

Abstract
Two new acylated flavonol glycosides named kaempferol-3-O-(2-O-sinapoyl)-β-d-galactopyranosyl-(1→2)-β-d-glucopyranoside-7-O-α-l-rhamnopyranoside (1) and quercetin-3-O-(6-O-benzoyl)-β-d-glucopyranosyl-(1→3)-β-d-galactopyranoside-7-O-α-l-rhamnopyranoside (2), were isolated together with six known compounds from the seeds of L. sativum. Their structures were elucidated on the basis of spectroscopic analysis and chemical methods. In vitro 1 and 2 inhibited nitric oxide production in lipopolysaccharide (LPS)-stimulated RAW 264.7 cells, with IC50 values of 25.36 and 25.08 µM, respectively.
Keywords: Lepidium sativum, flavonol glycoside, anti-inflammatory activity
1. Introduction
Lepidium sativum L. (Cruciferae), also known as “garden cress”, is a fast-growing annual herb popularly used for its wide therapeutic application including anti-inflammatory [1], hypoglycemic [2], antihypertensive [3], fracture healing activities [4] and efficacy in gastrointestinal diseases [5]. In some regions, seedlings of L. sativum are also used in salads because of their pungent taste. Previous phytochemical investigations disclosed the presence of sinapic acid, alkaloids [6], flavonoids [7], steryl ester [8] and terpenes [9]. In the course of our search for novel bioactive agents, two new acylated flavonol glycosides, kaempferol-3-O-(2-O-sinapoyl)-β-d-galactopyranosyl-(1→2)-β-d-glucopyranoside-7-O-α-l-rhamnopyranoside (1) and quercetin-3-O-(6-O-benzoyl)-β-d-glucopyranosyl-(1→3)-β-d-galacto-pyranoside-7-O-α-l-rhamnopyranoside (2), were isolated from the seeds of L. sativum along with six known compounds. Herein, we report the isolation and characterization of new compounds, as well as their inhibitory activities against NO production induced by LPS and α-glucosidase.
2. Results and Discussion
Compound 1 was isolated as a yellow amorphous powder. Its UV spectrum exhibited a characteristic flavonol absorption band at 268 nm. The HRESIMS spectrum showed a quasi-molecular ion at m/z 985.2630 (calc. for C44H50O24Na, 985.2589), from which in conjunction with NMR data the molecular formula was established as C44H50O24, suggesting twenty indices of hydrogen deficiency. The 1H-NMR spectrum (Table 1) indicated the presence of kaempferol as an aglycone [δH 6.32 (1H, d, J = 2.4 Hz, H-6), 6.63 (1H, d, J = 2.4 Hz, H-8), 6.89 (2H, d, J = 9.0 Hz, H-3ꞌ, 5ꞌ), 8.01 (2H, d, J = 9.0 Hz, H-2ꞌ, 6ꞌ)], two methoxyl groups at δH 3.72 (6H, s), two benzene protons at δH 6.70 (2H, s), and trans-olefinic protons at δH 6.29 and δH 7.37 (each d, J = 16.2 Hz), and three anomeric protons at δH 5.50, 5.81, 5.08 . Except for carbon signals of an kaempferol, The 13C-NMR spectrum showed 29 carbon signals, including one carbonyl (δC 165.7), six benzene carbons [δC 105.4 (×2), 124.3, 137.9, 147.7 (×2)], two olefinic carbons (δC 115.2, 144.5), and two methoxy groups [δC 55.8 (×2)] ascribed to a sinapoyl group, three anomeric carbon signals at δC 97.0, 98.3, 98.5. NMR data indicated that 1 is an acylated kaempferol glycoside. All the 1H- and 13C-NMR data of 1 were assigned by TOCSY, HSQC, and HMBC experiments. Acid hydrolysis of 1 afforded kaempferol, glucose, rhamnose and galactose, identified by direct TLC comparison with authentic samples. The absolute configurations of the sugars were determined by GC analysis to be d- for glucose and galactose, and l- for rhamnose. Unequivocal assignment could be achieved by 2D-NMR spectra. The HMBC spectrum showed correlations between δH 5.50 (Rha H-1) and 161.50 (C-7), 5.81(H-1glc) and 133.13 (C-3), 5.08 (H-1gal) and 77.42 (C-2glc), 3.77 (H-2glc) and 98.28 (C-1gal), 4.69 (H-2gal) and 165.66 (C-9sinapoyl) (Figure 1). Detailed analyses of the 1H- (δ 5.81, d, J = 7.8 Hz, H-1''') and 13C-NMR (δ 97.0, 77.4, 75.6, 68.1, 72.0, 61.0) suggested glucopyranose as the sugar moiety, the C-2 (glu) was shifted downfield at δ 77.4, indicating that glycosylation of the galactose unit by the glucopyranosyl was on the 2-hydroxyl. A downfield shift of C-2'''' was from δ C 71.2 to 73.5, and an upfield shift of C-1'''' was from δ C 103.4 to 98.3, which were in accordance with the acylation of C-2ꞌꞌꞌꞌ of the galactose moiety [10]. Moreover, the downfield shift of H-2ꞌꞌꞌꞌ to 4.69 (dd, J = 9.6, 9.0 Hz) further confirmed the presence of a C-2ꞌꞌꞌꞌ sinapoyl in compound 1 [11]. The β configuration of the anomeric carbon of glucose and aglycone were inferred from the coupling constant of H-1ꞌꞌꞌ (J = 7.8 Hz) observed in the 1H-NMR spectrum [12]. The coupling constant of H-1ꞌꞌꞌꞌ (J = 7.8 Hz) demonstrated that the galactose was in the β-orientation, while the smaller coupling constant value (J = 1.2 Hz) indicated that the rhamnosyl group was α-linked to the aglycone. Thus, 1 was a new compound identified as kaempferol-3-O-(2-O-sinapoyl)-β-d-galactopyranosyl-(1→2)-β-d-glucopyranoside-7-O-α-l-rhamnopyranoside.
Table 1.
1H and 13C-NMR data for compounds 1 and 2 (600 and 150 MHz, DMSO-d6, δ ppm).
| No. | 1 | 2 | ||||
|---|---|---|---|---|---|---|
| δC | δH (mult, J in Hz) | HMBC | δC | δH (mult, J in Hz) | HMBC | |
| 2 | 155.7 | 155.7 | ||||
| 3 | 133.1 | 133.3 | ||||
| 4 | 177.4 | 177.4 | ||||
| 5 | 160.7 | OH (12.66) | 160.9 | 12.65 (OH) | ||
| 6 | 99.3 | 6.32(d, 2.4) | C-5, C-7, C-8 | 99.2 | 6.38 (d, 1.8) | C-5, 8, 10 |
| 7 | 161.5 | 161.4 | ||||
| 8 | 93.8 | 6.63(d, 2.4) | C-7, C-9, C-10 | 94.1 | 6.69 (d, 1.8) | C-6, 7, 9, 10 |
| 9 | 155.7 | 155.6 | ||||
| 10 | 105.5 | 105.5 | ||||
| 1ꞌ | 120.9 | 122.2 | ||||
| 2ꞌ | 130.9 | 8.01 (d, 9) | C-2, C-4ꞌ | 115.8 | 7.50 (d, 1.8) | C-1ꞌ, 3ꞌ, 4ꞌ, 2 |
| 3ꞌ | 115.4 | 6.89 (d, 9 ) | C-4ꞌ | 144.9 | ||
| 4ꞌ | 156.0 | OH (s, 10.18) | 148.8 | |||
| 5ꞌ | 115.4 | 6.89 (d, 9) | C-4ꞌ | 115.2 | 6.82 (d, 8.4) | C-3ꞌ, 4ꞌ, 6ꞌ |
| 6ꞌ | 130.9 | 8.01 (d, 9) | C-2ꞌ | 120.7 | 7.65 (dd, 8.4, 1.8) | C-2ꞌ, 4ꞌ, 5ꞌ, 2 |
| 1ꞌꞌ | 98.5 | 5.50 (d, 1.2) | C-7, 3ꞌꞌ | 98.4 | 5.54 (s) | C-7, 2ꞌꞌ |
| 2ꞌꞌ | 69.8 | 3.87 (br. m) | C-3ꞌꞌ, 4ꞌꞌ | 69.8 | 3.86 (br. s) | C-5ꞌꞌ |
| 3ꞌꞌ | 70.2 | 3.63 (m) | 70.3 | 3.65 (dd, 3.0, 9.0 ) | ||
| 4ꞌꞌ | 71.6 | 3.31 (m) | C-3ꞌꞌ | 71.6 | 3.33 (m) | C-2ꞌꞌ, 6ꞌꞌ |
| 5ꞌꞌ | 70.0 | 3.43 (m) | C-4ꞌꞌ, 6ꞌꞌ | 70.1 | 3.45 (m) | C-3ꞌꞌ, 4ꞌꞌ |
| 6ꞌꞌ | 18.0 | 1.13 (3H, d, 6.0) | C-4ꞌꞌ, 5ꞌꞌ | 17.9 | 1.14 (d, 6.0) | C-4ꞌꞌ, 5ꞌꞌ |
| 1ꞌꞌꞌ | 97.0 | 5.81 (d, J = 7.8) | C-3, 2ꞌꞌꞌ, 5ꞌꞌꞌ | 98.5 | 5.59 (d, 7.8 ) | C-3 |
| 2ꞌꞌꞌ | 77.4 | 3.77 (dd, 9.6, 7.8) | C-5ꞌꞌꞌ, 1ꞌꞌꞌ | 72.9 | 3.61 (dd, 9.0, 7.8) | C-3ꞌꞌꞌ |
| 3ꞌꞌꞌ | 75.6 | 3.34 (m) | C-2ꞌꞌꞌ | 81.9 | 3.76 (dd, 9.0, 4.0) | C-1ꞌꞌꞌꞌ, C-1ꞌꞌꞌ, C-2ꞌꞌꞌ |
| 4ꞌꞌꞌ | 68.1 | 3.65 (m) | C-5ꞌꞌꞌ | 67.5 | 3.68 (dd, 4.0, 3.0) | C-ꞌꞌꞌ, 3ꞌꞌꞌ |
| 5ꞌꞌꞌ | 72.0 | 3.62 (m) | C-6ꞌꞌꞌ | 75.8 | 3.30 (m) | C-1ꞌꞌꞌ, 4ꞌꞌꞌ, 6ꞌꞌꞌ |
| 6ꞌꞌꞌ | 61.0 | 3.72 (m) 3.51 (m) |
59.9 | 3.23 (m), 3.38 (m) | C-4ꞌꞌꞌ, 5ꞌꞌꞌ | |
| 1ꞌꞌꞌꞌ | 98.3 | 5.08 (d, 7.8) | C-2ꞌꞌꞌ | 105.0 | 4.59 (d, 7.8 ) | C-3ꞌꞌꞌ, 2ꞌꞌꞌꞌ, 5ꞌꞌꞌꞌ |
| 2ꞌꞌꞌꞌ | 73.5 | 4.69 (dd, 9.0, 7.8) | C-1ꞌꞌꞌꞌ, 3ꞌꞌꞌꞌ, 1ꞌꞌꞌꞌꞌ | 74.8 | 3.15 (dd, 9.0, 7.8 ) | C-1ꞌꞌꞌꞌ, 5ꞌꞌꞌꞌ |
| 3ꞌꞌꞌꞌ | 74.4 | 3.45 (m) | C-2ꞌꞌꞌꞌ, 4ꞌꞌꞌꞌ | 74.0 | 3.56 (dd, 9.0, 7.2) | C-4ꞌꞌꞌꞌ |
| 4ꞌꞌꞌꞌ | 70.4 | 3.23 (m) | C-3ꞌꞌꞌꞌ, 5ꞌꞌꞌꞌ | 70.0 | 3.23 (m) | C-5ꞌꞌꞌꞌ |
| 5ꞌꞌꞌꞌ | 76.6 | 3.28 (m) | C-4ꞌꞌꞌꞌ | 76.0 | 3.30 | C-4ꞌꞌꞌꞌ |
| 6ꞌꞌꞌꞌ | 59.9 | 3.23 (m) 3.36 (m) |
C-3ꞌꞌꞌꞌ, 5ꞌꞌꞌꞌ | 64.2 | 4.30 (dd, 6.0, 12.0) 4.40 (br. d, 12.0) |
C-1ꞌꞌꞌꞌꞌ, C-3ꞌꞌꞌꞌ C-1ꞌꞌꞌꞌꞌ, 3ꞌꞌꞌꞌ |
| 1ꞌꞌꞌꞌꞌ | 165.7 | 165.4 | ||||
| 2ꞌꞌꞌꞌꞌ | 115.2 | 6.31 (d, 16.2) | C-4ꞌꞌꞌꞌꞌ, C-1ꞌꞌꞌꞌꞌ | 129.4 | ||
| 3ꞌꞌꞌꞌꞌ | 144.5 | 7.37 (d, 16.2) | C-1ꞌꞌꞌꞌꞌ, 2ꞌꞌꞌꞌꞌ, 4ꞌꞌꞌꞌꞌ, 5ꞌꞌꞌꞌꞌ, 9ꞌꞌꞌꞌꞌ | 128.7 | 7.69 (d, 7.2) | C-1ꞌꞌꞌꞌꞌ, 5ꞌꞌꞌꞌꞌ |
| 4ꞌꞌꞌꞌꞌ | 124.3 | 128.1 | 7.20 (t, 7.2) | C-1ꞌꞌꞌꞌꞌ, 3ꞌꞌꞌꞌꞌ, 7ꞌꞌꞌꞌꞌ, 5ꞌꞌꞌꞌꞌ | ||
| 5ꞌꞌꞌꞌꞌ | 105.4 | 6.70 (s) | C-3ꞌꞌꞌꞌꞌ, 4ꞌꞌꞌꞌꞌ 6ꞌꞌꞌꞌꞌ, 7ꞌꞌꞌꞌꞌ, 8ꞌꞌꞌꞌꞌ | 132.7 | 7.38 (t, 7.2) | C-3ꞌꞌꞌꞌꞌ, 7ꞌꞌꞌꞌꞌ |
| 6ꞌꞌꞌꞌꞌ | 147.7 | 128.1 | 7.20 (t, 7.2 ) | C-1ꞌꞌꞌꞌꞌ, 3ꞌꞌꞌꞌꞌ, 7ꞌꞌꞌꞌꞌ, 5ꞌꞌꞌꞌꞌ | ||
| 7ꞌꞌꞌꞌꞌ | 137.9 | 8.75 (OH) | 128.7 | 7.69 (d, 7.2) | C-1ꞌꞌꞌꞌꞌ, 5ꞌꞌꞌꞌꞌ | |
| 8ꞌꞌꞌꞌꞌ | 147.7 | |||||
| 9ꞌꞌꞌꞌꞌ | 105.4 | 6.70 (s) | C-3ꞌꞌꞌꞌꞌ, 4ꞌꞌꞌꞌꞌ, 6ꞌꞌꞌꞌꞌ, 7ꞌꞌꞌꞌꞌ, 8ꞌꞌꞌꞌꞌ | |||
| CH3O | 55.8 | 3.72 (s) × 2 | C-5ꞌꞌꞌꞌꞌ, 6ꞌꞌꞌꞌꞌ, 8ꞌꞌꞌꞌꞌ, 9ꞌꞌꞌꞌꞌ | |||
Figure 1.

Chemical structures of 1 and 2.
Compound 2 was also obtained as a yellow amorphous powder with a molecular formula C40H44O22 ([M+Na]+ m/z 899.2222) on HRESIMS. The UV spectrum was similar to that of 1, suggesting a flavonol glycoside structure. Acid hydrolysis and GC analysis showed that the sugar units of 2 were the same as 1. Comparison of NMR spectroscopic data of 2 (Table 1) with those of 1 indicated that the difference of both compounds were the aglycone and acyl moiety, the kaempferol and sinapoyl group in 1 being replaced by quercetin and benzoyl in 2, respectively. The higher frequency signals in 1H-NMR at 7.69 (2H, d, J = 7.2 Hz), 7.20 (2H, t, J = 7.2 Hz) and 7.38 (t, J = 7.2 Hz) indicated the presence of a benzoyl moiety in 2. The higher frequency chemical shift of hydroxymethylene (C-6ꞌꞌꞌꞌ at δ 64.2 revealed the attachment of a benzoyl moiety at glucose. Detailed comparison of the 13C-NMR and HMBC spectra between the two compounds indicated that the difference was in the link location and link order of the glycoside moiety. The carbon signals at δC 98.5 (C-1ꞌꞌꞌ), 72.9 (C-2ꞌꞌꞌ), 81.9 (C-3ꞌꞌꞌ), 67.5 (C-4ꞌꞌꞌ), 75.8 (C-5ꞌꞌꞌ) and 59.9 (C-6ꞌꞌꞌ), together with an HMBC correlation between C-3 and H-1ꞌꞌꞌ (5.59, J = 7.8 Hz) revealed a galactopyranoside moiety was located on C-3 [13]. The HMBC experiment indicated correlations between δH 5.54 (H-1rha) and δC 161.38 (C-7), δH 5.60 (H-1gal) and δC 133.26 (C-3), δH 4.60 (H-1glc) and δC 81.94 (C-3gal), δH 3.76 (H-3gal) and δC 105.03 (C-1glc), δH 4.30 (H-6glc) and δH 165.40 (C-1benzoyl) (Figure 1). Consequently, 2 was a new compound and identified as quercetin-3-O-(6-O-benzoyl)-β-d-glucopyranosyl-(1→3)-β-d-galactopyranoside-7-O-α-l-rhamnopyranoside.
The known compounds were identified as isorhamnetin (3), quercetin (4), kaempferol (5), osthole (6), protocatechuic acid (7), and staphylionosides A (8), respectively, on the basis of their spectroscopic data and by comparison of their spectroscopic data with previously published values [14,15,16,17,18,19].
Considering the medical applications of L. sativum, 1 and 2 were evaluated for their inhibitory effects on NO release in the lipopolysaccharide stimulated RAW 264.7 macrophage cell line and their α-glucosidase inhibitory activity. The results showed that compounds 1 and 2 inhibited NO production in LPS-stimulated RAW 264.7 cells with IC50 values of 24.40 µg/mL and 21.97 µg/mL, respectively. Compounds 1 and 2 also exhibited α-glucosidase inhibitory activity at 20 µg/mL and the inhibitory activity was 10.50 and 8.93, respectively. The inhibitory rate (%) of compound 1 at 6.50 µM (6.25 µg/mL), 13.00 µM (12.5 µg/mL), 26.00 µM (25 µg/mL) and 52.00 µM (50 µg/mL) were 20.82, 31.67, 43.99 and 96.77, respectively. The inhibitory rate (%) of compound 2 at 7.13 µM (6.25 µg/mL), 14.27 µM (12.5 µg/mL), 28.54 µM (25 µg/mL) and 56.97 µM (50 µg/mL) were 24.93, 31.96, 67.74 and 84.46, respectively. Dexamethasone (1.27 µM) showed 27.1% inhibition [20]. Compound 1 and 2 also exhibited α-glucosidase inhibitory activity at 20.79 µM (20 µg/mL) and 22.78 µM (20 µg/mL) and the inhibitory rate (%) was 10.50 and 8.93, respectively, while the IC50 of acarbose was 200 μg/mL [21].
3. Experimental
3.1. General Procedures
Optical rotations were obtained on a Perkin-Elmer 341 digital polarimeter (Waltham, MA, USA). UV spectra were recorded on Shimadzu UV2550 (Tokyo, Japan). NMR spectra were obtained with a Bruker AV β 600 NMR spectrometer (chemical shift values are presented as δ values with TMS as the internal standard; Munich, Germany). HR-ESI-MS spectra were performed on a LTQ-Orbitrap XL spectrometer. GC analysis was carried out on a GC-7890: column, DB-5 (30 m × 0.32 mm × 0.25 mm); detector, FID-6850 (Agilent, Santa Clara, CA, USA). ODS gel (50 µm, YMC, Kyoto, Japan), Sephadex LH-20 (Pharmacia, Uppsala, Sweden), and MDS gel (Beijing Medicine Technology Center, Beijing, China) were used for column chromatography. HPLC separations were performed using a Waters 2535 series pump equipped with a PDA detector and a YMC (250 × 10 mm, 5 μm) semi-preparative column. TLC was carried out on silica gel GF254 (Yantai Chemical Inst., Yantai, China) plates, and spots were visualized under UV light (254 or 365 nm) or by spraying with 5% H2SO4 in 95% EtOH followed by heating.
3.2. Plant Material
The seeds of L. sativum were purchased from the Xinjiang Uygur Autonomous Region in August 2010. The plant material was authenticated by one of the authors (B.-L. Guo). A voucher specimen is deposited at the Natural Medicine Research Center of Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences and Peking Union Medical College.
3.3. Extraction and Isolation
The dried seeds of L. sativum (45 kg) were chopped and extracted with 95% EtOH (270 L) three times (each time 8 h) under percolation and then concentrated under vacuum. The residue was extracted three times under reflux by 50% EtOH for 1.5 h. The dried 95% EtOH extract was further suspended in water and partitioned successively with petroleum ether, CHCl3, EtOAc and n-BuOH (50 L, 80 L, 100 L, 100 L). After concentration the n-BuOH layer (400 g) was subjected to column chromatography on MDS gel column (15 cm × 47 cm, 75–150 μm) eluted with a gradient of MeOH–H2O (5:95, 10:90, 15:85, 20:80, 30:70, 50:50, 70:30, 100:0, v/v) to give seven factions (Fr.1–Fr.7) according to TLC analyses. Fr.6 (MeOH–H2O 1:1, v/v) was subjected to chromatography on a ODS gel (5 cm × 50 cm, 50 μm) column with gradient elution (MeOH–H2O, 10:90, 20:80, 30:70, 40:60, 50:50, 70:30, 100:0, v/v), to give seven subfractions (Fr.6a–Fr.6g). From Fr.6c, compound 1 (343 mg) was obtained by repeated Sephadex LH-20 (MeOH–H2O, 1:1, v/v) chromatography. Compounds 2 (25 mg) and 3 (5 mg) was obtained by semi-preparative HPLC (MeOH–H2O, 2:3, v/v) from Fr.6d. Fr.5 (MeOH–H2O 3:7) was subjected to ODS gel column chromatography (3.2 cm × 50 cm, 50 μm, MeOH–H2O, 20:80, 30:70, 35:65, 40:60, 100:0 v/v) to afford five subfractions (Fr.5a–Fr.5e). From fraction Fr.5b compound 4 (13 mg) was isolated using semi-preparative HPLC (MeOH–H2O, 2:3). Fraction Fr.5c was further purified by semi-preparative HPLC with MeOH–H2O (2:3) to afford compound 5 (15 mg).
The 50% EtOH extract which was dissolved in water and chromatographed on a D101 macroporous adsorptive resin column eluting with a gradient of EtOH–H2O (0:100, 10:90, 20:80, 30:70, 50:50, 95:5, v/v), the eluates were concentrated under reduced pressure to dryness and six fractions were obtained.
The 30% ethanol eluate was subjected to MDS-gel chromatography (5 cm × 50 cm, 75–150 μm, MeOH–H2O, 20:80, 25:75, 30:70, 35:65, 40:60, 100:0 v/v), and the fourth fraction (MeOH–H2O, 35:65 ) was separated by ODS (5 cm × 50 cm, 50 μm, MeOH–H2O, 38:62, isocratic elution) and Sephadex LH-20 (MeOH ), to yield compounds 6–8 (15 mg, 13 mg, 5 mg, respectively).
3.4. Spectroscopic Data
Compound 1, Yellow amorphous powder; 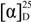 = −127.7° (c = 0.065, MeOH); UV λmax (MeOH) nm: 224, 268, and 331 nm; IR νmax (KBr) cm−1: 3259, 1660, 1595, 1520. 1H- and 13C-NMR data, see Table 1; HR-ESI-MS m/z 985.2630 [M+Na]+, (calc for C44H50O24Na, 985.2589).
= −127.7° (c = 0.065, MeOH); UV λmax (MeOH) nm: 224, 268, and 331 nm; IR νmax (KBr) cm−1: 3259, 1660, 1595, 1520. 1H- and 13C-NMR data, see Table 1; HR-ESI-MS m/z 985.2630 [M+Na]+, (calc for C44H50O24Na, 985.2589).
Compound 2, Yellow amorphous powder; = −82.8° (c = 0.064, MeOH); UV (MeOH) λmax 203, 257, and 358 nm; IR νmax (KBr) cm−1: 3254, 1656, 1590, 1513. 1H- and 13C-NMR data, see Table 1; HR-ESI-MS m/z 899.2222 [M+Na]+, (calc. for C40H44O22Na: 899.2221).
3.5. Determination of Sugar Components
Compounds 1 (5 mg) and 2 (3 mg) were hydrolyzed with 2N TFA (5.0 mL) for 6 h in a boiling water bath. After extraction three times with CH2Cl2, the remaining aqueous layer was concentrated and identified by TLC (CHCl3/MeOH/H2O = 8:5:1) comparison with authentic samples. Spots were detected by spraying with 1% anisaldehyde (in EtOH) followed by heating. The absolute configuration of monosaccharides from each aqueous layer was determined by GC-MS of their trimethylsilylated derivatives. Column temperature: 180–250 °C, programmed increase: 15 °C/min, carrier gas: N2 (1 mL/min); injection temperature: 250 °C, injection volume: 1 mL. By comparing with authentic samples, l-rhamnose, d-glucose, and d-galactose were detected from 1 and 2 at tR: 5.815, 6.313; 7.013, and 7.713; 7.228, 7.916 min, respectively.
3.6. NO Inhibition Assay
Inhibition of NO production and cell viability of LPS-stimulated RAW 264.7 macrophage cells were determined. The NO production assay was carried out according to the method described before [22]. The murine monocytic RAW 264.7 macrophages were dispensed into 96-well plates (2 × 105 cells/well) containing RPMI 1640 medium (Hyclone, Logan, UT, USA) with 10% FBS under humidified atmosphere of 5% CO2 at 37 °C. After 24 h of preincubation, cells were treated with serial dilutions of compounds 1 and 2 with the maximum concentration of 50 µM in the presence of 1 µg/mL LPS for 18 h. Each compound (purity > 95%) was dissolved in DMSO and further diluted in the medium to produce different concentrations. NO production in each well was assessed by adding 100 µL of Griess reagents A and B to 100 µL of each supernatant from LPS or the compound-treated cells in triplicate. After 5 min of incubation, the absorbance was measured at 570 nm with a 2104 Envision multilabel plate reader (Perkin-Elmer Life Sciences, Inc., Rowville, Victoria, Australia).
3.7. α-Glucosidase Inhibition Assay
Compounds 1 and 2 have been screened for α-glucosidase inhibitory activity with a microplate-based screening method with reference to previous literature [23]. A total of 100 μL of reaction mixture contained 25 μL of 0.1 mol/L phosphate buffer (pH 6.8), 25 μL of substratesolution (2.5 mmol/L pNPG in 0.1 mol/L phosphate buffer), 25 μL varying concentration of experimental drugs, and 25 μL of α-glucosidase solution (0.2 U/mL α-glucosidase in 0.1 mol/L phosphate buffer). After incubation of the plates at 37 °C for 15 min, 25 μL of Na2CO3 (0.2 mol/L) was added to each well to stop the reaction. The absorption was measured at 405 nm using Multiskan plate reader (Thermo Labsystems, Basingstoke, UK). The inhibitory rate (%) was calculated according to the formula: {[1 − (ODtest − ODblank)]/(control ODtest − control ODblank)} × 100%.
4. Conclusions
From the seeds of L. sativum, two new acylated flavonol glycosides, named kaempferol-3-O-(2-O-sinapoyl)-β-d-galactopyranosyl-(1→2)-β-d-glucopyranoside-7-O-α-l-rhamnopyranoside and quercetin-3-O-(6-O-benzoyl)-β-d-glucopyranosyl-(1→3)-β-d-galactopyranoside-7-O-α-l-rhamnopyranoside, were isolated. The NO production and α-glucosidase inhibitory activity were assayed in vitro. The two compounds showed significantly active against NO production induced by LPS. The current research demonstrates that L. sativum might be a great source for potential bioactive flavonol glycosides.
Acknowledgements
We gratefully acknowledge financial support of this work by Program for Innovative Research Team in IMPLAD (PIRTI).
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/19/8/11341/s1.
Supplementary Files
Author Contributions
Qing-Lu Fan, Wen-Hua Huang, Yun Qi and Bao-Lin Guo designed research; Qing-Lu Fan, Yin-Di Zhu and Yun Qi performed research and analyzed the data; Qing-Lu Fan and Yin-Di Zhu wrote the paper. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 1–8 are available from the authors.
References
- 1.Al-Yahya M.A., Mossa J.S., Ageel A.M., Rafatullah S. Pharmacological and safety evaluation studies on Lepidium sativum L., Seeds. Phytomedicine. 1994;1:155–159. doi: 10.1016/S0944-7113(11)80035-8. [DOI] [PubMed] [Google Scholar]
- 2.Eddouks M., Maghrani M., Zeggwagh N.A., Michel J.B. Study of the hypoglycaemic activity of Lepidium sativum L. aqueous extract in normal and diabetic rats. J. Ethnopharmacol. 2005;97:391–395. doi: 10.1016/j.jep.2004.11.030. [DOI] [PubMed] [Google Scholar]
- 3.Maghrani M., Zeggwagh N.A., Michel J.B., Eddouks M. Antihypertensive effect of Lepidium sativum L. in spontaneously hypertensive rats. J. Ethnopharmacol. 2005;100:193–197. doi: 10.1016/j.jep.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 4.Agarwal J., Verma D.L. Antioxidant activity-guided fractionation of aqueous extracts from Lepidium sativum and identification of active flavonol glycosides. Acad. Arena. 2011;3:14–18. [Google Scholar]
- 5.Najeeb Ur R., Mehmood M.H., Alkharfy K.M., Gilani A.H. Prokinetic and laxative activities of Lepidium sativum seed extract with species and tissue selective gut stimulatory actions. J. Ethnopharmacol. 2011;134:878–883. doi: 10.1016/j.jep.2011.01.047. [DOI] [PubMed] [Google Scholar]
- 6.Maier U.H., Gundlach H., Zenk M.H. Seven imidazole alkaloids from Lepidium sativum. Phytochemistry. 1998;49:1791–1795. doi: 10.1016/S0031-9422(98)00275-1. [DOI] [PubMed] [Google Scholar]
- 7.Agarwal J., Verma D.L. Antioxidative activity and flavonoid composition from Lepidium sativum. Nat. Sci. 2011;9:21–25. [Google Scholar]
- 8.Saba, Mughal M.H., Ali M., Iqbal M., Srivastava P.S. A steryl ester from Lepidium sativum. Phytochemistry. 1999;50:1375–1377. doi: 10.1016/S0031-9422(98)00704-3. [DOI] [Google Scholar]
- 9.Pande S.D., Ali M., Iqbal M., Srivastava P.S. Three new phytoconstituents from Lepidium sativum. Die Pharmazie. 1999;54:851–853. [Google Scholar]
- 10.Wang X.M., Wan C.P., Zhou S.R., Qiu Y. Two new flavonol glycosides from Sarcopyramis bodinieri var. delicate. Molecules. 2008;13:1399–1405. doi: 10.3390/molecules13061399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vitalini S., Braca A., Passarella D., Fico G. New flavonol glycosides from Aconitum burnatii Gayer and Aconitum variegatum L. Fitoterapia. 2010;81:940–947. doi: 10.1016/j.fitote.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 12.Agrawal P.K. NMR spectroscopy in the structural elucidation of oligosaccharides and glycosides. Phytochemistry. 1992;31:3307–3330. doi: 10.1016/0031-9422(92)83678-R. [DOI] [PubMed] [Google Scholar]
- 13.Hirayama C., Ono H., Meng Y., Shimada T., Daimon T. Flavonoids from the cocoon of Rondotia menciana. Phytochemistry. 2013;94:108–112. doi: 10.1016/j.phytochem.2013.05.023. [DOI] [PubMed] [Google Scholar]
- 14.Mousallami A.M., Afifi M.S., Hussein S.A. Acylated flavonol diglucosides from Lotus polyphyllos. Phytochemistry. 2002;60:807–811. doi: 10.1016/S0031-9422(02)00177-2. [DOI] [PubMed] [Google Scholar]
- 15.Wei X.H., Yang S.J., Liang N., Hu D.Y., Jin L.H., Xue W., Yang S. Chemical constituents of Caesalpinia decapetala (Roth) Alston. Molecules. 2013;18:1325–1336. doi: 10.3390/molecules18011325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H.M., Wang C.F., Shen S.M., Wang G.L., Liu P., Liu Z.M., Wang Y.Y., Du S.S., Liu Z.L., Deng Z.W. Antioxidant phenolic compounds from Pu-erh tea. Molecules. 2012;17:14037–14045. doi: 10.3390/molecules171214037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu R., Feng L., Sun A., Kong L. Preparative isolation and purification of coumarins from Cnidium monnieri (L.) Cusson by high-speed counter-current chromatography. J. Chromatorg. A. 2004;1055:71–76. doi: 10.1016/j.chroma.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 18.Yuan H.E., Zhou X.D., Meng L.J., Qin F.M., Zhou G.X. Chemical constituents from Commelina communis. China J. Chin. Mater. Med. 2013;38:3304–3308. [PubMed] [Google Scholar]
- 19.Yu Q., Matsunami K., Otsuka H., Takeda Y. Staphylionosides A-K: Megastigmane glucosides from the leaves of Staphylea bumalda DC. Chem. Pharm. Bull. 2005;53:800–807. doi: 10.1248/cpb.53.800. [DOI] [PubMed] [Google Scholar]
- 20.Wang S., Wang W., Luo M., Zhao X., Zu Y., Zho Y. Regulating effect of compound glycyrrhizin on LPS-induced inflammatory factor release from murine RAW 264.7 cells. Prog. Pharm. Sci. 2012;36:465–470. [Google Scholar]
- 21.Zhang J., Zhao S., Yin P., Yan L., Han J., Shi L., Zhou X., Liu Y., Ma C. Alpha-glucosidase inhibitory activity of polyphenols from the burs of Castanea mollissima Blume. Molecules. 2014;19:8373–8386. doi: 10.3390/molecules19068373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao X., He J., Wu X.D., Peng L.Y., Dong L.B., Deng X., Li Y., Cheng X., Zhao Q.S. Further lignans from Saururus chinensis. Planta Med. 2013;79:1720–1723. doi: 10.1055/s-0033-1351053. [DOI] [PubMed] [Google Scholar]
- 23.Hua J., Qi J., Yu B.Y. Iridoid and phenylpropanoid glycosides from Scrophularia ningpoensis Hemsl. and their alpha-glucosidase inhibitory activities. Fitoterapia. 2014;93:67–73. doi: 10.1016/j.fitote.2013.11.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.