

Abstract
1-Indanones have been successfully prepared by means of three different non-conventional techniques, namely microwaves, high-intensity ultrasound and a Q-tube™ reactor. A library of differently substituted 1-indanones has been prepared via one-pot intramolecular Friedel-Crafts acylation and their efficiency and “greenness” have been compared.
Keywords: 1-indanones, microwaves, ultrasound, Q-tube™; Friedel-Crafts acylation
1. Introduction
In the last two decades, environmental issues linked to the chemical and associated industries, such as the pharmaceutical industry, have become increasingly pertinent. Many classical synthetic methodologies require large amounts of natural resources and generate copious amounts of waste [1]. Thus, the need to incorporate green chemistry into the synthesis of active pharmaceutical ingredients and intermediates is of primary importance for the pharmaceutical industry [2].
Indanones and related compounds are important bioactive molecules. These compounds have exhibited biological activity against cancer cells and Alzheimer’s disease; moreover they can be used as synthetic intermediates for several drugs and as precursors to natural products [3,4,5,6,7,8,9,10]. Other applications include their use as ligands in olefinic polymerization catalysts [11,12,13,14,15,16,17,18,19,20] and as discotic liquid crystals [21]. The most famous drug which bears an indanone moiety is probably donepezil hydrochloride, which has been approved by the United States Food and Drug Administration (US-FDA) for the treatment of mild-moderate Alzheimer’s disease [22]. The intramolecular Friedel-Crafts cyclization reaction of 3-arylpropionic acids or chlorides is one of the most common methods for the preparation of 1-indanones [23,24,25,26,27,28,29]. Although the direct dehydrative cyclization of 3-arylpropionic acids is more difficult than cyclization via acid chlorides, it is preferable because of the environmental benefits it provides. In fact, the “one-step reaction” produces water as the only by-product while the “two-step reaction” generates a large amount of toxic and corrosive compounds. Nevertheless, direct cyclization usually requires an excess of protic acids (even as solvents) such as sulfuric acid, hydrogen fluoride [30], polyphosphoric acid [31], methanesulfonic acid (MSA) [32], a mixture of MSA and P2O5 [33], or Lewis acids such as AlCl3 and SnCl4 [30]. Some lanthanide triflates, in particular Tb(OTf)3, were reported to be useful catalysts for the dehydrative cyclization of 3-arylpropionic acids to form 1-indanones [34] in o-chlorobenzene at very high temperatures. We report herein an improved method for the synthesis of 1-indanone derivatives which proceeds via the superacid-catalyzed intramolecular Friedel-Crafts acylation of 3-arylpropionic acids.
As mentioned above, the examples of direct intramolecular Friedel-Crafts acylation of 3-arylpropionic acids to produce 1-indanone derivatives are very far from satisfying many green chemistry principles since they are performed under extremely drastic experimental conditions, such as elevated reaction temperatures (250 °C in chlorobenzene) and require very long reaction times [23,24,25,26,27,28,29,30,31,32,33,34]. In light of the enormous diffusion and application of non-conventional techniques in the implementation of synthetic green processes [35,36,37,38], and our own experience in this field [39,40,41,42,43,44], we decided to explore the use of microwave irradiation (MW), high-intensity ultrasound (US) and high-pressure conditions to develop a greener synthesis of 1-indanone derivatives via the intramolecular Friedel-Crafts acylation of 3-arylpropionic acids.
2. Results and Discussion
To start our investigation, it was decided to study the cyclization of 3-(4-methoxyphenyl)propionic acid (1) in different solvents and experimental conditions using Tb(OTf)3 as the Lewis acid, the best reported in the literature [34], and triflic acid [27] (Scheme 1). All attempts to perform the MW-assisted Tb(OTf)3 catalyzed reaction in environmentally benign solvents such as PEG, n-butanol, ethyl lactate or water, or even in slightly activated aromatic solvents failed (Table 1, entries 1–6).
Scheme 1.

Synthesis of 1-indanone 1a via the cyclization of 3-(4-methoxyphenyl) propionic acid 1 under US or MW irradiation.
Table 1.
MW vs. US in the intramolecular Friedel-Craft acylation of 3-(4-methoxyphenyl) propionic acid (1).
| Entry | Catalyst (%mol) | Solvent | T (°C) | Method | Time (min) | Conv. (%) | Yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | Tb(OTf)3 (20) | PEG a | 180 | MW | 30 | - | - |
| 2 | Tb(OTf)3 (20) | n-BuOH a | 180 | MW | 30 | - | - |
| 3 | Tb(OTf)3 (20) | Ethyl lactate a | 180 | MW | 30 | - | - |
| 4 | Tb(OTf)3 (20) | H2O | 180 | MW | 30 | - | - |
| 5 | Tb(OTf)3 (20) | Toluene b | 180 | MW c | 30 | 10 | 10 |
| 6 | Tb(OTf)3 (20) | Xylene b | 180 | MW c | 30 | - | - |
| 7 | Tb(OTf)3 (20) | Isooctane c | 250 | MW c | 10 | - | - |
| 8 | Tb(OTf)3 (20) | Cl-benzene | 250 | MW | 60 | 85 | 33 d |
| 9 | TfOH (10 eq.) | CH2Cl2 (dry) | 25 | r.t. | 1440 | 85 | 61 d |
| 10 | TfOH (3 eq.) | CH2Cl2 (dry) | 80 | MW | 60 | 100 | 100 |
| 11 | TfOH (1 eq.) | CH2Cl2 (dry) | 80 | MW | 120 | 50 | 20 d |
| 12 | TfOH (2 eq.) | CH2Cl2 (dry) | 80 | MW | 60 | 75 | 53 d |
| 13 | TfOH (3 eq.) | CH2Cl2 (dry) | 110 | MW | 30 | 100 | 100 |
| 14 | TfOH (3 eq.) | CH2Cl2 (dry) | 40 | US | 120 | - | - |
| 15 | TfOH (5 eq.) | CH2Cl2 (dry) | 40 | US | 1260 | 80 | 80 |
| 16 | TfOH (10 eq.) | CH2Cl2 (dry) | 40 | US | 150 | 100 | 100 |
| 17 | TfOH-SiO2 (30) | CH2Cl2 (dry) | 110 | MW | 60 | - | - |
| 18 | TfOH-SiO2 (30) | CH2Cl2 (dry) | 40 | US | 60 | - | - |
a Formation of side-esterification or trans-esterification by-products; b Formation of intermolecular Friedel-Craft acylation by-products; c Reaction conducted in the presence of a SiC tablet used as a MW inert absorbent; d Formation of by-products.
A negative result was also observed when the reaction was performed in inert high boiling solvents such as isooctane (Table 1, entry 7) and only in chlorobenzene a good conversion of substrate was observed after only 60 min, albeit affording a low yield of product 1a (Table 1, entry 8).
Clearly improved results were obtained when the reaction was catalyzed by a large excess of triflic acid (TfOH) in CH2Cl2. As previously reported in the literature [27], a 61% yield of 1a was observed at room temperature, but only after a very long reaction time (Table 1, entry 9), whereas complete conversion of substrate 1 to indanone 1a was observed after only 60 min when the reaction was performed under MW at 80 °C using three equivalents of TfOH (Table 1, entry 10).
Lower amounts of catalyst gave poorer yields due to the considerable number of by-products formed (Table 1, entries 11 and 12). The same positive result was registered in only 30 min when the reaction temperature was raised to 110 °C (Table 1, entry 13). Comparable results were registered when the same reaction was accomplished under US, however, the complete conversion of the substrate 1 to indanone 1a was observed in an acceptable reaction time only when a large excess of triflic acid was used in CH2Cl2 at 40 °C (Table 1, entries 14–16). Triflic acid is a highly corrosive and fuming liquid. It is the strongest Brønsted acid, having a Ho value of −14.1. Unmodified chromatographic silica gel-supported triflic acid has very recently been proposed as an efficient and recyclable catalyst under solvent free conditions [45]. Thus, in an attempt to add this more environmentally friendly way to use this superacid to our synthetic protocol, the test reaction reported in Scheme 1 was also performed using silica gel supported triflic acid (TfOH-SiO2). Unfortunately, no conversion (of substrate 1) was observed after 60 min under MW or US (Table 1, entries 17 and 18). In a last test, the MW- and US-assisted reactions were performed in significantly lower reaction times in dry CH2Cl2, to give quantitative yields of product 1a and avoiding the formation of by-products. All the attempts to use more environmentally acceptable solvents gave scarce results, leaving dry CH2Cl2 as the best choice for performing the reaction reported in Scheme 1.
Performing the reaction in dry CH2Cl2 permitted the total conversion of the substrate 1 into 1a in 60 min at 80 °C using only 3 equivalents of TfOH (Table 1 entry 10). On the other hand, in the US-assisted protocol, the same reaction was performed at lower temperature with a cleaner reaction profile and simplified work-up, despite the significantly higher amount of catalyst necessary to obtain the quantitative conversion of 1 into 1a.
In the attempt to unify the advantages of both assisted methods, the reaction pictured in Scheme 1 was performed in the Q-tube™ which is a safe pressure reactor which features a patent pending pressure-release and reseal system that prevents accidental explosions due to over–pressurization. Q-tube™ is an affordable alternative to expensive and cumbersome MW synthesizers; this system enables a reaction to be carried out at higher temperature than the boiling point of solvents and reagents, which will increase the reaction rate.
The results reported in Table 2, clearly confirm the efficiency of Q-tube™ as a valid alternative technique (Table 2, entry 1), which provides a cleaner reaction profile very similar to what is observed in the US-assisted protocol. Longer reaction times did not improve the reaction performance and only traces of product 1a were obtained at 150 °C (Table 2, entries 2 and 3). It is of interest to know if Tb(OTf)3, the most active Lewis acid reported in literature for this process, can be used as catalyst in the Q-tube experiments. A poor result was registered when the reaction was carried out in chlorobenzene as reported [34] (Table 2, entry 4), whereas a good yield of product 1a was obtained when the reaction was performed in toluene (Table 2, entry 5).
Table 2.
Q-Tube method in the intramolecular Friedel-Craft acylation of 3-(4-methoxyphenyl) propionic acid 1.
| Entry | Catalyst (%mol) | Solvent | T (°C) | Time (min) | Conv. (%) | Yield (%) a |
|---|---|---|---|---|---|---|
| 1 | TfOH (3 eq.) | CH2Cl2 (dry) | 80 | 60 | 100 | 100 |
| 2 | TfOH (3 eq.) | CH2Cl2(dry) | 110 | 30 | 100 | 96 |
| 3 | TfOH (3 eq.) | CH2Cl2(dry) | 150 | 10 | 100 | trace b |
| 4 | Tb(OTf)3 (10) | C6H5Cl | 180 | 180 | 100 | 40 |
| 5 | Tb(OTf)3 (10) | toluene | 150 | 180 | 100 | 86 c |
| 6 | Tb(OTf)3 (10) | n-C7H14 | 180 | 240 | - | - |
| 7 | Tb(OTf)3 (20) | n-C7H14 | 250 | 120 | 18 | 45 |
| 8 | Tb(OTf)3 (10) | isooctane | 250 | 240 | 32 | 20 |
| 9 | TfOH-SiO2(30 ) | CH2Cl2 (dry) | 25 | 120 | - | - |
| 10 | TfOH-SiO2 (30) | CH2Cl2 (dry) | 180 | 180 | - | - |
a Isolated yields; b The high temperature broke the Teflon septum; c Formation of intermolecular Friedel-craft acylation by-products.
Very poor results were obtained when the reaction was performed in high-boiling point hydrocarbon solvents and at very high reaction temperatures (Table 2, entries 7 and 8). Finally, the endeavor to carry out the reaction again using silica gel-supported triflic acid in CH2Cl2 gave a negative result (Table 2, entries 9 and 10) leaving the following three experimental procedures as the best choices for the reaction depicted in Scheme 1; triflic acid is added at 0 °C to a solution of 0.5 mmol of substrate in dry CH2Cl2, then the solution is heated to room temperature and the reaction is conducted under MW- or US-assistance or in the Q-tube™ (Table 1 entries 10, 16 and Table 2 entry 1 respectively). The best experimental conditions obtained with MW, US- and Q-tubeTM protocols were applied to several substrates (Scheme 2) with the aim of testing the applicability of the methods and comparing their efficiency and versatility (Table 3).
Scheme 2.

MW-, US-assisted and Q-tube™ protocols for 1-indanone synthesis applied to several aryl-3-propionic acids.
Table 3.
MW vs. US method in the intramolecular Friedel-Craft acylation of phenyl propionic acids 1–7.
| Entry | Product | US-Assisted Reaction a | MW-Assisted Reaction a | Q-Tube-Assisted Reaction a | ||||||
| Time (min) | Conv (%) | Yield (%) | Time (min) | Conv (%) | Yield(%) | Time (min) | Conv (%) | Yield(%) | ||
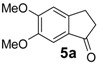
| 1 |
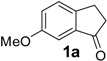
|
150 | 100 | 100 | 60 | 100 | 100 | 60 | 100 | 100 |
| 210 b | 100 | 100 | 90 b | 100 | 100 | 90 b | 100 | 100 | ||
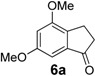
| 2 |
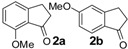
|
60 | 100 | 88/12 | 60 | 100 | 90/10 | 60 | 100 | 90/10 |
| (2b)/(2a) | (2b)/(2a) | (2b)/(2a) | ||||||||
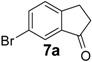
| 3 |
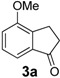
|
120 | 100 | - c | 180 | 100 | - c | 180 | 100 | - c |
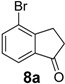
| 4 |
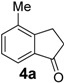
|
60 | 100 | 100 | 60 | 100 | 100 | 60 | 100 | 100 |
| 5 |
|
60 | 100 | 100 | 60 | 100 | 100 | 60 | 100 | 100 |
| 6 |
|
360 | - | - | 180 | - | - | 180 | - | - |
| 7 |
|
360 | 100 | 100 | 180 d | 42 | 33 | 180 d | 54 | 36 |
| 60 e | 100 | 100 | 60 e | 100 | 100 | |||||
| 8 |
|
1200 | 90 | 90 | 180 d | 58 | 48 | 180 d | 44 | 43 |
| 60 e | 100 | 100 | 60 e | 100 | 100 | |||||
| 9 |
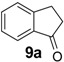
|
60 | 100 | 100 | 180 | 100 | 100 | 180 | 100 | 100 |
| 10 |
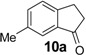
|
60 | 100 | 100 | 60 | 100 | 100 | 60 | 100 | 100 |
| 11 |
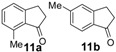
|
60 | 100 | 85/15 | 60 | 100 | 88/12 | 60 | 100 | 88/12 |
| (11b)/(11a) | (11b)/(11a) | (11b)/(11a) | ||||||||
| 12 |
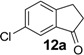
|
400 | 100 | 100 | 180 d | 38 | 30 | 180 d | 45 | 33 |
| 60 e | 100 | 100 | 60 e | 100 | 100 | |||||
| 13 |
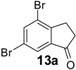
|
460 | 100 | 100 | 180 d | 32 | 28 | 180 d | 40 | 36 |
| 120 e | 100 | 100 | 120 e | 100 | 100 | |||||
| 14 |
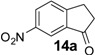
|
1200 d | - | - | 360 e | - | - | 360 e | - | - |
a All new products were characterized by GC/MS and 1H-NMR spectroscopy; all the known products were characterized by comparison with the spectral data in the literature; b Reaction conducted with 5.0 mmol of reactant; c Formation of poli-ketones by polymerization; d Reaction conducted in presence of 5 eq. of TfOH; e Reaction conducted in presence of 10 equivalents of TfOH.
In the case of 3-(2-methoxyphenyl)propionic acid (3), the only available cyclization site is the non-activated 6-position on the aromatic ring, so that no product formation was observed and only polymerized products were detected (Table 3, entry 3). Moreover, an unfavorable electronic effect can be invoked to explain the failure of the intramolecular cyclization in substrate 3. As depicted in Figure 1, an H-bond can constrain the acidic OH to form a transient stable ring and keep the carboxylic moiety distant from the aromatic ring. This hypothesis is confirmed by the results observed for 3-(2-methylphenyl)propionic acid where the absence of the ether oxygen prevents the formation of the transient ring through the O-H interaction leaving the carboxylic group free to attack the non-activated 6-position on the aromatic ring and furnishing a quantitative yield of the 1-indanone derivative 4a (Table 3, entry 4).
Figure 1.
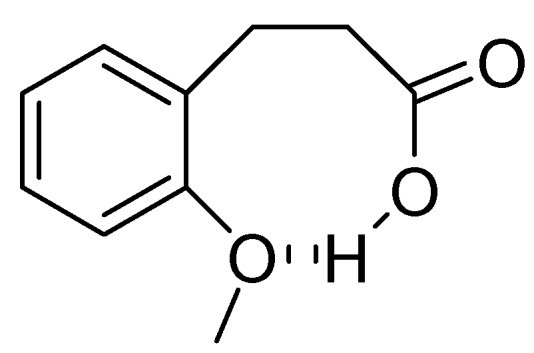
Proposed unfavourable electronic effect involved in the failure of the 3-(2-methylphenyl)propionic acid 3 intramolecular cyclization.
Similarly, the unfavourable electronic effect associated with greater steric hindrance can explain the extremely poor reactivity in the case of 3-(2,4-dimethoxyphenyl) propionic acid (6) where only the starting material was recovered in all three reaction systems, even after prolonged reaction times (Table 3, entry 6). In all the other reported examples quantitative product yields were observed for all three activation methodologies in very short reaction times and without significant by-product formation (Table 3, entries 1, 2, 5, 8–13). It is worth noting that the dis-activated halo-substituted phenylpropionic acids 7, 8, 12 and 13 gave quantitative yields for the direct intramolecular cyclization which, to the best of our knowledge, has never reported before. However, no conversion was observed in the case of the highly deactivated 3-(4-nitrophenyl)propionic acid (14) under all the experimental conditions adopted (Table 3, entry 14), even after very prolonged reaction times and using higher amounts of catalyst.
Finally, in order to test the scalability of the protocol, a scaled-up reaction was realized on 5.0 mmol of 3-(4-methoxyphenyl)propionic acid (1) (Table 3, entry 1, line 2), giving rise to the same results in terms of conversion, yield and reaction profile, but after a slightly longer reaction time. All the synthetic methodologies tested in this work enable much shorter reaction times and use of an acceptable amount of triflic acid [27]. o-Chlorobenzene was replaced by CH2Cl2 [34]. The comparison of the three techniques reported in Table 3 shows that MW-assisted reactions could be performed using only 3 equivalents of triflic acid in shorter reaction times than the other protocols. On the other hand, US-assistance allows the reaction to be conducted at lower temperature and with a significantly cleaner profile, simplifying the reaction work-up despite the higher amount of triflic acid required. The Q-tube™ technology gave the same results as the MW-assisted protocol but with cleaner reaction profiles, offering a good alternative to MW or US technologies in terms of yield, safety and efficiency.
3. Experimental
3.1. General Information
All chemicals were obtained from Sigma-Aldrich or Acros Organics (Geel, Belgium) and used as received. All solvents were distilled using standard methods before use. All reactions were carried out in flame-dried glassware, under a dry nitrogen atmosphere. MW-assisted reactions were performed on a Synthos 3000 instrument from Anton Paar (Torino, Italy), equipped with a 64MG5 Rotor and an IR probe used for external temperature control. US-assisted reactions were performed in a high-power US-bath (19.6 kHz) made by Danacamerini (Torino, Italy). Q-Tube assisted reactions were performed in a Q-tube™ safe pressure reactor from Q Labtech (distributed by Sigma-Aldrich), equipped with a cap/sleeve, a pressure adapter (120 psi), a needle adapter/needle, a borosilicate glass tube, a Teflon septum and a catch bottle. Reactions were monitored using a GC–MS Thermo Fisher Scientific workstation, composed of a Focus GC (Thermo TR, Waltham, MA, USA- 5 ms SQC 15 m × 0.25 mm ID × 0.25 µm, working on split mode, 1.2 mL/min using He as the carrier gas) and a DSQ II mass detector. TLC were performed using Kielsegel 60-F264 on aluminium plates, commercially available from Merck (Darmstadt, Germany). Liquid flash chromatography was performed on a Supelco VERSA FLASH HTFP station (distributed by Sigma-Aldrich) using silica cartridges commercially available from Supelco. 1H-NMR spectra were recorded on a Bruker WM 300 instrument (Milano, Italy) on samples dissolved in CDCl3. Chemical shifts are given in parts per million (ppm) from tetramethylsilane as the internal standard (0.0 ppm). All products in this report are known and were characterized by standard techniques (1H- and 13C-NMR, GC/MS) and the data were compared with those reported in the literature [33,46,47,48] for identification.
3.2. General US-Assisted Procedure
Trifluoromethane sulfonic acid (10 eq.) was gently added to a cooled (0 °C) solution of a 3-phenyl propionic acid (0.5 mmol) in dry CH2Cl2 (0.5 mL) in a two-necked round bottom flask. The temperature was raised to room temperature. The mixture was reacted in a high-power US-bath (19.6 kHz) at 40 °C. The reaction was monitored by TLC and GC/MS until the reactant disappeared. The mixture was poured into ice and extracted three times with CH2Cl2. The organic phase collected was dried over Na2SO4, filtered and concentrated under vacuum. The desired pure product was separated from the crude by flash chromatography.
3.3. General MW-Assisted Procedure
Trifluoromethanesulfonic acid (3 eq.) was gently added to a cooled (0 °C) solution of a 3-phenyl propionic acid (0.5 mmol) in dry CH2Cl2 (1.0 mL) in 3 mL glass vial using a Synthos 3000 microwave oven (Anton-Paar). The temperature was raised to room temperature. Appropriate Teflon and screw caps were placed on the top of the vial. The mixture was heated in the MW reactor at 80 °C in “power-controlled mode” for the appropriate time. The reaction was monitored by TLC and GC/MS until the reactant disappeared. The mixture was poured onto ice and extracted three times with CH2Cl2. The collected organic phase was dried on Na2SO4, filtered and concentrated under vacuum. The desired pure product was separated from the crude by flash chromatography.
3.4. General Q-tubetm-Assisted Procedure
Trifluoromethane sulfonic acid (3 eq.) was gently added to a cooled (0 °C) solution of a 3-Phenyl propionic acid (0.5 mmol) in dry CH2Cl2 (1.0 mL) in a 12 mL Q-tube™ pressure tube, furnished by Q Labtech. The temperature was raised to room temperature. A Teflon septum was placed on the top of the tube and the appropriate cap and pressure adapter were used. The mixture was heated in an oil bath at 80 °C. The reaction was monitored by TLC and GC/MS until the reactant disappeared. The mixture was poured into ice and extracted three times with CH2Cl2. The organic phase collected was dried on Na2SO4, filtered and concentrated under vacuum. The desired pure product was separated from the crude by flash chromatography.
4. Conclusions
We have reported the application of three different non-conventional techniques to the synthesis of a library of substituted 1-indanones, via the direct Friedel-Crafts intramolecular cyclization of arylpropionic acids. The comparison between three alternative reaction methodologies, namely MW, US and Q-Tube™ assisted reactions, showed that Q-tube™ equipment can be proposed as a valid alternative to monomode MW and US technologies in term of efficiency, safety (virtually eliminating the risk of pressure explosions) and a cleaner reaction profile.
Acknowledgments
This study was supported by the following grants: PON a3_00359 CUP F61D11000120007 “Potenziamento Strutturale”.
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/19/5/5599/s1.
Author Contributions
The main part of this work was realized by Manuela Oliverio (planning, synthesis and purification of products) with the practical contribution of Paola Costanzo and Luca Cariati. Monica Nardi performed compound characterization, while Salvatore Vincenzo Giofrè contributed to the theoretical choice of indanone moieties. Giancarlo Cravotto contributed on setting US-reactor conditions. The whole work was coordinated and written by Antonio Procopio.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds are available from the authors.
References
- 1.Anastas P.T., Warner J.C. Green Chemistry: Theory and Practice. Oxford University Press; Oxford, UK: 1998. [Google Scholar]
- 2.Gawande M.B., Bonifácio V.D.B., Luque R., Branco P.S., Varma R.S. Benign by design: Catalyst-free in-water, on-water green chemical methodologies in organic synthesis. Chem. Soc. Rev. 2013;42:5522–5551. doi: 10.1039/c3cs60025d. [DOI] [PubMed] [Google Scholar]
- 3.Wu C., Nakamura H., Murai A., Inouye S. Chemical studies on the chiral indanone derivatives as the inhibitor of Renilla luciferase. Tetrahedron. 2001;57:9575–9583. doi: 10.1016/S0040-4020(01)00980-2. [DOI] [Google Scholar]
- 4.Kerr D.J., Hamel E., Jung M.K., Flynn B.L. The concise synthesis of chalcone, indanone and indenone analogues of combretastatin A4. Bioorg. Med. Chem. 2007;15:3290–3298. doi: 10.1016/j.bmc.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Bansal R., Narang G., Zimmer C., Hartmann R.W. Synthesis of some imidazolyl-substituted 2-benzylidene indanone derivatives as potent aromatase inhibitors for breast cancer therapy. Med. Chem. Res. 2011;20:661–669. doi: 10.1007/s00044-010-9368-4. [DOI] [Google Scholar]
- 6.Byrne A.J., Barlow J.W., Walsh J.J. Synthesis and pharmacological evaluation of the individual stereoisomers of 3-[methyl(1,2,3,4-tetrahydro-2-naphthalenyl)amino]-1-indanone, a potent mast cell stabilising agent. Bioorg. Med. Chem. Lett. 2011;21:1191–1194. doi: 10.1016/j.bmcl.2010.12.095. [DOI] [PubMed] [Google Scholar]
- 7.Saxena H.O., Faridi U., Srivastava S., Kumar J.K., Darokar M.P., Luqman S., Chanotiya C.S., Krishna V., Negi A.S., Khanuja S.P.S. Gallic acid-based indanone derivatives as anticancer agents. Biorg. Med. Chem. Lett. 2008;18:3914–3918. doi: 10.1016/j.bmcl.2008.06.039. [DOI] [PubMed] [Google Scholar]
- 8.Schumann H., Stenzel O., Girgsdies F. (–)-2-Menthylindenyl and (–)-2-menthyl-4,7-dimethylindenyl complexes of rhodium, iridium, cobalt, and molybdenum. Organometallics. 2001;20:1743–1751. doi: 10.1021/om001074m. [DOI] [Google Scholar]
- 9.Herzog M.N., Chien J.C.W., Rausch M.D. Novel 2-indenyl ansa-zirconocenes for the polymerization of α-olefins. J. Organomet. Chem. 2002;654:29–35. doi: 10.1016/S0022-328X(02)01333-5. [DOI] [Google Scholar]
- 10.Leoni L.M., Hamel E., Genini D., Shih H., Carrera C.J., Cottam H.M., Carson D.A. Indanocine, a microtubule-binding indanone and a selective inducer of apoptosis in multidrug-resistantm cancer cells. J. Natl. Cancer Inst. 2000;92:217–224. doi: 10.1093/jnci/92.3.217. [DOI] [PubMed] [Google Scholar]
- 11.Beukes D.R., Davies-Coleman M.T., Kelly-Borges M., Harper M.K., Faulkner D.J., Dilemmaones A.-C. Dilemmaones A−C, unusual indole alkaloids from a mixed collection of South African Sponges. J. Nat. Prod. 1998;61:699–701. doi: 10.1021/np970580m. [DOI] [PubMed] [Google Scholar]
- 12.Pellissier H. Recent developments in the Nazarov process. Tetrahedron. 2005;61:6479–6517. doi: 10.1016/j.tet.2005.04.014. [DOI] [Google Scholar]
- 13.Frontier A.J., Collinson C. The Nazarov cyclization in organic synthesis. Recent advances. Tetrahedron. 2005;61:7577–7606. doi: 10.1016/j.tet.2005.05.019. [DOI] [Google Scholar]
- 14.Coyanis E.M., Panayides J.L., Fernandes M.A., de Koning C.B., van Ottoerlo W.A.L. Ring-closing metathesis for the synthesis of substituted indenols, indenones, indanones and indenes: Tandem RCM-dehydrogenative oxidation and RCM-formal redox isomerization. J. Organomet. Chem. 2006;691:5222–5224. doi: 10.1016/j.jorganchem.2006.08.074. [DOI] [Google Scholar]
- 15.Clark W.M., Tickner-Eldridge A.M., Huang G.K., Pridgen L.N., Olsen M.A., Mills R.J., Lantos I., Baine N.H. A catalytic enantioselective synthesis of the endothelin receptor antagonists SB-209670 and SB-217242. A base-catalyzed stereospecific formal 1,3-hydrogen transfer of a chiral 3-arylindenol. J. Am. Chem. Soc. 1998;120:4550–4551. doi: 10.1021/ja973882j. [DOI] [Google Scholar]
- 16.Wang G., Zheng C., Zhao G. Asymmetric reduction of substituted indanones and tetralones catalyzed by chiral dendrimer and its application to the synthesis of (+)-sertraline. Tetrahedron: Asymmetry. 2006;17:2074–2081. doi: 10.1016/j.tetasy.2006.07.010. [DOI] [Google Scholar]
- 17.Kajiro H., Mitamura S., Mori A., Hiyama T. Enantioselective synthesis of 2-hydroxy-1-indanone, a key precursor of enantiomerically pure 1-amino-2-indanol. Tetrahedron: Asymmetry. 1998;9:907–910. doi: 10.1016/S0957-4166(98)00050-0. [DOI] [Google Scholar]
- 18.Torisawa Y., Aki S., Minamikawa J. Conversion of indanone oximes into isocarbostyrils. Bioorg. Med. Chem. Lett. 2007;17:453–455. doi: 10.1016/j.bmcl.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 19.Yadav J.S., Reddy G.S.K.K., Sabitha G., Krishna A.D., Prasad A.R., Rahaman H.R.R., Rao K.V., Rao A.B. Daucus carota and baker’s yeast mediated bio-reduction of prochiral ketones. Tetrahedron: Asymmetry. 2007;18:717–723. doi: 10.1016/j.tetasy.2007.03.009. [DOI] [Google Scholar]
- 20.Sharma A.K., Subramani A.V., Gorman C.B. Efficient synthesis of halo indanones via chlorosulfonic acid mediated Friedel-Crafts cyclization of aryl propionic acids and their use in alkylation reactions. Tetrahedron. 2007;63:389–395. doi: 10.1016/j.tet.2006.10.065. [DOI] [Google Scholar]
- 21.Fukuoka M., Yoshihira K., Natori S., Mihashi K., Nishi M. Carbon-13 nuclear magnetic resonance spectra of pterosin-sesquiterpenes and related indan-1-one derivatives. Chem. Pharm. Bull. 1983;31:3113–3128. doi: 10.1248/cpb.31.3113. [DOI] [Google Scholar]
- 22.Sugimoto H. Structure-activity relationships of acetylcholinesterase inhibitors: Donepezil hydrochloride for the treatment of Alzheimer’s Disease. Pure Appl. Chem. 1999;71:2031–2037. doi: 10.1351/pac199971112031. [DOI] [Google Scholar]
- 23.Bhattacharya A., Segmuller B., Ybarra A. Preparation of acrylophenones and 2-alkyl indanones utilizing hexamethylenetetramine as an inexpensive mannich reagent. Synth. Commun. 1996;26:1775–1784. doi: 10.1080/00397919608002617. [DOI] [Google Scholar]
- 24.De Castro C., Primo J., Corma A. Heteropolyacids and large-pore zeolites as catalysts in acylation reactions using α,β-unsaturated organic acids as acylating agents. J. Mol. Catal. A-Chem. 1998;134:215–222. doi: 10.1016/S1381-1169(98)00038-7. [DOI] [Google Scholar]
- 25.Gevorgyan V., Quan L.G., Yamamoto Y. Synthesis of indenols and indanones via catalytic cyclic vinylpalladation of aromatic aldehydes. Tetrahedron Lett. 1999;40:4089–4092. doi: 10.1016/S0040-4039(99)00656-5. [DOI] [Google Scholar]
- 26.Rudler H., Denise B. Copper(II)-catalyzed aerobic oxidation of indane in the presence of aldehydes: Intermediate formation of hydroperoxides. J. Mol. Catal. A-Chem. 2000;154:277–279. doi: 10.1016/S1381-1169(00)00044-3. [DOI] [Google Scholar]
- 27.Prakash G.K.S., Yan P., Torok B., Olah G.A. Gallium(III) trifluoro-methanesulfonate: A Water tolerant, reusable lewis acid catalyst for Friedel-Crafts reactions. Catal. Lett. 2003;87:109–112. doi: 10.1023/A:1023482904174. [DOI] [Google Scholar]
- 28.Gagnier S.V., Larock R.C. Palladium-catalyzed carbonylative cyclization of unsaturated aryl iodides and dienyl triflates, iodides, and bromides to indanones and 2-cyclopentenones. J. Am. Chem. Soc. 2003;125:4804–4807. doi: 10.1021/ja0212009. [DOI] [PubMed] [Google Scholar]
- 29.Fillion E., Fishlock D. Convenient access to polysubstituted 1-indanones by Sc(OTf)3-Catalyzed intramolecular Friedel-Crafts acylation of benzyl meldrum’s acid derivatives. Org. Lett. 2003;5:4653–4656. doi: 10.1021/ol035817m. [DOI] [PubMed] [Google Scholar]
- 30.Johnson W.S. The formation of cyclic ketones by intramolecular acylation. Org. React. 1949;2:114–177. [Google Scholar]
- 31.Popp F.D., McEwen W.E. Polyphosphoric acids as a reagent in organic chemistry. Chem. Rev. 1958;58:321–401. doi: 10.1021/cr50020a004. [DOI] [Google Scholar]
- 32.Miller R.B., Frincke J.M. Stereospecific total synthesis of (.+-.)-vetiselinenol. J. Org. Chem. 1981;46:2972–2974. doi: 10.1021/jo00327a027. [DOI] [Google Scholar]
- 33.Eaton P.E., Carlson G.R., Lee J.T. Phosphorus pentoxide-methanesulfonic acid. Convenient alternative to polyphosphoric acid. J. Org. Chem. 1973;38:4071–4073. doi: 10.1021/jo00987a028. [DOI] [Google Scholar]
- 34.Cui D.M., Zhang C., Kawamura M., Shimada S. Synthesis of 1-indanones by intramolecular Friedel-Crafts reaction of 3-arylpropionic acids catalyzed by Tb(OTf)3. Tetrahedron Lett. 2004;45:1741–1745. doi: 10.1016/j.tetlet.2003.12.085. [DOI] [Google Scholar]
- 35.Mason T.J. Ultrasound in synthetic organic chemistry. Chem. Soc. Rev. 1997;26:443–451. doi: 10.1039/cs9972600443. [DOI] [Google Scholar]
- 36.Cravotto G., Nano G.M., Palmisano G., Tagliapietra S., Demetri A., Penoni A. The aldol reaction under high-intensity ultrasound: A novel approach to an old reaction. Eur. J. Org. Chem. 2003;22:4438–4444. [Google Scholar]
- 37.Kappe C.O., Dallinger D., Murphree S.S. Practical Microwave Synthesis for Organic Chemists. 1st ed. Wiley-VCH; Weinheim, Germany: 2009. [Google Scholar]
- 38.Kappe C.O. Controlled microwave heating in modern organic synthesis. Angew. Chem. Int. Ed. 2004;43:6250–6284. doi: 10.1002/anie.200400655. [DOI] [PubMed] [Google Scholar]
- 39.Procopio A., Gaspari M., Nardi M., Oliverio M., Tagarelli A., Sindona G. Simple and efficient MW-assisted cleavage of acetals and ketals in pure water. Tetrahedron. Lett. 2007;48:8623–8627. [Google Scholar]
- 40.Procopio A., de Luca G., Nardi M., Oliverio M., Paonessa R. General MW-assisted grafting of MCM-41: Study of the dependence on time dielectric heating and solvent. Green Chem. 2009;11:770–773. doi: 10.1039/b820417a. [DOI] [Google Scholar]
- 41.Procopio A., Costanzo P., Dalpozzo R., Maiuolo L., Nardi M., Oliverio M. Efficient ring opening of epoxides with trimethylsilyl azide and cyanide catalyzed by erbium (III) triflate. Tetrahedron Lett. 2010;51:5150–5153. doi: 10.1016/j.tetlet.2010.07.123. [DOI] [Google Scholar]
- 42.Procopio A., Cravotto G., Oliverio M., Costanzo P., Nardi M., Paonessa R. An eco-sustainable erbium(III)-catalysed method for formation/cleavage of O-tert-butoxy carbonates. Green Chem. 2011;13:436–443. doi: 10.1039/c0gc00728e. [DOI] [Google Scholar]
- 43.Nardi M., Cozza A., Maiuolo L., Oliverio M., Procopio A. 1,5-Benzoheteroazepines through eco-friendly general condensation reactions. Tetrahedron Lett. 2011;52:4827–4834. doi: 10.1016/j.tetlet.2011.06.029. [DOI] [Google Scholar]
- 44.Cravotto G., Procopio A., Oliverio M., Orio L., Carnaroglio D. Simple sonochemical protocols for fast and repeatable Grignard reactions. Green Chem. 2011;13:2806–2809. doi: 10.1039/c1gc15756f. [DOI] [Google Scholar]
- 45.Liu P.N., Xia F., Wang Q.W., Ren Y.J., Chen J.Q. Triflic acid adsorbed on silica gel as an efficient and recyclable catalyst for the addition of β-dicarbonyl compounds to alcohols and alkenes. Green Chem. 2010;12:1049–1055. [Google Scholar]
- 46.Duddeck H., Toth G., Simon A. Chemical Shifts for Oxygen-Springer. Verlag Berlin; Heidelberg, Germany: 2002. [Google Scholar]
- 47.Gomez-Lor B., Frutos O.D., Ceballos P.A., Granier T., Echavarren A.M. Synthesis of new C3h and C3v Truxene derivatives. Eur. J. Org. Chem. 2001;11:2107–2114. [Google Scholar]
- 48.Takeuchi R., Yasue H. Rhodium complex-catalyzed desilylative cyclocarbonylation of 1-aryl-2-(trimethylsilyl)acetylenes: A new route to 2,3-dihydro-1H-inden-1-ones. J. Org. Chem. 1993;58:5386–5392. doi: 10.1021/jo00072a020. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.