

Abstract
Promoter DNA methylation, which occurs on cytosine nucleotides across CpG islands, results in gene silencing and represents a major epigenetic alteration in human cancer. Methylation‐specific PCR can amplify these modifications as markers in cancer cells. In the present work, we rigorously review the published literatures describing DNA methylation in the promoters of critical tumor suppressor genes; detection of promoter DNA methylation in various body fluids permits early detection of cancer cells during perioperative courses of clinical treatment. The latest whole‐genome comprehensive explorations identified excellent epigenetic biomarkers that could be detected at high frequency with high specificity; these biomarkers, which are designated highly relevant methylation genes (HRMG), permit the discrimination of tumor tissues from the corresponding normal tissues; these markers are also associated with unique cancer phenotypes, including dismal prognosis. In humans, HRMG include the CDO1,GSHR,RASSF1 and SFRP1 genes, with these markers permitting discrimination depending on the organs tested. The combination of several HRMG increased the early detection of cancer and exhibited reliable surveillance potential in human body fluids. Cancer clinics using such epigenetic biomarkers are entering a new era of enhanced decision‐making with the potential for improved cancer prognosis.
Keywords: body fluid, cancer, CDO1, methylation, tumor suppressor gene
1. INTRODUCTION
Epigenetic alterations are found in primary human cancers, and such aberrations are composed of DNA methylation and its linked histone modification.1 DNA methylation occurs in cytosine residues among the CpG islands of the promoter regions of individual genes; methylated cytosine has been referred to as the “5th nucleotide,” in addition to the 4 canonical DNA bases (adenine, cytosine, thymine and guanine). Methylated cytosines are totally different from unmethylated cytosines from a phenotypic point of view. Methylated cytosines can be bound by methyl‐CpG‐binding protein 2 (MeCP2), and the resulting protein‐nucleotide can be incorporated into protein complexes that include histone modification enzymes (eg, histone deacetylase complex [HDAC])2 leading to dynamic changes in chromatin structure (Figure 1A).3 As a result, DNA methylation can result in gene silencing due to impaired access of transcription factors through condensed and closed chromatin (Figure 1B).
Figure 1.
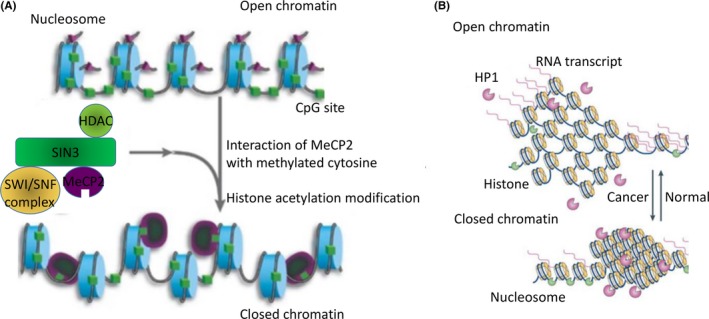
A, Methylated cytosines (green boxes) can bind to MeCP2 protein, which is itself a complex composed of proteins with SWI/SNF and HDAC activities, leading, in turn, to alterations of chromatin remodeling and histone acetylation (respectively). These activities regulate the state (open or closed) of chromatin. B, Open chromatin facilitates access by the various transcription factors, permitting transcription to generate RNA. Heterochromatin protein 1 (HP1) functions include gene repression by heterochromatin formation
Promoter DNA methylation of tumor suppressor genes (TSG) is accompanied by gene silencing in human cancers, and combination treatments using demethylating agents (5‐aza‐2′‐deoxycytidine) and HDAC inhibitors in cancer cell lines reactivate silenced TSG expression. Techniques such as pharmacological unmasking microarrays (PUM) have been used to identify novel, critical TSG candidates.4 Nevertheless, frequent cancer‐specific methylation is rare across the whole genome, and few methylated genes have been validated as sites of its frequent aberration with high specificity in human cancer. 5, 6, 7 Rigorous screening by PUM has repeatedly unveiled novel cancer‐prone methylation genes associated with tumor suppressive functions, such as the encoding homeobox only protein homeobox (HOPX) gene has been identified in various cancers,8, 9, 10, 11, 12 and HOPX expression has been independently revealed to be a biomarker representing differentiation or quiescent stem cell signatures in normal organ tissues.13, 14, 15, 16, 17 Hence, epigenetic changes in differentiation markers may be essential for the initiation of cancer cell growth.
DNA methylation in primary cancer tissues does not necessarily represent cancer‐specific methylation. For example, actual cancer specificity has been confirmed only in a very limited number of genes in primary gastric cancer.9 In a screen that used direct sequencing to distinguish the “wheat” (genes with cancer‐specific methylation) from the “chaff” (other methylated genes), HOPX was selected with the highest ranking (frequently methylated in 90% of primary tumors), followed by Reprimo (80%) and NMDAR2B (70%); high‐throughput analysis using quantitative methylation‐specific PCR (Q‐MSP) validated these priorities.9, 18 Q‐MSP can be used to screen for such cancer specificity because of its high‐throughput nature; recent searches for the best performance showed high (exceeding .9) area under curve (AUC) values in human cancer for genes like that encoding cysteine dioxygenase 1 (CDO1).19 Among many other promising genes that have also been reported to be methylated in primary tumor tissues, few have been validated for their cancer‐prone nature using the “gold standard” method, Q‐MSP.
Such candidate biomarkers for methylated genes are widely scattered across the entire genome, amounting to a total of 200‐300 genes.6, 8, 20 After rigorous validation, superior biomarker candidates representing cancer‐specific methylation are now considered ready for use in clinical decision‐making in the development of therapeutic strategies, with possible use extending even to cases where cancer surgery is indicated.21 In the present review article, the realistic potential of the epigenetic markers in cancer clinics is described.
2. METHYLATION ASSAY
2.1. Direct sequencing
Bisulfite treatment of DNA converts unmethylated cytosine to thymine without yielding a change to methylated cytosine. Following PCR amplification of the bisulfite‐treated sequences, direct sequencing (ie, without first cloning the fragment) permits “direct” visualization of either the cytosine or thymine. This method is appealing for discovery screening of novel methylation events that represent novel TSG in primary tumors,4, 5, 6, 7 as well as the clear differentiation of primary tumors from the corresponding normal tissues (Figure 2A). In addition to direct sequencing, cloned sequencing (ie, sequencing of individual fragments after cloning of the PCR products) can elucidate the fine‐scale mapping of methylation of the cytosines21, 22 (Figure 2B). However, cloned sequencing can be costly (in both time and money) and so is inappropriate for high‐throughput analysis.
Figure 2.
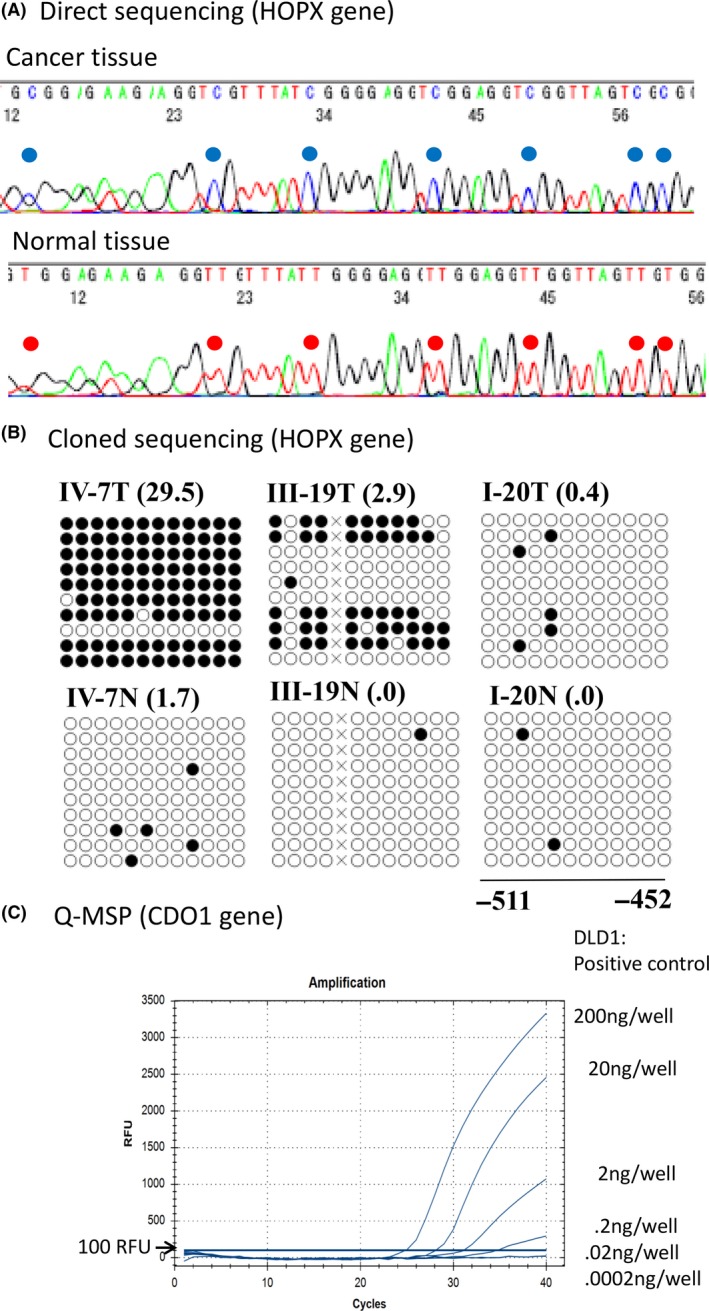
A, Direct sequencing revealed cytosine methylation (at the blue location) in primary tumor tissues (upper panel), while no cytosine methylation was detected (at the red locations) in the corresponding normal tissues. These cytosine methylation sites were consistent with CpG islands in the promoter DNA of the HOPX gene. B, Cloned sequencing after PCR amplification of products revealed finer‐scale status of the methylated cytosines of the CpG portions of the HOPX gene promoter. Black circles represent methylated residues, while white circles indicate residues that lacked methylation (as assessed by cloned sequencing). TaqMeth values by Q‐MSP (HOPX value/beta‐actin value × 100) are indicated within parentheses (right corners of each panel). C, Q‐MSP for CDO1 gene methylation was performed in DLD1 cells, revealing almost full methylation (93.3% of the CpG sites were methylated as judged by cloned sequencing) at various dilutions. PCR amplification detected signals at dilutions of 10‐, 100‐ and 1000‐fold, but not at 10 000‐ and 100 000‐fold dilutions
2.2. Real‐time methylation‐specific PCR
Methylation‐specific PCR (MSP) is appropriate for high‐throughput analysis of DNA methylation,23 and quantitative MSP using a TaqMan probe (Q‐MSP) permits the investigation of both the tumor tissues and the corresponding normal tissues of cancer patients in a high‐throughput manner; evaluation of the resulting receiver operating characteristic (ROC) curves permits discrimination of the tumor from the normal mucosa based on the most objective optimal criterion (optimal cut‐off value).9, 18 The AUC of the ROC curve is a “gold standard” for identifying excellent epigenetic cancer biomarkers. However, “the cut‐off value or below” does not necessarily represent non‐methylated status. It is more accurate to say that methylation values exceeding the cut‐off value represent relative hypermethylation. For example, in primary gastric cancer, the most optimal cut‐off value of the HOPX TaqMeth value (HOPX methylation value/beta‐actin methylation value × 100%) was calculated as 3.6; this value permitted discrimination of the tumor from the normal tissues by Q‐MSP.9 When this cut‐off value was used, HOPX hypermethylation was seen in 84% of the tissues defined as primary gastric tumors (based on classical histopathology), while HOPX hypermethylation was seen in 10% of the corresponding “normal” mucosa (again, as defined by classical histopathology). Intriguingly, this definition of hypermethylation by the Q‐MSP technique was consistent with the results (presence or absence of methylation) of direct sequencing in gastric cancer cell lines.9 The use of a Q‐MSP cut‐off value to discriminate cancer tissues from normal tissues, therefore, is highly consistent with the empirical results of direct sequencing. Representative methylation values of the HOPX gene based on the cloned sequencing are shown for primary cancer and normal mucosa tissues in Figure 2B.22 These results demonstrate that hypomethylation is not synonymous with complete unmethylation.
At best, Q‐MSP can detect methylation at dilution levels 1/1000 that of full methylation (Figure 2C), although Q‐MSP is unable to detect a 1/10 000 to 1/100 000 dilution of methylation, a detection level that would be equivalent to that of digital PCR technique that is the most sensitive system available at present.21, 24 However, there are currently few papers on the use of digital PCR for methylation analysis in cancer clinics.
3. THE BEST PERFORMANCE AS AN EPIGENETIC CANCER BIOMARKER: CDO1
Cancer‐specific methylation should be designated only after comparing methylation in tumor tissues with that of the corresponding non‐cancerous tissues. Nevertheless, many methylation genes have been so‐called without such validation. CpG island methylator phenotype (CIMP) was defined based on the methylation profiles of multiple well‐known tumor suppressor genes (e.g. p16, MLH1 and TIMP3) in the same tissue samples; however, the individual genes showed methylation frequency of <50% in most primary tumors.25, 26 Such methylation frequencies are rather low compared to the >60% methylation seen in human cancer loci recently identified as highly relevant methylation genes (HRMG).27 The relations of the CIMP phenotype to HRMG methylation status in human cancer have remained elusive, although several reports have suggested that HRMG are closely associated with CIMP. For example, methylation of RIZ1, an HRMG in gastric cancer (hypermethylated in 69% of primary tumors), was closely associated with the CIMP phenotype (P = .002).28 While HRMG differ in various organs (Table 1), and the CDO1 gene is one of the most common HRMG reported to data. The story thus begins at the CDO1 gene followed by other HRMG.
Table 1.
Methylation frequencies of highly relevant methylation genes (HRMG) in human cancers
| Organ | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Head and neck SCC | HOXA9 (.81, 60%) | NID2 (.79, 71%) | UCHL1 (.78, 66%) | DCC (.77, 75%) | KIF1A (.76, 72%) |
| Esophageal SCC | ZNF582 (.95, 86%) | NEFH (.93, 86%) | CDO1 (.91, 84%) | NMDAR2B (.91, 78%) | PAX1 (.89, 100%) |
| Esophageal adenocarcinoma | SFRP1 (96%) | CDO1 (95%) | APC (92%) | CDH1 (84%) | TIMP3 (74%) |
| Lung | GHSR (1.0) | CDO1 (.87, 92%) | HOXA9 (96%) | SHOX2 (94%) | TAC1 (87%) |
| Stomach | CDO1 (.95, 87%) | DLEC1 (.87, 93%) | HOPX (.85, 84%) | Reprimo (.77, 69%) | FLNC (.72, 93%) |
| Large intestine | CDO1 (.96, 91%) | SFRP1 (.96, 85%) | GFRA1 (.95, 89%) | SEPT9 (.94, 100%) | DCLK1 (.93, 82%) |
| Biliary tract | SFRP1 (.95, 84%) | OPCML (.93, 89%) | CDO1 (.91, 85%) | ZSCAN18 (.77, 65%) | DCLK1 (.75, 58%) |
| Gallbladder | SEPT9 (.82, 77%) | CDO1 (.74, 72%) | 14‐3‐3 sigma (90%) | 3‐OST‐2 (72%) | Maspin (70%) |
| Pancreas | GHSR (1.00) | CDO1 (.97, 94%) | HOPX (.85, 83%) | NPTX2 (100%) | UCHL1 (100%) |
| Breast | GHSR (.98, 92%) | CDO1 (.84, 79%) | MAL (95%) | 14‐3‐3 sigma (91%) | VGF (89%) |
| Uterus (cervical) | NKX6‐1 (.97, 93%*) | SOX9 (.96, 92%) | SOX1 (.95, 88%*) | ZNF516 (.92, 90%) | LMX1A (.9, 89%*) |
| Uterus (endometrial) | GALR1 (.97, 100%) | COL14A1 (.96, 92%) | ZNF177 (.95, 92%) | ZNF154 (.94, 82%) | TMEFF2 (.90, 65%) |
| Bladder | CDO1 (.87, 78%) | APAF‐1 (100%) | Twist1 (98%) | NID2 (96%) | PCDH17 (92%) |
| Prostate | RASSF1 (.99, 96%) | MDR1 (.98, 88%) | APC (.97, 90) | GHSR (.97) | GSTPI (.96, 93%) |
(Area under curve [AUC] of receiver operating characteristic [ROC] curve to differentiate the tumor from the normal counterpart, positive methylation frequency).
Asterisks were assessed by scrape samples.
Area under curve (AUC) could not always endowed in this table due to lack of data.
SCC, squamous cell carcinoma.
Cysteine metabolism plays a pivotal role in cell stemness through regulation of reactive oxygen species (ROS),29 and CDO1 can actually augment ROS generation to induce apoptosis.19, 30 The CDO1 protein can also bind peroxisome proliferation‐activated receptor (PPAR) gamma to activate the critical onco‐transcriptional factor CCAAT‐enhancer‐binding proteins (CEBP) alpha,31 which can inhibit transduction of the Wnt signal.32 Hence, CDO1 may play a critical tumor suppressive role in tumorigenesis.
Since 2010, the frequent hypermethylation of the CDO1 gene has been reported in primary breast,33, 34 lung,19 biliary tract,35 esophageal squamous cell carcinoma (SCC)36 and adenocarcinoma,37 gastric,19 colorectal,19, 38 bladder,19 penile (SCC),39 kidney,40 prostate,41 endometrial,42 pancreatic43 and gallbladder cancer.44 The frequencies of the aberrations in CDO1 methylation in these cancers are high (Figure 3A). Intriguingly, however, there are several unique cancers in which the CDO1 gene did not show an HRMG phenotype: for instance, renal cell clear carcinoma.28 The frequencies were largely determined by generating an ROC curve to determine the most optimal cut‐off value of methylation after comparing the tumor with the corresponding normal tissues in Q‐MSP; an example of such as analysis is shown in Figure 3B for pancreatic cancer.43
Figure 3.
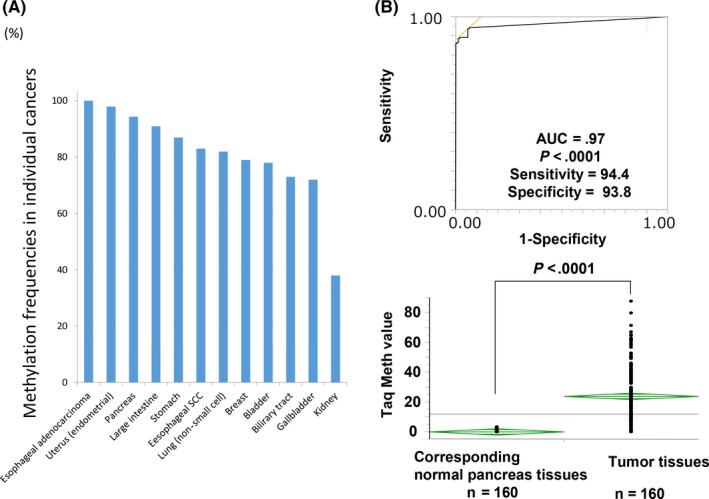
A, Methylation of the CDO1 gene is shown in order of the frequencies in various cancers of esophageal adenocarcinoma (100%), endometrial adenocarcinoma (98%), pancreatic adenocarcinoma (94%), large intestine adenocarcinoma (CRC; 91%), stomach (gastric) adenocarcinoma (87%), esophageal squamous cell carcinoma (SCC) (83%), lung (non‐small cell) cancer (82%), breast adenocarcinoma (72%), bladder cancer (78%), biliary tract cancer (73%), gallbladder cancer (72%) and kidney cancer (38%). B, Area under curve (AUC) of the receiver operating characteristic (ROC) curve to discriminate primary tumor tissues from the corresponding normal tissues is shown for pancreatic cancer. Quantitative methylation‐specific PCR (Q‐MSP) showed significant difference (P < .0001) in CDO1 methylation between the primary tumor tissues and the corresponding normal tissues
In contrast, the clinical relevance of CDO1 gene in primary tumor tissues has also been demonstrated in several cancers. Specifically in esophageal cancer, CDO1 methylation was significantly higher in advanced SCC tumors with cStage II/III than in the superficial tumors with cStage I,36 and was significantly higher in larger‐sized adenocarcinomas than in smaller‐sized adenocarcinomas.37 Aberrant methylation of the CDO1 gene accumulated as the tumor progressed, as demonstrated in colorectal38 and gallbladder tumorigenesis44 (Figure 4A,B). These findings indicate that promoter DNA methylation of the CDO1 gene commonly accumulates with progression in human cancers.
Figure 4.
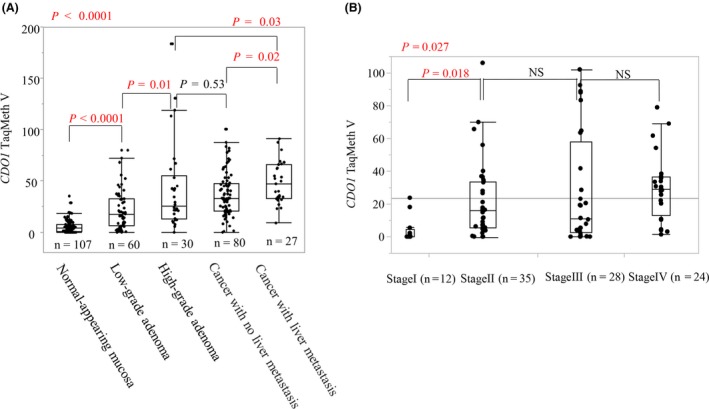
Methylation value increased as the tumor progressed. A, In colorectal carcinogenesis, the CDO1 methylation level increased significantly during the course of Vogelstein adenoma carcinoma sequence (P < .0001). B, In gallbladder carcinogenesis, the CDO1 methylation level increased significantly as the disease progressed from stage I to stage II
Cases with CDO1 hypermethylation also showed poorer prognosis than those with hypomethylation in primary breast,34 prostate,41 colorectal,38 gallbladder44 and esophageal cancers.36, 37 Interestingly, in primary breast cancer, CDO1 hypermethylation did not correlate significantly with markers of tumor progression such as TNM factors, while CDO1 hypermethylation was the strongest independent prognostic factor.34 Importantly, the prognostic relevance of CDO1 methylation was confirmed even in a prospective nationwide cohort study in the Netherlands of patients with renal cell carcinoma.40 These findings suggest that the methylation status of the CDO1 gene could be used as a prognostic marker in various human cancers. However, the cut‐off values of CDO1 methylation as prognostic markers were always higher than those used to discriminate the tumors from the corresponding normal tissues, suggesting that cancer progression resulting in dismal prognosis is associated with stronger epigenetic changes than those observed at initial tumorigenesis.
4. CANCER DETECTION MARKER OF CDO1 GENE METHYLATION IN HUMAN BODY FLUIDS OR CONVENTIONAL BIOPSY/CYTOLOGY TEST
Highly relevant methylation genes could also be used as detection markers for minute cancer cells, because of the cancer‐specific and prevalent nature of HRMG. Promoter DNA methylation of the CDO1 gene is one of the most relevant changes across the whole genome, so methylation of this gene's promoter could be a highly promising epigenetic cancer biomarker candidate, even in human body fluids. Q‐MSP can detect, at most, a 1/1000‐dilution level of the fully methylated genes (Figure 2C); however, this detection level would not provide satisfactory sensitivity for detection of the marker in the plasma DNA of colorectal cancer (CRC) patients (in whom the CDO1 promoter is seen in 40% of stage IV disease).45 Digital PCR for CDO1 methylation (which can detect a 1/100 000 dilution of fully methylated CDO1) might be sufficient to detect cancer cells in plasma, as shown for VIMENTIN (VIM) gene hypermethylation in patients with CRC.46 However, CDO1 gene methylation was reported to be readily detected in the plasma of patients with lung cancer,47, 48 where Q‐MSP detection of CDO1 methylation in plasma or serum showed 65% sensitivity with 74% specificity, and, when assessed in combination with the methylation of other HRMG, showed high sensitivity even in stage I disease. This extraordinarily high sensitivity of detection of cancer cells in plasma has been for the first time experienced in cancer clinics. Plasma surveillance will play a pivotal role in evaluating therapeutic efficacy during cancer management including surgery.
CDO1 hypermethylation could be useful for assisting preoperative disease diagnosis in critical body fluids other than plasma: for instance, through testing of endoscopic retrograde cholangio‐pancreatography (ERCP) brushing solution in biliary tract cancer49 and cervical scrapings in endometrial cancer.42 These body fluids are important for diagnosis, because inaccurate diagnosis would lead to unnecessary invasive surgery (false positive) or to missing the true disease (false negative). Cytology testing of ERCP brushing solution can be used to diagnose biliary tract cancer in approximately 60% of cases, while the CDO1 methylation cytology test showed a high AUC of .93 in predicting biliary tract cancer.49 Cervical scrapings were reported as a potential source of material for molecular testing in endometrial cancer, and CDO1 hypermethylation amazingly showed an 82% sensitivity with 94% specificity for detecting endometrial cancer in cervical scrapings.42
DNA testing using CDO1 hypermethylation may be useful for intraoperative diagnosis in gastric cancer surgery. The peritoneal DNA cytology test‐positive gene (CY1) is a critical prognostic marker of gastric cancer, and CY1‐positive cases represent stage IV disease. However, the conventional cytology test does not achieve sufficient sensitivity because peritoneal recurrences have been recognized in cytology‐negative (CY0) cases. DNA cytology testing using CDO1 methylation showed 2‐fold higher detection rates of the minimal residual disease than did conventional CY tests in the peritoneum, which was all positive for CY1 cases (Figure 5A), and can predict peritoneal recurrence accurately.50 DNA CY1 diagnosis in the peritoneum would be beneficial for planning future multimodal therapy in gastric cancer after curative surgery. Cytology tests targeted at the peritoneum are critical for other types of cancer, such as colorectal51 or ovarian cancer,52 and DNA CY1 information may be useful for decision‐making in the context of various abdominal cancers other than gastric cancer.
Figure 5.
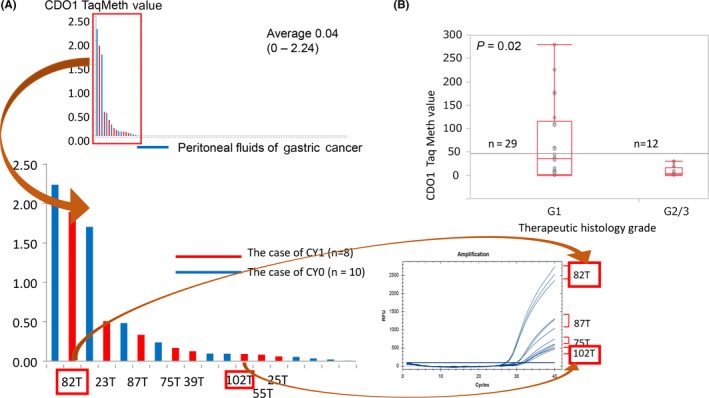
A, In gastric cancer, DNA cytology test using CDO1 methylation showed an approximately 2‐fold higher detection rate of minimal residual disease in the peritoneum, among cases that were positive for CY1 (n = 104). B, In esophageal squamous cell carcinoma (ESCC) with neoadjuvant chemotherapy showing histological grade 2/3, CDO1 methylation level was significantly lower than in other cases (n = 41)
CDO1 methylation could also be used to evaluate conventional biopsy samples evaluating tumor eradication after neoadjuvant therapy in esophageal cancer.36 In esophageal SCC showing histological grade 2/3 that has been treated with neoadjuvant chemotherapy, the CDO1 methylation level is significantly lower than in other cases (Figure 5B). These findings suggested that CDO1 methylation can reflect the presence of remnant cancer cells in the biopsies after treatment by neoadjuvant chemotherapy. If confirmed by rigorous validation, such information could affect surgical indications in the near future.
5. CLINICAL DECISION BY COMBINATION OF HIGHLY RELEVANT METHYLATION GENE EPIGENETIC BIOMARKERS
Epigenetic biomarkers using HRMG can affect the accuracy of: (i) preoperative diagnosis; (ii) intraoperative diagnosis; (iii) pathological diagnosis; and (iv) follow‐up surveillance on the therapeutic decision in cancer clinics.
5.1. Preoperative diagnosis
In hepato‐pancreato‐biliary (HPB) cancer, pancreaticoduodenectomy, which has a high mortality rate, is needed for cure, and surgeons would prefer to definitively confirm cancer preoperatively. However, biopsy samples of HPB tumors cannot be obtained directly without invasive procedures because tumors of this type are located off the luminal mucosa. Cytological testing of the ERCP wash solution is currently the only method in a less invasive manner diagnose the tumors preoperatively.
In pancreatic cancer, 4 sequential case‐control studies (discovery, technical validation, biological validation and clinical piloting) were conducted to demonstrate the diagnostic utility of HRMG analysis of CD1D (encoding a member of the CD1 family of transmembrane glycoproteins) methylation. Results of those studies showed that CD1D methylation in the pancreatic juice yielded an AUC value of .92 for patients with pancreatic cancer compared to patients with normal pancreas and chronic pancreatitis.53 CD1D methylation in the pancreatic juice detected pancreatic cancer with 75% sensitivity and 95% specificity. That work also reported that CD1D methylation showed an AUC value of .94 in primary pancreatic cancers compared to the corresponding normal tissues.53 Subsequent analyses identified HRMG with higher performance in pancreatic cancer than CD1D, including the GHSR 54 and CDO1 43 genes (Table 1). Liquid biopsy of pancreatic juice using such novel HRMG (or combinations thereof) holds great promise.
In biliary tract cancer (BTC), bile solution was used to detect cancer using CDO1/CNRIP1/SEPT9/VIM methylation.49 These genes were HRMG of BTC, and the combination detection in bile solution showed 85% sensitivity with 95% specificity. The AUC of the combination yielded an AUC of .94, but CDO1, the marker with the best performance, also exhibited an AUC of .93. Liquid biopsy using such HRMG showed superior diagnosis (73%‐91%) and high specificity (95%‐100%) compared to the conventional cytology test (58%‐63%). Recently, additional HRMG with high performance have been identified, including SFRP1,55 OPCML 55 and SHOX2 56 (Table 1), and additional optimization of the HRMG analyses may yield further improvements in performance.
Lung cancer also presents similar issues with regard to preoperative diagnosis. The conventional sputum cytology test shows low sensitivity; however, recent research using HRMG has shown excellent performance in lung cancer diagnosis. The TAC1/HOXA7/SOX17 methylation combination showed 88% sensitivity with 71% specificity (AUC, .89).47 These genes were HRMG in lung cancer and increased methylation was detected in the plasma of lung cancer, but the best discrete combination of the methylated genes (CDO1/TAC1/SOX17) exhibited the highest performance (91% sensitivity with 62% specificity).47 Relatively low specificity is a concern in lung cancer. Ooki et al48 selected a methylation panel of 6 genes (CDO1/HOXA9, AJAP1/PTGDR/UNCX/MARCH11) from the TOGA dataset. Even in serum samples from the stage IA subjects and population‐matched control subjects, the gene panel yielded a sensitivity of 72.1% and a specificity of 71.4%.
In contrast, in gastrointestinal cancer, screening by endoscopy provides excellent diagnosis, and direct biopsy can guarantee the accuracy of preoperative diagnosis. Therefore, a cytological test is being developed for the sake of supplemental information. The identification of HRMG in oral rinse or gastric rinse specimens may be beneficial for the purpose of disease screening. On the other hand, a fecal DNA test providing quantitative molecular assays for K‐ras mutations, aberrant NDRG4 and BMP3 methylation, with reference gene β‐actin was shown to increase the diagnostic accuracy of colonic neoplasms by adding HRMG analysis to the conventional fecal immunochemical test (FIT).57 Colonoscopy is an invasive procedure with a high risk of complications, so a fecal DNA test would be a promising tool for assessment prior to colonoscopy.
In oral cancer, a saliva test using HRMG such as HOXA9/NID2 detected 50% of cancers with 90% specificity.58 This sensitivity was not satisfactory as a liquid biopsy using HRMG, putatively due to inferior discriminating markers of oral SCC cancer (representing AUC of approximately .8 in primary tumors). However, saliva testing using another combination of HRMG (EDNRB/DCC) yielded improved diagnostic accuracy of oral neoplasms including precancerous lesions when combined with professional classification of clinical risk.59
In gynecological and urological cancer research, methylation markers were applied to the testing of cervical scrapings and urine specimens. Both tests are kinds of cytological assays, and various methylation markers showed superior clinical outcomes for diagnosis of the individual cancers as described below. This improved performance was especially notable for prostate cancer, where recurrence was defined as biochemical recurrence by serum PSA. These results demonstrate that biomarkers could have a significant role in disease surveillance, beyond the use of imaging modalities.
Screening by HRMG analysis of cervical scrapings using HRMG was applied to both cervical cancer and endometrial cancer. In cervical SCC, SOX1 and PAX1 methylation have been reported to have 74% sensitivity with 97% specificity in scrapings.60 In combination with the Pap test, the sensitivity reached 89%.61 Early detection of cancer would improve the clinical outcome of less invasive surgical treatments. However, CADM1/MAL combination analysis showed 92% sensitivity compared to CIN3 and 100% sensitivity for cancer.62
The HRMG profile of endometrial cancer is different from that of cervical SCC cancer; however, the combination of HRMG showed more successful performance in the former than in the latter. Specifically, the BHLHE22/CDO1/CELF4 combinations of methylation markers showed 92% sensitivity with 95% specificity in scrapings tests.42 Given that there have been no descriptions of methylation frequencies of primary tumors for assessing whether CDO1 is an HRMG in this disease, that gene was not included as an HRMG for endometrial cancer in Table 1.
In urological cancers as well as in gynecological cancers, DNA diagnosis in urine sediments has been rigorously explored using HRMG analysis. In bladder cancer, urine sediments using Twist1/NID2 methylation showed 90% sensitivity with 95% specificity, compared to 48% sensitivity in conventional urine cytology test.63 In bladder cancer, there is little information regarding the AUC values assessed for both tumor and corresponding normal tissues by Q‐MSP for genes such as CDO1.19
In prostate cancer and renal cell cancer, methylation biomarkers are likely to be promising for early detection of cancer cells. In prostate cancer, the combination of GSTP1/p16/ARF/MGMT methylation in urine sediments showed 87% sensitivity with 100% specificity.64 In prostate cancer, many excellent HRMG have been reported,54, 65 and further optimization is anticipated. The CDO1 methylation level is significantly higher in prostate cancer than in benign prostate hyperplasia or normal prostate tissues; however, neither AUC nor frequencies were reported in that study.41
In gynecological and urological cancer research, comparisons of tumor tissues with the corresponding normal tissues have seldom been investigated, but test samples such as urine sediments or scrapings have been examined directly. To date, it has not been proved whether methylation markers are actually HRMG, although in the present review, the HRMG was defined using AUC of scraping samples. Accurate diagnosis using such HRMG is expected to lead to appropriate treatment strategies including surgery.
5.2. Intraoperative diagnosis
In the abdominal oncology field, the intraoperative peritoneal cytology test is an emerging modality that is used in the staging system. However, histological findings are not sufficient to detect minimal residual disease; molecular markers are anticipated to have great potential in this context. Carcinoembryonic antigen (CEA) mRNA was initially applied to this cytology test, where the technique was able to predict poor prognosis;66 however, mRNA is unstable, and unlikely to be appropriate for the routine test. DNA markers are more stable than mRNA, but there has been (to date) no highly relevant marker.67 Nevertheless, a combination of methylation markers was tested for use in cancer detection in the intraoperative peritoneal cytology test. Testing of methylation at BNIP3/CHFR/CYP1B1/MINT25/SFRP2/RASSF2 showed that 7%‐20% of cases had no peritoneal dissemination, and 75% of those results were consistent with those of the peritoneal cytology test. This multiplex analysis predicted peritoneal dissemination in 33% of the DNA cytology test‐positive cases but in only 3% of the DNA cytology test‐negative cases. This finding suggested that DNA cytology test‐positive results represent viable cancer cell detection in the peritoneum. In short, the results of this analysis confirm the need to use multiplex assays (ie, using gene combinations), because the methylation frequency in the primary tumor tissues was not as high as that in individual genes, and there were cases that yielded conventional cytology test‐positive results but were DNA cytology test‐negative.
Using HRMG analysis, this point could be resolved using a peritoneal cytology test. CDO1 methylation is one of the most frequent aberrations in gastric cancer tissues. A DNA cytology test using CDO1 methylation detected cancer cells in all CY1 cases (100%), and, moreover, it detected cancer cells in 20% of all cases of gastric cancers, a rate that is twice that of the conventional cytology test.36 This DNA cytology test can predict peritoneal recurrence in gastric cancer with type III and type IV gastric cancer. A prospective trial (UMIN000026191) is currently being conducted to confirm the utility of a DNA cytology test using CDO1 methylation in 400 cases of gastric cancer. Table 2 provides a comparison between the clinical features of CDO1 methylation and those of CEA mRNA (cDNA) and methylation combinations in the peritoneal fluid washing cytology test.
Table 2.
Clinical features of CDO1 methylation compared with those of CEA and methylation combinations in peritoneal fluid washing cytology test
| mRNA | Non‐HRMG | CDO1 (HRMG) | |
|---|---|---|---|
| Sample stability | △ | 〇 | 〇 |
| Sensitivity | 23% | 25% | 20% |
| % of CY0 | 10% | 7%‐20% | 10% |
| % of CY1 | 69% | 75% | 100% |
| Marker | CEA | BNIP3/CHFR/CYP1B1/MINT25/SFRP2/RASSF2 | CDO1 |
| Reference | 66 | 67 | 36 |
HRMG, highly relevant methylation gene.
5.3. Pathological diagnosis
The most critical information representing cancer phenotypes must be obtained from the primary cancer tissues. Epigenetic information in the primary tumors should be considered in clinical decisions. Recent comprehensive exploration in large‐scale DNA methylome analyses identified many HRMG representing cancer phenotypes as described above. HRMG has often been linked to crucial cancer phenotypes, because these markers may reflect functional aspects of tumor aggressiveness. HRMG have been demonstrated to be associated with aggressive tumor phenotypes such as lymph node metastasis or distant metastasis and frequently were predictive of dismal prognosis.34, 36, 37, 38, 41, 44 Promoter DNA methylation accumulates with disease progression, and the groups with the highest methylation values showed the poorest prognosis. Among such candidates, several genes would affect therapeutic decisions after surgery.
Separately from methylation of the primary tumors, prognostic utility has been reported as a prognostic factor. This result was obtained in research that focused on minimal residual diseases in lymph nodes pathologically diagnosed as “negative.” Using HRMG for negative lymph nodes in stage I lung cancer, minimal residual disease in lymph nodes was detected, and such patients showed poorer prognosis than the other patients.68 This result indicated that the detected HRMG methylation may represent a micrometastasis of cancer cells in lymph nodes, which were not capable of being discerned by the conventional pathological searches. In stage I lung cancer, postoperative adjuvant chemotherapy was not indicated; however, methylation‐positive cases with the pathology‐negative lymph nodes showed a 70% survival rate, and this patient selection method may be appropriate in candidates for adjuvant chemotherapy. Such a therapeutic strategy could be applied to gastrointestinal cancer in pathology‐negative cases,69 given that there have been no reports describing prognostic significance of such molecular micrometastasis signatures in pathology‐negative lymph nodes.
Methylation genes harboring predictive value for anti‐cancer drug efficacy provide attractive information for use in the development of therapeutic strategies. MGMT methylation in brain tumors showed success as such a marker.70 MGMT methylation was able to predict the chemosensitivity to alkylating agents of brain tumors71, 72 and other cancers.73, 74 Brain tumor cases with MGMT hypermethylation exhibited better postoperative outcomes than those with MGMT hypomethylation. Specifically, patients with MGMT hypermethylation showed greater responsiveness to radiation therapy.75 In the brain tumor clinics, nanogram was useful including this epigenetic information.76
In esophageal cancer, CDO1 methylation was associated with histological grade of neoadjuvant chemotherapy.36 In tumors that were grade 2/3 after neoadjuvant chemotherapy, CDO1 methylation was not detected. The CDO1 gene is an HRMG in esophageal cancer, so the failure to detect CDO1 methylation in grade 2/3 tumors may represent cancer eradication by neoadjuvant chemotherapy. Issues of concern included cases in which genes exhibited hypomethylation before neoadjuvant chemotherapy, and those in which gene methylation status differed when compared before and after chemotherapy; these disparities will need to be investigated to clarify the clinical utility of screening biopsy samples for gene methylation. In this context, it would be beneficial to exclude patients in whom neoadjuvant chemotherapy was not effective, which would eliminate cases in which disease progresses despite neoadjuvant chemotherapy.
6. CONCLUSIONS
Recent research approaches have identified many HRMG in individual human cancers, and these novel emerging epigenetic biomarkers are ready for use in testing liquid biopsies in the clinic. Information about HRMG is expected to permit cancer surgeons to avoid unnecessary operations, to provide early detection of cancer occurrence and cancer recurrence, and to facilitate convenient surveillance with high accuracy in outpatients. These techniques are expected to permit the development of more sophisticated and optimized therapeutic strategies, yielding improved prognosis in cancer patients compared to current therapies. However, the optimization of screens employing known HRMG has not yet been achieved. The advantages of such markers are expected to depend on the specific clinical situations, and much research is ongoing to demonstrate the clinical significance of these approaches. Low levels of cancer‐derived DNA in body fluids can be detected using HRMG analysis with Q‐MSP; however, the use of digital PCR is expected to expand the clinical applicability of HRMG analysis to liquid biopsies. The surgical decisions that are expected to be most immediately affected by epigenetic markers are those relating to indications of adjuvant therapy for stage I lung cancer or glioblastoma, although cancer diagnosis and surveillance also will be improved by the epigenetic markers.
CONFLICT OF INTEREST
There is no conflict of interest in this study.
Yamashita K, Hosoda K, Nishizawa N, Katoh H, Watanabe M. Epigenetic biomarkers of promoter DNA methylation in the new era of cancer treatment. Cancer Sci. 2018;109:3695–3706. 10.1111/cas.13812
REFERENCES
- 1. Du J, Johnson LM, Jacobsen SE, Patel DJ. DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol. 2015;16:519‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nan X, Ng HH, Johnson CA, et al. Transcriptional repression by the methyl‐CpG‐binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386‐389. [DOI] [PubMed] [Google Scholar]
- 3. Harikrishnan KN, Chow MZ, Baker EK, et al. Brahma links the SWI/SNF chromatin‐remodeling complex with MeCP2‐dependent transcriptional silencing. Nat Genet. 2005;37:254‐264. [DOI] [PubMed] [Google Scholar]
- 4. Yamashita K, Upadhyay S, Osada M, et al. Pharmacologic unmasking of epigenetically silenced tumor suppressor genes in esophageal squamous cell carcinoma. Cancer Cell. 2002;2:485‐495. [DOI] [PubMed] [Google Scholar]
- 5. Tokumaru Y, Yamashita K, Osada M, et al. Inverse correlation between cyclin A1 hypermethylation and p53 mutation in head and neck cancer identified by reversal of epigenetic silencing. Cancer Res. 2004;64:5982‐5987. [DOI] [PubMed] [Google Scholar]
- 6. Kim MS, Yamashita K, Baek JH, et al. N‐methyl‐D‐aspartate receptor type 2B is epigenetically inactivated and exhibits tumor‐suppressive activity in human esophageal cancer. Cancer Res. 2006;66:3409‐3418. [DOI] [PubMed] [Google Scholar]
- 7. Yamashita K, Park HL, Kim MS, et al. PGP9.5 methylation in diffuse‐type gastric cancer. Cancer Res. 2006;66:3921‐3927. [DOI] [PubMed] [Google Scholar]
- 8. Yamashita K, Kim MS, Park HL, et al. HOP/OB1/NECC1 promoter DNA is frequently hypermethylated and involved in tumorigenic ability in esophageal squamous cell carcinoma. Mol Cancer Res. 2008;6:31‐41. [DOI] [PubMed] [Google Scholar]
- 9. Ooki A, Yamashita K, Kikuchi S, et al. Potential utility of HOP homeobox gene promoter methylation as a marker of tumor aggressiveness in gastric cancer. Oncogene. 2010;29:3263‐3275. [DOI] [PubMed] [Google Scholar]
- 10. Cheung WK, Zhao M, Liu Z, et al. Control of alveolar differentiation by the lineage transcription factors GATA6 and HOPX inhibits lung adenocarcinoma metastasis. Cancer Cell. 2013;23:725‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yamashita K, Katoh H, Watanabe M. The homeobox only protein homeobox (HOPX) and colorectal cancer. Int J Mol Sci. 2013;14:23231‐23243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ren X, Yang X, Cheng B, et al. HOPX hypermethylation promotes metastasis via activating SNAIL transcription in nasopharyngeal carcinoma. Nat Commun. 2017;8:14053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Takeda N, Jain R, LeBoeuf MR, Wang Q, Lu MM, Epstein JA. Interconversion between intestinal stem cell populations in distinct niches. Science. 2011;334:1420‐1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takeda N, Jain R, Leboeuf MR, et al. Hopx expression defines a subset of multipotent hair follicle stem cells and a progenitor population primed to give rise to K6 + niche cells. Development. 2013;140:1655‐1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barkauskas CE, Cronce MJ, Rackley CR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123:3025‐3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou X, Crow AL, Hartiala J, et al. The genetic landscape of hematopoietic stem cell frequency in mice. Stem Cell Reports. 2015;5:125‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Loh KM, Chen A, Koh PW, et al. Mapping the pairwise choices leading from pluripotency to human bone, heart, and other mesoderm cell types. Cell. 2016;166:451‐467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ooki A, Yamashita K, Yamaguchi K, Mondal A, Nishimiya H, Watanabe M. DNA damage‐inducible gene, reprimo functions as a tumor suppressor and is suppressed by promoter methylation in gastric cancer. Mol Cancer Res. 2013;11:1362‐1374. [DOI] [PubMed] [Google Scholar]
- 19. Brait M, Ling S, Nagpal JK, et al. Cysteine dioxygenase 1 is a tumor suppressor gene silenced by promoter methylation in multiple human cancers. PLoS One. 2012;7:e44951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hoque MO, Kim MS, Ostrow KL, et al. Genome‐wide promoter analysis uncovers portions of the cancer methylome. Cancer Res. 2008;68:2661‐2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yokoi K, Yamashita K, Watanabe M. Analysis of DNA methylation status in bodily fluids for early detection of cancer. Int J Mol Sci. 2017;18pii:E735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Katoh H, Yamashita K, Waraya M, et al. Epigenetic silencing of HOPX promotes cancer progression in colorectal cancer. Neoplasia. 2012;14:559‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation‐specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821‐9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Diehl F, Li M, Dressman D, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci USA. 2005;102:16368‐16373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Toyota M, Ahuja N, Ohe‐Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA. 1999;96:8681‐8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Toyota M, Ahuja N, Suzuki H, et al. Aberrant methylation in gastric cancer associated with the CpG island methylator phenotype. Cancer Res. 1999;59:5438‐5442. [PubMed] [Google Scholar]
- 27. Yamashita K, Sakuramoto S, Watanabe M. Genomic and epigenetic profiles of gastric cancer: potential diagnostic and therapeutic applications. Surg Today. 2011;41:24‐38. [DOI] [PubMed] [Google Scholar]
- 28. Oshimo Y, Oue N, Mitani Y, et al. Frequent epigenetic inactivation of RIZ1 by promoter hypermethylation in human gastric carcinoma. Int J Cancer. 2004;110:212‐218. [DOI] [PubMed] [Google Scholar]
- 29. Ishimoto T, Nagano O, Yae T, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(‐) and thereby promotes tumor growth. Cancer Cell. 2011;19:387‐400. [DOI] [PubMed] [Google Scholar]
- 30. Jeschke J, O'Hagan HM, Zhang W, et al. Frequent inactivation of cysteine dioxygenase type 1 contributes to survival of breast cancer cells and resistance to anthracyclines. Clin Cancer Res. 2013;19:3201‐3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deng P, Chen Y, Ji N, et al. Cysteine dioxygenase type 1 promotes adipogenesis via interaction with peroxisome proliferator‐activated receptor gamma. Biochem Biophys Res Commun. 2015;458:123‐127. [DOI] [PubMed] [Google Scholar]
- 32. Zhao X, Deng P, Feng J, et al. Cysteine dioxygenase type 1 inhibits osteogenesis by regulating Wnt signaling in primary mouse bone marrow stromal cells. Sci Rep. 2016;6:19296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dietrich D, Krispin M, Dietrich J, et al. CDO1 promoter methylation is a biomarker for outcome prediction of anthracycline treated, estrogen receptor‐positive, lymph node‐positive breast cancer patients. BMC Cancer. 2010;10:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Minatani N, Waraya M, Yamashita K, et al. Prognostic significance of promoter DNA hypermethylation of cysteine dioxygenase 1 (CDO1) gene in primary breast cancer. PLoS One. 2016;11:e0144862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Andresen K, Boberg KM, Vedeld HM, et al. Novel target genes and a valid biomarker panel identified for cholangiocarcinoma. Epigenetics. 2012;7:1249‐1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ushiku H, Yamashita K, Katoh H, et al. Promoter DNA methylation of CDO1 gene and its clinical significance in esophageal squamous cell carcinoma. Dis Esophagus. 2016;30:1‐9. [DOI] [PubMed] [Google Scholar]
- 37. Kojima K, Yamashita K, Ushiku H, et al. The clinical significance of cysteine dioxygenase type 1 methylation in Barrett esophagus adenocarcinoma. Dis Esophagus. 2017;30:1‐9. [DOI] [PubMed] [Google Scholar]
- 38. Kojima K, Nakamura T, Ohbu M, et al. Cysteine dioxygenase type 1 (CDO1) gene promoter methylation during adenoma‐carcinoma sequence in colorectal cancer. PLoS One. 2018;13:e0194785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Feber A, Arya M, de Winter P, et al. Epigenetics markers of metastasis and HPV‐induced tumorigenesis in penile cancer. Clin Cancer Res. 2015;21:1196‐1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Deckers IA, Schouten LJ, Van Neste L, et al. Promoter methylation of CDO1 identifies clear‐cell renal cell cancer patients with poor survival outcome. Clin Cancer Res. 2015;21:3492‐3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Meller S, Zipfel L, Gevensleben H, et al. CDO1 promoter methylation is associated with gene silencing and is a prognostic biomarker for biochemical recurrence‐free survival in prostate cancer patients. Epigenetics. 2016;11:871‐880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huang RL, Su PH, Liao YP, et al. Integrated epigenomics analysis reveals a DNA methylation panel for endometrial cancer detection using cervical scrapings. Clin Cancer Res. 2017;23:263‐272. [DOI] [PubMed] [Google Scholar]
- 43. Nishizawa N, Yamashita K, Ishii S, et al. Potential utility of cysteine dioxygenase 1 gene promoter methylation as a marker of tumor diagnosis in pancreatic adenocarcinoma. 2017. In: 70th Society for Surgical Oncology (SSO) Annual Cancer Meeting Symposium, PT170.
- 44. Igarashi K, Yamashita K, Kumomoto Y, et al. Prognostic significance of promoter DNA hypermethylation of cysteine dioxygenase 1 (CDO1) gene in primary gallbladder cancer and gallbladder diseases. PLoS One. 2017;12:e0188178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yamashita K, Waraya M, Kim MS, et al. Detection of methylated CDO1 in plasma of colorectal cancer; a PCR study. PLoS One. 2014;9:e113546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li M, Chen WD, Papadopoulos N, et al. Sensitive digital quantification of DNA methylation in clinical samples. Nat Biotechnol. 2009;27:858‐863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hulbert A, Jusue‐Torres I, Stark A, et al. Early detection of lung cancer using DNA promoter hypermethylation in plasma and sputum. Clin Cancer Res. 2016;23:1998‐2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ooki A, Maleki Z, Tsay JJ, et al. A panel of novel detection and prognostic methylated DNA markers in primary non‐small cell lung cancer and serum DNA. Clin Cancer Res. 2017;23:7141‐7152. [DOI] [PubMed] [Google Scholar]
- 49. Andresen K, Boberg KM, Vedeld HM, et al. Four DNA methylation biomarkers in biliary brush samples accurately identify the presence of cholangiocarcinoma. Hepatology. 2015;61:1651‐1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ushiku H, Yamashita K, Ema A, et al. DNA diagnosis of peritoneal fluid cytology test by CDO1 promoter DNA hypermethylation in gastric cancer. Gastric Cancer. 2017;20:784‐792. [DOI] [PubMed] [Google Scholar]
- 51. Katoh H, Yamashita K, Sato T, Ozawa H, Nakamura T, Watanabe M. Prognostic significance of peritoneal tumour cells identified at surgery for colorectal cancer. Br J Surg. 2009;96:769‐777. [DOI] [PubMed] [Google Scholar]
- 52. Simojoki M, Santala M, Vuopala S, Kauppila A. The prognostic value of peritoneal cytology in ovarian cancer. Eur J Gynaecol Oncol. 1999;20:357‐360. [PubMed] [Google Scholar]
- 53. Kisiel JB, Raimondo M, Taylor WR, et al. New DNA methylation markers for pancreatic cancer: discovery, tissue validation, and pilot testing in pancreatic juice. Clin Cancer Res. 2015;21:4473‐4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Moskalev EA, Jandaghi P, Fallah M, et al. GHSR DNA hypermethylation is a common epigenetic alteration of high diagnostic value in a broad spectrum of cancers. Oncotarget. 2015;6:4418‐4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Amornpisutt R, Proungvitaya S, Jearanaikoon P, Limpaiboon T. DNA methylation level of OPCML and SFRP1: a potential diagnostic biomarker of cholangiocarcinoma. Tumour Biol. 2015;36:4973‐4978. [DOI] [PubMed] [Google Scholar]
- 56. Branchi V, Schaefer P, Semaan A, et al. Promoter hypermethylation of SHOX2 and SEPT9 is a potential biomarker for minimally invasive diagnosis in adenocarcinomas of the biliary tract. Clin Epigenetics. 2016;8:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Quintero E, Castells A, Bujanda L, et al. Colonoscopy versus fecal immunochemical testing in colorectal‐cancer screening. N Engl J Med. 2012;366:697‐706. [DOI] [PubMed] [Google Scholar]
- 58. Guerrero‐Preston R, Soudry E, Acero J, et al. NID2 and HOXA9 promoter hypermethylation as biomarkers for prevention and early detection in oral cavity squamous cell carcinoma tissues and saliva. Cancer Prev Res (Phila). 2011;4:1061‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schussel J, Zhou XC, Zhang Z, et al. EDNRB and DCC salivary rinse hypermethylation has a similar performance as expert clinical examination in discrimination of oral cancer/dysplasia versus benign lesions. Clin Cancer Res. 2013;19:3268‐3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lai HC, Lin YW, Huang RL, et al. Quantitative DNA methylation analysis detects cervical intraepithelial neoplasms type 3 and worse. Cancer. 2010;116:4266‐4274. [DOI] [PubMed] [Google Scholar]
- 61. Kan YY, Liou YL, Wang HJ, et al. PAX1 methylation as a potential biomarker for cervical cancer screening. Int J Gynecol Cancer. 2014;24:928‐934. [DOI] [PubMed] [Google Scholar]
- 62. van Baars R, van der Marel J, Snijders PJ, et al. CADM1 and MAL methylation status in cervical scrapes is representative of the most severe underlying lesion in women with multiple cervical biopsies. Int J Cancer. 2016;138:463‐471. [DOI] [PubMed] [Google Scholar]
- 63. Renard I, Joniau S, van Cleynenbreugel B, et al. Identification and validation of the methylated TWIST1 and NID2 genes through real‐time methylation‐specific polymerase chain reaction assays for the noninvasive detection of primary bladder cancer in urine samples. Eur Urol. 2010;58:96‐104. [DOI] [PubMed] [Google Scholar]
- 64. Hoque MO, Topaloglu O, Begum S, et al. Quantitative methylation‐specific polymerase chain reaction gene patterns in urine sediment distinguish prostate cancer patients from control subjects. J Clin Oncol. 2005;23:6569‐6575. [DOI] [PubMed] [Google Scholar]
- 65. Yegnasubramanian S, Kowalski J, Gonzalgo ML, et al. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res. 2004;64:1975‐1986. [DOI] [PubMed] [Google Scholar]
- 66. Kodera Y, Nakanishi H, Ito S, et al. Quantitative detection of disseminated free cancer cells in peritoneal washes with real‐time reverse transcriptase‐polymerase chain reaction: a sensitive predictor of outcome for patients with gastric carcinoma. Ann Surg. 2002;235:499‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hiraki M, Kitajima Y, Koga Y, et al. Aberrant gene methylation is a biomarker for the detection of cancer cells in peritoneal wash samples from advanced gastric cancer patients. Ann Surg Oncol. 2011;18:3013‐3019. [DOI] [PubMed] [Google Scholar]
- 68. Brock MV, Hooker CM, Ota‐Machida E, et al. DNA methylation markers and early recurrence in stage I lung cancer. N Engl J Med. 2008;358:1118‐1128. [DOI] [PubMed] [Google Scholar]
- 69. Hiraki M, Kitajima Y, Sato S, et al. Aberrant gene methylation in the lymph nodes provides a possible marker for diagnosing micrometastasis in gastric cancer. Ann Surg Oncol. 2010;17:1177‐1186. [DOI] [PubMed] [Google Scholar]
- 70. Esteller M, Garcia‐Foncillas J, Andion E, et al. Inactivation of the DNA‐repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343:1350‐1354. [DOI] [PubMed] [Google Scholar]
- 71. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997‐1003. [DOI] [PubMed] [Google Scholar]
- 72. Wick W, Platten M, Meisner C, et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA‐08 randomised, phase 3 trial. Lancet Oncol. 2012;13:707‐715. [DOI] [PubMed] [Google Scholar]
- 73. Amatu A, Sartore‐Bianchi A, Moutinho C, et al. Promoter CpG island hypermethylation of the DNA repair enzyme MGMT predicts clinical response to dacarbazine in a phase II study for metastatic colorectal cancer. Clin Cancer Res. 2013;19:2265‐2272. [DOI] [PubMed] [Google Scholar]
- 74. Barault L, Amatu A, Bleeker FE, et al. Digital PCR quantification of MGMT methylation refines prediction of clinical benefit from alkylating agents in glioblastoma and metastatic colorectal cancer. Ann Oncol. 2015;26:1994‐1999. [DOI] [PubMed] [Google Scholar]
- 75. Stupp R, Hegi M, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5‐year analysis of the EORTC‐NCIC trial. Lancet Oncol. 2009;10:459‐466. [DOI] [PubMed] [Google Scholar]
- 76. Gorlia T, van den Bent MJ, Hegi ME, et al. Nomograms for predicting survival of patients with newly diagnosed glioblastoma: prognostic factor analysis of EORTC and NCIC trial 26981‐22981/CE.3. Lancet Oncol. 2008;9:29‐38. [DOI] [PubMed] [Google Scholar]