

Abstract
The heterobimetallic R2lox protein binds both manganese and iron ions in a site-selective fashion and activates oxygen, ultimately performing C-H bond oxidation to generate a tyrosine-valine crosslink near the active site. In this work, we demonstrate that following assembly, R2lox undergoes photoinduced changes to the active site geometry and metal coordination motif. Through spectroscopic, structural, and mass spectrometric characterization, the photoconverted species is found to consist of a tyrosinate-bound iron center following light-induced decarboxylation of a coordinating glutamate residue and cleavage of the tyrosine-valine crosslink. This process occurs with high quantum efficiencies (Φ = 3%) using violet and near-ultraviolet light, suggesting that the photodecarboxylation is initiated via ligand-to-metal charge transfer excitation. Site-directed mutagenesis and structural analysis suggest that the crosslinked tyrosine-162 is the coordinating residue. One primary product is observed following irradiation, indicating potential use of this class of proteins, which contains a putative substrate channel, for controlled photoinduced decarboxylation processes, with relevance for in vivo functionality of R2lox as well as application in environmental remediation.
Graphical Abstract
Introduction
The high levels of complexity seen in nature can in part be attributed to the advent of systems that harness solar energy to drive biological processes. Light-driven proteins are responsible for many of the key reactions underlying multicellular life on this planet. These systems include the light-harvesting complexes of green plants and cyanobacteria that perform photosynthesis, ultraviolet (UV) resistance proteins that shield DNA from photodamage, light-sensing receptors such as rhodopsin and G-protein-coupled ion channels, FAD-containing BLUF domains to drive DNA repair and circadian rhythms, and photoactive yellow protein (PYP) to control phototactic responses, among others.1–10 The light-responsive components of these proteins are typically made up of organic cofactors, though the active sites of metalloproteins can also be affected by light. One example of this is seen in the [NiFe] hydrogenases, in which the Ni-L state, now recognized as important for catalysis, can be generated by low-temperature irradiation of the metal-hydride-bearing Ni-C species.11 However, photochemical efficiencies are typically low for biologically relevant, first-row transition metals due to rapid deactivation of the excited state.12–16 These limitations also hamper the use of these earth-abundant elements for solar energy applications.17–19 Thus, identifying molecular mechanisms by which the photochemical efficiencies of first-row transition metals can be increased is relevant for understanding biological systems as well as in alternative energy research.
Two examples of efficient, photoinduced chemical reactions that utilize a first-row transition metal are the photo-Fenton and photodecarboxylation reactions.20–22 The former process relies on an FeIII-OH moiety, either in solution or as part of a nanoparticle assembly (e.g., rust). The Fe-O bond is cleaved upon photoexcitation to form Fe(II) and a hydroxyl radical (OH•), of which the latter can oxidize a wide variety of organic compounds owing to its high reduction potential (~1.9 V vs. NHE).23 Photodecarboxylation reactions of an FeIII-carboxylate moiety are observed to occur in small model complexes as well as natural siderophores, with relevance for iron release and cycling in marine environments.24–28 Both sets of reactions are driven by violet (400–430 nm) and UV (250–400 nm) light with quantum efficiencies that can approach values as high as 20%, depending on the reaction conditions.22,24 Due to the low costs and minimal toxicity of iron along with the potential to initiate these reactions using ambient sunlight, these processes are under investigation for use in environmental remediation.29 However, early reports indicate that substrate specificity and product selectivity remain low.22,30 As such, the ability to access this chemistry in a controlled fashion is of great interest.
Biological systems are known to impart high levels of selectivity due to the precise primary and secondary sphere control present within the active site of a protein. For example, the cytochrome P450 family catalyzes a vast array of selective oxidation reactions, each dependent on a specific substrate and protein-derived redox partner.31–33 However, these reactions are thermally or chemically activated and thus lack the temporal and spatial control that a light-driven process would confer. Combining the favorable aspects of photo-Fenton or photodecarboxylation chemistry with the advantages of a protein scaffold would be valuable, resulting in a biocompatible “green” catalyst for chemical conversion. Unfortunately, there are only a few proteins with the appropriate electronic and geometric configuration to carry out these light-driven processes.
The R2-like ligand-binding oxidases (R2lox) represent a new class of metalloproteins that, in the presence of manganese, spontaneously assemble a heterobimetallic Mn/Fe active site.34–36 In the absence of Mn, R2lox can also bind two Fe ions to generate a standard diiron cofactor,37 though both sites show relatively weak binding affinities for divalent metals.38,39 The in vivo functions of R2lox remain unclear at this point. The protein family was discovered in Mycobacterium tuberculosis and identified as one of the ten most upregulated proteins in the virulent H37Rv strain relative to the avirulent BCG vaccine strain,40 rendering it a potential virulence factor and therapeutic target. Organisms encoding R2lox proteins include bacterial and archaeal extremophiles as well as other Mycobacterium pathogens, motivating studies of these and related systems from a human health perspective.41 Additionally, the cambialistic nature of R2lox offers an opportunity to explore fundamental properties of heterobimetallic Mn/Fe cofactors relative to the canonical diiron sites.
From a structural standpoint, R2lox is a member of the large ferritin-like superfamily, with a mixed carboxylate-histidine bimetallic active site.42 More specifically, R2lox shares similarities both with the R2 subunit of class 1c ribonucleotide reductases (RNRs), a heterobimetallic Mn/Fe enzyme in which the high-valent MnIV/FeIII resting state acts as a one-electron oxidant to initiate DNA synthesis, as well as the bacterial multicomponent monooxygenases (BMMs), which perform two-electron oxidation reactions on a diverse array of substrates.42 Consistent with this comparison, R2lox has been shown to form an unprecedented intramolecular tyrosine-valine crosslink between Y162 and V72 upon metal binding and oxygen activation, reflecting a two-electron oxidation of the β-carbon of V72.34,35 The role of this ether crosslink has been postulated to be structural, poising the active site in the proper configuration for subsequent catalysis,43,44 and the mechanism of crosslink formation remains under investigation.38 Following assembly, R2lox adopts a MnIII/FeIII resting state with two carboxylate bridges and a single “hydroxo” (µ-OH) ligand between the two metals (Figure 1).
Figure 1.
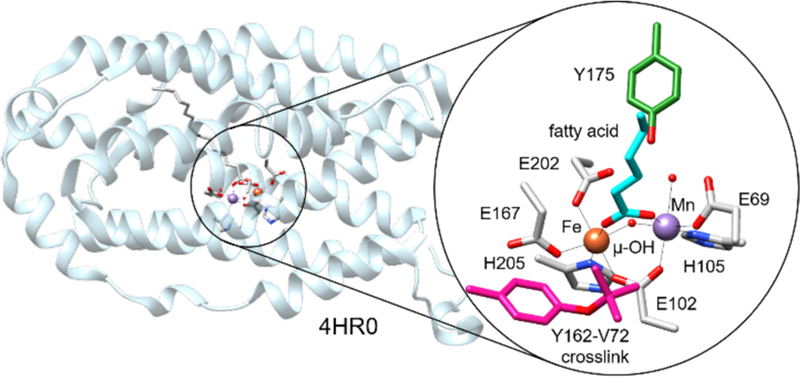
Cartoon diagram of R2lox with the active site highlighted. Key residues in the primary and secondary coordination spheres indicated in inset.35
This protonation state has been characterized using EPR and X-ray spectroscopy, as it is unique among this class of proteins; related diiron carboxylate proteins typically feature at least one deprotonated “oxo” (µ-O) ligand between the two metals.35,36,45,46 This difference may be relevant to the chemistry performed by the protein, as the protonation state of the bridging ligand plays a key role in tuning the reduction potential of the active site.47 Further, this proton may also participate in photoinduced chemical processes, as the metal cluster now closely resembles those implicated in iron-mediated photo-Fenton and photodecarboxylation reactions. In fact, a prior study revealed that cryogenic, visible-light photolysis of R2lox generates a new species that exhibits EPR spectral parameters consistent with intramolecular deprotonation of the hydroxide bridge.36 Upon warming, the resting state is recovered.
In the present work, we have expanded upon these initial observations. We find that photoexcitation of aerobically assembled R2lox at room temperature generates another new species with distinct spectral properties. This state features a broad visible absorption band and resonance Raman spectra with distinct high frequency modes from 1000–1600 cm−1. The optical and vibrational features of this new species are characteristic of proteins with metal-phenolate coordination, such as the purple acid phosphatases and a variant of RNR that auto-hydroxylates an active site phenylalanine.48,49 Herein, we present multiple lines of spectroscopic, structural, and mass spectrometric evidence for photoinduced rearrangement of the R2lox active site and propose a reaction mechanism that resembles the photode-carboxylation processes found in siderophores, which are responsible for mediating Fe release.25 Understanding the parameters governing the high efficiency (Φ ~ 3%) of this transformation has implications for developing molecular and enzymatic systems containing first-row transition metals that can effectively participate in controlled photochemical reactions. Additionally, these results expand the scope of potential reactivity for R2lox, furthering our understanding of the chemistry accessible to a heterobimetallic Mn/Fe cofactor.
Materials and Methods
Materials
All materials were obtained from Fisher Scientific or VWR unless otherwise stated. Optical elements were purchased from Newport Corporation or Thorlabs unless otherwise noted. All solutions were prepared using deionized water (ELGA Flex II).
Protein production and purification
R2lox was produced and purified as previously reported.35,38 Briefly, a 300 mL culture of E. coli strain DE3* (New England Biolabs, Cambridge, MA) containing the plasmid encoding the Geobacillus kaustophilus R2lox I gene was shaken at 200 rpm in TB media (Formedium, Norfolk, United Kingdom) at 37 ˚C for 15 hours. The cells were then diluted into 1 L of fresh culture and shaken at 200 rpm at 37 ˚C until reaching an OD of ~1.0. Cell growth was arrested by storage at 4 ˚C for up to 6 hours. Prior to induction, an aliquot of EDTA at pH 7.0 was added to the cell culture to a final concentration of 0.5 mM and shaken for 20 min to eliminate any free metals from the culture media. Protein expression was induced by addition of an aliquot of IPTG (GoldBio, St. Louis, MO) to a final concentration of 0.5 mM to the cell culture. Protein expression was allowed to continue for 15 h at 18 ˚C while shaking at 200 rpm. Finally, cells were collected by centrifugation at 5900xg and 4 ˚C. Pellets were frozen at −80 ˚C until purification of R2lox.
Cells were lysed by sonication on ice for 2 min in 15 sec increments followed by 1.75 min rest periods at a power level of 25% (Model Q700, Qsonica, Newtown, CT). The lysate was then centrifuged at 39,000xg for 30 min followed by a 10 minute heat treatment at 60 ˚C and a subsequent centrifugation under the same conditions. The lysate was then loaded in 5–10 mL aliquots onto 4 mL Ni-NTA columns (McLab, San Francisco, CA) that were equilibrated with a solution containing 25 mM HEPES, 20 mM imidazole, 300 mM NaCl, and 0.5 mM EDTA at pH 7.0. The lysate-loaded column was first washed with 25 mL of a solution containing 25 mM HEPES, 40 mM imidazole, 300 mM NaCl, and 0.5 mM EDTA at pH 7.0 followed by 50 mL of a solution containing 25 mM HEPES, 40 mM imidazole, and 300 mM NaCl at pH 7.0 to remove weakly bound proteins. Pure apo-R2lox was eluted with 25 mL of a solution containing 25 mM HEPES, 300 mM NaCl, and 250 mM imidazole at pH 7.0. Samples were exchanged into 25 mM HEPES, 50 mM NaCl at pH 7.0 using a stirred cell concentrator equipped with a 3 kDa molecular-weight cutoff filter (Amicon, Millipore Sigma, Billerica, MA). The protein was concentrated to a final concentration of ~1 mM, flash-frozen in liquid nitrogen, and stored in aliquots at −80 ˚C until use. Protein concentrations were determined using previously published extinction coefficients (ε280 = 47757 M−1cm−1).36 Protein purity was verified using SDS-PAGE (Figure S1).
Point mutations were introduced into the plasmid encoding full-length Geobacillus kaustophilus R2loxI by site-directed mutagenesis using the QuikChange Lightning kit (Agilent, Santa Clara, CA) and verified by DNA sequencing (Genewiz, South Plainfield, NJ). All point mutants were produced and purified in metal-free form according to the same protocols as wild-type (WT) R2lox.
Sample preparation
To prepare metallated samples, apo-R2lox was diluted to a concentration of 0.1 mM in 25 mM HEPES, 50 mM NaCl at pH 7.0. Protein was then rapidly mixed with either one equivalent each of MnCl2•4H2O (Amresco, Solon, OH) and (NH4)2Fe(SO4)2•6H2O (Sigma Aldrich, St. Louis, MO) or two equivalents of (NH4)2Fe(SO4)2•6H2O, both prepared in deionized water, to generate Mn/Fe or Fe/Fe R2lox, respectively. Metallation was monitored using optical spectroscopy over the timescale of 1 h.38 Deuterated samples were prepared in buffers made with D2O (99.9%, Cambridge Isotope Laboratories, Cambridge, MA). Protein samples were allowed to equilibrate in deuterated buffers at 4 ˚C for 16 h prior to metallation to promote complete exchange of protons. After metallation, holo-R2lox was run through a HiTrap desalting column (GE Amersham, Stockholm, SE) to separate the protein from any unbound metal ions. To incorporate 18O into the bridge position, samples were incubated for 20 hrs in 96% 18O-enriched buffer (98.5%, Isoflex, San Francisco, CA). Photoconversion for resonance Raman and EPR spectroscopy was accomplished using irradiation from the 457.9 nm line (P = 30 mW) of a Kr/Ar mixed gas laser (Coherent Innova Spectrum I-70C, Santa Clara, CA) for 20 min.
Iron quantification assay
The amount of iron bound was quantified using 2,2-bipyridine as a colorimetric indicator.50 To denature the protein and reduce the bound metals, R2lox was shaken at 200 rpm in solution with 143 mM sodium dithionite and 0.107 mM of 2,2-bipyridine at 37 ˚C for 90 min. The mixture was centrifuged at 17,900xg for 30 min to remove precipitate, and the supernatant was analyzed by UV-Vis absorption spectroscopy. The concentration of iron was determined by monitoring the absorbance of [FeII(bpy)3]2+ at 522 nm (ε = 8650 M−1cm−1).51 Metallation percentages were determined by comparison to the initial protein concentration.
Resonance Raman spectroscopy
Metallated protein samples were concentrated to ~1 mM and placed in flame-sealed 0.8 mm ID borosilicate capillaries (Kimble). Laser excitation wavelengths of 457.9 nm, 488.0 nm, 514.5 nm, and 568.2 nm were generated by a Kr/Ar laser (Coherent Innova Spectrum 70-C, Santa Clara, CA). The appropriate interference filters (Semrock, MaxLine, Rochester, NY) were used to remove plasma lines. The resonance Raman setup was similar to that described previously.52 Briefly, the beam was focused onto the sample at a 135˚ backscattering angle using an f/4 parabolic focusing mirror. The scattered light was collimated by an f/0.8 UV-fused silica aspheric lens (Edmund Optics, Barrington, NJ), focused onto the 100 µm slit of an f/4.6 single-grating spectrograph equipped with a 1200 gr/mm grating blazed at 500 nm (Princeton Instruments Isoplane 320, Trenton, NJ), and imaged onto a Peltier-cooled CCD (Princeton Instruments Pixis 100B, Trenton, NJ). Rayleigh scattering was rejected by the appropriate edge filter (Semrock, RazorEdge, Rochester, NY). A 50/50 (%v/v) mixture of toluene and acetonitrile was used to calibrate the spectrograph to within ±1 cm−1. Individual spectra were collected in 1 min increments and summed together to minimize laser-induced damage. The buffer was then subtracted from the protein and the resulting spectra were baseline-corrected to account for any intrinsic fluorescence or scatter off the sample holder. Relative resonance Raman enhancement profiles were generated by normalizing all intensities to the non-resonant phenylalanine peak at 1005 cm−1. Spectra were exported and analyzed in Igor Pro 6 (Wavemetrics, Portland, OR).
Electronic paramagnetic resonance (EPR) spectroscopy
Freshly metallated and desalted samples of both dark-conditioned and irradiated Mn/Fe R2lox were frozen in Suprasil EPR sample tubes (727-SQ-250M Wilmad Glass, Vineland, NJ) using liquid nitrogen at a concentration of ~0.75 mM. EPR measurements were made at the Ohio Advanced EPR Center (Miami University, Oxford, OH). EPR spectra were measured at 5 K at X-band frequencies (νµw = 9.6340 GHz; Pµw = 2 mW) on a Bruker CW-EPR instrument (Bruker E500, Billerica, MA) equipped with an Oxford ITC-500 cryostat.
Quantitative photochemical experiments
Photochemical action spectra were obtained with 200 µL samples of 20 μM Mn/Fe and Fe/Fe R2lox within 4 × 10 mm fluorescence cuvettes (SF-Q-10, Starna Cells Inc, Atascadero, CA). The samples were irradiated for a fixed amount of time within a Fluoromax-4 fluorimeter (JY Horiba, Kyoto, Japan) at 25 ˚C using a bandwidth of 10 nm. Measurements were made in 40-nm increments from 300 to 700 nm. Absorbance spectra were measured before and after irradiation. Fresh samples were used for each wavelength. The difference in absorption at 550 nm (Fe/Fe R2lox) or 540 nm (Mn/Fe R2lox) due to photolysis was normalized to the integrated lamp intensity over the irradiation range at each excitation wavelength (Figure S2).
Extinction coefficient determination
The extinction coefficient of the photoconverted product was obtained by irradiating the sample with 402 nm light until the absorption features of the photoproduct were no longer changing. To determine the concentration of protein present, an iron quantification assay was performed on the photoproduct. For the Fe/Fe R2lox samples, homobimetallic cofactor formation was assumed. From this assumption, 80 ± 3% of the protein was found to be metallated. For Mn/Fe R2lox, the concentration of metallated cofactor present was estimated from the iron quantification assay by assuming a similar proportion of metals bound (~80%), this time as the heterobimetallic cofactor.
Quantum yield measurements
A potassium ferrioxalate actinometer was used to quantify the flux of the fluorimeter lamp for the quantum yield experiments. The actinometry protocol was adapted from Murov et al. and Ma et al.53,54 Briefly, a solution of 0.15 M potassium ferrioxalate trihydrate (Strem Chemicals, Inc., Boston, MA) in 0.05 M H2SO4 was prepared along with a 0.2% solution of 1,10-phenanthroline (Sigma Aldrich, St. Louis, MO) in 1.64 M sodium acetate and 0.5 M H2SO4. A 0.2 mL sample of the potassium ferrioxalate solution was irradiated in the fluorimeter at 402 nm (3 nm bandwidth). Following irradiation, 0.4 mL of the phenanthroline solution was added to the potassium ferrioxalate solution. The solution was allowed to develop in the dark for 10 min at 25 ˚C, after which time the absorbance at 510 nm was measured (ε510 = 11,000 M−1cm−1). The flux of the fluorimeter lamp was determined based on the concentration of [Fe(phen)3]2+ using eq. 13–2 in Murov et al.53 The measurement was repeated in triplicate for three different irradiation periods (10, 20, and 30 seconds).
To quantify the R2lox photoconversion efficiencies, a sample of 20 μM Fe/Fe or Mn/Fe R2lox was irradiated in the fluorimeter at 402 nm (3 nm bandwidth) for a given amount of time. The absorption spectrum was measured following irradiation, and the concentration of the photogenerated product was found using the previously determined extinction coefficient. To calculate the quantum yield of the process, the rate of formation of photoconverted R2lox was divided by the photon flux and fraction of photons absorbed.53,54 Spectra of a representative sample and corresponding calculations are shown in the Supporting Information (Figure S2). New samples were used for each independent measurement.
Crystallization, data collection and structure determination
Apo-R2lox was crystallized in metal-free form by vapor diffusion in hanging drops at 22 ˚C in 25–27.5% (w/v) PEG 1500, 100 mM HEPES-Na pH 7.3–7.5. To reconstitute the oxidized resting state Fe/Fe cofactor, crystals were transferred into a drop of mother liquor additionally containing 5 mM (NH4)2Fe(SO4)2 for 2.5–4 h under aerobic conditions. Fesoaked crystals were then photoconverted in the sealed hanging drop by positioning a 455 nm high-powered LED with a focusing lens at a distance of ~1 cm above the drop and irradiating the drop for 10 min, a duration sufficient to cause the complete photoconversion of Fe-reconstituted R2lox in solution. Both irradiated and non-irradiated Fesoaked crystals were briefly washed in 40% (w/v) PEG 1500, 100 mM HEPES-Na pH 7.3–7.5 before flash-cooling in liquid nitrogen. Data were collected at 100 K at beamline X06DA of the Swiss Light Source (Villigen, Switzerland).
Data were processed with XDS.55 The structures of the “dark state” and “photoconverted” R2lox were solved by Fourier synthesis using the structure of the wild-type protein in the oxidized state not containing any ligands as a starting model.43 Refinement was carried out with phenix.refine and iterated with rebuilding in Coot.56–58 Refinement included bulk solvent corrections, individual atomic coordinate and isotropic B factor refinement, and occupancy refinement for alternate conformations. Metal-ligand bond lengths were restrained. Solvent molecules were added with phenix.refine and manually. Hydrogens were added to the models in the later stages of refinement. Structures were validated using MolProbity.59 Data and refinement statistics are given in Tables S1 and S2. All figures were prepared with PyMOL (version 1.6.0.0, Schrodinger, LLC). Models and structure factors were deposited in the Protein Data Bank under accession codes 5OMK (dark state) and 5OMJ (photoconverted state).
Mass spectrometry analysis
Three replicates of 200 µM apoprotein were incubated with 3 equivalents (per monomer) of (NH4)2Fe(SO4)2 in 100 mM HEPES-Na pH 7.0, 50 mM NaCl for 1 h at room temperature in the dark. Samples were protected from light as much as possible during processing. Excess metal ions were removed by passing the samples through a HiTrap Desalting column (GE Healthcare) equilibrated in 25 mM HEPES-Na pH 7.0, 50 mM NaCl. The reconstituted protein was concentrated to 2–3 mM, as determined by its absorbance at 280 nm. Each sample was split into two aliquots. One aliquot was kept dark, while the other was irradiated in a clear Eppendorf tube with a 455 nm high-powered LED equipped with a focusing lens until the UV/Vis spectrum was no longer changing. UV/Vis spectra of aliquots taken from the dark samples confirmed that no conversion to the purple species had taken place in these samples. From each sample, 2 μL of solution (i.e. approx. 150–200 μg of total protein in each sample) were taken and diluted in 200 μL of phosphate buffer (50 mM, pH 7.6) with 0.5 M urea. Each resulting solution was applied to a 10 kDa molecular weight filter (Millipore, Billerica, MA) previously washed with water and the urea-phosphate buffer and centrifuged at 14000xg. After a buffer wash step using an additional 200 μL aliquot of the urea-phosphate buffer, digestion was finally carried out on the filter with 100 μL of Glu-C protease (3 μg used per sample, Promega, Madison, WI) in urea-phosphate buffer by incubating at 37 ˚C in the dark for 16 h with mild shaking (60 rpm). In phosphate buffer, proteins are cleaved by Glu-C at the C-terminal side of glutamic and aspartic acid residues. The digested sample was collected from the flow-through and cleaned by Strong Cation Exchange Solid Phase Extraction (Strata-X-C Columns P/N 8B-S029-TAK-TN, Phenomenex, Torrance, CA) and dried in vacuo. Samples were then dissolved in solvent A (see below) at a concentration of approx. 0.4 μg/μL. The auto sampler of a HPLC 1200 system (Agilent Technologies) injected 0.5 μL (200 ng of peptides) into a C18 guard desalting column (Zorbax 300SB-C18, 5 × 0.3 mm, 5-μm bead size; Agilent). A 15-cm-long C18 picofrit column (100-μm internal diameter, 5-μm bead size; Nikkyo Technos) installed onto the nano-electrospray ionization source was then used. Solvent A was 97% water, 3% acetonitrile (ACN), and 0.1% formic acid (FA); solvent B was 5% water, 95% ACN, and 0.1% FA. At a constant flow of 0.4 μL/min, a linear gradient went from 2% B up to 40% B in 45 min, followed by a steep increase to 100% B in 5 min. Online liquid chromatography-MS was performed using a LTQ Orbitrap Velos Pro mass spectrometer (Thermo Scientific). Fourier transform MS master scans (AGC target of 1e6) were acquired with a resolution of 30000 in mass range 300–1700 m/z, and were followed by data-dependent MS/MS (AGC target of 1e5) at a resolution of 15000. In d-d MS/MS, the top three ions from the master scan were selected for both collision induced dissociation (CID at 35% collision energy) and higher energy collision dissociation (HCD at 35% energy). Precursors were isolated with a 2 m/z window. Dynamic exclusion was used with 2 repeat counts and 30 s duration. In addition to the sample preparation triplicates, LC-MS technical triplicates were also run, resulting in a total of 9 “dark” plus 9 “light” LC-MS runs.
To assist in peptide identification, searches of all MS/MS spectra by Sequest were performed using the software platform Proteome Discoverer (v1.4, Thermo Scientific) against the R2lox primary sequence. A precursor mass tolerance of 10 ppm and product mass tolerance of 0.02 Da were used. The variable modifications considered were decarboxylation ((C(−1) O(−2)) of glutamic acid; oxidation (O) of methionine, valine, tyrosine, leucine or isoleucine; and dehydrogenation (H(−2)) of valine, tyrosine, leucine or isoleucine. The proteolysis enzyme used was Glu-C, with up to 2 missed cleavages allowed.
Results and Discussion
Exposure to light changes the absorption spectra of R2lox
Upon violet and near-UV photoexcitation of desalted solutions of either Mn/Fe or Fe/Fe R2lox, a broad absorption band centered around 550 nm appears in the optical spectrum (Figure 2). Similar features and timescales of conversion are seen for both the Fe/Fe and Mn/Fe cofactors (Figure 2, inset). However, the absorption intensities of the new species appear to be metal-dependent, with the maximum extinction coefficients estimated at 2600 M−1cm−1 (550 nm) for Fe/Fe R2lox and only 1200 M−1cm−1 (540 nm) for Mn/Fe R2lox. These values were calculated assuming comparable levels of metallation (~80%) for both cofactors and quantitative conversion after 4 hrs, which may underestimate the extinction coefficient. Metallated protein stored in the dark did not undergo this change (Figure S3).
Figure 2.
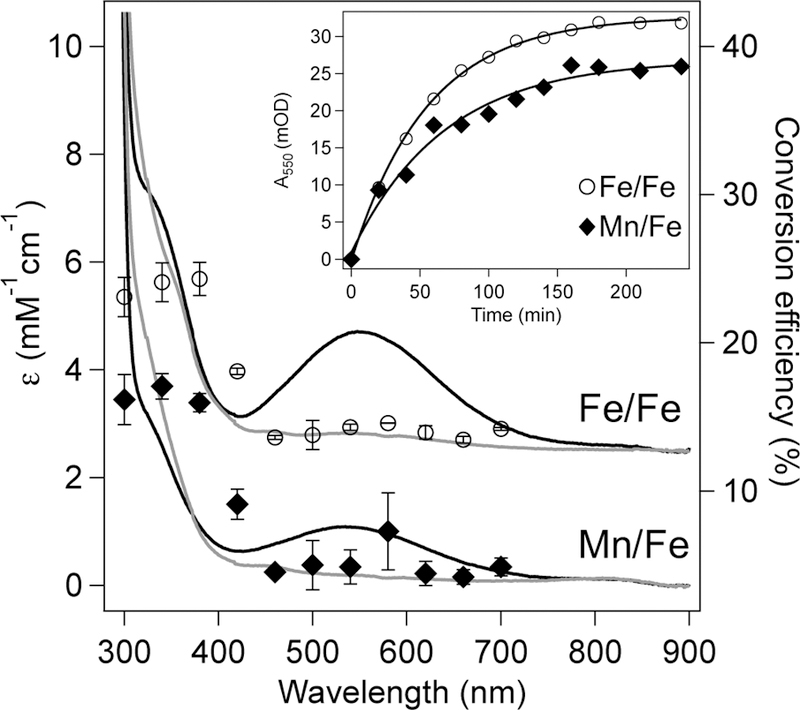
Absorption spectra (left hand axis) of Fe/Fe (top) and Mn/Fe (bottom) R2lox prior to (grey) and following (black) irradiation overlaid with the action spectrum of each species as indicated (right hand axis). Error bars reflect the standard deviation from three independent trials. Spectra of Fe/Fe R2lox are offset vertically by +3000 M−1 cm−1 for clarity. (Inset) Photoconversion of Fe/Fe (open circles) and Mn/Fe (filled diamonds) R2lox as a function of time. Solid traces show a single exponential fit to the data with τ = 55.3 ± 1.55 and 67.5 ± 9.32 min for Fe/Fe and Mn/Fe R2lox, respectively.
To quantify the wavelength dependence of this transformation, the action spectrum was measured (Figure 2, right hand axis). Overall, the photoconversion efficiencies follow the absorption spectrum of metallated R2lox, suggesting this process derives from the protein itself and is not simply an artifact of irradiation. Both Mn/Fe and Fe/Fe cofactors exhibit low efficiencies in the violet region of the spectrum (~400 nm), likely due to excitation into the weakly absorbing feature at 465 nm, with higher conversion efficiencies seen in the near-UV from 300 – 360 nm. Irradiation wavelengths longer than 460 nm do not induce this transition in either cofactor to any appreciable amount. Using actinometry, the quantum yields for this process at 402 nm were found to be 2.3 ± 0.090% for Fe/Fe R2lox and 3.1 ± 0.26 % for Mn/Fe R2lox. That these values are quite similar indicates that the metal in site 1 exerts only a minor influence on this process. The quantum efficiencies for conversion are relatively high for a protein-based photochemical process mediated by a first-row transition metal; for comparison, the aforementioned Ni-C to Ni-L conversion in the [NiFe] hydrogenase, in which a proton is transferred from nickel to a sulfur thiolate, has an estimated quantum yield of 0.07%.60
Resonance Raman spectroscopy of photoconverted R2lox
Further characterization of the photoconverted R2lox samples was carried out using resonance Raman spectroscopy, with excitation into the new visible absorption band (Figure 3). Interestingly, the photoconverted R2lox samples exhibit intense vibrational bands at 1170 cm−1, 1288 cm−1, 1501 cm−1, and 1601 cm−1, which are much higher in energy than typically observed in non-heme diiron enzymes. Additional bands are observed at 590 cm−1 and 800 cm−1, with similar spectra obtained for both the Fe/Fe and Mn/Fe cofactors. To explore the degree of coupling between the active site vibrations and exchangeable protons, solvent isotope-dependent experiments were performed by exchanging R2lox into deuterated buffers prior to metallation and carrying out all experiments in D2O (Figure S4A). While only minor changes are seen in the high-frequency region of the spectrum, a pronounced upshift to higher frequencies can be resolved in the difference spectra of both photoconverted Mn/Fe and Fe/Fe R2lox for the band at 586 cm−1, with differential peak shifts of +14 cm−1 and +12 cm−1. Resonance Raman excitation profiles indicate that the pronounced vibrational bands all derive from a single species via excitation into one electronic transition (Figure S4B).61 Attempts to obtain resonance Raman spectra of R2lox prior to photoconversion were unsuccessful, as even low photon fluxes caused this change to occur during spectral collection, and the vast amounts of sample that would be required for a single-pass experiment are prohibitive.
Figure 3.
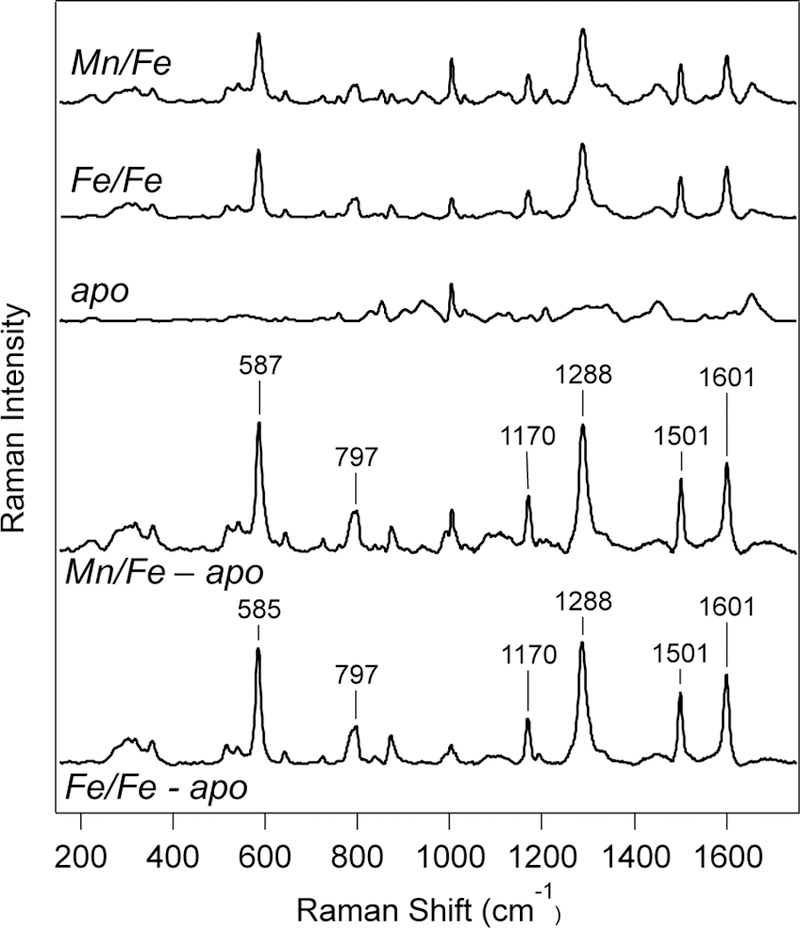
Resonance Raman spectra (λex = 457.9 nm; P = 20 mW; T = 298 K) of (top) photoconverted Mn/Fe, Fe/Fe, and apo-R2lox. (Bottom) Resonance Raman spectra of photoconverted Mn/Fe and Fe/Fe R2lox after subtraction of apo-R2lox spectrum to eliminate contributions from protein-derived bands.
The electronic structure of Mn/Fe R2lox does not change significantly upon photoconversion
Electron paramagnetic resonance (EPR) spectroscopy is a highly sensitive reporter of local electronic structure, particularly for paramagnetic metalloproteins. While Fe/Fe R2lox has an overall S=0 ground state and does not exhibit an EPR spectrum at X-band frequencies, the MnIII/FeIII R2lox resting state has a characteristic spectrum with ~7 peaks split by ~250 MHz, attributed to anisotropic 55Mn hyperfine coupling along the Jahn-Teller axis.35 The spectrum spans approximately 65 mT at X-band frequencies and has previously been studied in great detail.35,36 To investigate whether photolysis induces an oxidation state change or significant electronic rearrangement at the metal center, EPR spectra of photoconverted Mn/Fe R2lox were measured (Figure 4). While slight changes in the relative intensities of peaks are observed, the overall spectral shape, width, and 55Mn hyperfine splitting remains approximately the same. This provides strong evidence that the oxidation states and electronic coupling between the two metal centers are largely unchanged upon photoconversion. For comparison, the EPR spectrum of a MnIV/FeIV intermediate in the related class Ic RNRs is significantly more narrow, spanning only 50 mT, due to decreased geometric distortion imposed by the MnIV oxidation state.62 By contrast, the previously identified photoactive state of Mn/Fe R2lox generated using cryogenic photolysis shows greater apparent 55Mn hyperfine coupling and reduced g-tensor anisotropy, resulting in a more narrow spectrum with greater spacing between peaks. This previous species was tentatively assigned to a MnIII(µ-O)FeIII state caused by photoinduced deprotonation of the bridging hydroxide ligand.36 These prior observations indicate that significant changes to the EPR spectrum would be expected if the primary coordination sphere was significantly perturbed.
Figure 4.
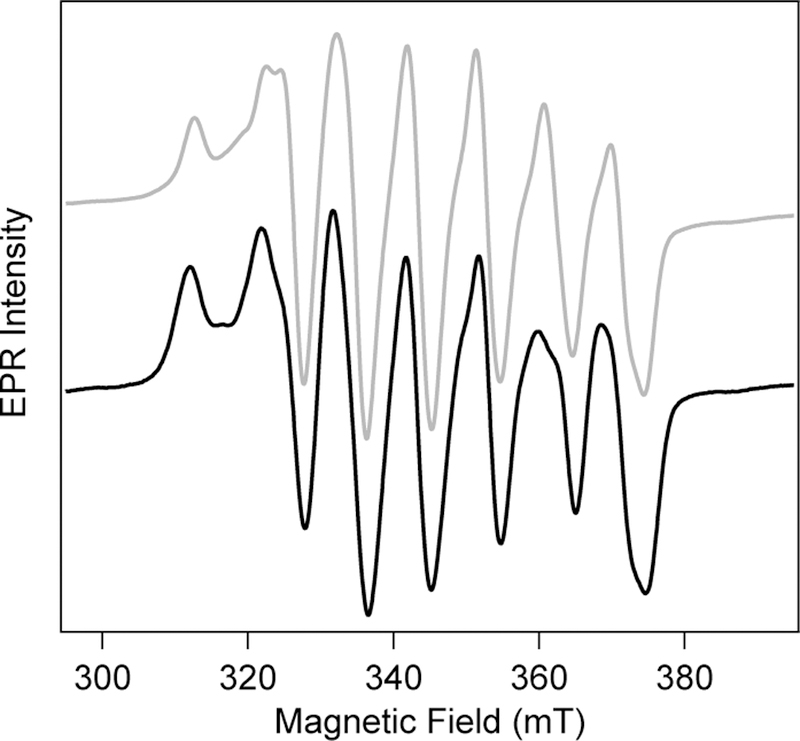
CW X-band EPR spectra of MnIII/FeIII R2lox (T = 5 K) prior to (grey) and following (black) complete photoconversion.
Spectroscopic evidence suggests R2lox undergoes photoinduced rearrangement to yield tyrosinate coordination
The spectra of photoconverted R2lox bear close resemblance to the purple acid phosphatases (PAPs) and related metal-tyrosinate proteins and model compounds.48,63 In addition to hydrolysis reactions, PAPs have also been implicated in peroxidase catalysis,64 another potential point of functional comparison to R2lox. These species feature phenolate-to-FeIII LMCT bands centered around 550 nm, which confer the characteristic purple color associated with these enzymes.65,66 The phenolate ligands in these systems are typically bound to the FeIII center in a terminal fashion. The striking similarity in optical spectra between the photoconverted Mn/Fe and Fe/Fe cofactors suggests that the dominant ion contributing to the LMCT transition is the FeIII center in site 2. The observed variation in extinction coefficient may be attributed to differences in the site 1 metal identity, though the various metallation states of PAPs (FeIII/FeIII, FeIII/ZnII, and FeIII/MnII) do not show significant differences in their extinction coefficients (Table S3).65,67,68 Additionally, small-molecule MIII-phenolate compounds show an increase in transition energy and decrease in extinction coefficient when MnIII is used rather than FeIII for a given ligand.69,70 Thus, the origin of the different extinction coefficients for photoconverted Mn/Fe and Fe/Fe R2lox may also stem from differences in metallation yield.
This model of tyrosinate-iron coordination is further supported by the resonance Raman spectra of photoconverted R2lox, which also share a number of similarities with the spectra of purple acid phosphatases and metal-phenolate complexes. Typical RR bands of PAP include a low frequency mode between 580–600 cm−1 that is dominated by iron-oxygen stretching motion. Additionally, the spectra of PAPs all feature four distinctive, high-frequency bands reflecting the in-plane tyrosine C-H bending motion (Y9a), C-O stretch (Y7a’), and ring-stretching modes (Y19a and Y8b) between 1150 cm−1 and 1600 cm−1.71 The frequencies of these bands do not change significantly upon deuteration, suggesting there are no exchangeable protons directly coupled into these vibrations.
The notable change observed in the Fe-O stretching mode of photoconverted R2lox upon deuteration, mentioned above, does suggest coupling of this mode to nearby protons, with the shift to higher frequencies for the heavier isotope indicative of hydrogen bonding between the ligand and other exchangeable sites.72,73 However, upon incubation with H218O buffer, no prominent isotope-dependent bands are observed that would indicate an enhanced vibrational mode due to an exchangeable µ-OH bridge between the two metals (Figure S5), as has been seen previously for incubation of R2lox with H217O buffer.74 This suggests the Fe-O stretch observed originates from an endogenous, rather than solvent-derived, oxygen atom. All of the peaks described above are absent in the apo-R2lox Raman spectrum (Figures 3 and S5).
Unlike the optical and resonance Raman spectra, the EPR signatures of Mn/Fe R2lox remain virtually unchanged upon room temperature photoconversion. This indicates little variation in the primary coordination sphere or protonation state of the bridging ligand, as either would be expected to significantly perturb the magnetic coupling between the metals. However, like carboxylate and hydroxide ligands, phenolate species are monoanionic.75 In fact, phenolate functional groups are often used to generate multimetallic model compounds in place of carboxylate or hydroxide species due to synthetic accessibility.76,77 The EPR spectra of a Mn/Fe system with a phenolate ligand would therefore be expected to resemble that containing a carboxylate or hydroxide ligand. Taken together, the multiple lines of spectroscopic evidence suggest that the active site of photoconverted R2lox features a coordinated tyrosinate ligand.
Mutation studies suggest Y162 is the coordinating tyrosinate ligand
With the nature of the photoinduced change identified, it becomes necessary to identify the coordinating tyrosine residue. As R2lox features the aforementioned tyrosine-valine crosslink near the active site, with the phenolic oxygen -only ~5 Å from the Fe center in the WT crystal structure, it was hypothesized that this moiety may be responsible for the light-induced transition seen. Thus, photolysis experiments were also performed on R2lox mutants with changes in the residues that are involved in the ether crosslink, including the V72A, V72L, V72I, and Y162F variants. With the exception of isoleucine, which features a tertiary carbon at the same position as the native valine, these mutants are unlikely to form the ether crosslink upon metal binding and oxygen activation due to decreased stability of the resultant alkyl radical that would be formed.38 However, both V72A and V72L undergo photoconversion upon irradiation with efficiencies that are indistinguishable from the native protein (Table S4), suggesting the presence of the crosslink has no impact on the photoconversion process. The quantum yield for photoconversion of the V72I Mn/Fe protein was increased by a factor of ~2.5, perhaps owing to increased steric bulk of the larger isoleucine sidechain. On the other hand, the Y162F mutant assembles with both Fe/Fe and Mn/Fe cofactors to give optical spectra that are similar to the WTresting state species (Figure S6), but samples of this variant do not turn purple, even after prolonged irradiation. However, dramatic perturbation to the EPR spectrum occurs upon irradiation of Mn/Fe Y162F R2lox, indicating a different process occurs upon illumination (Figure S7). Consistent with this observation, the resonance Raman spectra of photoconverted Y162F Fe/Fe R2lox show a prominent band at 517 cm−1 that shifts to 498 cm−1 when H218O buffer is used, indicative of a solvent-derived ν (Fe-O) stretching mode (Figure S8).78 This may indicate the presence of a deprotonated (µ-O) bridge between the metal centers in photoconverted Y162F R2lox, though this structure is under further investigation. Together, these data suggest that Y162 is the tyrosine residue that coordinates to the metal center in the photoconverted WT R2lox species.
We also considered whether multiple species could be contributing to the observed spectroscopic signals. Like the related RNR and BMM enzymes, R2lox contains a number of tyrosine residues near the active site.79–81 The second closest tyrosine, Y175, is only ~5 Å away from the Mn center and ~5.5 Å away from the Fe center and participates in hydrogen bonding interactions with the Mn-bound water ligand and/or the coordinating E202.46 Because of this proximity to the active site, Y175 was also explored as a candidate for photoinduced modification. Upon reconstitution with metals under aerobic conditions, the Y175F mutant develops optical and EPR spectra characteristic of the oxidized MnIII/FeIII protein, suggesting this residue is not critical for assembly. Photoexcitation of Y175F with violet or near-UV light results in a purple product that is spectrally indistinguishable from photoconverted WT R2lox (Figure S6), indicating Y175 does not coordinate to the metal center upon irradiation. However, the quantum yield for photoconversion is decreased by approximately 50% (Table S4), suggesting this residue is relevant for the efficiency of the process.
Structural characterization of photoconverted R2lox
To further characterize the structure of photoconverted R2lox, X-ray crystallography was employed. Crystals of apo-R2lox were grown, and the protein was metallated with Fe by crystal soaking. Irradiation with blue light was carried out in crystallo using settings determined to cause complete photoconversion of the metallated protein in solution.
Conversion to the photolyzed product was evident by visual inspection of the crystals. Data were also collected on Fesoaked R2lox crystals from the same batch harvested before photoconversion to ensure that the normal oxidized resting state structure, including the tyrosine-valine ether crosslink, had been formed prior to light exposure. The structure of “dark state” Fe/Fe R2lox from these crystals is indeed indistinguishable from the previously determined structure.37 The electron density of irradiated R2lox crystals, however, indicated a number of drastic changes around the metal center, as suggested by the spectroscopic results (Figure 5). The tyrosine-valine crosslink is no longer evident. The valine residue appears to be intact, though present in multiple conformations. Y162 coordinates to the Fe center via the phenolate oxygen in place of E167. Moreover, the electron density for E167 extends only to cover the beta-carbon of the sidechain. While this is not uncommon and usually simply due to structural flexibility of the sidechain, the new position of Y162 and the clear electron density of surrounding residues are not sterically compatible with the full E167 sidechain being present. For this reason, we investigated the possibility that E167 was truncated. As discussed further below, mass spectrometry shows that E167 is indeed decarboxylated to homoalanine (alpha-aminobutyric acid) in the irradiated protein (Figure 6). These dramatic changes around the active site seem to only influence the site 2 Fe center, as the site 1 metal coordination sphere is unperturbed. This is consistent with the EPR spectra, which show little change upon photoconversion.
Figure 5.
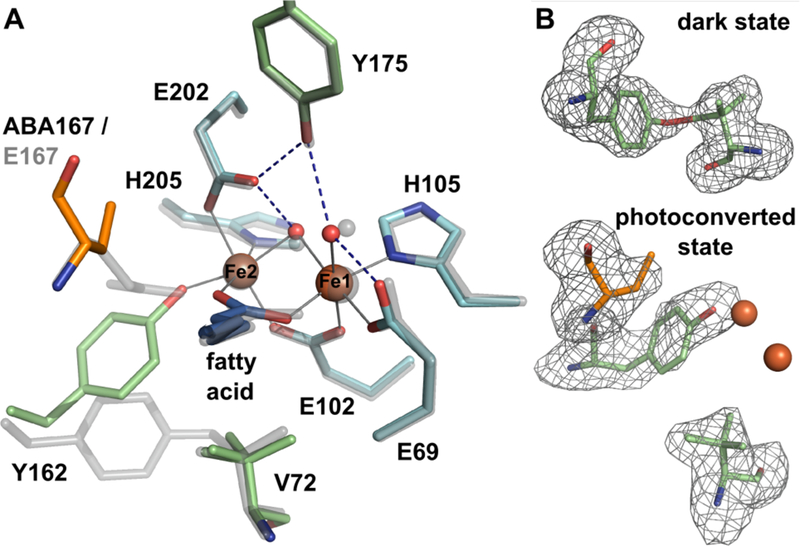
(A) Active site structure of photoconverted Fe/Fe R2lox (color), superimposed with the oxidized, dark state structure (transparent grey). The Y162-V72 ether crosslink has been broken, the site 2 metal ligand E167 has been decarboxylated to alpha-aminobutyric acid (ABA), and Y162 coordinates to Fe2 instead. V72 is present in two different rotamers. The rest of the active site and the global structure are unaffected. Metal-ligand bonds in the photoconverted state are indicated by grey lines, hydrogen bonds by dashed blue lines. (B) mFo – DFc omit electron density for residues Y162 and V72 of dark state Fe/Fe-R2lox (top) and Y162, V72, and ABA167 of photoconverted Fe/Fe-R2lox (bottom), contoured at 3σ, shown with the final model. The crosslink is formed in dark state Fe/Fe-R2lox, but only partially, leading to poor refinement results when the ether bond is restrained. The bond was therefore not modeled in the final structure, but is shown here for clarity.
Figure 6.
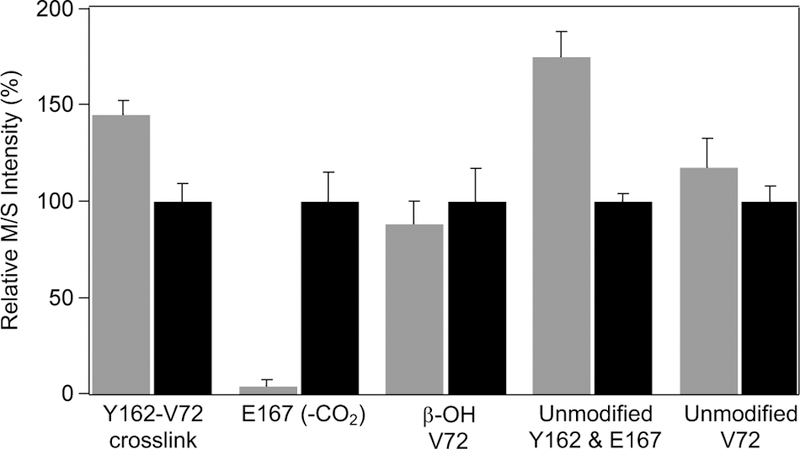
Mass spectrometry quantification of changes in peptide levels for samples that have been dark-conditioned (grey) versus samples following photoconversion (black). Intensities have been normalized to the photoconverted values for each peptide. Values reflect average and standard deviation of three sample replicates times three technical replicates.
Mass spectrometry quantitates changes in peptide structure upon irradiation
To validate the implied changes in the relevant amino acid residues and quantify the effects of illumination on the primary structure of the protein, liquid chromatography-mass spectrometry (LC-MS) was performed on the peptide mixtures resulting from Glu-C digestion of dark-conditioned or irradiated R2lox (Figure 6). Owing to differences in ionization efficiencies, only relative amounts of the same peptide can be directly compared.
The amount of Glu-C peptide containing the Y-V crosslink (AVIRAATVYNMIVE-AVTLD) decreased by 30% following irradiation, indicative of photoinduced crosslink cleavage.35 Additionally, a large peak attributed to the non-crosslinked peptide with a decarboxylated E167 sidechain (AVIRAATVYNMIVE(-CO2)GTLAE, see Figure S9 for MS2 spectral annotation) was observed only after light exposure. Glu-C peptides could also be identified containing the unmodified Y162 and V72 residues of the protein. The peptide containing simultaneously both unmodified Y162 and E167 residues decreased upon illumination, likely due to the extensive glutamate decarboxylation reaction taking place, while the unmodified Val-containing peptide remained the same, within error. A small amount of hydroxylated valine was also observed, likely reflecting a side reaction upon oxygen activation. The amount of this peptide was unchanged by irradiation, suggesting little contribution of a photo-Fenton process to the chemistry observed. No additional modifications of protein sidechains were observed in the mass spectrometry data to a significant extent following irradiation.
Taken together, the spectroscopic, structural, and mass spectrometry data suggest that irradiation causes decarboxylation of the coordinating E167 side chain and cleavage of the tyrosine-valine crosslink, ultimately resulting in coordination of the Fe center by the liberated Y162 residue.
Putative mechanism for photoconversion in R2lox
The proposed mechanism for the light-induced conversion process resembles those invoked for photoinduced decarboxylation reactions of FeIII-OOCR species (Scheme 1). These light-driven processes have been identified in FeIII-carboxylate moieties in siderophores, playing an important role in mediating iron release from chelators, as well as in model compounds and atmospheric processes.25,28 LMCT excitation of the metal-carboxylate species in the resting state of R2lox generates an FeII-carboxylate radical species. This carboxylate radical rearranges to lose CO2, leaving a primary radical on the γ-carbon of E167. This radical abstracts both an electron and a proton from the nearby metallocofactor, resulting in the formation of homoalanine. The pentacoordinate FeIII site binds to the ether moiety of the Tyr-Val crosslink, initiating homolysis to form an Fe-tyrosinate bond and regenerate an octahedral geometry around the Fe center. The cofactor then rearranges to a MIV(µ-O)FeIII state. In the absence of the crosslink, as in the V72A and V72L variants, the carboxylate radical may instead directly oxidize the nearby tyrosine residue, inducing coordination (Figure S10). It is this Fe-tyrosinate bond that is observed optically, via resonance Raman spectroscopy, and in the crystal structure.
Scheme 1.

Proposed mechanism for photoconversion process in R2lox.
The nearby Y175 residue can donate both a proton and an electron to the metal centers, restoring the MIII(µ-OH)FeIII cofactor. The Y175 radical is in close proximity to a solvent-exposed tyrosine residue, Y176, which can donate an electron and proton to oxidized Y175• and then be regenerated by solvent or buffer molecules. This mechanism is consistent with the significantly lower photoconversion yields seen for the Y175F mutant, which would not be able to donate an electron or proton to the oxidized cofactor. As the mass spectrometry experiments demonstrate both homoalanine and valine are formed, rather than the dehydrogenated counterparts, it is likely that a second equivalent of H• is donated to V72 from a nearby aliphatic residue, such as I76, again propagating the oxidizing equivalent to solvent and regenerating the closed-shell residues. No dehydrogenated aliphatic residues were observed in the mass spectrometry data, suggesting that solvent or the HEPES buffer may serve as the ultimate sacrificial reductant.82
This hypothesis is consistent with the observations made in this study, though alternative mechanisms or multiple pathways operating in parallel cannot be excluded. To the best of our knowledge, this reactivity represents the first example of light-driven cofactor rearrangement in a member of the ferritin-like superfamily. R2lox is relatively unique among this class of proteins in that the bridging, oxygen-derived ligand in the resting state of the protein is protonated; most well-characterized members of this family feature deprotonated (µ-O) ligands.47,83–86 This proton may be critical for the photochemistry observed, localizing the LMCT excited states onto the carboxylate ligand rather than the bridging oxo moiety.87 This process resembles that seen in iron-bound siderophores, in which metal-carboxylate excitation drives the decarboxylation process.25–27 The exceptionally high quantum yields seen for this process are also reminiscent of those previously noted for photodriven decarboxylation of FeIII-EDTA and FeIII-NTA compounds.22,88–90 A unique aspect of this reaction is that a single protein species, as characterized crystallographically and through mass spectrometry, is formed after excitation, suggesting controlled reactivity that is localized to only one of the carboxylate ligands.30,91–93 Moreover, the R2lox protein features a deep, hydrophobic ligand-binding channel that can be harnessed for substrate binding and manipulation. Photoinduced iron-mediated decarboxylation reactions have also been used to generate new carbon-carbon bonds through radical coupling processes, an underexplored area of research for enzymatic catalysis. These initial studies on R2lox may provide a foundation for engineering metalloproteins that perform controlled light-activated decarboxylation reactions, which may ultimately lead to formation of unique chemical species.
Conclusions
This work demonstrates that R2lox, a member of the ferritin-like superfamily capable of binding either an Fe/Fe or a Mn/Fe cofactor, undergoes a photoinduced transition to generate a new state of the protein. The optical, resonance Raman, and EPR spectra of this distinct species suggest the rearranged active site features a MnIII/FeIII or FeIII/FeIII cofactor with a bound tyrosinate ligand. Photochemical investigations revealed that the photoconversion is initiated by UV and violet light with remarkably high efficiency for an Fe-mediated process. Structural characterization coupled with mass spectrometric analyses indicate that light-driven decarboxylation of a terminal carboxylate ligand is coupled to cleavage of the unique tyrosine-valine crosslink, resulting in coordination of Y162 to the Fe center. A mechanism consistent with these data has been proposed. Further studies are underway to probe the structure, mechanism, and potential utility of this photoconversion process.
Supplementary Material
ACKNOWLEDGMENT
We would like to thank Matthew Bennett for construction of the blue-light LED setup used for photoconversion in crystals, as well as Hugo Lebrette, Vivek Srinivas and the staff at beamline X06DA/SLS for assistance with X-ray data collection. P. T. M., E. K. M., Z. R. S., and H. S. S. would like to acknowledge the support of The Ohio State University Department of Chemistry and Biochemistry.
Funding Sources
P. T. M. was funded in part by the Cellular, Molecular, and Biochemical Training Grant (T32 GM-086252). This work was supported in part by the National Science Foundation (CHE-1454289) grant to H.S.S. M.H. acknowledges the Swedish Research Council (2017-04018), the European Research Council (HIGH-GEAR 724394), and the Knut and Alice Wallenberg Foundation (Wallenberg Academy Fellows). J.J.G. acknowledges the support of the Swedish Research Council (2016-03770). Support from the Swedish National Infrastructure for Biological Mass Spectrometry (BioMS), funded by the Swedish Research Council, is gratefully acknowledged by R.M.B.
REFERENCES
- (1).Kühlbrandt W; Wang DN; Fujiyoshi Y Nature 1994, 367, 614–621. [DOI] [PubMed] [Google Scholar]
- (2).Wu D; Hu Q; Yan Z; Chen W; Yan C; Huang X; Zhang J; Yang P; Deng H; Wang J; Deng X, Shi Y Nature 2012, 484, 214–U96. [DOI] [PubMed] [Google Scholar]
- (3).Jenkins GI Curr. Opin. Struct. Biol 2014, 29, 52–57. [DOI] [PubMed] [Google Scholar]
- (4).Dexheimer SL; Wang Q; Peteanu LA; Pollard WT; Mathies RA; Shank CV Chem. Phys. Lett 1992, 188, 61–66 [Google Scholar]
- (5).Luecke H; Schobert B; Richter HT; Cartailler JP; Lanyi JK J. Mol. Biol 1999, 291, 899–911. [DOI] [PubMed] [Google Scholar]
- (6).McCamant DW; Kukura P; Mathies RA J. Phys. Chem. B 2005, 109, 10449–10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Jung A; Domratcheva T; Tarutina M; Wu Q; Ko W; Shoeman RL; Gomelsky M; Gardner KH; Schlichting I Proc. Natl. Acad. Sci. U. S. A 2005, 102, 12350–12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Conrad KS; Manahan CC; Crane BR Nat. Chem. Biol 2014, 10, 801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Imamoto Y; Kataoka M Photochem. Photobiol 2007, 83, 40–49. [DOI] [PubMed] [Google Scholar]
- (10).Johansson LC; Arnlund D; Katona G; White TA; Barty A; DePonte DP; Shoeman RL; Wickstrand C; Sharma A; Williams GJ; Aquila A; Bogan MJ; Caleman C; Davidsson J; Doak RB; Frank M; Fromme R; Galli L; Grotjohann I; Hunter MS; Kassemeyer S; Kirian RA; Kupitz C; Liang M; Lomb L; Malmerberg E; Martin AV; Messerschmidt M; Nass K; Redecke L; Seibert MM; Sjöhamn J; Steinbrener J; Stellato F; Wang D; Wahlgren WY; Weierstall U; Westenhoff S; Zatsepin NA; Boutet S; Spence JCH; Schlichting I; Chapman HN; Fromme P; Neutze R Nat. Commun 2013, 4, 2911–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hidalgo R; Ash PA; Healy AJ; Vincent KA Angew. Chem. Int. Ed 2015, 54, 7110–7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).McCusker JK; Toftlund H; Rheingold AL; Hendrickson DN J. Am. Chem. Soc 1993, 115, 1797–1804. [Google Scholar]
- (13).Monat JE; McCusker JK J. Am. Chem. Soc 2000, 122, 4092–4097. [Google Scholar]
- (14).Cannizzo A; Milne CJ; Consani C; Gawelda W; Bressler C; van Mourik F; Chergui M Coord. Chem. Rev 2010, 254, 2677–2686. [Google Scholar]
- (15).Auböck G; Chergui M Nat. Chem 2015, 7, 629–633. [DOI] [PubMed] [Google Scholar]
- (16).Büldt LA; Guo X; Vogel R; Prescimone A; Wenger OS J. Am. Chem. Soc 2017, 139, 985–992. [DOI] [PubMed] [Google Scholar]
- (17).O’Regan B; Grätzel M Nature 1991, 353, 737–740. [Google Scholar]
- (18).McDaniel ND; Coughlin FJ; Tinker LL; Bernhard SJ Am. Chem. Soc 2008, 130, 210–217. [DOI] [PubMed] [Google Scholar]
- (19).Roger I; Symes MD J Mater Chem A 2016, 4, 6724–6741. [Google Scholar]
- (20).Haag WR; Yao CC D. Environ. Sci. Technol 1992, 26, 1005–1013. [Google Scholar]
- (21).Ruppert G; Bauer R; Heisler G; Novalic S Chemosphere 1993, 27, 1339–1347. [Google Scholar]
- (22).Feng W; Nansheng D Chemosphere 2000, 41, 1137–1147. [DOI] [PubMed] [Google Scholar]
- (23).Warren JJ; Tronic TA; Mayer JM Chem. Rev 2010, 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Grabo JE; Chrisman MA; Webb LM; Baldwin MJ In-org. Chem 2014, 53, 5781–5787. [DOI] [PubMed] [Google Scholar]
- (25).Butler A; Theisen RM Coord. Chem. Rev 2010, 254, 288–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Barbeau K; Rue EL; Bruland KW; Butler A Nature 2001, 413, 409–413. [DOI] [PubMed] [Google Scholar]
- (27).Küpper FC; Carrano CJ; Kuhn J-U; Butler A Inorg. Chem 2006, 45, 6028–6033. [DOI] [PubMed] [Google Scholar]
- (28).Barbeau K; Zhang G; Live DH; Butler AJ Am. Chem. Soc 2002, 124, 378–379. [DOI] [PubMed] [Google Scholar]
- (29).Jack RS; Ayoko GA; Adebajo MO; Frost RL Environ. Sci. Pollut. Res 2015, 22, 7439–7449. [DOI] [PubMed] [Google Scholar]
- (30).Kavitha V; Palanivelu K Chemosphere 2004, 55, 1235–1243. [DOI] [PubMed] [Google Scholar]
- (31).Lewis DFV; Hlavica P Biochim. Biophys. Acta BBA Bio-energ 2000, 1460, 353–374. [DOI] [PubMed] [Google Scholar]
- (32).Guengerich FP; Munro AW J. Biol. Chem 2013, 288, 17065–17073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).De Montellano PRO Cytochrome P450: Structure, Mechanism, and Biochemistry; Springer Science & Business Media, New York, NY; 2005. [Google Scholar]
- (34).Andersson CS; Högbom M Proc. Natl. Acad. Sci 2009, 106, 5633–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Griese JJ; Roos K; Cox N; Shafaat HS; Branca RMM; Lehtiö J; Gräslund A; Lubitz W; Siegbahn PEM; Högbom M Proc. Natl. Acad. Sci 2013, 110, 17189–17194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Shafaat HS; Griese JJ; Pantazis DA; Roos K; Andersson CS; Popović-Bijelić A; Gräslund A; Siegbahn PEM; Neese F; Lubitz W; Högbom M; Cox NJ Am. Chem. Soc 2014, 136, 13399–13409. [DOI] [PubMed] [Google Scholar]
- (37).Griese JJ; Kositzki R; Schrapers P; Branca RMM; Nordström A; Lehtiö J; Haumann M; Högbom MJ Biol. Chem 2015, 290, 25254–25272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Miller EK; Trivelas NE; Maugeri PT; Blaesi EJ; Shafaat HS Biochemistry 2017, 56, 3369–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kutin Y; Srinivas V; Fritz M; Kositzki R; Shafaat HS; Birrell J; Bill E; Haumann M; Lubitz W; Högbom M; Griese JJ; Cox NJ Inorg. Biochem 2016, 162, 164–177. [DOI] [PubMed] [Google Scholar]
- (40).Schmidt F; Donahoe S; Hagens K; Mattow J; Schaible UE; Kaufmann SHE; Aebersold R; Jungblut PR Mol. Cell. Proteomics 2004, 3, 24–42. [DOI] [PubMed] [Google Scholar]
- (41).Hogbom MJ Biol. Inorg. Chem 2010, 15, 339–349. [DOI] [PubMed] [Google Scholar]
- (42).Lundin D; Poole AM; Sjoberg B-M; Hogbom MJ Biol. Chem 2012, 287, 20565–20575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Cooley RB; Rhoads TW; Arp DJ; Karplus PA Science 2011, 332, 929–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Cooley RB; Arp DJ; Karplus PA J. Mol. Biol 2011, 413, 177–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Solomon EI; Brunold TC; Davis MI; Kemsley JN; Lee S-K; Lehnert N; Neese F; Skulan AJ; Yang Y-S; Zhou J Chem. Rev 2000, 100, 235–350. [DOI] [PubMed] [Google Scholar]
- (46).Kositzki R; Mebs S; Marx J; Griese JJ; Schuth N; Högbom M; Schünemann V; Haumann M Inorg. Chem 2016, 55, 9869–9885. [DOI] [PubMed] [Google Scholar]
- (47).Ray K; Pfaff FF; Wang B; Nam WJ Am. Chem. Soc 2014, 136, 13942–13958. [DOI] [PubMed] [Google Scholar]
- (48).Averill BA; Davis JC; Burman S; Zirino T; Sanders-Loehr J; Loehr TM; Sage JT; Debrunner PG J. Am. Chem. Soc 1987, 109, 3760–3767. [Google Scholar]
- (49).Baldwin J; Voegtli WC; Khidekel N; Moënne-Loccoz P; Krebs C; Pereira AS; Ley BA; Huynh BH; Loehr TM; Riggs-Gelasco PJ; Rosenzweig AC; Bollinger JM Jr. J. Am. Chem. Soc 2001, 123, 7017–7030. [DOI] [PubMed] [Google Scholar]
- (50).Moss ML; Mellon MG Ind. Eng. Chem. Anal. Ed 1942, 14, 862–865. [Google Scholar]
- (51).Braterman PS; Song JI; Peacock RD Inorg. Chem 1992, 31, 555–559. [Google Scholar]
- (52).Slater JW; Marguet SC; Cirino SL; Maugeri PT; Shafaat HS Inorg. Chem 2017, 56, 3926–3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Murov SL; Carmichael I; Hug GL Handbook of Photo-chemistry, 2nd ed.; Marcel Dekker, Inc.: New York, NY, 1993. [Google Scholar]
- (54).Ma X; Tian H Photochemistry and Photophysics. Concepts, Research, Applications; Wiley-VCH Verlag: Weinheim, Germany, 2014. [Google Scholar]
- (55).Kabsch W Acta Crystallogr. D Biol. Crystallogr 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Afonine PV; Grosse-Kunstleve RW; Echols N; Headd JJ; Moriarty NW; Mustyakimov M; Terwilliger TC; Urzhumtsev A; Zwart PH; Adams PD Acta Crystallogr. D Biol. Crystallogr 2012, 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH Acta Crystallogr. D Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Emsley P; Lohkamp B; Scott WG; Cowtan K Acta Crystallogr. D Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Chen VB; Arendall WB III; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC Acta Crystallogr. Sect. D 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Fichtner C; van Gastel M; Lubitz W Phys. Chem. Chem. Phys 2003, 5, 5507–5513. [Google Scholar]
- (61).Wang X; Valverde-Aguilar G; Weaver MN; Nelsen SF; Zink JI J. Phys. Chem. A 2007, 111, 5441–5447. [DOI] [PubMed] [Google Scholar]
- (62).Jiang W; Hoffart LM; Krebs C; Bollinger JM Biochemistry 2007, 46, 8709–8716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Gaber BP; Sheridan JP; Bazer FW; Roberts RM J. Biol. Chem 1979, 254, 8340–8342. [PubMed] [Google Scholar]
- (64).Schenk G; Mitić N; Gahan LR; Ollis DL; McGeary RP; Guddat LW Acc. Chem. Res 2012, 45, 1593–1603. [DOI] [PubMed] [Google Scholar]
- (65).Schenk G; Boutchard CL; Carrington LE; Noble CJ; Moubaraki B; Murray KS; Jersey J. de; Hanson GR; Hamilton S J. Biol. Chem 2001, 276, 19084–19088. [DOI] [PubMed] [Google Scholar]
- (66).Schenk G; Ge Y; Carrington LE; Wynne CJ; Searle IR; Carroll BJ; Hamilton S; de Jersey J Arch. Biochem.Biophys 1999, 370, 183–189. [DOI] [PubMed] [Google Scholar]
- (67).Antanaitis BC; Strekas T; Aisen PJ Biol. Chem 1982, 257, 3766–3770. [PubMed] [Google Scholar]
- (68).Beck JL; McConachie LA; Summors AC; Arnold WN; Dejersey J; Zerner B Biochim. Biophys. Acta 1986, 869, 61–68. [Google Scholar]
- (69).Neves A; Erthal SMD; Vencato I; Ceccato AS; Mascarenhas YP; Nascimento OR; Hörner M; Batista AA Inorg. Chem 1992, 31, 4749–4755. [Google Scholar]
- (70).Setyawati IA; Rettig SJ; Orvig C Can. J. Chem 1999, 77, 2033–2038. [Google Scholar]
- (71).Que L Coord. Chem. Rev 1983, 50, 73–108. [Google Scholar]
- (72).Creswell CJ; Allred AL J. Am. Chem. Soc 1962, 84, 3966–3967. [Google Scholar]
- (73).Rao CNR J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1975, 71, 980–980. [Google Scholar]
- (74).Rapatskiy L; Ames WM; Pérez-Navarro M; Savitsky A; Griese JJ; Weyhermüller T; Shafaat HS; Högbom M; Neese F; Pantazis DA; Cox NJ Phys. Chem. B 2015, 119, 13904–13921. [DOI] [PubMed] [Google Scholar]
- (75).Tshuva EY; Lippard SJ Chem. Rev 2004, 104, 987–1012. [DOI] [PubMed] [Google Scholar]
- (76).Lambert E; Chabut B; Chardon-Noblat S; Deronzier A; Chottard G; Bousseksou A; Tuchagues J-P; Laugier J; Bardet M; Latour J-MJ Am. Chem. Soc 1997, 119, 9424–9437. [Google Scholar]
- (77).Than R; Feldmann A; Krebs B Coord. Chem. Rev 1999, 182, 211–241. [Google Scholar]
- (78).Zheng H; Zang Y; Dong Y; Young VG; Que LJ Am. Chem. Soc 1999, 121, 2226–2235. [Google Scholar]
- (79).Winkler JR; Gray HB Philos. Trans. R. Soc. Math. Phys. Eng. Sci 2015, 373, 20140178–20140178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Winkler JR; Gray HB Q. Rev. Biophys 2015, 48, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Warren JJ; Herrera N; Hill MG; Winkler JR; Gray HB J. Am. Chem. Soc 2013, 135, 11151–11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Brown KA; Harris DF; Wilker MB; Rasmussen A; Khadka N; Hamby H; Keable S; Dukovic G; Peters JW; Seefeldt LC; King PW Science 2016, 352, 448–450. [DOI] [PubMed] [Google Scholar]
- (83).Fox BG; Sureus KK; Munck E; Lipscomb JD J. Biol. Chem 1998, 263, 10553–10556. [PubMed] [Google Scholar]
- (84).Baik M-H; Newcomb M; Friesner RA; Lippard SJ Chem. Rev 2003, 103, 2385–2420. [DOI] [PubMed] [Google Scholar]
- (85).Fox BG; Shanklin J; Ai J; Loehr TM; Sanders-Loehr J Biochemistry 1994, 33, 12776–12786. [DOI] [PubMed] [Google Scholar]
- (86).Solomon EI; Light KM; Liu LV; Srnec M; Wong SD Acc. Chem. Res 2013, 46, 2725–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Ensing B; Buda F; Baerends EJ J. Phys. Chem. A 2003, 107, 5722–5731. [Google Scholar]
- (88).Carey JH; Langford CH Can. J. Chem 1973, 51, 3665–3670. [Google Scholar]
- (89).Kocot P; Szaciłowski K; Stasicka ZJ Photochem. Photobiol. Chem 2007, 188, 128–134. [Google Scholar]
- (90).Budac D; Wan PJ Photochem. Photobiol. Chem 1992, 67, 135–166. [Google Scholar]
- (91).Stadtman ER; Berlett BS J. Biol. Chem 1991, 266, 17201–17211. [PubMed] [Google Scholar]
- (92).Berlett BS; Stadtman ER J. Biol. Chem 1997, 272, 20313–20316. [DOI] [PubMed] [Google Scholar]
- (93).Stadtman ER; Levine RL Amino Acids 2003, 25, 207–218. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.