

Abstract
The new mulinane diterpenoids 1 and 2 were isolated from the EtOAc extract of Mulinum crassifolium, while the rearranged mulinane 5, which was isolated for the first time from a natural source, was isolated from Azorella compacta. Compounds 1–2 were prepared by semi-synthesis thorough acetylation of the diterpene 17-acetoxymulinic acid (3). A mechanism of reaction was proposed, while the structures of the new compounds were elucidated on the basis of comprehensive spectroscopic analysis and computational methods.
Keywords: mulinanes, azorellanes, new diterpenoids, Chilean native plants, semisynthesis
1. Introduction
Mulinum crassifolium is a 15 cm small cushion shrub, which grows in the north of Chile at altitudes above 4,000 m. This plant (Figure 1 left), commonly known as “chuquican” or “espinilla” is used in folk medicine, principally against diabetes and bronchial and intestinal disorders [1]. Mulinum crassifolium is well recognized as an important source of diterpenes bearing the mulinane skeleton, while Azorella compacta, another cushion shrub known as “llareta” (Figure 1 right) is a source of diterpenoids with both the mulinane and azorellane skeletons [2,3,4]. There is a phylogenetic relationship between these two shrubs, both belonging to the group Azorella (Apiaceae). This group includes six genera (Azorella, Laretia, Schizeilema, Mulinum, Huanaca, and Stilbocarpa) with Austral-Antarctic affinities and a geographic distribution that extends from Oceania to South America. The “Mulinum clade” is further divided into six subclades, which collectively include 23 of 26 species of Azorella, the monotypic Laretia and all species of Mulinum [5]. The rare diterpenoids isolated from these genera have displayed a wide variety of interesting biological activities, including trypanosomicidal [6], trichomonicidal [7], toxoplasmocidal [8], antiplasmodial [9], antibacterial [10], spermicidal [11], antihyperglycemic [12], antitubercular [13], antiinflamatory and analgesic activities [14,15]. In our continuing efforts in the search for the structurally diverse and biologically important metabolites from the genus Azorella [4,7,16] and Mulinum [4], two new mulinane diterpenoids, named mulinones a-b (compounds 1 and 2, Figure 2) were isolated from M. crassifolium together with 17-acetoxymulinic acid (3) and mulinolic acid (4). The rearranged mulinane 5, which is a new natural product but was previously reported as a reaction product [15], together with the known compounds azorellanol (6), and 13,14-dihydroxymulin-11-en-20-oic acid (7), were isolated from A. compacta. Compounds 1, 2 and 5 [14,15] were synthesized from their precursor natural products.
Figure 1.

Pictures of Mulinum crassifolium (left) and Azorella compacta (right), from Northern Chile. (Taken by Jorge Bórquez and Mario J. Simirgiotis, on March 2011).
Figure 2.

Structures of the diterpenoids isolated from Mulinum crassifolium and Azorella compacta.
This paper deals with the isolation and structural elucidation of the new compounds. Furthermore the semi-synthesis of compounds 1, 2 from the parent compound 3 (Figure 2) and the proposed mechanism of reaction were addressed.
2. Results and Discussion
2.1. Isolation and Structural Elucidation of the New Diterpenoids
From the ethyl acetate extracts of the aerial parts of the Chilean cushion shrubs Mulinum crassifolium and Azorella compacta two new diterpenoids (compounds 1 and 2, Figure 2) were isolated together with five known compounds (compounds 3–7, Figure 2, for details please see the experimental part). These new compounds along with the new natural product 5 were semi-synthesized; compounds 1–2 from 17-acetoxymulinic acid 3, while compound 5, which is isolated for the first time from a plant, was previously synthesized from the azorellane diterpenoid 6 [15]. This fact confirmed their precursor natural products. The compounds isolated were proved not to be artifacts of the extraction procedure with ethyl acetate by extracting a portion of the plant material in the presence of calcium carbonate (0.1%). The same natural products were detected and isolated using this procedure. The structural elucidation of the new compounds and proposed mechanism of reaction is explained below.
HREIMS of 1 showed an ion consistent with a molecular formula of C24H34O7 (required: m/z 434.2304, found 434.2284). The total of 24 carbons suggested the presence of a diacetylated diterpene. The 1H-NMR and 13C-NMR spectral data were easily assigned by comparison with NMR data of related compounds and in particular with the parent compound 17-acetoxymulinic acid [9,17,18] (Table 1 and Table 2, Figures S1–S8, Supplementary Material). The chemical shift values of the cyclopentane and cyclohexane rings were practically identical, confirming the configuration of the stereogenic centers and substitution pattern. The remaining signals indicated the presence of a trisubstituted double bond in the seven membered ring (δC-12 127.1, d, and δC-13 150.5, s), together with the corresponding signal of one vinylic proton (δH-12 5.84 brs), a secondary acetoxyl group (δH-14 5.75, dd, J = 11.3, 3.8; δC-14 71.5, d) and a ketone (δC-11 202.6, s). HMBC cross-peaks of the proton at δH 2.63 (H-9) with C-11 (202.6) and C-12 (127.1) suggested the presence of an α,β-unsaturated ketone. The methyl group CH3-16 showed HMBC correlations to three carbons (C-12, C-13 and C-14) which confirmed the position of the double bond. The methyl group at δH 2.11 (3H, s, H-22) showed cross-peaks with C-21 and C-14, while H-14 (δH 5.75) showed cross peaks with C-12, C-13 and C-15, which further confirmed the position of the additional acetyl group on C-14. The main results from phase sensitive NOESY spectra (Table 1, Figure S7) suggested that 1 had the stereochemistry shown in Figure 2. Proton H-14 showed dipolar correlations with H-17 and H-9 protons, both with β configuration; this fact confirmed the α-orientation of the acetyl group on C-14. All the spectroscopic evidence stated above proved that 1 has the relative stereochemistry as proposed.
Table 1.
1H-NMR data, HMBC and NOE correlations for compounds 1 and 2 in CDCl3 (J in Hz in parentheses).
| 1 | 2 | |||||
|---|---|---|---|---|---|---|
| Proton | δH mult. (J in Hz) | HMBC (H→C) | NOE | δH mult. (J in Hz) | HMBC (H→C) | NOE |
| 1α | 1.24 m | C-5, 9 | 1.83 m | - | - | |
| 1β | 1.84 dd (11.4, 4.7) | - | 1.37 m | - | - | |
| 2α | 1.97 m | - | 1.79 m | - | - | |
| 2β | 1.58 m | - | 1.48 m | - | - | |
| 3α | 1.48 m | - | 1.35 m | C-2, 4, 5, 18, 19 | H-10α | |
| 4 | 1.54 m | - | 1.45 m | - | - | |
| 6α | 1.43 m | - | 1.22 m | - | H-10α | |
| 6β | 1.52 m | - | 2.45 dt (7.2, 4.0) | C-5, 8 | H-7β, Me-19 | |
| 7α | 2.56 dt (13.3, 3.2) | C-15, 9, 17 | 1.80 dd (13.6, 4.5) | - | - | |
| 7β | 1.59 m | - | 1.71 dt (14.3, 4.3) | C-8, 9, 15 | H-6β, H-7β, | |
| 9β | 2.63 m | C-7, 15, 10, 11 | 2.23 m | C-8, 10, 11, 12, 15 | H-11β, H-17β | |
| 10α | 2.22 m | C-20, 9, 5, 8, 6 | H-1α, H-6α, H-3α | 1.51 m | C-1, 5, 9 | H-6α, H-3α |
| 11β | - | - | 5.94 br d (2.4) | C-8, 9, 10, 12, 21 | H-9β, H-17β | |
| 12 | 5.84 br s | C-9, 13, 14 | 6.20 br s | C-9, 13, 14, 16 | H-11, H-16 | |
| 14 | 5.75 dd (11.3, 3.8) | C-12, 13, 15 | H-17β, H-15β, H-9β | - | - | - |
| 15α | 261 dd (14.1, 11.3) | C-14, 7, 17, 13 | 2.62 d (12.7) | C-14, 7, 17, 13, 8 | - | |
| 15β | 1.68 dd (14.1, 3.8) | - | 2.50 d (12.7) | - | - | |
| 16 | 1.85 br s | C-13, 12, 14 | 1.87 br s | C-13, 12, 14 | - | |
| 17α | 3.85 d (11.3) | C-9, 7, 15 | 3.94 d (11.1) | C-8, 9, 15 | - | |
| 17β | 4.17 d (11.3) | - | H-14β, H-15β | 4.00 d (11.1) | - | H-9β, H-11β |
| 18 | 0.85 d (5.5) | C-4, 3 | 1.99 d (6.3) | C-4, 3 | - | |
| 19 | 1.02 d (5.5) | C-4, 3 | 0.82 d (6.3) | C-4, 3 | - | |
| 22 | 2.11 s | C-21, 14 | 2.09 s | - | - | |
| 24 | 1.99 s | C-23, 17 | 2.04 s | C-23, 17 | - |
Table 2.
13C-NMR data (100.25 MHz) for the new compounds 1 and 2.
| C# | 1 | 2 | C# | 1 | 2 |
|---|---|---|---|---|---|
| 1 | 24.2 t | 25.7 t | 13 | 150.5 s | 138.1 s |
| 2 | 28.4 t | 28.5 t | 14 | 71.5 d | 201.7 s |
| 3 | 57.5 d | 57.3 d | 15 | 36.2 t | 52.5 t |
| 4 | 31.6 d | 31.6 d | 16 | 28.4 q | 18.5 q |
| 5 | 57.0 s | 56.1 s | 17 | 71.6 t | 71.0 t |
| 6 | 31.6 t | 32.7 t | 18 | 22.7 q | 22.6 q |
| 7 | 36.0 t | 34.3 t | 19 | 22.3 q | 22.3 q |
| 8 | 46.8 s | 37.9 s | 20 | 178.4 s | 178.1 s |
| 9 | 57.4 d | 45.9 d | 21 | 170.5 s | 170.7 s |
| 10 | 46.9 d | 47.3 d | 22 | 20.9 q | 21.2 q |
| 11 | 202.6 s | 72.2 d | 23 | 169.5 s | 169.9 s |
| 12 | 127.1 d | 139.2 d | 24 | 20.5 q | 20.8 q |
The diterpenoid 2 had the same molecular formula (C24H34O7, (required: m/z 434.2304, found 434.2348), as 1, and showed a similar IR spectrum. The 13C-NMR spectrum exhibited 24 peaks (Table 2) while the DEPT-135 showed evidence for the same numbers of methyl, methylene, methine and quaternary (including four carbonyls) carbons, as compound 1. However, in the 1H-NMR spectra the simplification in the coupling pattern of the methylene protons (δH15α 2.62, d, J = 12.7 and δH15β 2.50, d, J = 12.7) of the cycloheptane ring as well as the strong deshielding of the beta oriented one which appeared as a broad doublet (δH11 5.94, brd, J = 2.4) suggested that the location of the keto carbonyl group was at C-14. Moreover, in the 13C-NMR spectrum of compound 2, C-14 showed a big upfield shift δC14 = 201.7), whereas C-11 showed a downfield shift (δC11 = 72.2), this suggested that the keto group is placed at C-14 while the acyl group is at C-11 (See Figures S9–S16). The structure of 2 was confirmed by careful analysis of the HMBC spectra (Table 1, Figure S16). Strong correlations were observed between the signal at δH 1.87 (3H, s) assigned to CH3-16, and signals at δC 139.2 (d, olefin C-12), δC 138.1 (s, olefin C-13) and δC 201.7 (s, carbonyl C-14). The signals at δH 2.62 and 2.50 assigned to H-15α and H-15β correlated with the signal at δC 201.7 (s, carbonyl C-14), δC 138.1 (s, olefinic C-13) and 71.0 (t, CH2OAc, C-17). These facts confirmed the location of the keto group at C-14 and the assignment of a double bond between C-12 and C-13. The location of the acetyl groups at C-17 and C-11 was confirmed by the correlation of the protons CH2OAc (C-17, δH 3.94 and 4.00, both, d, J = 11.1 Hz) with C-8, C-9 and C-15 and the proton CHOAc (C-11, δH 5.94, brd, J = 2.4 Hz) with C-8, C-9, C-10 and C-12. Hence, the relative stereochemistry of 2 corresponds to mulin-12-en-11α,17-diacetoxy-14-one-20-oic acid (Figure 2).
2.2. Semisynthetic Procedures and Proposed Mechanisms of Reaction
Compounds 1, 2 and 5 were prepared from their precursors 3 and 6. The reactions are explained below.
2.2.1. Semi-Synthesis of Compounds 1 and 2
Treatment of 3, with pyridine-Ac2O, at 25 °C under inert atmosphere yielded compounds 1 and 2. The proposed mechanism of the reaction is shown in Figure 3. The two reaction pathways a–b shown for compound 3 can be explained by a mechanism in which the base pyridine abstracts a proton from either the a or b sides (H11 or H14, respectively, Figure 3) of the mulinane skeleton leading to the opening of the endoperoxide ring with simultaneous formation of an α-β-unsaturated carbonyl followed, by acylation of the remaining oxygen anion with Ac2O leading to the mulinane compounds 1 and 2 (79.2% and 13% yield, respectively).
Figure 3.

Proposed rearrangement of 3 with Ac2O in pyridine at 25 °C.
2.2.2. Computational Analyses and Mechanism of Reaction
To explain the difference in the reaction yields for compounds 1 and 2, two possible transition states TSa y TSb were designed according to the mechanism proposed (Figure 3) to investigate the activation energy for the reactions pathways a and b.
The geometries of the reactant (compound 3), and transition states (TSa and TSb) for the two reaction pathways (Figure 3), were optimized using the Gaussian software version 09 software package at the B3LYP/3-21G* level [19]. The structures and imaginary frequencies of transition states were confirmed by the vibration analysis and the intrinsic reaction coordinate (IRC) method at the same level. In pathway a, the activation energy barrier TSa is 18.27 kJ/mol, while in pathway b the activation energy barrier TSb is 20.26 kJ/mol (Figure 4). This difference can explain why the base pyridine takes a proton from both a or b sides in parallel to produce compounds 1 and 2. However, the lower energy of TSa would reflect a hindered approach of pyridine to the proton H14 in side b and could explain the higher yield for compound 1 (pathway a). Furthermore the higher acidity of leaving proton H11 of side a compared to the acidity of leaving proton H14 in side b (the calculated positive charge of H11 using the Gaussian 09 software is three times higher than H14) proved that the reaction step a was favored. This fact was further supported by the calculated transition states for the reaction of mulinane 3 with pyridine. Figure 5 shows the transition states for the subtraction of proton H-11 (TSa) or proton H-14 (TSb) by pyridine and breakage of the endoperoxide as postulated.
Figure 4.

Reaction coordinate (reaction steps a, and b) calculated (Gaussian 09) [19] for the reaction between mulinane 3 and pyridine leading to compounds 1 and 2.
Figure 5.

Transition states (TSa and TSb, for the reaction steps a, and b) calculated (Gaussian 09) [19] for the reaction between mulinane 3 and pyridine leading to compounds 1 and 2.
2.2.3. Semi-Synthesis of Compound 3
The new natural product 5 was also obtained by semi-synthesis from azorellane 6 by stirring this compound in CHCl3 at 25 °C for 48 h, until compound 6 was undetectable by TLC, undergoing the mulinane-azorellane rearrangement reported previously [10,14,15]. This reaction proves that the new natural product 5 can be produced from 6 in the plant.
3. Experimental
3.1. General Experimental Procedures
Infrared (IR) spectra were recorded on a 783 FTS 165 FT-IR spectrometer (Perkin Elmer, Waltham, MA, USA).1H and 13C-NMR spectra were recorded on an Avance DRX 500 spectrometer (Bruker Biospin Gmbh, Rheinstetten, Germany) using TMS as internal standard. EIMS and HREIMS spectra were obtained using a MAT 95 XL Mass Spectrometer (Thermo Finnigan, San Jose, CA, USA). Thin-layer chromatography was performed using aluminum-coated silica gel plates (Kieselgel F254, Merck, Darmstadt, Germany) with hexane/EtOAc (9:1) as solvent system. The chromatograms were visualized under UV light (254 nm) and then sprayed with 1% vanillin in EtOH (w/v) and heated (60 °C) to see the compound spots. Medium pressure column chromatography was performed using Kieselgel 60 H (Merck), 55 μm particle size, FMI QG 150 medium pressure lab pumps (Syosset, NY, USA) and Ace Glass Inc. medium pressure columns (Vineland, NJ, USA).
3.2. Plant Material
Mulinum crassifolium Phil. and Azorella compacta Phil. were collected in El Tatio, Antofagasta, Chile in March 2011. Voucher herbarium specimens are deposited at the Laboratory of Natural Products, University of Antofagasta with the numbers Ac-031511 and Mc-031511, respectively.
3.3. Extraction and Isolation
Dried and powdered aerial parts of Mulinum crassifolium (630 g) were defatted with n-hexane (3 times, 1 L each time, 1 day/extraction) and the remaining plant material was extracted with ethyl acetate (3 times, 1 L each time, 1 day/extraction) at room temperature for one day each. After filtration, the solvent was evaporated in vacuo yielding a gum (17 g). The concentrated extract was fractionated on a medium pressure silica gel column (5 cm × 20 cm) with hexane-ethyl acetate mixtures of increasing polarity as elution solvents to give six fractions (1–6). Fraction 3 (355 mg) was further separated and purified by medium pressure silica gel chromatography (2 cm × 20 cm) to give 72.2 mg of 17-acetoxymulinic acid (3, Figure S17). Fraction 4 (266 mg) was rechromatographed as above to give compounds 1 (95.6 mg) and 2 (15.86 mg). Fraction 5 (327 mg) was re-chromatographed as above to give 42.6 mg of mulinolic acid (4) [17]. Dried aerial parts of Azorella compacta (745 g) were defatted with n-hexane (3 times, 1 L each time, 1 day/extraction) and the remaining plant material was extracted with ethyl acetate (3 times, 1 L each time, 1 day/extraction) at room temperature for one day each and concentrated as stated above to yield 7.2 g of a brown gum. The mulinane diterpenoid 5 (25.6 mg), together with the known compounds azorellanol (6, 293.4 mg) and 13,14-dihydroxy-mulin-11-en-20-oic acid (7, 74.2 mg) were isolated from this extract by medium pressure column chromatography (Kieselgel 60 H, 5 cm × 20 cm) using the solvent system mentioned above. The structures of the new and known compounds were elucidated by MS and 1D-2D NMR and comparison with literature data [4,9,17,18].
3.4. Semisynthesis of 14α,17-Diacetoxymulin-12-ene-11-one-20-oic acid (1) and 11α,17-Diacetoxy-mulin-12-en-14-one-20-oic Acid (2)
A portion (50 mg, 0.128 mmol) of 3 was dissolved in a mixture of Ac2O/Py (1:1, 2 mL); the mixture was stirred at room temperature for 24 h under N2. Afterwards Et2O (10 mL) was added, and the Et2O fraction was washed with copper sulphate solution (4 × 10 mL) and dried over anh. Na2SO4. Solvent was evaporated and the remaining reaction products purified by silica gel column chromatography to afford 1 (39.6 mg, 79.2%) and 2 (6.5 mg, 13.0%).
14α,17-Diacetoxymulin-12-ene-11-one-20-oic acid (mulinone a, 1). White crystals, mp 177–178 °C; 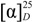 = + 35°, (c = 0.200, CHCl3); IR (liquid film) νmax (cm−1): 3000–3100 (br); 1650, 1700, 1740, 1220; EIMS m/z (rel. int.): 434 [M+] (10), 392 [M+–COCH3] (100), 374 [M+–AcOH] (7), 314 [M+–2 AcOH] (45), 286 (44), 243 (35), 91 (34); HREIMS m/z: found 434.2284 (calcd for C24H34O7, 434.2304); 1H-NMR (500.13 MHz) and 13C-NMR (125.76 MHz), data (CDCl3) see Table 1 and Supplementary Material.
= + 35°, (c = 0.200, CHCl3); IR (liquid film) νmax (cm−1): 3000–3100 (br); 1650, 1700, 1740, 1220; EIMS m/z (rel. int.): 434 [M+] (10), 392 [M+–COCH3] (100), 374 [M+–AcOH] (7), 314 [M+–2 AcOH] (45), 286 (44), 243 (35), 91 (34); HREIMS m/z: found 434.2284 (calcd for C24H34O7, 434.2304); 1H-NMR (500.13 MHz) and 13C-NMR (125.76 MHz), data (CDCl3) see Table 1 and Supplementary Material.
11α,17-Diacetoxymulin-12-ene-14-one-20-oic acid (mulinone b, 2). White crystals, mp 185–186 °C; = −28.11°, (c = 0.280, CHCl3); IR (liquid film) νmax (cm−1): 3000 (br), 1700, 1710, 1730, 1230; EIMS m/z (rel. int.): 434 [M+] (10), 392 (100), 350 (10), 331 (5), 286 (44), 169 (17), 105 (18), 91 (25), 55 (38); HREIMS m/z: found 434.2349 (calcd for C24H34O7, 434.2304); 1H-NMR (500.13 MHz) and 13C-NMR (125.76 MHz), data (CDCl3) see Table 1 and Supplementary Material.
4. Conclusions
Based on full spectroscopic evidence, 1 and 2 are new compounds which were named mulinones a-b. Compounds 1 and 2 were synthesized thorough acetylation from 3 in one step. The mechanism of reaction proposed for the semisynthesis of these diterpenoids was explained by computational analyses. This is the first report of the occurrence of diterpenoid 5 in a plant source. These new compounds were semi-synthesized from their precursor natural products—compounds 1–2 from 17-acetoxymulinic acid 3 and compound 5 from the azorellane diterpenoid 6—proving the relationship between them and their parent natural products.
Acknowledgments
Financial support by FONDECYT (Grant 1140178), CODEI (University of Antofagasta, Grant 5383) and EULADIV Alfa Project are gratefully acknowledged.
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/19/4/3898/s1.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds and plant extracts are available from the authors.
References
- 1.Munizaga C., Gunkel H. Notas etnobotánicas del pueblo Atacameño de Socaire. [(accessed on 24 March 2014)]. Available online: http://www.memoriachilena.cl/archivos2/pdfs/MC0038217.pdf.
- 2.Chiaramello A.I., Ardanaz C.E., Garcia E.E., Rossomando P.C. Mulinane-type diterpenoids from mulinum spinosum. Phytochemistry. 2003;63:883–886. doi: 10.1016/S0031-9422(03)00344-3. [DOI] [PubMed] [Google Scholar]
- 3.Colloca C.B., Pappano D.B., Bustos D.A., Sosa V.E., Baggio R.F., Garland M.T., Gil R.R. Azorellane diterpenes from Azorella cryptantha. Phytochemistry. 2004;65:2085–2089. doi: 10.1016/j.phytochem.2004.03.038. [DOI] [PubMed] [Google Scholar]
- 4.Loyola L.A., Borquez J., Morales G., Martin A.S. Diterpenoids from Azorella compacta. Phytochemistry. 1997;44:649–651. doi: 10.1016/S0031-9422(96)00603-6. [DOI] [Google Scholar]
- 5.Nicolas A.N., Plunkett G.M. Untangling generic limits in Azorella, Laretia, and Mulinum (Apiaceae: Azorelloideae): Insights from phylogenetics and biogeography. Taxon. 2012;61:826–840. [Google Scholar]
- 6.Neira I., Pobleta L., Porcille P., Silva P., Araya J., Bórquez J., Morales G., Loyola L.A., Sagua H. Activity of diterpenoids isolated from Azorella compacta (Llareta) on Trypanosoma cruzi amastigotes. Bol. Chil. Parasitol. 1998;53:9–13. [PubMed] [Google Scholar]
- 7.Loyola L.A., Borquez J., Morales G., Araya J., Gonzalez J., Neira I., Sagua H., San-Martín A. Diterpenoids from Azorella yareta and their trichomonicidal activities. Phytochemistry. 2001;56:177–180. doi: 10.1016/S0031-9422(00)00380-0. [DOI] [PubMed] [Google Scholar]
- 8.Loyola L.A., Borquez J., Morales G., Araya J., Gonzalez J., Neira I., Sagua H., San-Martín A. Azorellane diterpenoids from Laretia acaulis and its toxoplamacidal activity. Bol. Soc. Chil. Quím. 2001;46:9–13. [Google Scholar]
- 9.Loyola L.A., Borquez J., Morales G., San-Martin A., Darias J., Flores N., Gimenez A. Muliane-type diterpenoids from Azorella compacta display antiplasmodial activity. Phytochemistry. 2004;65:1931–1935. doi: 10.1016/j.phytochem.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 10.Wächter G.A., Matooq G., Hoffmann J.J., Maiese W.M., Singh M.P., Montenegro G., Timmermann B.N. Antibacterial diterpenoid acids from Azorella madreporica. J. Nat. Prod. 1999;62:1319–1321. doi: 10.1021/np990134u. [DOI] [PubMed] [Google Scholar]
- 11.Morales P., Kong M., Pizarro E., Pasten C., Morales G., Borquez J., Loyola L.A. Effect of azorellanone, a diterpene from Azorella yareta Hauman on human sperm physiology. J. Androl. 2003;24:364–370. doi: 10.1002/j.1939-4640.2003.tb02684.x. [DOI] [PubMed] [Google Scholar]
- 12.Fuentes N.L., Sagua H., Morales G., Borquez J., San-Martín A., Soto J., Loyola L.A. Experimental antihyperglycemic effect of diterpenoids of llareta Azorella compacta (Umbelliferae) Phil in rats. Phytother. Res. 2005;19:713–716. doi: 10.1002/ptr.1740. [DOI] [PubMed] [Google Scholar]
- 13.Wächter G.A., Franzblau S.G., Montenegro G., Suarez E., Fortunato R.H., Saavedra E., Timmermann B.N. A new antitubercular mulinane diterpenoid from Azorella madreporica Clos. J. Nat. Prod. 1998;61:965–968. doi: 10.1021/np980066w. [DOI] [PubMed] [Google Scholar]
- 14.Delporte C., Backhouse N., Salinas P., San-Martin A., Borquez J. Pharmacotoxicological study of diterpenoids. Bioorg. Med. Chem. 2003;11:1187–1190. doi: 10.1016/S0968-0896(02)00645-4. [DOI] [PubMed] [Google Scholar]
- 15.Bórquez J., Loyola L.A., Morales G., San-Martín A., Roldan R., Márquez N., Muñoz E. Azorellane diterpenoids from Laretia acaulis inhibit nuclear factor-kappa B activity. Phytother. Res. 2007;21:1082–1086. doi: 10.1002/ptr.2218. [DOI] [PubMed] [Google Scholar]
- 16.Molina-Salinas G.M., Bórquez J., Ardiles A., Said-Fernández S., Loyola L.A., San-Martín A., González-Collado I., Peña-Rodríguez L.M. Antituberculosis activity of natural and semisynthetic azorellane and mulinane diterpenoids. Fitoterapia. 2010;81:50–54. doi: 10.1016/j.fitote.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 17.Loyola L.A., Bórquez J., Morales G., San-Martín A. Mulinolic acid, a diterpenoid from Mulinum crassifolium. Phytochemistry. 1996;43:165–168. doi: 10.1016/0031-9422(96)00159-8. [DOI] [Google Scholar]
- 18.Loyola L.A., Bórquez J., Morales G., San-Martín A. A new diterpenoid from Mulinum crassifolium. Bol. Soc. Chil. Quím. 1997;42:311–315. [Google Scholar]
- 19.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., et al. Gaussian software. Gaussian Inc.; Wallingford, CT, USA: 2009. version 09 revision D01. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.