biomol. Chem. 2010;8:3868–3873. doi: 10.1039/c005048b. [DOI] [PubMed] [Google Scholar]
- 25.Izquierdo C., Luis-Barrera J., Fraile A., Alemán J. 1,4-Michael additions of cyclic-β-ketoesters catalyzed by DNA in aqueous media. Catal. Commu. 2014;44:10–14. doi: 10.1016/j.catcom.2013.08.015. [DOI] [Google Scholar]
- 26.Lindstrom U.M. Stereoselective organic reactions in water. Chem. Rev. 2002;102:2751–2772. doi: 10.1021/cr010122p. [DOI] [PubMed] [Google Scholar]
- 27.Habibi A., Tarameshloo Z. A new and convenient method for synthesis of barbituric acid derivatives. J. Iran. Chem. Soc. 2011;8:287–291. doi: 10.1007/BF03246226. [DOI] [Google Scholar]
- 28.Rastaldo R., Penna C., Pagliaro P. Comparison between the effects of pentobarbital or ketamine/nitrous oxide anesthesia on metabolic and endothelial components of coronary reactive hyperemia. Life Sci. 2001;69:729–738. doi: 10.1016/S0024-3205(01)01161-4. [DOI] [PubMed] [Google Scholar]
- 29.Jursic B.S., Stevens D.E. Transition metal free reductive dimerization of nitrogen containing barbituric acid benzylidenes. J. Heterocycl. Chem. 2003;40:701–706. doi: 10.1002/jhet.5570400423. [DOI] [Google Scholar]
- 30.Wolff M.E. Burger’s Medicinal Chemistry and Drug Discovery. Wiley; New York, NY, USA: 1997. [Google Scholar]
- 31.Holtkamp M., Meierkord H. Anticonvulsant, antiepileptogenic, and antiictogenic pharmacostrategies. Cell. Mol. Life Sci. 2007;64:2023–2041. doi: 10.1007/s00018-007-7021-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kotha S., Dep A., Kumar R. Design and synthesis of spiro-annulated barbituric acid derivatives and its analogs by ring-closing metathesis reaction as key steps. Bioorg. Med. Chem. Lett. 2005;15:1039–1043. doi: 10.1016/j.bmcl.2004.12.034. [DOI] [PubMed] [Google Scholar]
- 33.Barakat A., Al Majid A.M.A., Shahidul Islam M., Al-Othman Z.A. Highly enantioselective Friedel−Crafts alkylations of indoles with α,β-unsaturated ketones under Cu(II)-simple oxazoline-imidazoline catalysts. Tetrahedron. 2013;69:5185–5192. doi: 10.1016/j.tet.2013.04.063. [DOI] [Google Scholar]
- 34.Barakat A., Al-Majid A.M. Synthesis, structural characterization of monodentate phosphite ligands and phosphite ruthenium complexes derived from d-mannitol. Arab. J. Chem. 2013 in press. [Google Scholar]
- 35.Al-Majid A.M.A., Barakat A., Mabkhot Y.N., Islam M.S. Synthesis and characterization of privileged monodentatephosphoramidite ligands and chiral brønsted acid derived from d-mannitol. Int. J. Mol. Sci. 2012;13:2727–2743. doi: 10.3390/ijms13032727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Al-Majid A.M., Barakat A., AL-Najjar H.J., Mabkhot Y.N., Ghabbour H.A., Fun H.K. Tandem Aldol-Michael reactions in aqueous diethylamine medium: A greener and efficient approach to bis-pyrimidine derivatives. Int. J. Mol. Sci. 2013;14:23762–23773. doi: 10.3390/ijms141223762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barakat A., Al-Majid A.M., AL-Najjar H.J., Mabkhot Y.N., Ghabbour H.A., Fun H.K. An efficient and green procedure for synthesis of rhodanine derivatives by Aldol-thia-Michael protocol using aqueous diethylamine. RSC Adv. 2014;4:4909–4916. doi: 10.1039/c3ra46551a. [DOI] [Google Scholar]
- 38.Abaee M.S., Cheraghi S., Navidipoor S., Mojtahedi M.M., Forghani S. An efficient tandem aldol condensation-thia-Michael addition process. Tetrahedron Lett. 2012;53:4405–4408. doi: 10.1016/j.tetlet.2012.06.040. [DOI] [Google Scholar]
- 39.Gruttadauria M., Giacalone F., Marculesco A.M., Meo P.L., Riela S., Noto R. Hydrophobically directed Aldol reactions: Polystyrene-supported l-proline as a recyclable catalyst for direct asymmetric Aldol reactions in the presence of water. Eur. J. Org. Chem. 2007;2007:4688–4698. [Google Scholar]
- 40.Breslow R. Determining the geometries of transition states by use of antihydrophobic additives in water. Acc. Chem. Res. 2004;37:471–478. doi: 10.1021/ar040001m. [DOI] [PubMed] [Google Scholar]
- 41.Blackmond D.G., Armstrong A., Coombe V., Wells A. Water in organocatalytic processes: Debunking the myths. Angew. Chem. Int. Ed. Engl. 2007;46:3798–3800. doi: 10.1002/anie.200604952. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.