

Abstract
Despite the broad antitumor spectrum of cisplatin, its therapeutic efficacy in cancer treatment is compromised by the development of drug resistance in tumor cells and systemic side effects. A close correlation has been drawn between cisplatin resistance in tumor cells and increased levels of intracellular thiol-containing species, especially glutathione (GSH). The construction of a unique nanoparticle (NP) platform composed of poly(disulfide amide) polymers with a high disulfide density for the effective delivery of Pt(IV) prodrugs capable of reversing cisplatin resistance through the disulfide-group-based GSH-scavenging process, as described herein, is a promising route by which to overcome limitations associated with tumor resistance. Following systematic screening, the optimized NPs (referred to as CP5 NPs) showed a small particle size (76.2 nm), high loading of Pt(IV) prodrugs (15.50% Pt), a sharp response to GSH, the rapid release of platinum (Pt) ions, and notable apoptosis of cisplatin-resistant A2780cis cells. CP5 NPs also exhibited long blood circulation and high tumor accumulation after intravenous injection. Moreover, in vivo efficacy and safety results showed that CP5 NPs effectively inhibited the growth of cisplatin-resistant xenograft tumors with an inhibition rate of 83.32% while alleviating serious side effects associated with cisplatin. The GSH-scavenging nanoplatform is therefore a promising route by which to enhance the therapeutic index of Pt drugs used currently in cancer treatment.
Keywords: Cancer, cisplatin resistance, glutathione, nanoparticles, Pt(IV) prodrugs
Graphical Abstract
As a DNA cross-linking molecule with remarkable antitumor efficacy, cisplatin has become one of the most widely used chemotherapeutics in cancer treatment.1 The development of cisplatin resistance for some primary tumors and most recurrent tumors has, however, seriously challenged its clinical benefits.2 Multiple mechanisms of cisplatin resistance have been proposed, including the reduced accumulation of platinum (Pt) ions by decreased transport and increased effiux, elevated levels of thiol-containing molecules, activated translesion DNA synthesis, down-regulated mismatch repair, aberrant apoptotic signals, and up-regulated nucleotide excision repair.3–8 Among them, the sensitivity of tumor cells to cisplatin can be greatly increased by reducing the concentrations of over-expressed thiol-containing species, especially glutathione (GSH).9,10 The detoxification of cisplatin in the presence of thiols has been attributed to the avid binding of biological nucleophiles with Pt ions.11 Furthermore, the conjugation of Pt with GSH catalyzed by glutathione S-transferases12,13 expedites the export of Pt from cells via ATP-dependent glutathione S-conjugate pumps.11,14 Different strategies have therefore been proposed to protect Pt against GSH deactivation. For example, picoplatin, a Pt(II) complex designed with a methyl group in the ortho position of the pyridine ring, sterically blocked the attack of its Pt center by GSH during in vitro studies. In clinical trials, a significant reduction in neurotoxicity was reported for picoplatin, but the expected improvement in anticancer properties in cisplatin-resistant tumors was not observed.15 Similarly, the Pt(IV) prodrugs, ormaplatin and iproplatin, which were designed to consume intracellular GSH, also showed disappointing results in clinical trials.16,17 The phase II trial of ormaplatin was halted owing to several toxicity concerns attributed to the rapid biological reduction of prodrugs in the blood.18 In contrast, iproplatin was less prone to reduction, which, in turn, contributed to its modest efficacy in the phase III trial.19,20 New strategies that target the GSH pathway and restore cisplatin sensitivity in tumor cells are therefore highly sought after as routes to deliver more effective anticancer agents that do not suffer from current limitations associated with the development of drug resistance.
Nanoparticle (NP) technologies have shown promise in cancer therapy, potentially offering the safer and more-efficient delivery of therapeutic agents to tumors.21–29 A variety of nanoplatforms have been developed to improve blood circulation, decrease adverse reactions, and enhance the efficacy of Pt drugs.30–43 Recently, several GSH-sensitive NPs have also been reported as methods to trigger Pt drug release:44–47 GSH-responsive Pt(II) prodrug micelles conjugated with folate ligands have shown promise for the treatment of cervical carcinoma,46 and GSH-responsive albumin NPs loaded with cisplatin displayed improved biosafety and promising efficacy in preliminary in vitro studies with medulloblastoma cells.47 Nevertheless, none of these GSH-sensitive nanoplatforms has been explored as a method by which to reverse cisplatin resistance. In light of the aforementioned link between GSH and tumor resistance, we hypothesized that the intracellular GSH-scavenging process by NPs with a high density of disulfide groups could be beneficial for reversing drug resistance, thus improving the sensitivity of tumor cells to Pt drugs.
Here, we report the synthesis of cysteine-based poly-(disulfide amide) (Cys-PDSA) polymers that readily react with GSH via disulfide-mediated reduction and their combination with a series of Pt(IV) prodrugs having tunable hydrophobicity. These Cys-PDSA polymers and Pt(IV) prodrugs were formulated together with lipid-PEG to generate a library of Pt(IV) prodrug-loaded Cys-PDSA NPs by nanoprecipitation (Figure 1A). After screening and optimization, we identified the optimal NP formulation (referred to as CP5 NPs). Initial experiments confirmed that CP5 NPs have small particle size (76.2 nm) and high Pt loading efficiency (15.50%), while the intravenous injection of CP5 NPs indicated that they had long blood circulation and high tumor-accumulation properties. Subsequent experimental data supported our hypothesis that, upon tumor cell uptake, CP5 NPs rapidly disassemble and release Pt drugs in response to intracellular GSH while simultaneously consuming GSH to restore Pt sensitivity in cisplatin-resistant tumor cells (Figure 1B). Both in vitro and in vivo results with CP5 NPs support the growth inhibition of cisplatin-resistant A2780cis xenograft tumors. CP5 NPs also induce negligible systemic toxicities. Mechanistic studies with CP5 NPs were performed to better understand the observed reversal of Pt resistance.
Figure 1.
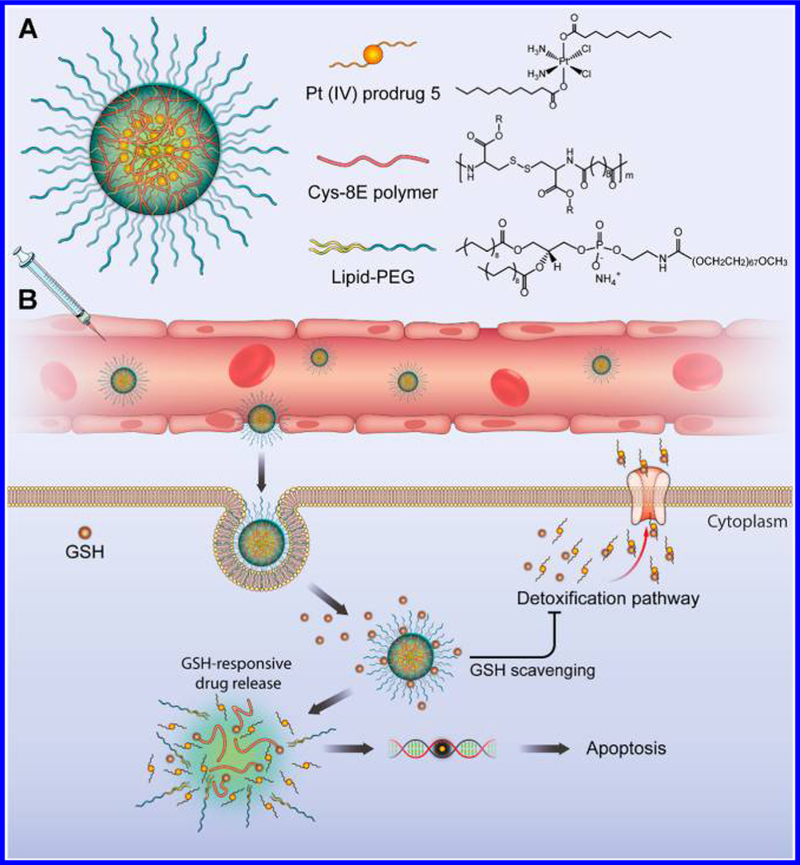
(A) Illustration of the redox-responsive nanoplatform, composed of Pt(IV) prodrug 5, Cys-8E polymer, and lipid-PEG, for the in vivo Pt delivery and treatment of cisplatin-resistant tumors. (B) CP5 NPs coated with lipid-PEG were designed to achieve long blood circulation, leading to high tumor accumulation via the enhanced permeability and retention (EPR) effect. Following cellular uptake, high levels of GSH in the cytosol promoted the rapid disintegration of CP5 NPs and release of Pt(IV) prodrugs. The Cys-8E polymer-mediated GSH-scavenging process was expected to minimize the GSH-induced detoxification pathway, decreasing the likelihood of released Pt drugs being deactivated and enabling them to diffuse into nuclei, where they would bind covalently with purine bases of DNA and ultimately trigger apoptosis.
Results and Discussion.
Cys-PDSA polymers were prepared as previously reported via a one-step rapid polycondensation of two nontoxic building blocks: L-cysteine ester and versatile fatty diacids (Figure S1).48 These polymers were denoted Cys-nE (n = 2, 4, 6, 8, or 10), with n representing the number of methylene groups in the diacid repeating unit and E indicating the methyl ester of carboxylic acid on the side chain. Pt(IV) prodrugs were synthesized as previously described by first oxidizing cisplatin with H2O2, then reacting intermediates with desired anhydride to obtain cis,cis,trans-[Pt(NH3)2Cl2(OOCR)2], where R was methyl (1), propyl (2), pentyl (3), heptyl (4), nonyl (5), phenyl (6), 2,4,6-trimethylphenyl (7) or 4-tert-butyl-phenyl (8) (Figures S2–18 and Table S1).49 The length of alkyl chain and the type of aromatic functionality were varied to regulate the hydrophobicity of prodrugs.
Pt(IV) prodrug-loaded Cys-PDSA NPs were first formulated by nanoprecipitation of Cys-nE polymers with Pt(IV) prodrug 5 and then coated with lipid-PEG (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-3000] (ammonium salt), DSPE-PEG 3000) (Table S2). Initial studies examining the effect that varying the length of methylene linkers in Cys-nE polymers had on particle size and Pt loading identified polymer Cys-8E as a suitable candidate for formulation with all Pt(IV) prodrugs.
NPs designated CP1–8 were thus prepared with the Cys-8E polymer and Pt(IV) prodrugs containing variable R groups, as described above (Table S3). The alkyl chain length (1–5) and the type of aromatic functionality (6–8) in the prodrugs controlled the particle size and Pt loading of the NPs. As the hydrocarbon chain increased from methyl to phenyl, the particle size of corresponding NPs increased systematically from 64.8 to 76.2 nm and the Pt loading from 1.44% to 15.50%. The addition of trimethyl and tertiary butyl group substituents onto the aromatic ring enlarged the particle size but had only a modest effect on Pt loading. All formulations exhibited negative ζ potentials owing to the exterior DSPE-PEG 3000 layer. Finally, CP5 NPs were chosen for further evaluation because of their small particle size (76.2 nm) and high Pt loading (15.50%), desirable properties for a Pt drug-delivery platform.
For comparison, the redox potentials for reduction of Pt(IV) prodrug 5 and the Cys-8E polymer were determined (Figure S19). Pt(IV) prodrug 5 displayed an irreversible cyclic voltammetric response for the Pt(IV)/Pt(II) couple near −0.61 V versus Ag/AgCl, whereas that of the Cys-8E polymer is approximately −0.36 V versus Ag/AgCl. These results indicate that the Cys-8E polymer is more easily reduced than Pt(IV) prodrug 5 upon exposure to intracellular GSH. In addition, the reduction kinetics of Pt(IV) prodrug 5 and Cys-8E polymer were measured (Figure S20A,B). The linear plot of pseudo-first-order rate constants versus different concentrations of dithiothreitol (DTT, a model thiol-reductant with a reduction potential similar to that of GSH)50,51 indicated that the redox reaction followed a second-order rate law (Figure S20C,D). In comparison to Pt(IV) prodrug 5, which has a moderate reduction rate (k ≈ 0.15 M−1 s−1), Cys-8E polymer with k ≈ 0.38 M−1 s−1 had a much higher reduction rate, suggesting the stronger potential of the polymer in scavenging intracellular GSH. Note that a spectrophotometric investigation of the reduction of Pt(IV) prodrug 5 and Cys-8E polymer by GSH was not feasible because of the insolubility of GSH in organic solvents (e.g., DMSO and DMF) in which the prodrug and polymer were dissolved; thus, DTT was chosen here. To further demonstrate the GSH-scavenging capability of Cys-8E polymer, control NPs consisting of Cys-8E polymer without Pt(IV) prodrug 5 were incubated with GSH at 37 °C in PBS, and the GSH level was quantitated as a function of time. (Figure S21). A rapid decline in GSH content was observed, confirming the GSH-scavenging capability of Cys-8E polymer. Incubation of CP5 NPs with GSH in PBS resulted in a little precipitation, which might have been induced by the substitution of chloride in the Pt(II) reduction product with GSH/GS−..52–54
In vitro disintegration of NPs in response to DTT was monitored using TEM (Figure 2A,B). Following the incubation of CP5 NPs with 10 mM DTT for 72 h, the rapid degradation of spherical NPs into irregularly shaped debris was observed. In contrast, CP5 NPs were found to be colloidally stable in PBS over the course of a week (Figure S22). Platinum released from NPs incubated with PBS containing 0, 1, or 10 mM DTT was also measured by graphite furnace atomic absorption spectrometry (GFAAS) (Figure 2C). Approximately 80% of the total Pt loading was released from CP5 NPs over the course of 72 h following incubation in a 10 mM DTT solution. In contrast, approximately 30% of the payload was released when CP5 NPs were incubated in a 1 mM DTT solution, and less than 10% was released as CP5 NPs were incubated in PBS alone. These results support the reduction-promoted disassembly of CP5 NPs with concomitant release of Pt drugs.
Figure 2.
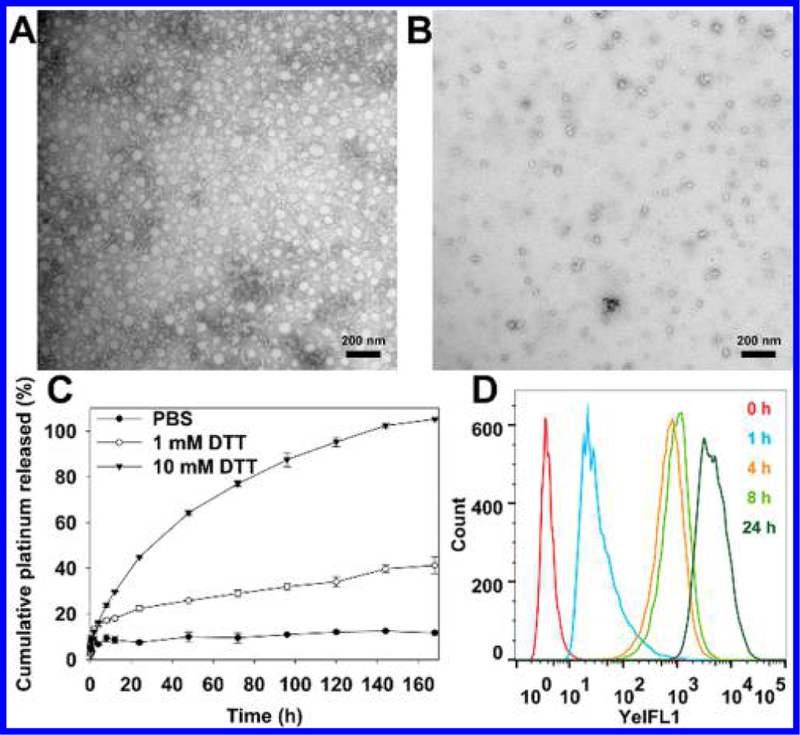
Representative TEM images of CP5 NPs stored in (A) water or (B) 10 mM DTT for 72 h (scale bar: 200 nm). (C) Pt release profiles of CP5 NPs measured by GFAAS. (D) Cellular uptake of Dil-loaded CP5 NPs detected by flow cytometry.
To evaluate the internalization of NPs, A2780 and A2780cis cells were incubated with CP5 NPs for 0–24 h using Dil as the fluorescent probe (Figures 2D and S23). Flow cytometry analysis confirmed that CP5 NPs entered tumor cells in a time-dependent manner. The intracellular disintegration of CP5 NPs was monitored by Förster resonance energy transfer (FRET) probes (Figures 3A, S24, and 25). The donor chromophore, coumarin 6, initially in its electronic excited state (410 nm), transfers energy to the acceptor chromophore, Nile red, through nonradiative dipole–dipole coupling (but only if the chromophore pair is in close proximity, 1–10 nm) and then fluoresces at 590 nm. Once the FRET pair is completely separated, the green fluorescence of coumarin 6 is restored, and Nile red stops fluorescing.55 For normal A2780 cells, an obvious FRET image could be seen after 4 h of incubation, whereas for A2780cis cells, a much-weaker FRET image was captured. We attribute these results to the up-regulated GSH level in cisplatin-resistant A2780cis cells, which is postulated to promote the faster disassembly of CP5 NPs co-loaded with Nile red and Coumarin 6 and therefore increase the separation rate of FRET pair. After 18 h, only green fluorescence was observed, indicating the complete decom-position of CP5 NPs in both cell lines. However, when both cells were pretreated with N-ethylmaleimide (NEM), we observed the persistent FRET imaging, owing to NEM-mediated inhibition of GSH activity,56 further confirming the interactions of intracellular GSH with CP5 NPs.
Figure 3.
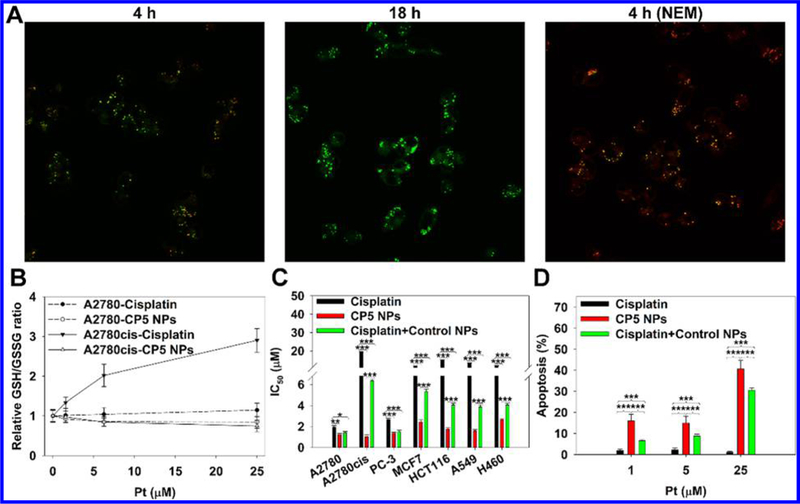
(A) Confocal fluorescence images of A2780cis cells incubated with Nile red and Coumarin 6-co-loaded CP5 NPs for 4 and 18 h (60× objective). To investigate the effect of GSH on NP disassembly, cells were also pretreated with NEM to consume intracellular GSH. (B) Relative GSH-to-GSSH ratio of A2780 and A2780cis cells treated with cisplatin or CP5 NPs. (C) In vitro cytotoxicity of cells treated with cisplatin, CP5 NPs, or cisplatin + control NPs for 48 h. (D) In vitro apoptosis of A2780cis cells treated with cisplatin, CP5 NPs, or cisplatin + control NPs for 24 h.
As Pt(IV) drugs enter tumor cells, GSH and other intracellular reductants such as ascorbate will reduce the Pt(IV) to form Pt(II) ions. Rapid GSH binding to the Pt(II) center, cisplatin, will provide a route for detoxification. This behavior results in the sequential up-regulation of mRNA expression of γ-glutamylcysteine synthetase and γ-glutamyl transpeptidase and, ultimately, the restoration of GSH levels.10 To investigate the cytosolic reduction of CP5 NPs, the relative ratio of GSH to its oxidized form, glutathione disulfide (GSSG), was measured (Figure 3B). As shown in the figure, a remarkable increase in relative GSH-to-GSSG ratio caused by cisplatin was observed for Pt concentrations within the range from 0 to 25 μM. The sustained relative ratio increase is proposed to be a consequence to the up-regulated GSH biosynthesis in response to the consumption of GSH during detoxification. Meanwhile, the increase in relative GSH-to-GSSG ratio was more striking in A2780cis cells, which increased from 1.00 to 2.90, while an increase of 1.00 to 1.14 was recorded for A2780 cells, confirming that the biological response to cisplatin (or GSH biosynthesis) was much more sensitive in A2780cis cells than A2780 cells.57 Conversely, CP5 NPs successfully suppressed the relative ratio increase (from 1.00 to 0.84 for 2780, from 1.00 to 0.75 for 2780cis) by consuming intracellular GSH and generating GSSG. Moreover, the GSH level in A2780cis cells (3.85 ± 0.05 wt %) was more than two times higher than that in A2780 cells (1.63 ± 0.03 wt %), supporting the above results from FRET experiments, as well as those previously reported.57
We next extensively examined the cytotoxicity of NPs in multiple cancer cell lines (A2780, A2780cis, PC-3, MCF7, HCT116, A549, and H460). IC50 values and cell viability results for each cell line are presented in Figures 3C, S26, and 27. Compared with CP5 NPs that displayed the lowest IC50 values across the cell lines investigated, cisplatin displayed limited toxicity against the cancer cells evaluated except A2780 and PC-3. When both cisplatin and control NPs were incubated together with the above cells, IC50 values decreased noticeably compared with those reported for cisplatin alone, indicating that Cys-8E polymer might contribute to the high residue of Pt ions in the cytosol through a GSH-scavenging effect, thus enhancing the cytotoxicity. Consistently, CP5 NPs induced enhanced apoptosis in both A2780 and A2780cis cells (Figures 3D, S28, and 29). Taken together, we postulated that these promising anticancer properties arose from high-efficiency cellular uptake of CP5 NPs that liberated a large amount of Pt ions upon the consumption of thiol-containing species, particularly GSH. The drug release strategy thus served a dual purpose, both delivering the active Pt anticancer agent and depleting the GSH concentration attributed to cisplatin-resistant cell lines.
After verifying in vitro antitumor efficiency, the potential of NPs for in vivo therapy was assessed. First, the pharmacokinetics of CP5 NPs loaded with lipophilic dye, DID, were tested. Figure 4A shows that, unlike rapid elimination of free DID, DID-loaded CP5 NPs produced a much more stable and mild decline curve, exposing the nature of longer retention. A noncompartment model was applied using a Phoenix WinNonlin 6.3 Program to calculate pharmacokinetic parameters (Table S4). AUC0→inf and AUMC0→inf of NPs were 4-fold higher, while CL and Vss decreased by ~99%, reflecting the enhanced bioavailability and delayed clearance. Next, the biodistribution of NPs was determined with A2780cis tumor-bearing athymic nude mice by real-time imaging (Figure 4B). DID-loaded CP5 NPs were observed to enrich in tumors and plasma, indicating significantly improved distribution behavior of the dye as compared with free DID. Moreover, CP5 NPs primarily bypassed the mononuclear phagocyte system (MPS), resulting in minimal accumulation in liver, spleen and lung (Figure 4C,D).58 While in tumor tissue, it was observed that NPs could readily extravasate microvessels and permeate tumor parenchyma.
Figure 4.
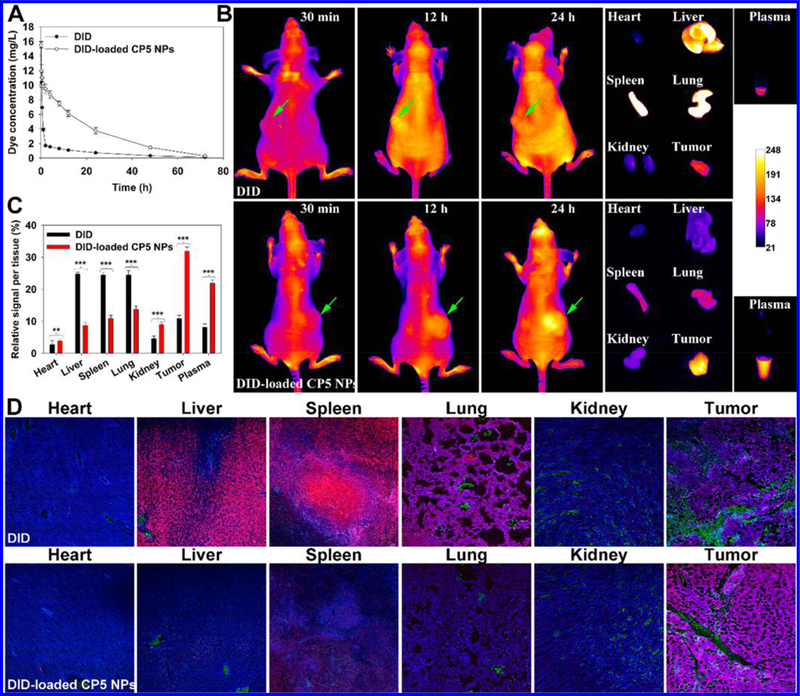
(A) Pharmacokinetics of DID or DID-loaded CP5 NPs in healthy BALB/c mice (n = 5). (B) Biodistribution of DID or DID-loaded CP5 NPs in A2780cis tumor-bearing athymic nude mice (n = 3). (C) Relative fluorescence signal per tissue as quantified from panel B. (D) The co-localization of dyes in organs and tumors with microvessels stained with anti-CD31 antibody (green) and nuclei stained with DAPI (blue) (20× objective).
To test the antitumor efficacy of NPs, A2780cis tumor-bearing athymic nude mice were intravenously injected with PBS, free cisplatin, CP5 NPs, or control NPs. The tumor volumes of control and cisplatin groups grew sharply, confirming that cisplatin was ineffective in altering the natural progression of Pt-resistant ovarian tumors (Figures 5A,B and S30A). By comparison, mice treated with CP5 NPs displayed decelerated tumor growth, with tumor inhibition rates (TIR) recorded as 83.32 ± 5.80% versus 1.46 ± 1.29% for cisplatin and 1.48 ± 0.53% for control NPs, respectively. Upon the termination of the in vivo efficacy study, tumors were collected and stained by H&E, and their pathology was evaluated (Figure 5C). Spherical or spindle tumor cells from control and cisplatin groups contained more chromatin and caryosomes, revealing extensive proliferation. However, tumors collected from mice treated with CP5 NPs displayed shrunken or fragmented cells, concentrated chromatin, and pyknotic nuclei undergoing karyorrhexis or karyolysis, indicative of tumor necrosis.59 Tumors from mice treated with cisplatin also displayed some of same markers of necrosis, but these were typically less severe than those reported with CP5 NPs, further reflecting the improved therapeutic effect of CP5 NPs over established cisplatin treatment. Additionally, weight loss, a commonly reported side effect of cisplatin, was not observed for mice treated with either control NPs or CP5 NPs, and in fact, mice treated with NP formulations were reported to have a slightly increased body weight over the duration of this study (Figure S30B).
Figure 5.
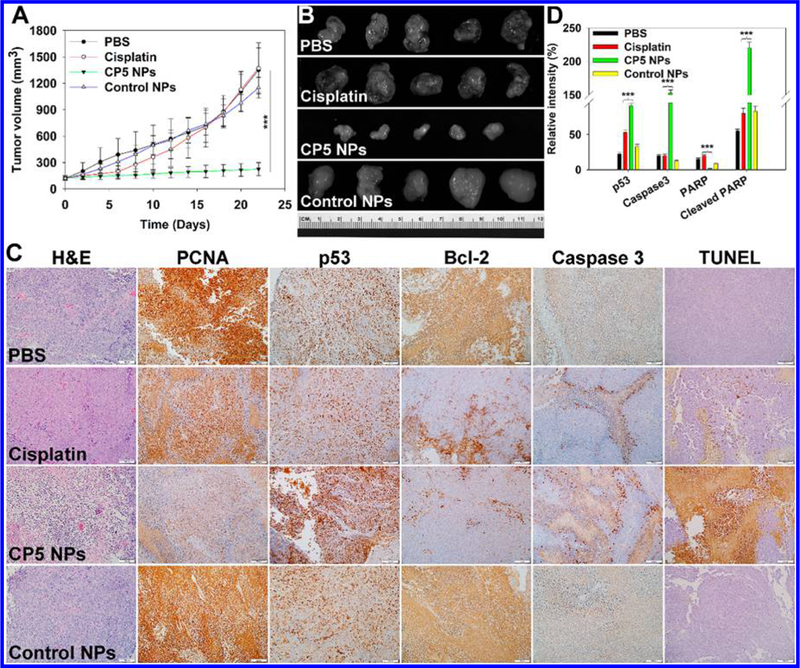
(A) Tumor volumes of A2780cis tumor-bearing athymic nude mice during chemotherapy (n = 5). CP5 NPs showed statistically significant growth suppression compared to cisplatin. (B) Harvested tumors after systemic treatment captured using the Maestro 2 in vivo imaging system. (C) H&E, IHC, and TUNEL images for tumors after treatment with PBS, cisplatin, CP5 NPs, or control NPs. (D) Western blot quantification of p53, Caspase 3, PARP, and cleaved PARP for tumors after treatment with PBS, cisplatin, CP5 NPs, or control NPs (three asterisks indicate p < 0.001 compared with cisplatin).
To investigate the molecular mechanism of in vivo apoptosis, A2780cis tumor-bearing athymic nude mice were dosed with several different chemotherapeutic agents. After that, tumors were collected for immunoblotting studies. Once inside the cell, cisplatin undergoes aquation and reacts with the primary biological target, DNA, to form Pt-DNA adducts. Subsequently, p53 becomes activated in response to DNA damage induced by Pt-DNA adducts. The reinforced p53 destabilizes the balance between proapoptotic and antiapoptotic regulators and then activates the apoptotic executor Caspase 3, which promotes PARP cleavage-mediated cellular decomposition.60,61 Western blotting data (Figures 5D and S31) supported a significant increase in expression of p53, caspase 3, and cleaved PARP, especially in mice treated with CP5 NPs, supporting the proposed mitochondria control of apoptosis.
We further tested whether other mechanisms attributable to Pt resistance might be interrupted by NP treatment (Figure 5C). After Pt-DNA adducts have been formed, cellular survival (or tumor resistance) could occur by several tolerance mechanisms, such as enhanced translesion DNA synthesis and aberrant apoptotic signals.3–8 Certain DNA polymerases, i.e., β and η, can bypass Pt-DNA adducts via translesion synthesis, thereby maintaining the proliferative ability of tumor cells. As a processivity factor for the above DNA polymerases,62 proliferating cell nuclear antigen (PCNA) expression was tested. Evaluation of tumors harvested from mice dosed with CP5 NPs indicated that the lowest PCNA level was present, which was consistent with successful inhibition of cisplatin-resistant tumor proliferation. Moreover, drug resistance commonly occurs to most chemotherapeutics including cisplatin through decreased expression or loss of apoptotic signaling pathways. As mentioned above, being triggered by DNA damage, p53 induces BAX transcription to neutralize antiapoptotic Bcl-2, which goes on to result in mitochondria-control of apoptosis.63 CP5 NPs greatly increased p53, decreased Bcl-2, and increased caspase 3, demonstrating the activation of apoptotic signals. Besides, as indicated by the TUNEL assay, CP5 NPs were found to induce more-extensive DNA fragmentation than either cisplatin or control NPs, further confirming CP5 NPs treatment resulted in significant apoptosis in cisplatin-resistant tumors.
Finally, we also evaluated the in vivo safety of the NPs. Hepatotoxicity and nephrotoxicity were evaluated by measuring ALP, ALT, AST, BUN, and Scr in plasma taken from mice that had been treated with PBS, cisplatin, CP5 NPs, or control NPs (Figure S32A–E). Cisplatin, known for its harsh renal side effect profile, displayed upped levels of BUN and Scr. CP5 NPs, conversely, had negligible effect on all tested biomarkers and, thus, would be predicted to display significantly reduced nephrotoxicity and hepatotoxicity. Accordingly, it was evident that cisplatin treatment induced renal toxicity, while CP5 NPs inflicted limited adverse reactions.
In addition, hemolysis was quantified based on the concentration of hemoglobin released from red blood cells (Figure S32F). Both CP5 NPs and free cisplatin exhibited minimal hemolysis (<5%) compared with Tween 80 controls across all tested concentrations, suggesting CP5 NPs have good hemocompatibility. Pathological examination revealed no obvious changes in any of organs tested from mice treated with CP5 NPs (Figure S33). In comparison, cisplatin-treated mice showed strong systemic toxicities, resulting in liver damage with vacuolar degeneration, incomplete spleen structure with shrunken white pulp and lymphocyte depletion, alveolar hemorrhage, and renal tubular epithelial cells with hydropic or ballooning degeneration.59
Conclusions.
In summary, a family of Pt(IV) prodrugs with tunable hydrophobicity were synthesized and formulated with a library of Cys-PDSA polymers with a high disulfide density to form Pt(IV) prodrug-loaded Cys-PDSA NPs. After optimization, redox-responsive CP5 NPs with small particle size, high Pt loading, and good stability were chosen for the treatment of cisplatin-resistant tumors. Unlike cisplatin that was hindered by intracellular thiol-containing species, especially GSH, CP5 NPs protected the cargo from detoxification through a Cys-8E polymer-mediated GSH-scavenging process, which simultaneously triggered the release of Pt ions. Data acquired from in vivo studies supported our hypothesis that the poly(disulfide amide) NPs should offer a novel route for Pt anticancer agent delivery while limiting toxic side effects and the development of cisplatin resistance. Thus, the GSH-scavenging polymeric NP technology reported herein could provide a unique strategy for improving the therapeutic efficacy of current Pt drugs.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the National Institutes of Health grants HL127464 (O.C.F.), CA200900 (J.S.), and CA034992 (S.J.L.); the David H. Koch-PCF Program in Cancer Nanotherapeutics (O.C.F.); and the National Research Foundation of Korea Grant No. K1A1A2048701 (O.C.F.); the Science and Technology Planning Project of Guangdong Province no. 2016A010103015 (J.W.); and the Science and Technology Planning Project of Shenzhen City no. JCYJ20170307141438157 (J.W.).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nanolett.8b01924.
Synthesis and characterization of Cys-PDSA polymers and Pt(IV) prodrugs, cyclic voltammograms, reduction kinetics, GSH consumption by poly(disulfide amide) NPs, stability, cellular uptake, intracellular disintegration, cytotoxicity and apoptosis, tumor volume and body weight of mice, Western blot, blood chemistry and blood compatibility, histology, particle size, zeta potential and Pt loading of NPs, and pharmacokinetic parameters. (PDF)
Notes
The authors declare the following competing financial interest(s): O.C.F. has financial interests in Selecta Biosciences, Tarveda Therapeutics, and Placon Therapeutics.
REFERENCES
- (1).Wheate NJ; Walker S; Craig GE; Oun R Dalton Trans 2010, 39 (35), 8113–27. [DOI] [PubMed] [Google Scholar]
- (2).Kartalou M; Essigmann JM Mutat. Res., Fundam. Mol. Mech. Mutagen 2001, 478 (1–2), 23–43. [DOI] [PubMed] [Google Scholar]
- (3).Akiyama S; Chen ZS; Sumizawa T; Furukawa T Anti-Cancer Drug Des 1999, 14 (2), 143–51. [PubMed] [Google Scholar]
- (4).Siddik ZH Oncogene 2003, 22 (47), 7265–79. [DOI] [PubMed] [Google Scholar]
- (5).Rabik CA; Dolan ME Cancer Treat. Rev 2007, 33 (1), 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Galluzzi L; Senovilla L; Vitale I; Michels J; Martins I; Kepp O; Castedo M; Kroemer G Oncogene 2012, 31 (15), 1869–83. [DOI] [PubMed] [Google Scholar]
- (7).Xu X; Xie K; Zhang XQ; Pridgen EM; Park GY; Cui DS; Shi J; Wu J; Kantoff PW; Lippard SJ; Langer R; Walker GC; Farokhzad OC Proc. Natl. Acad. Sci. U. S. A 2013, 110 (46), 18638–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Xie K; Doles J; Hemann MT; Walker GC Proc. Natl. Acad. Sci. U. S. A 2010, 107 (48), 20792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kuppusamy P; Li H; Ilangovan G; Cardounel AJ; Zweier JL; Yamada K; Krishna MC; Mitchell JB Cancer Res 2002, 62 (1), 307–12. [PubMed] [Google Scholar]
- (10).Godwin AK; Meister A; O’Dwyer PJ; Huang CS; Hamilton TC; Anderson ME Proc. Natl. Acad. Sci. U. S. A 1992, 89 (7), 3070–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kelland L Nat. Rev. Cancer 2007, 7 (8), 573–84. [DOI] [PubMed] [Google Scholar]
- (12).Goto S; Iida T; Cho S; Oka M; Kohno S; Kondo T Free Radical Res 1999, 31 (6), 549–58. [DOI] [PubMed] [Google Scholar]
- (13).Chen HH; Kuo MT Met.-Based Drugs 2010, 2010 (35), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kurokawa H; Ishida T; Nishio K; Arioka H; Sata M; Fukumoto H; Miura M; Saijo N Biochem. Biophys. Res. Commun 1995, 216 (1), 258–64. [DOI] [PubMed] [Google Scholar]
- (15).Ndagi U; Mhlongo N; Soliman ME Drug Des., Dev. Ther 2017, 11, 599–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Johnstone TC; Suntharalingam K; Lippard SJ Chem. Rev 2016, 116 (5), 3436–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kenny RG; Chuah SW; Crawford A; Marmion CJ Eur. J. Inorg. Chem 2017, 2017 (12), 1596–1612. [Google Scholar]
- (18).Gibbons GR; Wyrick S; Chaney SG Cancer Res 1989, 49 (6), 1402–7. [PubMed] [Google Scholar]
- (19).Bramwell VH; Crowther D; O’Malley S; Swindell R; Johnson R; Cooper EH; Thatcher N; Howell A Cancer Treat. Rep 1985, 69 (4), 409–16. [PubMed] [Google Scholar]
- (20).Sessa C; Vermorken J; Renard J; Kaye S; Smith D; ten Bokkel Huinink W; Cavalli F; Pinedo HJ Clin. Oncol 1988, 6 (1), 98–105. [DOI] [PubMed] [Google Scholar]
- (21).Farokhzad OC; Langer R Adv. Drug Delivery Rev 2006, 58 (14), 1456–9. [DOI] [PubMed] [Google Scholar]
- (22).Chu C; Lin H; Liu H; Wang X; Wang J; Zhang P; Gao H; Huang C; Zeng Y; Tan Y; Liu G; Chen X Adv. Mater 2017, 29, (23). DOI: 10.1002/adma.201770164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Qian C; Yu J; Chen Y; Hu Q; Xiao X; Sun W; Wang C; Feng P; Shen QD; Gu Z Adv. Mater 2016, 28 (17), 3313–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kong SD; Sartor M; Hu CMD; Zhang W; Zhang L; Jin S Acta Biomater 2013, 9 (3), 5447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kim S; Park JW; Kim D; Kim D; Lee IH; Jon S Angew. Chem., Int. Ed 2009, 48 (23), 4138–41. [DOI] [PubMed] [Google Scholar]
- (26).Shi J; Kantoff PW; Wooster R; Farokhzad OC Nat. Rev. Cancer 2017, 17 (1), 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Czapar AE; Zheng YR; Riddell IA; Shukla S; Awuah SG; Lippard SJ; Steinmetz NF ACS Nano 2016, 10 (4), 4119–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lu Y; Aimetti AA; Langer R; Gu Z Nat. Rev. Mater 2017, 2 (1), 17. [Google Scholar]
- (29).Liu M; Shen S; Wen D; Li M; Li T; Chen X; Gu Z; Mo R Nano Lett 2018, 18 (4), 2294–2303. [DOI] [PubMed] [Google Scholar]
- (30).Ling X; Shen Y; Sun R; Zhang M; Li C; Mao J; Xing J; Sun C; Tu J Polym. Chem 2015, 6 (9), 1541–52. [Google Scholar]
- (31).Aryal S; Hu CM; Zhang L Chem. Commun. (Cambridge, U. K.) 2012, 48 (20), 2630–2. [DOI] [PubMed] [Google Scholar]
- (32).Lee DY; Kim JY; Lee Y; Lee S; Miao W; Kim HS; Min JJ; Jon S Angew. Chem., Int. Ed 2017, 56 (44), 13684–8. [DOI] [PubMed] [Google Scholar]
- (33).Xiao H; Qi R; Li T; Awuah SG; Zheng Y; Wei W; Kang X; Song H; Wang Y; Yu Y; Bird MA; Jing X; Yaffe MB; Birrer MJ; Ghoroghchian PP J. Am. Chem. Soc 2017, 139 (8), 3033–44. [DOI] [PubMed] [Google Scholar]
- (34).Miller MA; Zheng YR; Gadde S; Pfirschke C; Zope H; Engblom C; Kohler RH; Iwamoto Y; Yang KS; Askevold B; Kolishetti N; Pittet M; Lippard SJ; Farokhzad OC; Weissleder R Nat. Commun 2015, 6, 8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ma P; Xiao H; Yu C; Liu J; Cheng Z; Song H; Zhang X; Li C; Wang J; Gu Z; Lin J Nano Lett 2017, 17 (2), 928–37. [DOI] [PubMed] [Google Scholar]
- (36).Zhang L; Laug L; Munchgesang W; Pippel E; Gosele U; Brandsch M; Knez M Nano Lett 2010, 10 (1), 219–23. [DOI] [PubMed] [Google Scholar]
- (37).Dhar S; Gu FX; Langer R; Farokhzad OC; Lippard SJ Proc. Natl. Acad. Sci. U. S. A 2008, 105 (45), 17356–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Dhar S; Kolishetti N; Lippard SJ; Farokhzad OC Proc. Natl. Acad. Sci. U. S. A 2011, 108 (5), 1850–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kolishetti N; Dhar S; Valencia PM; Lin LQ; Karnik R; Lippard SJ; Langer R; Farokhzad OC Proc. Natl. Acad. Sci. U. S. A 2010, 107 (42), 17939–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Dhar S; Daniel WL; Giljohann DA; Mirkin CA; Lippard SJ J. Am. Chem. Soc 2009, 131 (41), 14652–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Feazell RP; Nakayama-Ratchford N; Dai H; Lippard SJ J. Am. Chem. Soc 2007, 129 (27), 8438–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Dai Y; Cheng S; Wang Z; Zhang R; Yang Z; Wang J; Yung BC; Wang Z; Jacobson O; Xu C; Ni Q; Yu G; Zhou Z; Chen X ACS Nano 2018, 12 (1), 455–63. [DOI] [PubMed] [Google Scholar]
- (43).Xiao H; Song H; Zhang Y; Qi R; Wang R; Xie Z; Huang Y; Li Y; Wu Y; Jing X Biomaterials 2012, 33 (33), 8657–69. [DOI] [PubMed] [Google Scholar]
- (44).Han Y; Li JJ; Zan MH; Luo SZ; Ge ZS; Liu SY Polym. Chem 2014, 5 (11), 3707–18. [Google Scholar]
- (45).Li Y; Li Y; Zhang X; Xu X; Zhang Z; Hu C; He Y; Gu Z Theranostics 2016, 6 (9), 1293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Tabatabaei Rezaei S. J.; Amani V; Nabid MR; Safari N; Niknejad H Polym. Chem 2015, 6 (15), 2844–53. [Google Scholar]
- (47).Catanzaro G; Curcio M; Cirillo G; Spizzirri UG; Besharat ZM; Abballe L; Vacca A; Iemma F; Picci N; Ferretti E Int. J. Pharm 2017, 517 (1–2), 168–74. [DOI] [PubMed] [Google Scholar]
- (48).Wu J; Zhao L; Xu X; Bertrand N; Choi WI; Yameen B; Shi J; Shah V; Mulvale M; MacLean JL; Farokhzad OC Angew. Chem., Int. Ed 2015, 54 (32), 9218–23. [DOI] [PubMed] [Google Scholar]
- (49).Johnstone TC; Lippard SJ Inorg. Chem 2013, 52 (17), 9915–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Rost J; Rapoport S Nature 1964, 201, 185. [DOI] [PubMed] [Google Scholar]
- (51).Cleland WW Biochemistry 1964, 3 (4), 480–2. [DOI] [PubMed] [Google Scholar]
- (52).Berners-Price SJ; Kuchel PW J. Inorg. Biochem 1990, 38 (4), 305–26. [DOI] [PubMed] [Google Scholar]
- (53).Murdoch P.d. S.; Kratochwil NA; Parkinson JA; Patriarca M; Sadler PJ Angew. Chem., Int. Ed 1999, 38 (19), 2949–51. [DOI] [PubMed] [Google Scholar]
- (54).Appleton TG; Connor JW; Hall JR; Prenzler PD Inorg. Chem 1989, 28 (11), 2030–37. [Google Scholar]
- (55).Zheng J, Spectroscopy-Based Quantitative Fluorescence Resonance Energy Transfer Analysis. In Ion Channels: Methods and Protocols, Stockand, J. D.; Shapiro MS, Eds.; Humana Press: Totowa, NJ, 2006; pp 65–77. [DOI] [PubMed] [Google Scholar]
- (56).Yang J; Chen H; Vlahov IR; Cheng JX; Low PS Proc. Natl. Acad. Sci. U. S. A 2006, 103 (37), 13872–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Lewis AD; Hayes JD; Wolf CR Carcinogenesis 1988, 9 (7), 1283–7. [DOI] [PubMed] [Google Scholar]
- (58).Hume DA Curr. Opin. Immunol 2006, 18 (1), 49–53. [DOI] [PubMed] [Google Scholar]
- (59).Shackelford C; Long G; Wolf J; Okerberg C; Herbert R Toxicol. Pathol 2002, 30 (1), 93–6. [DOI] [PubMed] [Google Scholar]
- (60).Carson DA; Lois A Lancet 1995, 346 (8981), 1009–11. [DOI] [PubMed] [Google Scholar]
- (61).Cleary ML; Smith SD; Sklar J Cell 1986, 47 (1), 19–28. [DOI] [PubMed] [Google Scholar]
- (62).Moldovan GL; Pfander B; Jentsch S Cell 2007, 129 (4), 665–79. [DOI] [PubMed] [Google Scholar]
- (63).Tsujimoto Y; Finger LR; Yunis J; Nowell PC; Croce CM Science 1984, 226 (4678), 1097–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.