

Abstract
Langerhans cell histiocytosis (LCH) is a rare systemic disorder characterized by the accumulation of CD1a+/Langerin+ LCH cells and wide‐ranging organ involvement. Langerhans cell histiocytosis was formerly referred to as histiocytosis X, until it was renamed in 1987. Langerhans cell histiocytosis β was named for its morphological similarity to skin Langerhans cells. Studies have shown that LCH cells originate from myeloid dendritic cells rather than skin Langerhans cells. There has been significant debate regarding whether LCH should be defined as an immune disorder or a neoplasm. A breakthrough in understanding the pathogenesis of LCH occurred in 2010 when a gain‐of‐function mutation in BRAF (V600E) was identified in more than half of LCH patient samples. Studies have since reported that 100% of LCH cases show ERK phosphorylation, indicating that LCH is likely to be a clonally expanding myeloid neoplasm. Langerhans cell histiocytosis is now defined as an inflammatory myeloid neoplasm in the revised 2016 Histiocyte Society classification. Randomized trials and novel approaches have led to improved outcomes for pediatric patients, but no well‐defined treatments for adult patients have been developed to date. Although LCH is not fatal in all cases, delayed diagnosis or treatment can result in serious impairment of organ function and decreased quality of life. This study summarizes recent advances in the pathophysiology and treatment of adult LCH, to raise awareness of this “orphan disease”.
Keywords: adult, histiocytosis, Langerhans cell, mitogen‐activated protein kinase, proto‐oncogene protein BRAF
1. OVERVIEW OF LANGERHANS CELL HISTIOCYTOSIS
The Langerhans cell was first described by Paul Langerhans Jr. in 1868.1 The first definitive case of Langerhans cell histiocytosis (LCH) was reported in 1893 by Alfred Hand.2 The patient was a 3‐year‐old boy with polyuria, exophthalmos, and hepatosplenomegaly; similar cases were reported by Henry Christian and Arthur Schuller.3, 4 The disease was named Hand‐Schuller‐Christian disease. Based on similar histopathological patterns, Hand‐Schuller‐Christian disease, Letterer‐Siwe disease, and eosinophilic granuloma were unified as histiocytosis X in 1953 and renamed LCH in 1985.5 Histopathologically, LCH is generally defined by CD1a+/Langerin+ Langerhans‐like cells. The specific origin of LCH cells has yet to be identified. The incidence of LCH is reported to be 3‐5/million; the majority of patients are children younger than 3 years and the incidence in adults is approximately 1‐2/million.6 Incidence appears to be higher in Caucasians in northern European countries, and lower in Asia and Africa.7
The clinical manifestation of LCH varies from a single‐organ disease that could spontaneously go into remission, to a systemic and aggressive disease that can lead to death. Any organ can be involved, individually or in combination, but bone and skin have a higher frequency of involvement. Diabetes insipidus (DI) is the most common initial sign of central nervous system (CNS) involvement in LCH. Isolated pulmonary LCH (PLCH), which is the most common manifestation in adult patients, is considered a specific type of LCH. Although PLCH cells have similar histopathologic characteristics, the majority of cell proliferation is polyclonal.8 Pulmonary LCH is strongly related to smoking; more than 90% of patients are smokers and quitting smoking could lead to remission without treatment.9, 10
Because LCH varies in its clinical presentation, particularly among adult patients, nonhematological medical professionals are likely to be involved in clinical settings, which could result in improper treatment procedures, including surgical excision and irradiation. It should be noted that LCH is categorized as a myeloid neoplasm, and requires accurate diagnosis and appropriate treatment as necessary.7, 11
2. ALTERATIONS OF THE MAPK PATHWAY IN LCH
The identification of an oncogenic BRAF ‐V600E mutation in more than half of all LCH cases represented a major advance in our understanding of the pathogenesis of LCH.12 The BRAF protein is a member of the serine/threonine kinase RAF family, and is a key component of the MAPK (RAS‐RAF‐MEK‐ERK) signaling pathway that leads to the activation of transcription factors essential for cell growth and proliferation. The V600E (c.1799T>A transversion in exon 15) is the most common mutation in BRAF, and is one of the major drivers of human malignancies that lead to downstream constitutive activation of MEK and ERK. The frequency of BRAF‐V600E in LCH ranges from 25% to 65%.12, 13, 14, 15, 16 Studies have reported that BRAF‐V600E is more commonly observed in multisystem LCH patients than in patients with isolated disease.17 It is associated with an increased risk of relapse.13
BRAF‐V600E mutations have been detected in circulating cell‐free DNA extracted from peripheral blood plasma in LCH patients using allele‐specific real‐time PCR or digital droplet PCR.18, 19 This noninvasive diagnostic procedure, a so‐called “liquid biopsy”, is a powerful tool for early detection of various cancers. It can be used to analyze cell‐free DNA, circulating tumor cells, and tumor‐derived extracellular vesicles. Langerhans cell histiocytosis can involve organs and tissues not readily accessible for biopsy, thereby delaying diagnosis. Liquid biopsy can mitigate this difficulty, and is also expected to be a potential biomarker in high‐risk LCH.
The introduction of next‐generation sequencing analyses, such as whole exome/genome sequencing and targeted sequencing, has revealed several BRAF mutations in addition to V600E, including V600D, 600DLAT in‐flame insertion, germ line T599A mutation, deletions in the β3‐αC loop of the kinase domain (485_490LNVTAP>F, NVTAP486del, and 486_491NVTAPT>K), and a splicing mutation at the end of exon 12 (R506_K507insLLR) (Figure 1).20, 21, 22 The kinase activity of these mutations has not been fully evaluated, but they might function similarly to BRAF‐V600E, resulting in constitutive activation of BRAF. Additionally, BRAF‐G466R, a missense mutation in the kinase domain of BRAF, has been described in LCH, although its function is also unknown.23 In non‐LCH histiocytosis, a BRAF‐F595L mutation was identified in histiocytic sarcoma,24 and an in‐flame deletion‐insertion (BRAF‐T599_V600delinsRE) was identified in Erdheim‐Chester disease (ECD).25 Erdheim‐Chester disease and LCH have different morphological and immunohistochemical characteristics; ECD cells have a foamy cytoplasm and lack CD1a staining. The relationship between ECD and LCH is not fully understood, and ECD and LCH occasionally overlap.26 RNA sequencing has uncovered recurrent BRAF fusions, such as PACSIN2‐BRAF and FAM73A‐BRAF.27, 28 These two mutations are both in‐flame fusions involving a wide range of exons, and lead to constitutive activation of BRAF.
Figure 1.
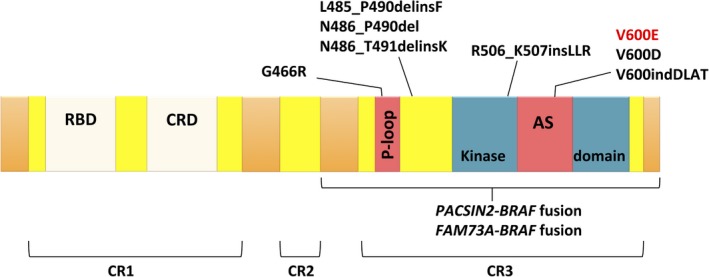
Schematic representation of BRAF structure and the location of reported mutations in Langerhans cell histiocytosis (LCH). AS, activation segment; CR, conserved region; CRD, cystine‐rich domain; RBD, RAS binding domain
The second most commonly mutated gene in LCH is MAP2K1, which is also a member of the MAPK pathway. The dual‐specificity kinase MAP2K1, also known as MEK1, is located downstream of BRAF and contributes to the activation of ERK1 and ERK2. MAP2K1 mutation is reported to occur in ~50% of LCH patients with wild‐type BRAF.29, 30 Studies have indicated that BRAF and MAP2K1 in LCH are mutually exclusive.30, 31
Nelson et al32 identified an activating ARAF mutation in the kinase domain (F351L) and an in‐flame deletion (Q347_A348del). Chakraborty et al30 identified an ARAF mutation (T70M) that co‐occurred with BRAF‐V600E in a patient with ECD/LCH overlap. Non‐LCH histiocytosis shows higher frequencies of ARAF mutations.33 Two somatic frameshift mutations in MAP3K1 (T799 fs and L1481 fs) have been reported.31 Mutations in tyrosine kinase receptor ERBB3 (P921Q) have also been reported in ECD/LCH patients.30 Approximately half of ECD patients harbor BRAF‐V600E as well as LCH, and ~30% carry MAP2K1 mutations. Whereas 10%‐20% of ECD are positive for NRAS or KRAS mutations, only one activating KRAS mutation in non‐PLCH has been reported to date.23 The effects of these recently identified mutations on ERK activation are still unclear. Mutations in MAPK pathway genes have been confirmed in nearly 80% of LCH patients. Additional MAPK mutations could be discovered because the ERK pathway is activated in 100% of LCH patients (Figure 2).12, 30
Figure 2.
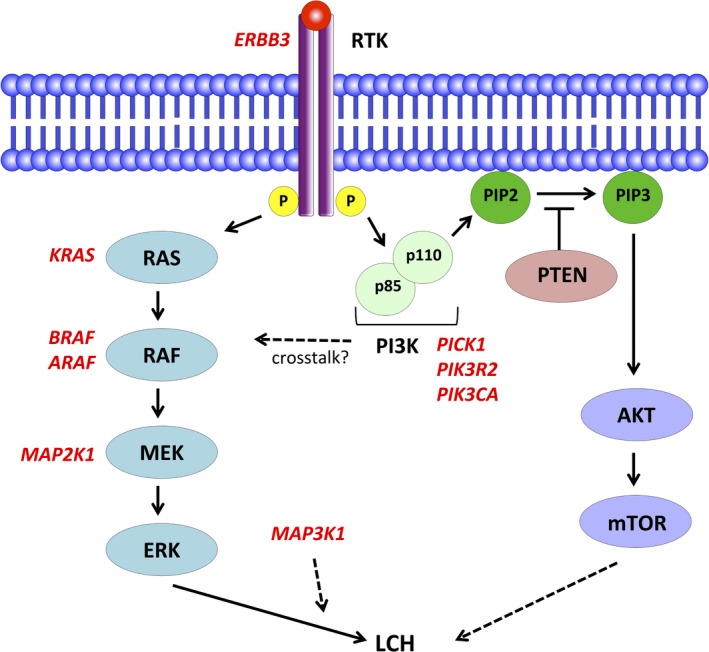
Diagram of the MAPK and PI3K pathways. The reported mutations in Langerhans cell histiocytosis (LCH) are indicated in red. The dotted arrow represents an effect that has not been fully evaluated. PIP1, phosphatidylinositol 3‐phosphate; PIP3, phosphatidylinositol‐3,4,5‐triphosphate; PTEN, phosphatase and tensin homolog deleted on chromosome 10; RTK receptor tyrosine kinase
3. OTHER POSSIBLE DRIVERS OF LCH
In addition to those noted above, a number of other mutations have been reported in LCH, although their role in its pathogenesis is unclear. One of the few possible drivers outside of MAPK is the PI3K (PI3K‐AKT‐mTOR) pathway. The PI3K pathway is an intracellular signaling pathway that regulates the cell cycle; activation of this pathway is a key driver of carcinogenesis. To date, three PI3K pathway alterations have been reported: PICK1, PIK3R2, and PIK3CA (Figure 2).33, 34 Of these, PIK3CA (E542K) is the only known activating somatic mutation, leading to constitutive activation of the PI3K pathway. Activating PIK3CA mutation is reported in 10%‐20% of ECD cases, whereas only a single case was reported in LCH.
4. THE CELL OF ORIGIN
As indicated by its name, LCH was believed to arise from skin Langerhans cells due to similarities in expression markers (CD1a+/CD207+) and histopathological findings with characteristic Birbeck granules. Results from gene expression analyses indicated that LCH cells were more similar to dendritic cell precursors derived from bone marrow, and did not originate from skin Langerhans cells.35 Berres et al13 confirmed BRAF‐V600E mutation in circulating CD11c+/CD14+ fractions, and in bone marrow CD34+ hematopoietic progenitor cells in high‐risk LCH patients. This finding indicated that LCH occurs as the result of a MAPK mutation of a hematopoietic progenitor cell line. Forced expression of BRAF‐V600E in CD11c+ dendritic progenitor cells caused LCH‐like lesions in mice. BRAF‐V600E has been shown to be present in hematopoietic stem cells and myeloid progenitors in the bone marrow of high‐risk LCH patients. Additionally, CD1c+ dendritic cells have been shown to acquire high CD1a and CD207 expression with granulocyte‐macrophage colony‐stimulating factor and transforming growth factor‐beta alone in vitro.36 In contrast, classical CD14+ monocytes required additional notch ligation. A recent study revealed the self‐renewing capacity of CD34+ cells, which drives the development of histiocytosis in xenograft transplantation assays in vivo.37 Constitutive activation of the MAPK pathway due to BRAF mutation plays a critical role in LCH. A previous study identified BRAF‐V600E in CD34+ cells in patients with hairy cell leukemia.38 Another study showed that BRAF‐V600E expression in murine hematopoietic stem/progenitor cells can cause hairy cell leukemia‐like disease.39 It remains unclear why the same mutation occurring in hematopoietic cells leads to distinctly different conditions, suggesting that mechanisms other than BRAF might be involved.
5. TREATMENT OPTIONS FOR ADULT LCH
There are no standard therapies for adult LCH, and no prospective trials have been undertaken on this population. Treatment of LCH varies due to its heterogeneous presentation; treatment recommendations are based on organ involvement and the extent of the disease (Figure 3). Langerhans cell histiocytosis is categorized as single‐system LCH (SS‐LCH) with multifocal or unifocal involvement, or as multisystem LCH (MS‐LCH) with multiple organ involvement with or without risk organ involvement. Organs at risk include the hematopoietic system (bone marrow), spleen, and liver.
Figure 3.
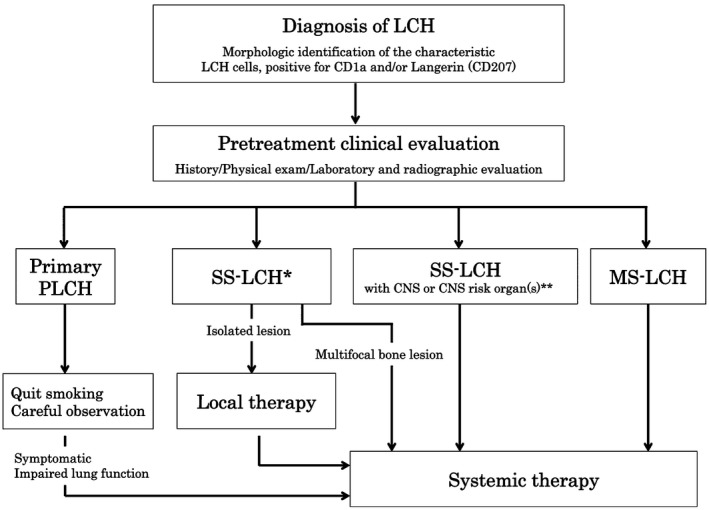
Treatment algorithm for adult Langerhans cell histiocytosis (LCH). Diagnosis of LCH is based on histological and immunophenotypic examination. The primary indicators are characteristic LCH cells and positivity for CD1a and/or Langerin (CD207) cells. Confirmation of cytoplasmic Birbeck granules by electron microscopy is no longer necessary. Complete patient history, including smoking history, must be determined, and specific physical examinations should be carried out, including neurological evaluation of central nervous system (CNS) and peripheral nerves. *In certain situations, such as involvement in vertebra or tissues near sensory organs, systemic therapy is recommended. **Involvement in craniofacial bones, eyes, ears, and oral cavity. MS, multisystem; PLCH, pulmonary LCH; SS, single system
6. TREATMENTS FOR PRIMARY PLCH
As mentioned, primary PLCH in adults has a strong relation with smoking and nearly all the patients are smokers. Therefore, smoking cessation should be the top priority in the treatment of primary adult PLCH. In non‐smokers or patients with progressive disease despite smoking cessation, systemic therapy of glucocorticoids or cladribine (2‐CdA) could be an option.10, 40, 41 It is important that radiation therapy is not useful in isolated PLCH. In patients with very aggressive PLCH with irreversible lung damage or severe pulmonary hypertension, lung transplantation might be considered.42
7. TREATMENTS FOR SS‐LCH
Local therapies are recommended for SS‐LCH with isolated skin or bone involvement. Surgical excision of the lesion should be undertaken in very limited cases of SS‐LCH with a solitary skin lesion. In skin‐only cases, weekly oral methotrexate (MTX) with or without 6‐mercaptopurine (or its prodrug azathioprine) is recommended.43, 44 Light therapies in skin‐only patients have been reported to be effective.45, 46 In contrast to pediatric patients, radiotherapy could be an option for SS‐LCH with bone lesions, with acceptable side‐effects.47, 48, 49 Recent reports have indicated that hydroxyurea with or without MTX or bisphosphonates might be effective in SS‐LCH with multifocal bone lesions.50, 51, 52, 53 However, in patients with multifocal bone involvement, systemic therapy equivalent to MS‐LCH is often undertaken as initial treatment.
8. TREATMENTS FOR MS‐LCH
Systemic therapy is strongly recommended for MS‐LCH or SS‐LCH with multifocal bone lesions. No standard treatments have been identified but a combination of vinblastine and prednisolone therapy could serve as the standard option, as in pediatric LCH (Table 1). Adult patients who undergo vinblastine + prednisolone treatment tend to show higher toxicity than children, resulting in a poor overall response. 54 , 55 , 56 , 57 Cytarabine or 2‐CdA might be preferred as first‐line treatments. 55 , 58 For patients with CNS lesions, or primary or relapsed/refractory cases, 2‐CdA and cytarabine are preferred because of their blood‐brain barrier penetration. Retrospective analyses of short‐course chemotherapy with MTX, doxorubicin, cyclophosphamide, vincristine, prednisone, and bleomycin in aggressive adult LCH have shown its efficacy. This intensive treatment should be reserved for very severe cases. 59 Allogeneic hematopoietic stem cell transplantation has been successful in some aggressive cases. 61 , 62
Table 1.
Reported case series of systemic therapies for adult Langerhans cell histiocytosis (LCH)
| Reference | No. of pts | Disease type | Median age, y (range) | Treatment | Overall response | Median follow‐up (y) |
|---|---|---|---|---|---|---|
| Saven and Burian 58 | 12 | 3 SS, 4 MS | 44 (19‐72) | 2‐CdA | 75% | 3.6 |
| McClain et al 57 | 7 | 7 MS | Not given | VBL, PSL followed by VBL, PSL, 6‐MP | 43% | 0.5 |
| Cantu et al55 | 58 | 58 SS with bone lesions | 32 (18‐72) |
VBL, PSL for 19 pts (1st 15, 2nd 4) 2‐CdA for 22 pts (1st 9, 2nd 9, 3rd 4) HDAC for 24 pts (1st 12, 2nd 5, 3rd 7) |
VBL, PSL 16%a
2‐CdA 31%a AraC 79%a |
8.5 |
| Morimoto et al56 | 14 | 4 SS, 10 MS | 43 (20‐70) | VBL, PSL, MTX, 6‐MP | 72% | 2.8 |
| Derenzini et al 59 | 11 | 6 SS, 5 MS | 27 (18‐62) | CPA, ADR, MTX, VCR, BLEO, PSL (MACOP‐B) | 100% | 6.7 |
| Duan et al 60 | 45 | 4 SS, 41 MS | 31 (18‐69) |
CPA, VDS, ETP, PSL (CEVP) for 31 pts VDS, PSL (VP) for 13 pts |
CEVP 70% VP 64% |
6.2 |
| Tazi et al54 | 35 | 7 SS, 28 MS | 33 (28‐42) | VBL, PSL | 72% | 7.6 |
Patients who relapsed within 1 year were excluded.
2‐CdA, cladribine; 6‐MP, 6‐mercaptopurine; ADR, adriamycin; AraC, cytarabine; BLEO, bleomycin; CEVP, cyclophosphamide, epirubicin, etoposide, and cis‐platinum; CPA, cyclophosphamide; ETP, etoposide; HDAC, high‐dose cytarabine; MACOP‐B, methotrexate, doxorubicin, cyclophosphamide, vincristine, prednisone, and bleomycin; MS, multisystem LCH; MTX, methotrexate; PSL, prednisolone; Pts, patients; SS, single system LCH; VBL, vinblastine; VCR, vincristine; VDS, vindesine; VP, vindesine and prednisone.
9. TREATMENTS FOR HYPOTHALAMIC‐PITUITARY MASS LESION
As mentioned, LCH can involve CNS and the most common manifestation is DI due to the hypothalamic‐pituitary mass lesion. Mass lesions in hypothalamus and/or pituitary glands are often treated with vinblastine/prednisone or 2‐CdA. 63 , 64 Regardless of the treatment, most reported cases have shown that symptoms of DI or hormone dysfunction could not be reversed and requires constitutive hormone replacement therapy.
10. TARGETED THERAPIES
Imatinib, a tyrosine kinase inhibitor targeting the receptors expressed in LCH, has been reported to be effective in refractory MS‐LCH cases. 65 , 66 Unfavorable outcomes have also been reported and few experts now recommend imatinib. 67 Vemurafenib, a BRAF inhibitor, has shown efficacy in several cases with BRAF‐V600E mutation. Vemurafenib is the first selective BRAF inhibitor approved by the US FDA for malignant melanomas, and was approved for ECD with BRAF‐V600E in 2017. Despite the efficacy of vemurafenib, severe adverse events, including the development of squamous cell carcinoma, have been reported and the majority of patients experienced early relapse after discontinuation of the therapy. 68 Diamond et al 69 recently reported 2‐year efficacy and safety data for vemurafenib in patients with ECD and LCH enrolled in the VE‐BASKET study, an open‐label, non‐randomized, histology‐independent evaluation of patients with nonmelanoma cancers harboring the BRAF‐V600E mutation. A total of 22 ECD and 4 LCH patients were treated with vemurafenib. The 2‐year progression‐free survival rate was 86% and the overall survival rate was 96%. No patient showed disease progression at the point of best response, and LCH patients showed higher efficacy than ECD patients. However, all patients had at least one adverse event that led to dose reduction and/or discontinuation of the therapy, including an instance of KRAS‐mutant papillary thyroid cancer that was considered treatment‐related. The optimal duration and dosage of vemurafenib require further investigation. A retrospective case series of refractory/relapsed ECD and LCH using dabrafenib, another BRAF inhibitor, indicated that it was efficacious and safe. 70 A total of 11 adult ECD patients (including 4 overlapping with LCH) carrying BRAF‐V600E were treated with a single dose of dabrafenib: 6 patients were pretreated with vemurafenib and treatment was discontinued due to toxicity; 1 experienced grade 3 toxicity (fever); and all achieved an at‐least partial metabolic response. Papapanagiotou et al 71 reported successful treatment of MS‐LCH with MEK1 (E102_I103delEI) mutation using trametinib, a MEK1/2 inhibitor. However, a cutaneous lesion reappeared after cessation of the treatment, which required retreatment.
11. CONCLUSION AND PERSPECTIVES
Since the discovery of the BRAF‐V600E mutation in LCH, advances in next‐generation sequencing technology have revealed several driver mutations and resolved the molecular pathophysiology of LCH. These results directly informed the application of targeted therapies, such as BRAF or MEK inhibitors. It is assumed that additional drivers in the MAPK pathway remain unidentified, and the role of structural alterations and gene fusion has yet to be elucidated. Although the expansion of treatment options has gradually improved outcomes, treatment of LCH remains challenging, particularly in adults. Clinical trials of combined and targeted therapies are ongoing, and the results are awaited with interest.
CONFLICT OF INTEREST
None.
Kobayashi M, Tojo A. Langerhans cell histiocytosis in adults: Advances in pathophysiology and treatment. Cancer Sci. 2018;109:3707–3713. 10.1111/cas.13817
REFERENCES
- 1. Langerhans P. Über die Nerven der menschlichen Haut. Arch Abl B Pathol. 1868;44:325‐337. [Google Scholar]
- 2. Hand AR. Polyuria and tuberculosis. Arch Pediatr. 1893;10:673‐675. [Google Scholar]
- 3. Schuller A. Uber eigenartige Schadeldefekte im Jugendalter. Fortschr Roentgenstr. 1915;23:12‐18. [Google Scholar]
- 4. Christian HA. Defects in membraneous bones, exophthalmos and diabetes insipidus: an unusual syndrome of dyspituitarism. Med Clin N Am. 1919;3:849‐871. [Google Scholar]
- 5. The Writing Group of the Histiocyte Society . Histiocytosis syndromes in children. Writing Group of the Histiocyte Society. Lancet. 1987;1(8526):208‐209. [PubMed] [Google Scholar]
- 6. Carstensen H, Ornvold K. The epidemiology of LCH in children in Denmark. Med Pediatr Oncol. 1993;21:387‐388. [Google Scholar]
- 7. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues In: Weiss LM, Jaffe R, Facchetti F, eds. WHO classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC; 2017:470‐472. [Google Scholar]
- 8. Yousem SA, Colby TV, Chen YY, et al. Pulmonary Langerhans’ cell histiocytosis : molecular analysis of clonality. Am J Surg Pathol. 2001;25:630‐636. [DOI] [PubMed] [Google Scholar]
- 9. Tazi A, de Margerie C, Naccache JM, et al. The natural history of adult pulmonary Langerhans cell histiocytosis: a prospective multicentre study. Orphanet J Rare Dis. 2015;10:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vassallo R, Ryu JH, Schroeder DR, Decker PA, Limper AH. Clinical outcomes of pulmonary Langerhans’‐cell histiocytosis in adults. N Engl J Med. 2002;346:484‐490. [DOI] [PubMed] [Google Scholar]
- 11. Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage‐dendritic cell lineages. Blood. 2016;127(22):2672‐2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Badalian‐Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919‐1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berres ML, Lim KP, Peters T, et al. BRAF‐V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med. 2014;211:669‐683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roden AC, Hu X, Kip S, et al. BRAF V600E expression in Langerhans cell histiocytosis: clinical and immunohistochemical study on 25 pulmonary and 54 extrapulmonary cases. Am J Surg Pathol. 2014;38(4):548‐551. [DOI] [PubMed] [Google Scholar]
- 15. Go H, Jeon YK, Huh J, et al. Frequent detection of BRAF(V600E) mutations in histiocytic and dendritic cell neoplasms. Histopathology. 2014;65(2):261‐272. [DOI] [PubMed] [Google Scholar]
- 16. Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF V600E mutations in Erdheim‐Chester disease but not in other non‐Langerhans cell histiocytoses. Blood. 2012;120(13):2700‐2703. [DOI] [PubMed] [Google Scholar]
- 17. Héritier S, Emile JF, Barkaoui MA, et al. BRAF mutation correlates with high‐risk langerhans cell histiocytosis and increased resistance to first‐line therapy. J Clin Oncol. 2016;34:3023‐3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kobayashi M, Tojo A. The BRAF‐V600E mutation in circulating cell‐free DNA is a promising biomarker of high‐risk adult Langerhans cell histiocytosis. Blood. 2014;124(16):2610‐2611. [DOI] [PubMed] [Google Scholar]
- 19. Héritier S, Hélias‐Rodzewicz Z, Lapillonne H, et al. Circulating cell‐free BRAFV600E as a biomarker in children with Langerhans cell histiocytosis. Br J Haematol. 2017;178(3):457‐467. [DOI] [PubMed] [Google Scholar]
- 20. Rollins BJ. Genomic alterations in langerhans cell histiocytosis. Hematol Oncol Clin North Am. 2015;29(5):839‐851. [DOI] [PubMed] [Google Scholar]
- 21. Satoh T, Smith A, Sarde A, et al. BRAF mutant alleles associated with Langerhans cell histiocytosis, a granulomatous pediatric disease. PLoS One. 2012;7(4):e33891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Héritier S, Hélias‐Rodzewicz Z, Chakraborty R, et al. New somatic BRAF splicing mutation in Langerhans cell histiocytosis. Mol Cancer. 2017;16(1):115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee LH, Gasilina A, Roychoudhury J, et al. Real‐time genomic profiling of histiocytoses identifies early‐kinase domain BRAF alterations while improving treatment outcomes. JCI Insight. 2017;2(3):e89473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kordes M, Röring M, Heining C, et al. Cooperation of BRAF(F595L) and mutant HRAS in histiocytic sarcoma provides new insights into oncogenic BRAF signaling. Leukemia. 2016;30(4):937‐946. [DOI] [PubMed] [Google Scholar]
- 25. Bentel JM, Thomas MA, Rodgers JJ, et al. Erdheim‐Chester disease associated with a novel, complex BRAF p.Thr599_Val600delinsArgGlu mutation. BMJ Case Rep. 2017;2017 10.1136/bcr-2017-219720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hervier B, Haroche J, Arnaud L, et al. Association of both Langerhans cell histiocytosis and Erdheim‐Chester disease linked to the BRAFV600E mutation. Blood. 2014;124(7):1119‐1126. [DOI] [PubMed] [Google Scholar]
- 27. Chakraborty R, Burke TM, Hampton OA, et al. Alternative genetic mechanisms of BRAF activation in Langerhans cell histiocytosis. Blood. 2016;128(21):2533‐2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zarnegar S, Durham BH, Khattar P, et al. Novel activating BRAF fusion identifies a recurrent alternative mechanism for ERK activation in pediatric Langerhans cell histiocytosis. Pediatr Blood Cancer. 2018;65(1). 10.1002/pbc.26699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brown NA, Furtado LV, Betz BL, et al. High prevalence of somatic MAP2K1 mutations in BRAF V600E‐negative Langerhans cell histiocytosis. Blood. 2014;124:1655‐1658. [DOI] [PubMed] [Google Scholar]
- 30. Chakraborty R, Hampton OA, Shen X, et al. Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood. 2014;124(19):3007‐3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nelson DS, van Halteren A, Quispel WT, et al. MAP2K1 and MAP3K1 mutations in Langerhans cell histiocytosis. Genes Chromosom Cancer. 2015;54:361‐368. [DOI] [PubMed] [Google Scholar]
- 32. Nelson DS, Quispel W, Badalian‐Very G, et al. Somatic activating ARAF mutations in Langerhans cell histiocytosis. Blood. 2014;123(20):3152‐3155. [DOI] [PubMed] [Google Scholar]
- 33. Diamond EL, Durham BH, Haroche J, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov. 2016;6(2):154‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Héritier S, Saffroy R, Radosevic‐Robin N, et al. Common cancer‐associated PIK3CA activating mutations rarely occur in Langerhans cell histiocytosis. Blood. 2015;125:2448‐2449. [DOI] [PubMed] [Google Scholar]
- 35. Allen CE, Li L, Peters TL, et al. Cell‐specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared with epidermal Langerhans cells. J Immunol. 2010;184(8):4557‐4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Milne P, Bigley V, Bacon CM, et al. Hematopoietic origin of Langerhans cell histiocytosis and Erdheim‐Chester disease in adults. Blood. 2017;130:167‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Durham BH, Roos‐Weil D, Baillou C, et al. Functional evidence for derivation of systemic histiocytic neoplasms from hematopoietic stem/progenitor cells. Blood. 2017;130:176‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy‐cell leukemia. N Engl J Med. 2011;364:2305‐2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chung SS, Kim E, Park JH, et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci Transl Med. 2014; 6(238): 238ra71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lazor R, Etienne‐Mastroianni B, Khouatra C, Tazi A, Cottin V, Cordier JF. Progressive diffuse pulmonary Langerhans cell histiocytosis improved by cladribine chemotherapy. Thorax. 2009;64(3):274‐275. [DOI] [PubMed] [Google Scholar]
- 41. Grobost V, Khouatra C, Lazor R, Cordier JF, Cottin V. Effectiveness of cladribine therapy in patients with pulmonary Langerhans cell histiocytosis. Orphanet J Rare Dis. 2014;9:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dauriat G, Mal H, Thabut G, et al. Lung transplantation for pulmonary langerhans’ cell histiocytosis: a multicenter analysis. Transplantation. 2006;81(5):746‐750. [DOI] [PubMed] [Google Scholar]
- 43. Hoeger PH, Nanduri VR, Harper JI, Atherton DA, Pritchard J. Long term follow up of topical mustine treatment for cutaneous langerhans cell histiocytosis. Arch Dis Child. 2000;82:483‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Steen AE, Steen KH, Bauer R, Bieber T. Successful treatment of cutaneous Langerhans cell histiocytosis with low‐dose methotrexate. Br J Dermatol. 2001;145:137‐140. [DOI] [PubMed] [Google Scholar]
- 45. Imafuku S, Shibata S, Tashiro A, Furue M. Cutaneous Langerhans cell histiocytosis in an elderly man success treated with narrowband ultraviolet B. Br J Dermatol. 2007;157:1277‐1279. [DOI] [PubMed] [Google Scholar]
- 46. Sakai H, Ibe M, Takahashi H, et al. Satisfactory remission achieved by PUVA therapy in Langerhans cell hisiocytosis in an elderly patient. J Dermatol. 1996;23:42‐46. [DOI] [PubMed] [Google Scholar]
- 47. Atalar B, Miller RC, Dincbas F, et al. Adult langerhans cell histiocytosis of bones : a rare cancer network study. Acta Orthop Belg. 2010;76:663‐668. [PubMed] [Google Scholar]
- 48. Heyd R, Strassmann G, Donnerstag F, Martin T, Zamboglou N. Radiotherapy in Langerhans‐cell histiocytosis. 2 case reports and review of the literature. Rontgenpraxis. 2000;53:51‐61. [PubMed] [Google Scholar]
- 49. Micke O, Seegenschmiedt MH. Consensus guidelines for radiation therapy of benign diseases: a multicenter approach in Germany. Int J Radiat Oncol Biol Phys. 2002;52:496‐513. [DOI] [PubMed] [Google Scholar]
- 50. Zinn DJ, Grimes AB, Lin H, Eckstein O, Allen CE, McClain KL. Hydroxyurea: a new old therapy for Langerhans cell histiocytosis. Blood. 2016;128:2462‐2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Arzoo K, Sadeghi S, Pullarkat V. Pamidronate for bone pain from osteolytic lesions in Langerhans’‐cell histiocytosis. N Engl J Med. 2001;345:225. [DOI] [PubMed] [Google Scholar]
- 52. Farran RP, Zaretski E, Egeler RM. Treatment of Langerhans cell histiocytosis with pamidronate. J Pediatr Hematol Oncol. 2001;23:54. [DOI] [PubMed] [Google Scholar]
- 53. Sivendran S, Harvey H, Lipton A, Drabick J. Treatment of Langerhans cell histiocytosis bone lesions with zoledronic acid: a case series. Int J Hematol. 2011;93:782. [DOI] [PubMed] [Google Scholar]
- 54. Tazi A, Lorillon G, Haroche J, et al. Vinblastine chemotherapy in adult patients with langerhans cell histiocytosis: a multicenter retrospective study. Orphanet J Rare Dis. 2017;12:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cantu MA, Lupo PJ, Bilgi M, Hicks MJ, Allen CE, McClain KL. Optimal therapy for adults with Langerhans cell histiocytosis bone lesions. PLoS One. 2012;7:e43257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Morimoto A, Shimazaki C, Takahashi S, et al. Therapeutic outcome of multifocal Langerhans cell histiocytosis in adults treated with the Special C regimen formulated by the Japan LCH Study Group. Int J Hematol. 2013;97:103‐108. [DOI] [PubMed] [Google Scholar]
- 57. McClain K, Allen C, Ebrahim S. Review of histiocytosis treatment and neurotoxicity in adult patients. Pediatr Blood Cancer. 2009;53:696. [Google Scholar]
- 58. Saven A, Burian C. Cladribine activity in adult langerhans‐cell histiocytosis. Blood. 1999;93:4125‐4130. [PubMed] [Google Scholar]
- 59. Derenzini E, Stefoni V, Pellegrini C, et al. High efficacy of the MACOP‐B regimen in the treatment of adult Langerhans cell histiocytosis, a 20 year experience. BMC Cancer. 2015;15:879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Duan MH, Han X, Li J, et al. Comparison of vindesine and prednisone and cyclophosphamide, etoposide, vindesine, and prednisone as first‐line treatment for adult Langerhans cell histiocytosis: a single‐center retrospective study. Leuk Res. 2016;42:43‐46. [DOI] [PubMed] [Google Scholar]
- 61. Ingram W, Desai SR, Gibbs JS, Mufti G. Reduced‐intensity conditioned allogeneic haematopoietic transplantation in an adult with Langerhans’ cell histiocytosis and thrombocytopenia with absent radii. Bone Marrow Transplant. 2006;37:713‐715. [DOI] [PubMed] [Google Scholar]
- 62. Xicoy B, Ribera JM, Batlle M, Feliu E. Sustained remission in an adult patient with Langerhans cell histiocytosis following T‐cell depleted allogenic cell transplantation. Med Clin (Barc). 2006;127:716. [DOI] [PubMed] [Google Scholar]
- 63. Ng Wing Tin S, Martin‐Duverneuil N, Idbaih A, et al. Efficacy of vinblastine in central nervous system Langerhans cell histiocytosis: a nationwide retrospective study. Orphanet J Rare Dis. 2011;6:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Imashuku S, Kudo N, Kaneda S, et al. Treatment of patients with hypothalamic‐pituitary lesions as adult‐onset Langerhans cell histiocytosis. Int J Hematol. 2011;94(6):556‐560. [DOI] [PubMed] [Google Scholar]
- 65. Janku F, Amin HM, Yang D, Garrido‐Laguna I, Trent JC, Kurzrock R. Response of histiocytoses to imatinib mesylate: fire to ashes. J Clin Oncol. 2010;28:e633‐e636. [DOI] [PubMed] [Google Scholar]
- 66. Montella L, Insabato L, Palmieri G. Imatinib mesylate for cerebral Langerhans cell histiocytosis. N Engl J Med. 2004;351:1034‐1035. [DOI] [PubMed] [Google Scholar]
- 67. Wagner C, Mohme H, Krömer‐Olbrisch T, et al. Langerhans cell histiocytosis: treatment failure with imatinib. Arch Dermatol. 2009;145:949. [DOI] [PubMed] [Google Scholar]
- 68. Haroche J, Cohen‐Aubart F, Emile JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim‐Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013;121:1495‐1500. [DOI] [PubMed] [Google Scholar]
- 69. Diamond EL, Subbiah V, Lockhart AC, et al. Histiocytosis: analysis of data from the histology‐independent, phase 2, open‐label VE‐BASKET Study. JAMA Oncol. 2018;4:384‐388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bhatia A, Ulaner G, Rampal R, et al. Single‐agent daBRAFenib for BRAFV600E‐mutated histiocytosis. Haematologica. 2018;103:e177‐e180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Papapanagiotou M, Griewank KG, Hillen U, et al. Trametinib‐induced remission of an MEK1‐mutated langerhans cell histiocytosis. JCO Precis Oncol. 2017;1:1‐5. [DOI] [PubMed] [Google Scholar]