

Abstract
Olaratumab, a monoclonal antibody targeting human platelet‐derived growth factor receptor α, plus doxorubicin significantly improved overall survival in patients with advanced soft‐tissue sarcoma (STS) in a prior phase 1b/2 randomized trial. Subsequent exposure‐response analysis suggested that higher olaratumab exposures earlier might improve outcomes in patients at risk of early disease progression. This phase 1 study (3 treatment cohorts; minimum 6 patients each) investigated the safety, pharmacokinetics and antitumor activity of olaratumab plus doxorubicin in Japanese patients with STS. Patients received olaratumab 15 mg/kg on Days 1 and 8 during each 21‐day cycle until disease progression. Patients in Cohort 3 received a 20 mg/kg loading dose of olaratumab in Cycle 1. Doxorubicin was administered for up to 6 cycles. Patients in Cohort 1 received doxorubicin 25 mg/m2 on Days 1, 2 and 3. Patients in Cohorts 2 and 3 received doxorubicin 75 mg/m2 on Day 1. One patient in Cohort 2 experienced a dose‐limiting toxicity of Grade 3 febrile neutropenia. Most treatment‐emergent adverse events were of mild and moderate severity, and were known doxorubicin toxicities. Olaratumab serum concentrations in Cohort 3 reached a steady‐state exceeding the target level in Cycle 1. Partial response was confirmed in 4 patients (2 each in Cohorts 2 and 3). Olaratumab plus doxorubicin had an acceptable safety profile in patients with STS. A loading dose of olaratumab 20 mg/kg was effective for achieving minimum serum concentrations above the target trough level in Cycle 1.
Keywords: Japanese, olaratumab, PDGFRα antagonist, phase 1, soft tissue sarcoma
1. INTRODUCTION
Soft‐tissue sarcoma (STS) is a rare form of malignant tumor that is associated with a poor prognosis. Specifically, estimates suggest that STS comprises approximately 1% of all solid malignancies1 and occurs at an incidence of 3 per 100 000 people in Japan.2 The current standard of care in STS is doxorubicin alone or in combination with other agents, even with recent FDA approvals of pazopanib, eribulin and trabectedin.3
Olaratumab is a recombinant human immunoglobulin G subclass 1 monoclonal antibody, which targets human platelet‐derived growth factor receptor α (PDGFRα).4 In a randomized phase 1b/2 study, olaratumab (15 mg/kg) plus doxorubicin (75 mg/m2) met the predefined primary endpoint for progression‐free survival (PFS) and significantly improved overall survival (OS) by 11.8 months vs doxorubicin alone in patients with advanced STS.5 The safety profile of this regimen was found to be acceptable and the benefit‐risk profile positive. Olaratumab has since received FDA accelerated approval for use with doxorubicin for treating advanced STS and conditional approval in the European Union, Canada and South Korea for use with doxorubicin for treating advanced STS.
To date, there is limited information available on the efficacy and safety of olaratumab in Japanese patients. In a phase 1 dose‐escalation study of Japanese patients with advanced/refractory solid malignancies, olaratumab monotherapy was found to have an acceptable safety profile and was well tolerated.6 The safety of olaratumab plus doxorubicin in Japanese patients with STS has not been reported in the published literature.
The primary objective of this phase 1 study was to evaluate the safety and tolerability of olaratumab plus doxorubicin in Japanese patients with advanced STS. The main secondary objective was to evaluate the pharmacokinetic (PK) profile of olaratumab following a 20 mg/kg loading dose.
2. MATERIALS AND METHODS
2.1. Study design
This open‐label, nonrandomized, multicenter phase 1 study evaluated olaratumab plus doxorubicin in Japanese patients with advanced STS (ClinicalTrials.gov Identifier: NCT02377752). This study also included a second part that evaluated olaratumab monotherapy; results for the second part of the study are not reported here.
All patients provided written informed consent, and the protocol and informed consent form were approved by ethics review boards at each site. The study was conducted in accordance with the Declaration of Helsinki, the Council for International Organizations of Medical Sciences International Ethical Guidelines and the International Conference on Harmonisation Good Clinical Practices Guideline (E6).
2.2. Study population
Key inclusion criteria were: age ≥20 years; histologically/cytologically confirmed advanced or metastatic solid tumor, in particular STS (other than Kaposi's sarcoma and gastrointestinal stromal tumors), not amenable to treatment with surgery or radiotherapy; the presence of measurable or nonmeasurable disease as defined by the Response Evaluation Criteria in Solid Tumors (version 1.1); Eastern Cooperative Oncology Group performance status score ≤1 at study entry; and left ventricular ejection fraction (assessed via echocardiography) ≥50%.
Key exclusion criteria were previous treatment with anthracyclines and previous radiotherapy to the mediastinal/pericardial area.
2.3. Study treatment
There were 3 treatment cohorts (minimum 6 patients each). All patients received olaratumab 15 mg/kg on Days 1 and 8 of each 21‐day cycle until progressive disease (PD) or other discontinuation criteria were met. Patients in Cohort 3 received a 20 mg/kg loading dose of olaratumab on Days 1 and 8 in Cycle 1. The loading dose was chosen on the basis of an exposure‐response analysis of the phase 1b/2 study, which suggested that clinical outcomes could be improved if therapeutic steady‐state serum levels were achieved earlier in treatment.7, 8, 9, 10 Specifically, the loading dose regimen is expected to exceed target trough levels (66 μg/mL) during the first cycle of treatment. Doxorubicin was administered for up to 6 cycles (or a cumulative dose of 500 mg/m2, whichever came later) until PD or other discontinuation criteria were met. Patients in Cohort 1 received doxorubicin 25 mg/m2 on Days 1, 2, and 3 of each cycle, whereas those in Cohorts 2 and 3 received doxorubicin 75 mg/m2 on Day 1 of each cycle.
No premedication, including steroids or diphenhydramine, was mandated prior to treatment with olaratumab. Supportive care with granulocyte colony stimulating factor (G‐CSF) was permitted at the investigator's discretion; however, primary prophylaxis with G‐CSF was not allowed. Dexrazoxane, a cardioprotectant for anthracycline‐induced cardiotoxicity, is not approved in Japan and was, therefore, not used in the study.
2.4. Safety assessments
Treatment‐emergent adverse events (TEAE) were summarized using the Medical Dictionary for Regulatory Activities (Version 19.1) and the Common Terminology Criteria for Adverse Events (Version 4.03). Dose‐limiting toxicities (DLT) were defined as any adverse event (AE) occurring during Cycle 1 that was possibly related to treatment and met 1 of the following criteria: febrile neutropenia; Grade 4 anemia; Grade 4 neutropenia lasting >1 week; Grade 3 or 4 thrombocytopenia requiring platelet transfusion; toxicities requiring dose reduction or omission of study drug(s) during Cycle 1; Grade ≥3 nonhematologic toxicity, except nausea, vomiting, diarrhea, constipation, or electrolyte abnormality that could be controlled with treatment, fatigue and anorexia, transient Grade 3 elevations of alanine aminotransferase (ALT) and/or aspartate aminotransferase without evidence of other hepatic injury. Infusion‐related reactions (IRR) were not to be considered a DLT. Echocardiogram scans were performed prior to the start of Cycles 5 and 7, and at the follow‐up visit.
2.5. Pharmacokinetics/immunogenicity assessments
Blood samples were collected throughout Cycles 1 and 3 to determine serum olaratumab concentrations using a validated ELISA, carried out by ICON Development Solutions (Whitesboro, NY). The lower limit of quantitation was 1 μg/mL and the upper limit of quantification was 100 μg/mL. Olaratumab PK parameters were calculated using a standard noncompartmental method of analysis. Blood samples were analyzed for the presence or absence of antidrug antibodies (ADA) using a validated ELISA, carried out by Pharmaceutical Product Development (Richmond, VA, USA). Samples identified as being ADA‐positive were further evaluated for the presence of neutralizing antibodies.
2.6. Antitumor activity assessments
Patients were assessed by computed tomography scan and/or MRI for tumor measurement according to the Response Evaluation Criteria in Solid Tumors (Version 1.1).11 The objective response rate (ORR), the disease control rate (DCR) and PFS were determined.
2.7. Statistical analysis
The sample size was determined by the study design, rather than a statistical power calculation. Six patients in each cohort were required to assess DLT.
Dose‐limiting toxicities‐related safety analyses included patients who completed Cycle 1 or who discontinued due to a DLT during Cycle 1. Other safety analyses and all efficacy analyses included patients who received ≥1 dose of study drug. PK analyses included patients who received ≥1 dose of study drug and had PK samples collected.
Pharmacokinetic parameters were calculated by standard noncompartmental methods of analysis using Phoenix WinNonlin version 6.4 (Certara, Princeton, USA).
All data management was carried out by ICON Plc (Dublin, Ireland). Statistical analyses were carried out by MacroStat Clinical Research (Shanghai, China) using the statistical packages SAS version 9.2 (SAS Institute, Cary, NC, USA).
3. RESULTS
3.1. Patient disposition and baseline characteristics
In total, 19 enrolled patients received the study drugs and were included in the safety and efficacy analyses (Figure 1). All patients had STS, with leiomyosarcoma and liposarcoma being the most common subtypes. One patient in Cohort 1 was replaced for DLT analysis because of a Grade 4 IRR in Cycle 1. The majority of patients discontinued study treatment because of PD.
Figure 1.
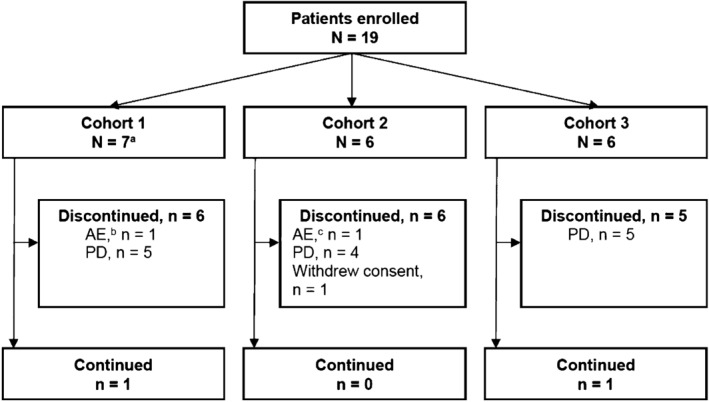
Patient disposition. aOne patient in Cohort 1 was replaced as per protocol because of an IRR; bDue to a treatment‐related adverse event of IRR during Cycle 1, Day 1; cDue to a treatment‐related adverse event of Grade 3 left ventricular dysfunction on Day 35 after completion of Cycle 6. AE, adverse event; IRR, infusion‐related reaction; PD, progressive disease
Baseline characteristics and extent of exposure are summarized in Tables 1 and 2, respectively.
Table 1.
Baseline characteristics
| Characteristic | Cohort 1 (N = 7) | Cohort 2 (N = 6) | Cohort 3 (N = 6) |
|---|---|---|---|
| Age, years, median (range) | 52.0 (30.0‐71.0) | 46.5 (33.0‐62.0) | 41.5 (26.0‐51.0) |
| Female, n (%) | 4 (57.1) | 2 (33.3) | 5 (83.3) |
| ECOG performance status, n (%) | |||
| 0 | 5 (71.4) | 3 (50.0) | 5 (83.3) |
| 1 | 2 (28.6) | 3 (50.0) | 1 (16.7) |
| Prior anticancer therapy, n (%) | |||
| Surgery | 5 (71.4) | 6 (100.0) | 4 (66.7) |
| Radiotherapy | 0 | 0 | 0 |
| Systemic therapy | 1 (14.3) | 1 (16.7) | 1 (16.7) |
| Tumor type | |||
| Leiomyosarcoma | 0 | 1 (16.7) | 4 (66.7) |
| Liposarcoma | 3 (42.9) | 2 (33.3) | 0 |
| Uterine adenosarcoma | 0 | 0 | 1 (16.7) |
| Embryonal sarcoma | 1 (14.3) | 0 | 0 |
| High‐grade undifferentiated pleomorphic sarcoma | 0 | 1 (16.7) | 0 |
| Malignant fibrous histiocytoma | 1 (14.3) | 0 | 0 |
| Malignant gastrointestinal neuroectodermal tumor | 0 | 1 (16.7) | 0 |
| Myxofibrosarcoma | 1 (14.3) | 0 | 0 |
| Myxoid/round cell liposarcoma | 0 | 1 (16.7) | 0 |
| Pleomorphic sarcoma | 0 | 0 | 1 (16.7) |
| Spindle cell sarcoma | 1 (14.3) | 0 | 0 |
ECOG, Eastern Cooperative Oncology Group.
Table 2.
Exposure to olaratumab and doxorubicin
| Exposurea | Cohort 1 (N = 7b) | Cohort 2 (N = 6) | Cohort 3 (N = 6) |
|---|---|---|---|
| Olaratumab | |||
| Number of cycles | 4.0 (1‐29) | 9.5 (1‐20) | 7.0 (2‐23) |
| Duration on therapy (wks) | 12.7 (3‐91) | 29.5 (3‐61) | 22.2 (6‐72) |
| Cumulative dose (mg/kg) | 119.9 (2‐868) | 220.3 (30‐578) | 225.2 (71‐686) |
| Relative dose intensity (%) | 95.6 (5.0‐100.9) | 91.5 (59.9‐101.1) | 96.8 (93.7‐100.9) |
| Doxorubicin | |||
| Number of cycles | 4.5 (1‐6) | 6.0 (1‐6) | 5.0 (2‐6) |
| Duration on therapy (wks) | 14.1 (3‐23) | 18.6 (3‐23) | 15.8 (6‐20) |
| Cumulative dose (mg/m2) | 337.6 (76‐456) | 426.1 (75‐455) | 375.6 (151‐460) |
| Relative dose intensity (%) | 96.0 (78‐101) | 87.6 (71‐100) | 95.4 (90‐100) |
Data are median (range).
N = 6 for doxorubicin exposure in Cohort 1 as 1 patient, who experienced an infusion‐related reaction, did not receive doxorubicin.
3.2. Safety and tolerability
There were no DLT in Cohorts 1 or 3. One patient in Cohort 2 experienced a DLT of Grade 3 febrile neutropenia, which led to doxorubicin dose reduction to 60 mg/m2 from Cycle 2 as per protocol.
All 19 patients experienced ≥1 TEAE during the study. The most common TEAE, regardless of causality, were nausea (Cohort 1 = 5, Cohort 2 = 6, Cohort 3 = 6), alopecia (4, 6, 6), increased ALT (3, 3, 4), decreased appetite (4, 2, 4), fatigue (3, 4, 3), decreased neutrophil count (2, 6, 1) and decreased white blood cell count (2, 6, 1). All Grade 3/4 TEAE (Table 3) occurred during the first 6 cycles (in combination with doxorubicin), except for 1 TEAE of Grade 3 hypophosphataemia that occurred after Cycle 7. A total of 3 patients received G‐CSF during the study due to neutropenia: 2 patients in Cohort 1 and 1 patient in Cohort 3. Two patients in Cohort 2 and 1 patient in Cohort 3 experienced Grade 3 ALT increases. No patient had a bilirubin level ≥2 times the upper normal limit at the time of ALT increase. No patient discontinued or had dose adjustment due to increased ALT, and all instances of increased ALT were observed within the first 4 cycles of treatment, suggesting that these ALT increases were likely due to the single dose of doxorubicin 75 mg/m2. In this study, Grade 1 musculoskeletal pain (consolidated term) was experienced by 1 patient (14.3%) each in Cohorts 1 and 2. Grade 2 musculoskeletal pain (consolidated term) was experienced by 2 patients (33.3%) in Cohort 3. There were no deaths during the study.
Table 3.
Summary of Grade 3/4 treatment‐emergent averse events (regardless of causality) occurring in ≥1 patient in any cohort
| TEAE | Cohort 1 (N = 7) | Cohort 2 (N = 6) | Cohort 3 (N = 6) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Any grade | Grade 3 | Grade 4 | Any grade | Grade 3 | Grade 4 | Any grade | Grade 3 | Grade 4 | |
| Patients with ≥1 TEAE | 7 (100.0) | 1 (14.3) | 2 (28.6) | 6 (100.0) | 2 (33.3) | 4 (66.7) | 6 (100.0) | 2 (33.3) | 0 |
| Anemia | 3 (42.9) | 0 | 0 | 3 (50.0) | 2 (33.3) | 0 | 1 (16.7) | 0 | 0 |
| Febrile neutropenia | 0 | 0 | 0 | 1 (16.7) | 1 (16.7) | 0 | 1 (16.7) | 1 (16.7) | 0 |
| LV dysfunction | 0 | 0 | 0 | 1 (16.7) | 1 (16.7) | 0 | 0 | 0 | 0 |
| Lung infection | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 1 (16.7) | 1 (16.7) | 0 |
| Meningitis | 0 | 0 | 0 | 1 (16.7) | 1 (16.7) | 0 | 0 | 0 | 0 |
| IRR | 1 (14.3) | 0 | 1 (14.3) | 0 | 0 | 0 | 0 | 0 | 0 |
| Hypophosphataemia | 0 | 0 | 0 | 1 (16.7) | 1 (16.7) | 0 | 0 | 0 | 0 |
| ALT increased | 3 (42.9) | 0 | 0 | 3 (50.0) | 2 (33.3) | 0 | 4 (66.7) | 1 (16.7) | 0 |
| ALP increased | 0 | 0 | 0 | 1 (16.7) | 1 (16.7) | 0 | 0 | 0 | 0 |
| Creatinine increased | 0 | 0 | 0 | 2 (33.3) | 1 (16.7) | 0 | 0 | 0 | 0 |
| GGT increased | 0 | 0 | 0 | 3 (50.0) | 1 (16.7) | 0 | 3 (50.0) | 0 | 0 |
| Lymphocyte count decreased | 0 | 0 | 0 | 1 (16.7) | 1 (16.7) | 0 | 0 | 0 | 0 |
| Neutrophil count decreased | 3 (42.9) | 1 (14.3) | 1 (14.3) | 6 (100.0) | 2 (33.3) | 4 (66.7) | 1 (16.7) | 0 | 0 |
| Platelet count decreased | 2 (28.6) | 0 | 0 | 2 (33.3) | 0 | 0 | 0 | 0 | 0 |
| WBC count decreased | 2 (28.6) | 1 (14.3) | 1 (14.3) | 6 (100.0) | 5 (83.3) | 0 | 1 (16.7) | 1 (16.7) | 0 |
ALP, alkaline phosphatase; ALT, alanine transaminase; GGT, gamma‐glutamyltransferase; IRR, infusion‐related reaction; LV, left ventricular; TEAE, treatment‐emergent adverse event; WBC, white blood cell.
Two patients discontinued study treatment because of a treatment‐related adverse event. One patient in Cohort 1 (a 64‐year‐old woman with no history of allergic reactions or hypersensitivity) discontinued study treatment and did not receive doxorubicin because of a Grade 4 IRR that occurred on Day 1 of Cycle 1. The patient experienced generalized pruritus and transient redness 2 minutes after the first olaratumab infusion was started, and subsequently developed hypotension and dyspnoea. The patient required continuous infusion of dopamine, due to persistent low blood pressure, until recovery the next morning. No other patients reported IRR. One patient in Cohort 2 (a 44‐year‐old woman with a normal pretreatment echocardiogram) discontinued study treatment because of Grade 3 left ventricular dysfunction. Her cumulative doxorubicin dose was 450 mg/m2. Three weeks after the last doxorubicin dose in Cycle 6, the patient complained of dyspnoea and palpitations; her ejection fraction (EF) had decreased from 70% to 48%. Subsequently, the patient required hospitalization for 1 month due to further deterioration of EF to 30%. This event was deemed to be a doxorubicin‐induced cardiotoxicity.
3.3. Pharmacokinetics
Olaratumab PK parameters are summarized in Table 4.
Table 4.
Olaratumab pharmacokinetic parameters during Cycle 1 and Cycle 3 after administration of olaratumab 15 mg/kg or 20 mg/kg on Day 1 and 8 in each cycle, combined with doxorubicin
| Parameter | Geometric mean (CV%) | |||||
|---|---|---|---|---|---|---|
| Cohort 1 (Olara 15 mg/kg + DOX 25 mg/m2) | Cohort 2 (Olara 15 mg/kg + DOX 75 mg/m2) | Cohort 3 (Olara 20 mg/kg [C1]/15 mg/kg [C2+] + DOX 75 mg/m2) | ||||
| Cycle 1 Day 1 | Cycle 1 Day 8 | Cycle 1 Day 1 | Cycle 1 Day 8 | Cycle 1 Day 1 | Cycle 1 Day 8 | |
| N | 6 | 6 | 6 | 6 | 6 | 6 |
| C max (μg/mL) | 274 (30) | 353(40) | 301 (25) | 351 (20) | 470 (27) | 610 (33) |
| t max (h)a | 2.70 (2.55‐24.97) | 1.59 (1.08‐2.82) | 1.09 (1.05‐2.57) | 2.08 (1.02‐2.17) | 1.12 (1.02‐2.50) | 1.11 (1.00‐2.10) |
| AUC(0‐t) (μg·h/mL) | 21700 (36) | 47300 (32) | 21900 (25) | 42000 (36) | 34300 (29) | 74100 (42) |
| C min (μg/mL) | — | 74.1, 57.2c | — | 57.5 (61) | — | 95.3 (83) |
| C av (μg/mL) | 129 (36) | 141 (32) | 130 (25) | 125 (36) | 204 (29) | 221 (42) |
| CLss (mL/h) | 41.3 (20) | 18.9 (17) | 45.7 (23) | 23.8 (33) | 33.5 (19) | 15.5 (35) |
| V ss (L) | 5.36 (22) | 5.50 (29) | 5.55 (28) | 5.86 (15) | 4.04 (23) | 3.91 (32) |
| t 1/2 (d)b | 3.86 (2.86‐4.52) | 8.91 (6.98‐11.3) | 3.64 (2.32‐5.79) | 7.84 (5.96‐11.6) | 3.64 (2.85‐4.66) | 7.88 (4.40‐12.3) |
| Cycle 3 Day 1 | Cycle 3 Day 8 | Cycle 3 Day 1 | Cycle 3 Day 8 | Cycle 3 Day 1 | Cycle 3 Day 8 | |
| N | 4 | 4 | 4 | 4 | 5 | 5 |
| C max (μg/mL) | 351 (28) | 415 (28) | 390 (13) | 474 (13) | 545 (34) | 541 (41) |
| t max (h)a | 2.74 (2.62‐2.82) | 2.09 (2.00‐2.15) | 2.59 (1.12‐2.67) | 2.06 (2.05‐2.08) | 2.53 (1.07‐2.58) | 1.10 (1.02‐2.12) |
| AUC(0‐t) (μg·h/mL) | 33600 (31) | 63000 (28) | 36000 (15) | 68400 (15) | 42500 (51) | 77700 (68)d |
| C min (μg/mL) | — | 94.3 (22) | — | 105 (33) | — | 103 (136)d |
| C av (μg/mL) | 200 (31) | 188 (28) | 214 (15) | 204 (15) | 253 (51) | 231 (46)d |
| CLss (mL/h) | 27.0 (15) | 14.4 (9) | 28.5 (14) | 15.0 (14) | 21.2 (34) | 11.7 (56)d |
| V ss (L) | 5.42 (19) | 4.22 (24) | 4.66 (13) | 4.62 (11) | 3.49 (33) | 3.10 (27)d |
| t 1/2 (d)b | 6.04 (5.26‐6.75) | 8.84 (7.27‐10.4) | 4.84 (4.46‐5.32) | 9.50 (7.86‐12.0) | 5.05 (3.43‐7.35) | 7.94 (5.04‐11.7)d |
Median (range) and is referenced to the start of infusion.
Geometric mean (range).
Individual values are given when N < 3.
N = 4.
AUC(0‐t), area under the serum concentration vs time curve from time zero to t hours (t is 168 and 336 for Day 1 and Day 8 in each cycle, respectively); C av, average serum concentration during the dosing interval; CLss, total body clearance at steady state; C max, observed maximum serum concentration; C min, observed serum concentration at 336 h postinfusion of olaratumab on Day 8 in each cycle; CV, coefficient of variation; DOX, doxorubicin; h, hour; Olara, olaratumab; N, number of patients; t 1/2, apparent terminal elimination half‐life; t max, time of observed maximum serum concentration; V ss, volume of distribution at steady state.
Serum olaratumab concentration data were available for 18 patients (Figure 2). The number of patients achieving olaratumab plasma concentration above the target trough level (>66 μg/mL) in Cycle 1 was 3/8 (37.5%) for Cohorts 1 and 2 combined, and 5/6 (83.3%) for Cohort 3.
Figure 2.
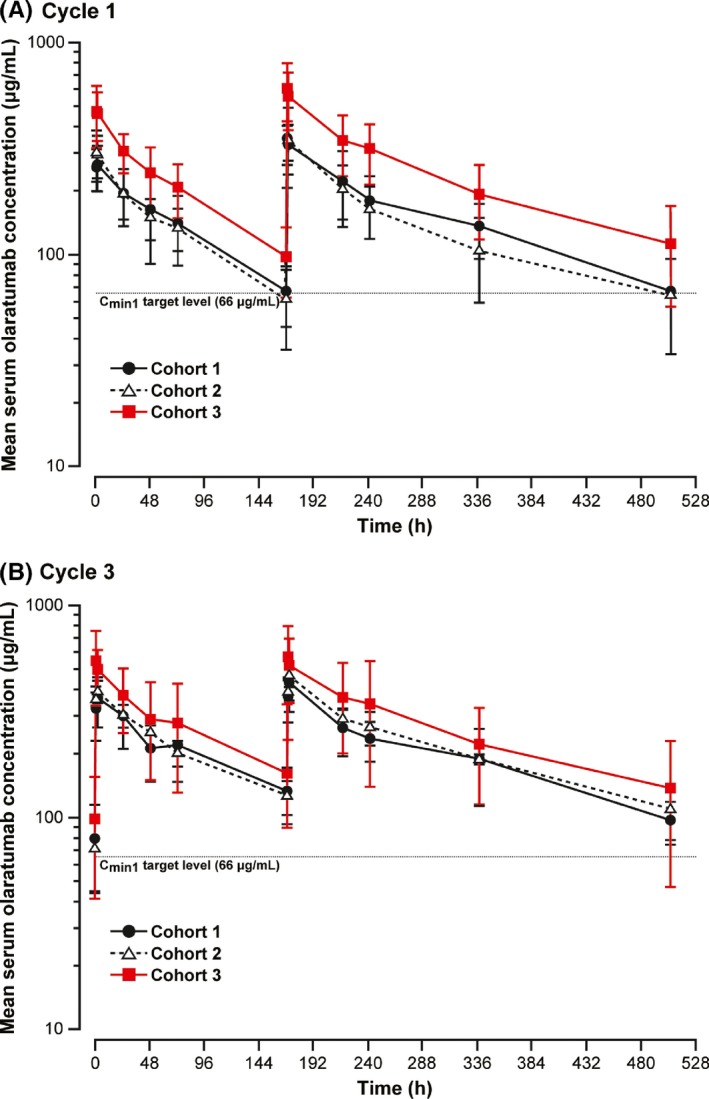
Arithmetic mean (±SD) serum olaratumab concentration‐time profiles in Cycle 1 (A) and Cycle 3 (B) after administration of olaratumab on Days 1 and 8. Patients in Cohorts 1 and 2 received olaratumab 15 mg/kg on Days 1 and 8 of each 21‐day cycle; patients in Cohort 3 received olaratumab 20 mg/kg on Days 1 and Day 8 of Cycle 1, and 15 mg/kg on Days 1 and 8 of subsequent cycles. Patients in Cohort 1 received doxorubicin 25 mg/m2 on Days 1, 2 and 3 of each 21‐day cycle; patients in Cohorts 2 and 3 received doxorubicin 75 mg/m2 on Day 1. C min1, minimum serum concentration after the first dose
3.4. Antitumor activity
A partial response was confirmed in 2 patients each in Cohorts 2 and 3 (Figure 3), giving an ORR of 33.3% in each of these cohorts (Table 5). No patient in Cohort 1 had a PR. The DCR were 42.9%, 83.3% and 66.7% in Cohorts 1, 2 and 3, respectively (Table 5).
Figure 3.
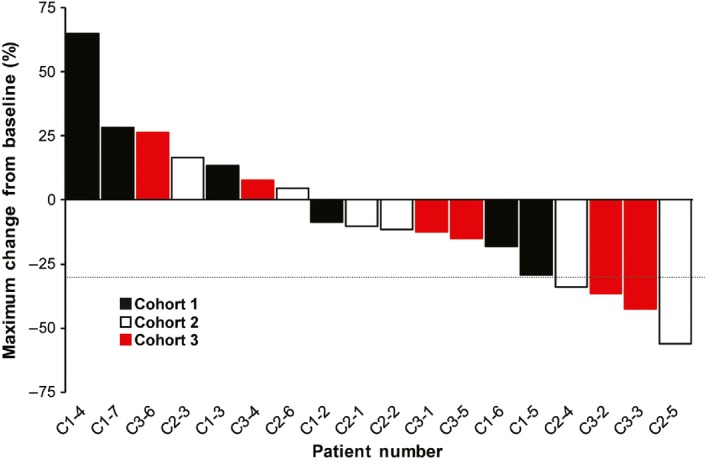
Waterfall plot of maximum changes from baseline in tumor size. Dotted line indicates a 30% tumor shrinkage
Table 5.
Summary of best overall response
| Response | Cohort 1 (N = 7) | Cohort 2 (N = 6) | Cohort 3 (N = 6) |
|---|---|---|---|
| Best overall response, n (%) | |||
| CR | 0 | 0 | 0 |
| PR | 0 | 2 (33.3) | 2 (33.3) |
| SD | 3 (42.9) | 3 (50.0) | 2 (33.3) |
| PD | 3 (42.9) | 1 (16.7) | 2 (33.3) |
| NE | 1 (14.3) | 0 | 0 |
| Objective response rate, n (%) | 0 | 2 (33.3) | 2 (33.3) |
| 95% CIa | 0, 35.4 | 9.7, 70.0 | 9.7, 70.0 |
| Disease control rate, n (%) | 3 (42.9) | 5 (83.3) | 4 (66.7) |
| 95% CIa | 15.8, 75.0 | 43.6, 97.0 | 30.0, 90.3 |
Calculated using the Wilson formula.
CI, confidence interval; CR, complete response; NE, not evaluable; PD, progressive disease; PR, partial response; SD, stable disease.
Two patients (1 with spindle cell sarcoma in Cohort 1 and 1 with leiomyosarcoma in Cohort 3) were continuing treatment as of the data cut‐off (Figure 4). Both patients had received ≥1 year of treatment with olaratumab and had stable disease (SD) at the last tumor assessment before the data cut‐off (Cohort 1: 588 days of treatment; Cohort 3: 478 days of treatment).
Figure 4.
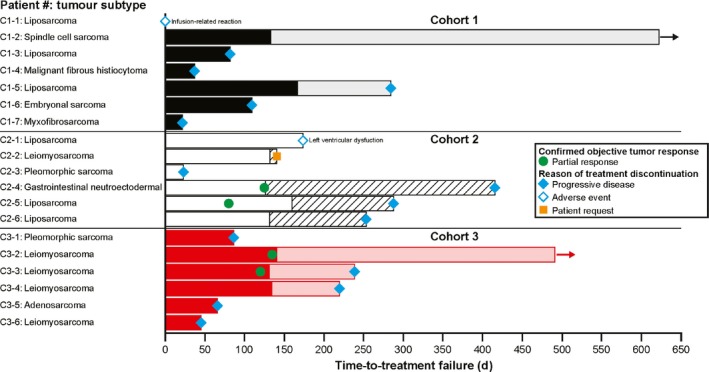
Time‐to‐treatment failure. Coloring of bars before Cycle 6: Cohort 1 = black, Cohort 2 = white, Cohort 3 = red; coloring of bars from Cycle 7 onwards = Cohort 1 = grey, Cohort 2 = diagonal lines, Cohort 3 = light red. Arrows indicate ongoing treatment
Median (95% confidence interval) PFS was: Cohort 1 = 9.1 months (0.7, not achieved); Cohort 2 = 9.2 months (0.8, 13.6); and Cohort 3 = 5.0 months (1.5, not achieved).
3.5. Immunogenicity
Anti‐drug antibodies were not detected in any patients, including the patient who experienced an IRR.
4. DISCUSSION
This is the first report on olaratumab plus doxorubicin in Japanese patients with advanced STS. Key findings from the study include that this combination had an acceptable safety profile, and that an olaratumab 20 mg/kg loading dose in Cycle 1 led to early achievement of steady‐state serum concentrations above the target trough level while preserving the safety profile of olaratumab observed with 15 mg/kg dose.
The safety and tolerability findings from this study are consistent with those from the phase 1b/2 study of olaratumab plus doxorubicin in, predominantly, Caucasian patients with advanced STS.5 Few patients discontinued due to TEAE and only 1 patient experienced a DLT of Grade 3 febrile neutropenia (this patient was able to continue study treatment on dose adjustment as per protocol without recurrence of the event). Furthermore, most Grade 3/4 TEAE occurred during the first 6 cycles of treatment in combination with doxorubicin; the frequency of these TEAE did not exceed that expected for toxicity related to doxorubicin alone. In the phase 1b/2 study of olaratumab plus doxorubicin, there was an imbalance between the olaratumab plus doxorubicin arm and the control (doxorubicin) arm in the frequency of musculoskeletal pain.5 In the current study, a total of 4 patients experienced Grade 1/2 musculoskeletal pain. One patient in the current study experienced an IRR, a known hypersensitivity reaction associated with monoclonal antibody treatment (note: premedication was not mandated in this study as per protocol). Infusion‐related reactions reported for olaratumab have predominantly been Grade 1/25 and severe IRR have only been reported during the first infusion of olaratumab. Therefore, all patients in ongoing olaratumab trials are receiving premedication with dexamethasone and an H1 antagonist in Cycle 1 and an H1 antagonist in the subsequent cycles. In Japan, doxorubicin 75 mg/m2 has traditionally been given over 3 consecutive days (ie, 25 mg/m2, Days 1‐3) rather than as a single dose, which is a common regimen elsewhere for the treatment of STS. In this study, 1 patient in Cohort 2 (doxorubicin 75 mg/m2 as a single dose) experienced Grade 3 cardiac dysfunction. The occurrence of this event is not unexpected given that the median cumulative dose doxorubicin at the time of the event was close to 450 mg/m2. Except for ALT increase, there were no notable increases in the prevalence of toxicities potentially related to doxorubicin in Cohort 2 and Cohort 3 compared with Cohort 1. Therefore, doxorubicin 75 mg/m2 given as a single dose had acceptable safety in Japanese patients. Overall, the safety findings from this study indicate that olaratumab plus doxorubicin has acceptable and manageable safety and tolerability profiles in Japanese patients with advanced STS. Although not required in this study, the current US label for olaratumab12 specifies premedication with intravenous diphenhydramine and dexamethasone, prior to olaratumab administration on Day 1 of Cycle 1.
A key finding of this study is that an olaratumab loading dose of 20 mg/kg led to the early achievement of steady‐state serum concentrations above the target trough level of >66 μg/mL identified in the phase 1b/2 study.7, 8, 9, 10 This finding supports the loading cycle of olaratumab 20 mg/kg as part of the olaratumab plus doxorubicin regimen in the treatment of advanced STS. Other PK parameters were similar to those reported in a previous US study of patients with STS.5
Olaratumab plus doxorubicin provided antitumor activity in Japanese patients with advanced STS. Of note, 4/19 (21%) patients had ≥30% tumor shrinkage and 8/19 (42.1%) achieved SD, while median PFS ranged from 5.0 to 9.2 months among the cohorts. These findings are in line with the antitumor activity demonstrated in the phase 1b/2 study.5
This study is limited by the small sample size, heterogeneity of disease and the lack of long‐term follow up for assessment of OS and late‐onset cardiotoxicity.
In conclusion, olaratumab plus doxorubicin given as a single 75 mg/m2 dose had an acceptable safety profile in Japanese patients with advanced STS and demonstrated antitumor activity. Notably, a loading dose of olaratumab 20 mg/kg achieved steady‐state serum concentrations above the target trough level as early as Cycle 1. An ongoing, multinational phase 3 study including Japanese STS patients (ANNOUNCE; NCT02451943) is evaluating the efficacy and safety of this regimen.
CONFLICTS OF INTEREST
This study was sponsored by Eli Lilly and Company, the manufacturer/licensee of olaratumab. Eli Lilly and Company was involved in the study design, funding, provision of study drugs, data collection, data analysis, and preparation of the manuscript. K. Yonemori has received daily allowances/honoraria from Eisai. M. Kodaira contributed to this work as an employee of the Department of Breast and Medical Oncology, National Cancer Center Hospital. The opinions expressed in this work are solely his and do not represent those of his current affiliation, Kodaira Hospital. T. Satoh has received: daily allowances/honoraria from Yakult Honsha, Chugai Pharmaceutical, Ono Pharmaceutical, Eli Lilly and Company, Bristol‐Myers Squibb, Merck‐Serono, Merck, Takeda Pharmaceutical, Daiichi‐Sankyo and Sanofi‐Genzyme; manuscript fees from Yakult Honsha, Chugai Pharmaceutical, Ono Pharmaceutical, Eli Lilly and Company, Bristol‐Myers Squibb, Merck‐Serono, Merck, Takeda Pharmaceutical, Daiichi‐Sankyo and Sanofi‐Genzyme; and research funds from Yakult Honsha, Chugai Pharmaceutical, Ono Pharmaceutical, Eli Lilly and Company, Bristol‐Myers Squibb, Merck‐Serono, Merck, Takeda Pharmaceutical, Daiichi‐Sankyo, Sanofi‐Genzyme and Gilead Sciences. T. Kudo belongs to a donated fund laboratory from Yakult Honsha, Chugai Pharmaceutical, and Ono Pharmaceutical. S. Takahashi has received daily allowances/honoraria from Daiichi‐Sankyo, Sanofi, Eisai and Bayer, and research funds from Eli Lilly Japan K.K., Daiichi‐Sankyo, Sanofi, Eisai, Bayer, Taiho Pharmaceutical, MSD, Novartis, Chugai Pharmaceutical and AstraZeneca. K. Nakano contributed to this work as an employee of the Department of Medical Oncology, Cancer Institute Hospital of the Japanese Foundation for Cancer Research. The opinions expressed in this work are solely his and do not represent those of his current affiliation, Pharmaceuticals and Medical Devices Agency (PMDA). Y. Ando has received daily allowances/honoraria from Chugai Pharmaceutical, and research funds from Geo Holdings Corporation, Mochida Pharmaceutical, Yakult Honsha, Chugai Pharmaceutical, Kyowa Hakko Kirin, Eisai, Ono Pharmaceutical, Eli Lilly Japan K.K. and Novartis Pharma K.K. K. Tamura has received clinical trial funds from Pfizer, Ono Pharmaceutical, Eli Lilly, Daiichi‐Sankyo and MSD. J. Mori, K. Inoue and S. Sakaguchi are employed by Eli Lilly Japan K.K. G. Oakley is employed by and owns shares in Eli Lilly and Company. T. Shimokata has no conflicts of interest to declare.
ACKNOWLEDGMENTS
We thank all patients, their families and their caregivers for participating in the study. We also thank Shinya Hirota, PhD (Eli Lilly Japan K.K.) for medical assistance. Medical writing assistance was provided by Hiroko Ebina, BPharm, Ph, MBAr, and Luke Carey, PhD, of ProScribe ‐ Envision Pharma Group, and was funded by Eli Lilly and Company. ProScribe's services complied with international guidelines for Good Publication Practice (GPP3).
Yonemori K, Kodaira M, Satoh T, et al. Phase 1 study of olaratumab plus doxorubicin in Japanese patients with advanced soft‐tissue sarcoma. Cancer Sci. 2018;109:3962–3970. 10.1111/cas.13846
Clinicaltrials.gov: NCT02377752
REFERENCES
- 1. Schoffski P, Cornillie J, Wozniak A, Li H, Hompes D. Soft tissue sarcoma: an update on systemic treatment options for patients with advanced disease. Oncol Res Treat. 2014;37:355‐362. [DOI] [PubMed] [Google Scholar]
- 2. Nakamura K, ed. “Passage” Series for Specialists of Clinical Orthopaedics (6). Practical Guide for Soft Tissue Tumors. Tokyo, Japan: Nakayama Shoten; 2011. [Google Scholar]
- 3. Deshpande HA, Cecchini M, Ni Choileain S, Jones R. Olaratumab for the treatment of soft tissue sarcoma. Drugs Today (Barc). 2017;53:247‐255. [DOI] [PubMed] [Google Scholar]
- 4. Loizos N, Xu Y, Huber J, et al. Targeting the platelet‐derived growth factor receptor alpha with a neutralizing human monoclonal antibody inhibits the growth of tumor xenografts: implications as a potential therapeutic target. Mol Cancer Ther. 2005;4:369‐379. [DOI] [PubMed] [Google Scholar]
- 5. Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft‐tissue sarcoma: an open‐label phase 1b and randomised phase 2 trial. Lancet. 2016;388:488‐497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Doi T, Ma Y, Dontabhaktuni A, Nippgen C, Nippgen J, Ohtsu A. Phase I study of olaratumab in Japanese patients with advanced solid tumors. Cancer Sci. 2014;105:862‐869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mo G, Jones R, Baldwin JR, et al. PK/PD modeling of overall and progression‐free survival in advanced soft tissue sarcoma patients treated with olaratumab in combination with doxorubicin. 25th Meeting of the Population Approach Group in Europe. Lisboa, Portugal, June 7‐10, 2016.
- 8. Jones R, Mo G, Baldwin JR, et al. Exposure‐response of olaratumab for survival outcomes and safety when combined with doxorubicin in soft tissue sarcoma (STS) patients. Ann Oncol. 2016;27(suppl 6):1402PD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. US Food and Drug Administration/Center for Drug Evaluation and Research . BLA Multi‐disciplinary Review and Evaluation of Lartruvo (olaratumab). 2016. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/761038Orig1s000MultiDisciplineR.pdf. Accessed August 15, 2018.
- 10. European Medicines Agency . European Public Assessment Report for Lartruvo (olaratumab). 2016. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004216/WC500216871.pdf. Accessed August 15, 2018.
- 11. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228‐247. [DOI] [PubMed] [Google Scholar]
- 12. Lartruvo (olaratumab) . [US Package Insert]. Indianapolis, IN: Eli Lilly and Company, 2017. http://pi.lilly.com/us/lartruvo-uspi.pdf. Accessed August 2018.