

Abstract
The p53‐inducible gene 3 (PIG3) is one of the p53‐induced genes at the onset of apoptosis, which plays an important role in cell apoptosis and DNA damage response. Our previous study reported an oncogenic role of PIG3 associated with tumor progression and metastasis in non‐small cell lung cancer (NSCLC). In this study, we further analyzed PIG3 mRNA expression in 504 lung adenocarcinoma (LUAD) and 501 lung squamous cell carcinoma (LUSC) tissues from The Cancer Genome Atlas database and we found that PIG3 expression was significantly higher in LUAD with lymph node metastasis than those without, while no difference was observed between samples with and without lymph node metastasis in LUSC. Gain and loss of function experiments were performed to confirm the metastatic role of PIG3 in vitro and to explore the mechanism involved in its oncogenic role in NSCLC metastasis. The results showed that PIG3 knockdown significantly inhibited the migration and invasion ability of NSCLC cells, and decreased paxillin, phospho‐focal adhesion kinase (FAK) and phospho‐Src kinase expression, while its overexpression resulted in the opposite effects. Blocking FAK with its inhibitor reverses PIG3 overexpression‐induced cell motility in NSCLC cells, indicating that PIG3 increased cell metastasis through the FAK/Src/paxillin pathway. Furthermore, PIG3 silencing sensitized NSCLC cells to FAK inhibitor. In conclusion, our data revealed a role for PIG3 in inducing LUAD metastasis, and its role as a new FAK regulator, suggesting that it could be considered as a novel prognostic biomarker or therapeutic target in the treatment of LUAD metastasis.
Keywords: focal adhesion kinase, invasion, migration, non‐small cell lung cancer, p53‐inducible gene 3
1. INTRODUCTION
Lung cancer is one of the most commonly diagnosed tumors and a leading cause of cancer‐related death globally. Approximately 222 500 newly diagnosed lung cancer cases and 155 870 lung cancer deaths in the United States were estimated in 2017,1 accounting for almost one‐seventh of all new cancer diagnoses and one‐quarter of all cancer deaths. In China, the situation is even worse due to severe air pollution since 2010.2 Indeed, lung cancer became the first cause of cancer death among both men and women in China in 2015, as predicted by the National Cancer Center.3 Non‐small cell lung cancer (NSCLC) is one of the most aggressive malignant diseases. The subtypes of NSCLC include lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC) and large cell carcinoma. It accounts for nearly 80% of lung cancer‐related death and is characterized by frequent and extensive metastasis. Despite significant therapeutic advances made in early diagnosis,4, 5 chemotherapy approaches and targeted therapeutics,6, 7 lung cancer patients’ 5‐year survival rate remains very low, especially for the invasive and metastatic NSCLC.8 Therefore, the identification of novel biomarkers that can predict NSCLC invasiveness and metastasis and the development of new strategies targeting cancer cell migration and invasion are urgently needed.
The p53‐inducible gene 3 (PIG3 or TP53I3) is one of the p53‐induced genes involved in apoptosis. PIG3 plays an important role in cell apoptosis through reactive oxygen species (ROS) generation and consequent oxidative stress induction.9 A recent study further revealed the role of PIG3 in DNA damage response: suppression of PIG3 expression sensitizes cells to DNA damage agents and reduces DNA repair.10 Given its relationship with cell apoptosis and DNA damage response, the role of PIG3 in cancer development and progression has attracted substantial attention in recent 10 years, and has become the subject of debate. For example, Zhang et al. report that PIG3 expression is positively correlated with the overall survival (OS) rate in breast cancer patients,11 while Xu J et al. demonstrate that PIG3 is aberrantly overexpressed and plays an oncogenic role in papillary thyroid carcinoma.12 Similarly, our previous research revealed that PIG3 expression was positively associated with tumor size, differentiation degree, pathological stage and distant metastasis, and negatively correlated with OS and disease‐free survival in NSCLC patients,13 suggesting an oncogenic role of PIG3 in NSCLC progression. Despite its role in cancer progression, the role of PIG3 in cancer metastasis remains controversial. Park et al. report that PIG3 silencing suppresses cell migration and invasion ability of HCT116 colon cancer cells,14 while in glioblastoma cells PIG3 inhibits cancer cell migration and invasion through matrix metalloproteinases inhibition.15 PIG3 also impairs tumor invasion in melanoma by suppressing Rho‐ROCK activity.16 Moreover, another group has reported that PIG3 deficiency promotes renal cancer cell migration through activation of the hypoxia‐inducible factor‐1‐vascular endothelial growth factor signal pathway.17 However, the effect of PIG3 on NSCLC invasion and metastasis is not yet fully understood.
Therefore, in the present study, we further examined the role of PIG3 in NSCLC metastasis in vitro and its mechanism of action. Our results showed that PIG3 expression was significantly higher in LUAD with lymph node metastasis than that in tumors without lymph node metastasis. To determine the role of PIG3 on NSCLC metastasis, gain and loss of function experiments were performed, showing that PIG3 knockdown significantly inhibited the migration and invasion ability of NSCLC cells, while its overexpression increased the above mentioned abilities. The underlying mechanism of PIG3 in NSCLC metastasis was further explored. Results showed that the phospho‐focal adhesion kinase (FAK)/phospho‐Src kinase/paxillin pathway was involved in PIG3‐mediated cell metastasis. Moreover, suppression of PIG3 increased the sensitivity of NSCLC cells to the FAK inhibitors PF‐573228 and PF‐562271.
2. MATERIALS AND METHODS
2.1. Patients and tissue specimens
Thirty‐seven fresh NSCLC tissue specimens were provided by the Nanjing Medical University Affiliated Suzhou Hospital (Suzhou, China). They were obtained immediately after surgical resection and stored at −80°C for further analysis. Clinicopathological parameters were collected and listed in Table S1. Among the 37 patients included in this study, 20 were male and 17 were female. Patients’ were aged from 35 to 79 years (mean age = 62.76 ± 12.02 years) at the time of operation. As regards lymph node metastasis of the NSCLC, 13 patients presented with lymph node metastasis while 24 did not show any lymph node metastasis. The present study was approved by the Ethics Committee of Nanjing Medical University Affiliated Suzhou Hospital. All patients signed a written informed consent.
2.2. RNA isolation and real‐time quantitative PCR assay
Total RNA from NSCLC samples was extracted using RNAiso Plus (TAKARA, Otsu, Japan) according to the manufacturer's protocol. RNA was quantified using NanoDrop ND‐1000 (Thermo Scientific, Waltham, MA, USA). cDNA was obtained using PrimeScript RT Reagent Kit (Perfect Real Time) (TAKARA, Otsu, Japan) following the manufacturer's instructions. SYBR Green Real‐Time PCR assay kit (TAKARA, Otsu, Japan) was used for cDNA amplification. PIG3 and GAPDH mRNA levels were measured by real‐time quantitative PCR (qPCR) in triplicate using a 7500 Real‐Time PCR System (Applied Biosystems, Foster City, CA, USA) and the following primers: PIG3, 5′‐ CCATGCAGGACTGAGTGGTG‐3′ (forward), 5′‐ CTGCTCCAAGCTTTTCTGCC‐3′ (reverse); GAPDH, 5′‐CAACTACATGGTCTACATGTTCC‐3′ (forward); 5′‐CAACCTGGTCCTCAGTGTAG‐3′ (reverse). GAPDH was used as the internal control. The relative PIG3 gene expression was calculated using the 2−ΔΔCT method.
2.3. PIG3 expression from human databases
PIG3 gene expression from 504 LUAD and 501 LUSC samples from The Cancer Genome Atlas (TCGA) database (http://cancergenome.nih.gov/) were used to analyze PIG3 expression in NSCLC. Normalized mRNA expression was analyzed as described in the subsection “Statistical analysis”.
2.4. Immunohistochemistry
PIG3 localization was evaluated by immunohistochemistry (IHC) as previously described.18 Rabbit anti‐human PIG3 polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used at a 100‐fold dilution.
2.5. Cell culture and transfection
NSCLC cell lines A549 and H1299 were kindly provided by Hongying Yang from the Medical College of Soochow University (Suzhou, China). Cells were maintained in RPMI‐1640 medium supplemented with 10% FBS (Biological Industries, Cromwell, CT, USA) in a humidified 5% CO2 incubator at 37°C.
To silence the expression of endogenous PIG3 in A549 cells, PIG3‐special siRNA (siPIG3) (#1: 5′‐AAAUGUUCAGGCUGGAGACUATT‐3′; #2: 5′‐GGAAGUCUGAUCACCAGUUTT‐3′) or non‐targeting control siRNA (siNC) (5′‐UUCUCCGAACGUGUCACGUTT‐3′) were synthesized by GenePharm (Shanghai, China). A549 cells were seeded in 3.5 cm culture dishes and transiently transfected using 20 μmol/L siPIG3 or siNC in Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) following the manufacturer's instructions. To overexpress PIG3 in H1299 cells, PIG3 constructs were generated by cloning PCR‐amplified full‐length human PIG3 cDNA into a pCMV‐TAG‐2B vector as described by Li B et al.19 H1299 cells were seeded in 3.5 cm culture dishes and transiently transfected with 5 μg PIG3 constructs (PIG3) or empty vector plasmids (pCMV) using Lipofectamine 3000. Cells were harvested 48 hours after transfection, and total cell protein was extracted to verify PIG3 overexpression by western blot analysis. To evaluate if FAK pathway is involved in the mechanism of PIG3‐mediated metastasis, FAK inhibitors such as PF‐573228 and PF‐562271 (Selleckchem, Houston, TX, USA) were used.
2.6. Protein extraction and western blot analysis
Protein extraction and western blot were performed according to published methods.18 The following primary antibodies were used: rabbit anti‐human PIG3 polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti‐human p‐FAK (Tyr397) monoclonal antibody, rabbit anti‐human p‐FAK (Tyr576) polyclonal antibody, rabbit anti‐human p‐FAK (Tyr925) polyclonal antibody, rabbit anti‐human total FAK monoclonal antibody, rabbit anti‐human p‐Src (Tyr416) monoclonal antibody, rabbit anti‐human total Src monoclonal antibody (Cell Signaling Technology, Beverly, MA, USA) and mouse anti‐human GAPDH monoclonal antibody (Beyotime Institute of Biotechnology, Haimen, China) as the loading control. All primary antibodies were used at a 1000‐fold dilution.
2.7. Wound‐healing assay
Non‐small cell lung cancer cells transfected with siRNA or PIG3 constructs were seeded into 6‐well plates. Confluent cell monolayers were scratched using a sterile 200‐μL pipette tip, followed by removal of the supernatant and addition of fresh medium containing 2% FBS. After 24 hours, cells that migrated into the wounded region were observed by an inverted microscope (Olympus, Tokyo, Japan) and photographed. The analysis of the wound‐healing assay was performed using the Image J software (National Institutes of Health, Bethesda, MD, USA).
2.8. Transwell assay
The transwell assay was carried out as previously described20 and performed using Transwell insert chambers (Corning Incorporated, Corning, NY, USA). Briefly, the top chambers were pre‐coated with 50 μL Matrigel for 1 hour, then 5 × 104 cells transfected with siRNA or PIG3 constructs were seeded into the top chambers. An amount of 500 μL RPMI‐1640 medium containing 10% FBS was added into the bottom chambers. After 24 hours, cells that invaded the bottom chambers were fixed in ice‐cold methanol for 30 minutes followed by staining with 2% crystal violet in methanol for 15 minutes. Finally, they were counted under an inverted microscope (Olympus, Tokyo, Japan) and photographed.
2.9. In vivo cell metastasis experiment
Six‐week‐old male NOD/SCID mice were purchased from Beijing Vital River Laboratory Animal Technology (Beijing, China), and maintained in the SPF animal laboratory, Soochow University. A549 cells stably expressing GFP (A549‐GFP) were generously provided by Dr Shitao Zou at Nanjing Medical University Affiliated Suzhou Hospital (Suzhou, China). A549‐GFP cells were transiently transfected with 20 μmol/L siPIG3 #1 or siNC using Lipofectamine 3000, harvested 24 hours post‐transfection and re‐suspended with normal saline at a concentration of 2 × 106 cells/mL. A volume of .1 mL of suspended cells was intravenously injected into the tail vein of each mouse (n = 5 per group). After 24 hours and 30 days, A549 cell colonization in the NOD/SCID mouse organs was photographed using the Kodak in vivo imaging system. All photos were taken under the same conditions. The intensity of the relative fluorescence in the tissues was measured using the Image J analysis software. The design and implementation of the study were approved by the Soochow University institutional Animal Care and Use Committee.
2.10. Immunofluorescence assay
Cells were plated on polylysine‐coated slides. After a specific culture time, cells were fixed in 4% paraformaldehyde/PBS at room temperature for 30 minutes. Then, the cells were permeabilized with .5% Triton X‐100/PBS at room temperature for 15 minutes. Next, slides were blocked with 1% BSA/PBS at room temperature for 30 minutes. Slides were incubated with rabbit anti‐human paxillin, rabbit anti‐human p‐FAK (Tyr397) or rabbit anti‐human p‐FAK (Tyr925) antibodies (Cell Signaling Technology, Beverly, MA, USA) diluted 1000‐fold for 4 hours at room temperature. After primary antibody incubation, slides were incubated with Alexa‐488 conjugated anti‐rabbit secondary antibodies (Invitrogen, Carlsbad, CA, USA) for 1 hour at 37°C. The mounting medium containing DAPI (Vector Laboratories, Burlingame, CA, USA) was used to visualize the nucleus. Images were obtained using a laser‐scanning confocal microscope (FV1200; Olympus, Tokyo, Japan).
2.11. Single cell tracking assay
A549 cells transfected with siPIG3 were seeded in confocal dishes. The single cell tracking assay was performed using the living cells workstation (Olympus, Tokyo, Japan). Cells were continually monitored for 6 hours.
2.12. Cell viability assay
Cell viability was evaluated using the Cell Counting Kit‐8 (CCK‐8; Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer's instructions. A549 cells transfected with siPIG3 were seeded in a 96‐well plate at a density of 2000 cells per well. Cells were then treated with different concentrations (0, 1.25, 2.5, 5, 10 and 20 μmol/L) of FAK inhibitors PF‐573228 or PF‐562271. After 72 hours incubation, 10 μL CCK‐8 solution was added into the 96‐well plate. Two hours later, absorbance at 450 nm was measured using a Microplate Reader (VersaMax; BioTek, Winooski, VT, USA).
2.13. Statistical analysis
The Mann‐Whitney U test was performed for analyzing the significance of the difference in PIG3 expression at different levels of lymph node metastasis. Spearman's test was performed for analyzing the correlation of PIG3 and lymph node metastasis. Student's t‐test was used for 2‐group comparison and one‐way ANOVA was performed for multiple comparisons. Statistical analyses were performed using SPSS 16.0 software (SPSS, Chicago, IL, USA). All tests were 2‐sided, and differences were considered statistically significant when P < .05.
3. RESULTS
3.1. PIG3 increased expression is associated with lymph node metastasis from non‐small cell lung cancer
To evaluate the potential role of PIG3 in NSCLC metastasis, PIG3 mRNA and protein expression in NSCLC tumor samples were detected by IHC staining and real‐time qPCR (Figure 1A and B). Consistent with our previous study, NSCLC patients with lymph node metastasis had higher PIG3 mRNA expression in comparison with those without (P = .039, Figure 1B). We further analyzed the expression of PIG3 mRNA in NSCLC tissues by using the publicly available database TCGA and found that PIG3 gene expression was increased in LUAD with lymph node metastasis compared with that without lymph node metastasis (P = .0034, Figure 1C), while it was not increased in LUSC with lymph node metastasis (P = .4054, Figure 1D). As the Mann‐Whitney U test and Spearman's test indicated, PIG3 expression was positively associated with lymph node metastasis from LUAD. In other words, LUAD patients with high PIG3 expression had a higher metastatic risk in comparison with those with low PIG3 expression (P = .001), suggesting that PIG3 might represent an auxiliary diagnostic element for lymph node metastasis in LUAD. Because PIG3 expression in lymph node metastasis from LUAD and LUSC was significantly different, PIG3 may be used as an additional diagnostic marker to discriminate between different NSCLC subtypes. Collectively, these findings suggested that PIG3 could be used to diagnose lymph node metastasis and to classify NSCLC subtypes carried by the patients.
Figure 1.
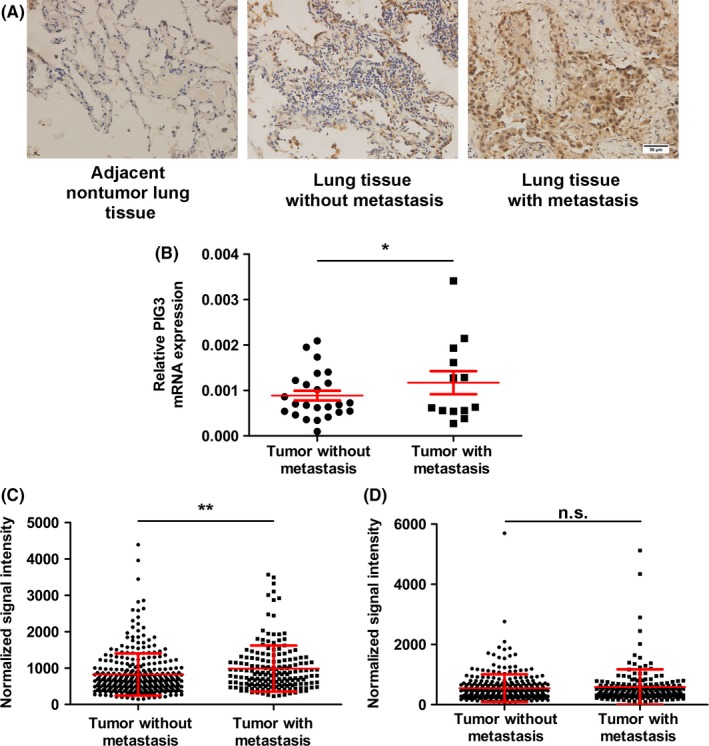
PIG3 is upregulated in samples from NSCLC patients with metastasis. A, Representative images of PIG3 expression in adjacent non‐tumor lung tissue and lung cancer tissue with or without metastasis detected by IHC. Scale bar = 50 μm. B, A dot plot showing PIG3 mRNA expression in NSCLC patients with (n = 13) or without (n = 24) lymph node metastasis detected by real‐time quantitative PCR. Data were presented as mean ± SEM (*P < .05). PIG3 expression in 504 lung adenocarcinoma (LUAD) (C) and 501 lung squamous cell carcinoma (LUSC) (D) tissues with or without metastasis using normalized PIG3 mRNA expression data from the TCGA database. Data were presented as mean ± SEM (**P < .01)
3.2. PIG3 dysregulation affects non‐small cell lung cancer cell migration
To determine the role of PIG3 on NSCLC metastasis, we performed the gain and loss of function experiments in vitro. Our preliminary results demonstrated that A549 cells possessed the highest PIG3 protein expression, while H1299 cells showed almost no PIG3 protein expression among all lung cancer cell lines we tested. Thus, we chose these 2 cell lines to perform the gain and loss of function experiments. Two different siRNA constructs targeting PIG3 and a negative control siRNA were synthesized and transfected into A549 cells. Western blot analysis demonstrated that siPIG3 markedly downregulated endogenous PIG3 protein expression compared with siNC (Figure 2A). A wound‐healing assay was performed to further explore the involvement of PIG3 in cell migration. PIG3 silencing significantly suppressed A549 cell migration to the scratched zone, showing 44% and 28% reduction in relative migration distance by siPIG3 #1 and siPIG3 #2 transfected cells, respectively, compared to corresponding siNC‐transfected cells (P < .05, Figure 2B and C). In addition, we continually monitored single cell migration for 6 hours using live image analysis. Representative cell migration tracks for siPIG3 #1 and siNC‐transfected cells are shown in Figure 2G. The mean migration distance of siPIG3‐transfected cells was much shorter than siNC‐transfected cells (P < .05, Figure 2H).
Figure 2.
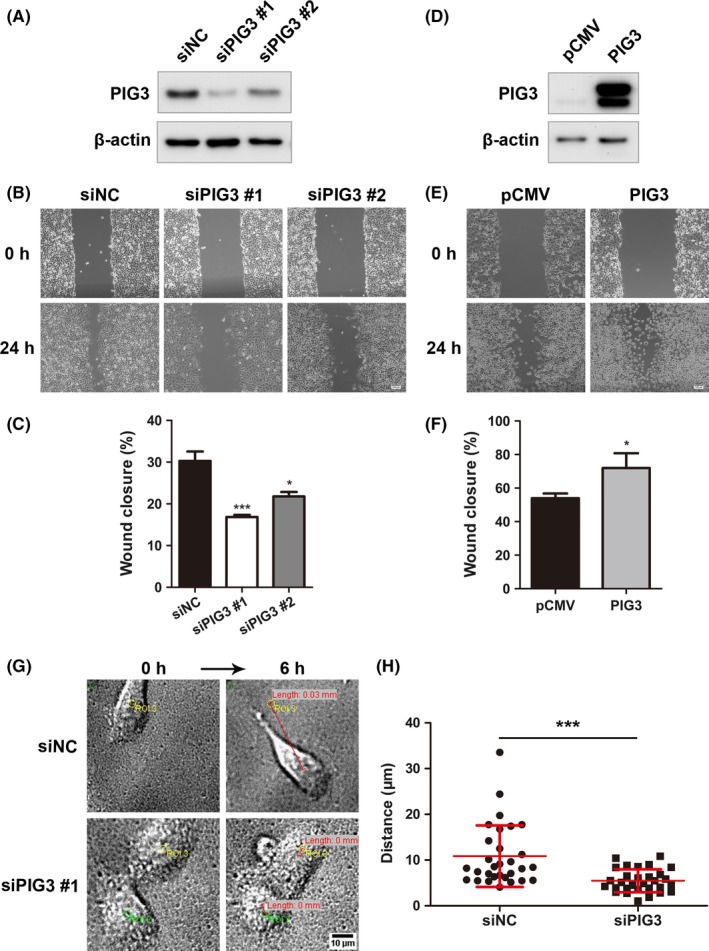
PIG3 promotes non‐small cell lung cancer (NSCLC) cell migration. PIG3 knockdown (A) and overexpression (D) were verified in A549 and H1299 cells by western blot. The cell migration of A549 cells transfected with PIG3‐special siRNA (siPIG3) #1, #2 or non‐targeting control siRNA (siNC) (B) and H1299 cells transfected with PIG3 constructs (PIG3) or empty vector (pCMV) (E) was determined as described in the Materials and Methods. Representative images of the migrated cells are shown. Scale bar = 100 μm. Histogram of relative migration distance of transfected A549 cells (C) and H1299 cells (F) determined by measuring the distance between the scratch. Data were presented as mean ± SD from 3 independent experiments. Compared with the corresponding control, *P < .05; ***P < .001. G, Representative images of single cell migration of A549 cells transfected with siPIG3 #1 or siNC. Scale bar = 10 μm. H, Migration distance of single A549 cell was measured using Image J software. Data were presented as mean ± SD from 3 independent experiments, with at least 30 cells measured in each experiment. ***P < .001 compared with siNC group
In contrast, H1299 cells were transfected with PIG3‐pCMV‐TAG‐2B plasmid and PIG3 overexpression in transfected H1299 cells was verified by western blot (Figure 2D). We further examined the effect of PIG3 overexpression on cell migration using a wound‐healing assay. The results showed that PIG3 upregulation significantly increased H1299 cell migration (53.98 ± 2.81% in pCMV transfected cells vs 71.96 ± 8.79% in PIG3‐pCMV transfected cells, P < .05, Figure 2E and F). These results indicated that PIG3 was crucial in promoting NSCLC cell migration.
3.3. PIG3 dysregulation affects invasion of non‐small cell lung cancer cells
A transwell assay was performed to explore the effect of PIG3 on cancer invasion. Only few siPIG3‐transfected cells were found in the Matrigel‐coated chambers compared to the amount of siNC‐transfected cells (P < .001, Figure 3A and B). In contrast, the invasiveness of H1299 cells was dramatically increased by PIG3 upregulation, as shown in Figure 3C and D (P < .001). These results indicated that PIG3 was essential in enhancing NSCLC cell invasion ability.
Figure 3.
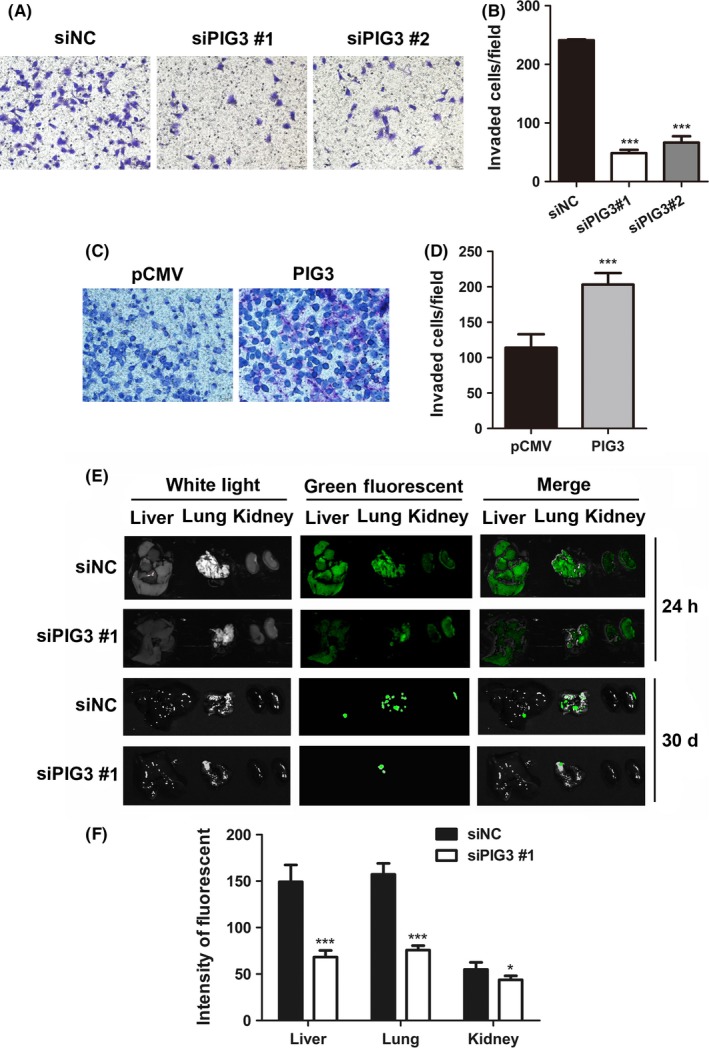
PIG3 promotes non‐small cell lung cancer (NSCLC) cell invasion in vitro and metastasis in vivo. The cell invasion of A549 cells transfected with PIG3‐special siRNA (siPIG3) #1, #2 or non‐targeting control siRNA (siNC) (A) and H1299 cells transfected with PIG3 constructs (PIG3) or empty vector (pCMV) (C) was determined as described in the Materials and Methods. Representative images of the invading cells are shown (200× magnification). Histogram of invading cells per field of transfected A549 cells (B) and H1299 cells (D) determined by counting the invading cells in 5 random fields. Data were presented as mean ± SD from 3 independent experiments (***P < .001). E, A549‐GFP cells were injected into NOD/SCID mice via the tail vein. After 24 h and 30 d, A549 cell colonization in the mice liver, lung and kidney was visualized using an in vivo imaging system. All photos were taken under the same conditions. F, The relative green fluorescence intensity in the indicated group of tissues was calculated using the Image J analysis software (*P < .05, ***P < .001)
3.4. PIG3 downregulation inhibits non‐small cell lung cancer cell metastasis in vivo
To validate the effect of PIG3 on the metastatic potential of A549 cells in vivo, A549‐GFP cells transfected with siPIG3 were injected into NOD/SCID mice via the tail vein. A549 cell colonization in different mouse organs was visualized using an in vivo imaging system at 24 hours and 30 days after inoculation. As shown in Figure 3E and F, PIG3 silenced cells exhibited a weaker green fluorescence signal in mice liver, lung and kidney compared with the fluorescence in the same organs of the control cells. The difference in green fluorescence levels, especially in the lung, was even more pronounced between the 2 groups of mice after 30 days inoculation. These in vivo data demonstrated that PIG3 was capable of promoting NSCLC cell metastasis in vivo.
3.5. PIG3 dysregulation affects focal adhesion
Cell invasion is promoted by focal adhesion, a cellular adhesion to the extracellular matrix (ECM).21 Because integrin‐mediated tethering of actin filaments forms the focal adhesion, we next addressed whether PIG3 affects the focal adhesion. Figure 4 shows the immunofluorescence staining with paxillin, which is a marker of focal adhesion, revealing the detection of focal adhesions around the cells as green foci. PIG3 silencing reduced the number of these foci as compared with siNC‐transfected A549 cells (P < .001, Figure 4A and B). In contrast, the number of stained foci in H1299 cells was dramatically increased after PIG3 upregulation, as shown in Figure 4C and D (P < .001). These observations suggested that PIG3 positively regulates cell invasion by promoting the effective assembly of focal adhesion complexes formation.
Figure 4.
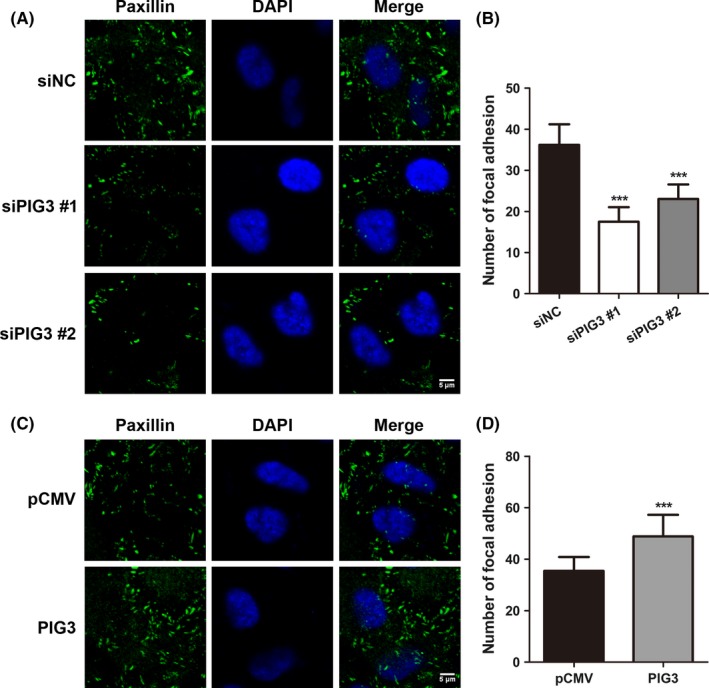
PIG3 positively regulates focal adhesion of non‐small cell lung cancer (NSCLC) cells. A549 cells transfected with control or PIG3 siRNA (A) and H1299 cells transfected with PIG3 constructs (PIG3) or empty vector (pCMV) (C) were stained with paxillin (green) antibody. DAPI (blue) was used to visualize the nucleus. Scale bar = 5 μm. B and D, Numbers of focal adhesions (paxillin dots) per cell were quantified. Data were presented as mean ± SD from 3 independent experiments (***P < .001)
3.6. FAK/Src signaling pathway is involved in PIG3‐promoting cell metastasis
Our results showed that PIG3 had the ability to promote cell metastasis in NSCLC cells. The FAK/Src pathway is known to play a role in tumor metastasis in many tumors types. To elucidate the involvement of the FAK/Src pathway in PIG3‐mediated cell metastasis, western blot analysis and immunofluorescence staining were performed. The results revealed that PIG3 overexpression increased the expression of phosphorylated FAK and phosphorylated Src but not total FAK and Src (Figure 5A and B). In contrast, PIG3 knockdown suppressed the expression of phosphorylated FAK and phosphorylated Src.
Figure 5.
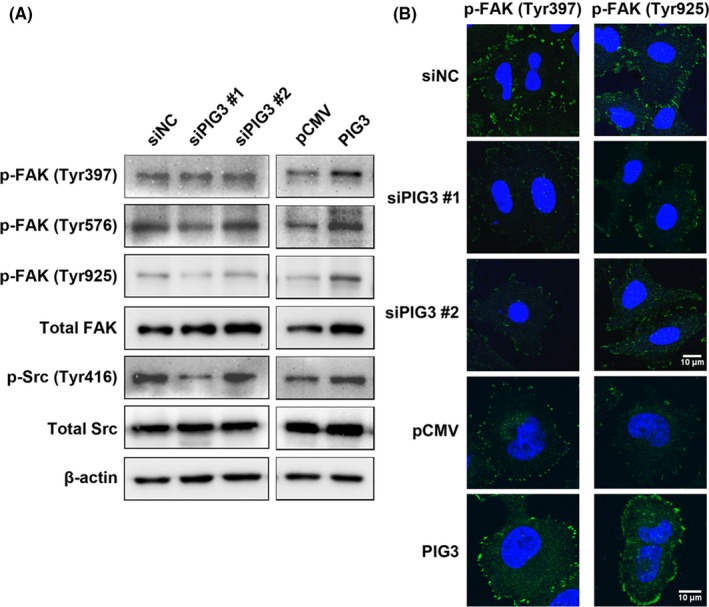
Fak/Src signaling is stimulated by PIG3. A, A549 cells were transfected with PIG3‐special siRNA (siPIG3) #1, #2 or non‐targeting control siRNA (siNC) and H1299 cells were transfected with PIG3 construct (PIG3) or empty vector (pCMV). Then the total cell lysates were subjected to western blot to analyze the phosphorylation of FAK and Src as described in the Materials and Methods. B, Transfected A549 and H1299 cells were fixed and subjected to immunofluorescence staining using p‐FAK (Tyr397) and p‐FAK (Tyr925) primary antibodies, followed by Alexa‐488 conjugated secondary antibodies (green), coupled with DAPI staining (for nucleus, blue). Scale bar = 10 μm
The involvement of FAK/Src pathway in PIG3‐mediated cell metastasis was further confirmed using the specific FAK inhibitor (PF‐573228). H1299 cells transfected with PIG3 constructs and treated with the FAK inhibitor PF‐573228 at different concentrations (0, 5, and 10 μmol/L) for 24 hours resulted in effective dose‐dependent inhibition of FAK phosphorylation (Figure 6A). This treatment and consequent effect on FAK reversed PIG3 overexpression‐induced migration and invasion in H1299 cells (Figure 6B‐E). These results indicated that PIG3 increasing NSCLC metastasis might be due to FAK/Src signaling pathway activation.
Figure 6.
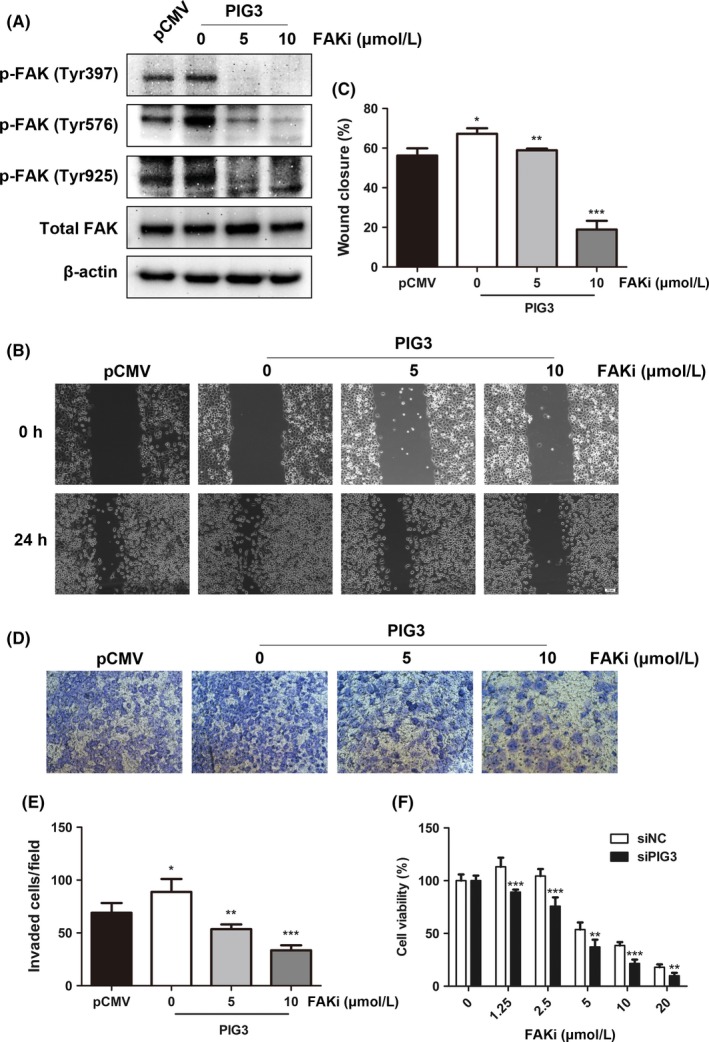
FAK inhibition reverses metastatic characteristics of non‐small cell lung cancer (NSCLC) cells induced by PIG3 overexpression. A, H1299 cells transfected with PIG3 constructs were treated with the FAK inhibitor (FAKi) PF‐573228 at different concentrations (0, 5, 10 μmol/L) for 24 h. Inhibition efficiency of PF‐573228 on FAK phosphorylation was examined by western blot. B, The wound‐healing assay of transfected H1299 cells treated with different concentrations of PF‐573228. Representative images of the migrated cells are shown. Scale bar = 100 μm. C, Histogram of relative migration distance of transfected H1299 cells determined by measuring the distance between the scratch. Data were presented as mean ± SD from 3 independent experiments. *P < .05; **P < .01; ***P < .001 compared with control cells. D, Transwell assay of transfected H1299 cells treated with PF‐573228 at different concentrations. Representative images of the invading cells are shown (200 × magnification). E, Histogram of invading cells per field of transfected H1299 cells determined by counting the invading cells in 5 random fields. Data were presented as mean ± SD from 3 independent experiments (*P < .05; **P < .01; ***P < .001). F, PIG3 silencing sensitizes A549 cells to PF‐573228. Twenty‐four hours after transfection with PIG3 and control siRNA, A549 cells were exposed to PF‐573228 at different concentrations (0, 1.25, 2.5, 5, 10, 20 μmol/L). Cell proliferation was determined by CCK8 assay 72 h post‐treatment. Data were presented as mean ± SD from 3 independent experiments (**P < .01; ***P < .001)
Furthermore, we evaluated whether PIG3 silencing might modulate the sensitivity of NSCLC cells to PF‐573228 and PF‐562271. The cell viability of siPIG3 and siNC‐transfected A549 cells was measured by CCK8 assay at 72 hours after PF‐573228 and PF‐562271 treatment. PIG3 silenced cells were more sensitive to PF‐573228 and PF‐562271 treatment compared with siNC‐transfected cells (Figure 6F and Figure S1).
4. DISCUSSION
Non‐small cell lung cancer patients have extremely high mortality rates. The main reason is that most of the NSCLC patients are already at an advanced metastatic stage when this cancer is diagnosed.22 Consequently, identifying specific biomarkers that predict the onset of metastasis is crucial for improving the outcomes in NSCLC patients. In this study, we found that PIG3 expression was positively associated with lymph node metastasis from LUAD. Moreover, there is a significant difference in the expression of PIG3 in different lymph node metastatic levels between LUAD and LUSC, indicating that PIG3 might be used in the diagnosis of lymph node metastasis and classification in NSCLC patients.
Lung adenocarcinoma is mostly found in young women, and its pathogenesis is not related to smoking, while LUSC is mostly found in men over 50 years old. The onset of LUSC is closely related to long‐term smoking. Our previous research showed that PIG3 expression is significantly and negatively correlated with smoking history (P < .05),13 indicating that non‐smokers are associated with high PIG3 expression while smokers are associated with low PIG3 expression. We speculated that a potential explanation to elucidate the difference in PIG3 expression in lymph node metastasis from LUAD and LUSC might be the smoking history of NSCLC patients. Because LUAD is typically found in young women who are mostly non‐smokers, metastasis can occur in early‐stage LUAD due to high PIG3 expression. LUSC is mostly found in men over 50 years old who are mostly smokers. The metastasis of LUSC patients occurred relatively late due to low PIG3 expression. The leading cause of metastasis in LUSC patients could also depend on other factors. Metastasis is a complex process influenced by many factors.
Our present data showed that both PIG3 mRNA and protein expressions are significantly higher in NSCLC with lymph node metastasis than the expression in the same tumor without lymph node metastasis. PIG3 expression is regulated by the tumor suppressor p53. The PIG3 promoter contains tandem repeats of pentanucleotides (TGYCC)n that is known as a p53 binding site.23 The induction of PIG3 by p53 is affected by mutation and post‐translational modification of p53. p53 mutations are present in many tumors, but different p53 mutants have different regulatory effects on PIG3. p53 point mutations mostly do not cause loss of transcriptional regulatory function. For example, the transcriptional activity of the p53 mutant C277Y isolated from a Ewing's sarcoma did not decrease but increased, and it is accompanied by high constitutive PIG3 expression.24 In contrast, Campomenosi et al. revealed that 16 of 77 tumor‐derived p53 mutants displayed a preferential binding to p21 but not to Bax or PIG3 promoters.25 Shajani‐Yi et al. show that 36% of NSCLC patients have one or more mutations in TP53.26 Eighty‐two percent of these mutations were found in the DNA binding region. Eleven percent were found in hotspot residues. The 2 most common mutations were V157 and R158 in their patient population. Therefore, regulation of PIG3 expression by p53 mutants in the context of NSCLC is still unclear. Besides p53, prohibitin and prohibiton are novel factors binding to the PIG3 promoter (TGYCC)n motif.27 Prohibitin and prohibiton promote the transcription of PIG3 independent of p53.27 Thus, high expression of PIG3 in NSCLC with lymph node metastasis might result from other factors such as prohibitin and prohibiton. Jiang et al. report that prohibitin1 expression is increased in NSCLC and prohibitin1 upregulation is associated with a clinically aggressive phenotype.28 Therefore, our future work will focus on the mechanism regulating PIG3 increase in NSCLC with lymph node metastasis.
Although PIG3‐mediated tumor dissemination is controversial, our results demonstrate a role of PIG3 in inducing NSCLC cell metastasis. Tumor metastasis is a multi‐step complex process involving a variety of signaling molecules. Tumor cells must adhere to the ECM and promote cell adhesion, motility and migration by promoting the ECM signal transduction dependent on the activity of protein tyrosine kinase. The FAK‐mediated signaling pathway is one of the most important cellular signaling pathways involved in cell metastasis. The FAK gene encodes a non‐receptor tyrosine kinase that is localized at focal adhesions representing the contact points between cells and ECM, and is activated by the cell surface receptor integrin or growth factor receptor (c‐Met, EGFR, PDGFR) or angiogenesis receptors.29 Here we demonstrate that PIG3 increased NSCLC cell migration and invasion by inducing FAK activation.
The relationship between p53 and FAK has been thoroughly studied. Golubovskaya et al. found that FAK promoter contains p53 binding sites, and that p53 negatively regulates FAK transcription both in vitro 30 and in vivo.31 FAK conversely inhibits p53‐induced apoptotic signaling by interacting with p53 itself32 and suppresses the transcriptional activity of p53 through its interaction with Mdm2, which mediates p53 ubiquitination and degradation.33 There is a feedback loop in FAK‐p53 regulation.29 In addition, p53 mutations that are frequently found in cancers can lead to upregulation and overexpression of FAK.34 p53 is inactivated in a significant number of NSCLC patients and contributes to metastatic spread.35 Thus, further investigation is needed to evaluate whether p53 mutation‐induced FAK upregulation is mediated by PIG3 in NSCLC.
Although the relationship between p53 and FAK is clear, it is still uncertain how PIG3 facilitates the activation of FAK. PIG3 belongs to the quinone oxidoreductase family, which is highly homologous to oxidoreductase TED2 in plant cells and oxidoreductase zeta‐crystalline in mammalian cells.9 It exhibits a NADPH‐dependent reductase activity with orthoquinones and contributes to p53‐induced cell apoptosis by promoting the formation of intracellular ROS.9, 36 Glutathione peroxidase 3 can also enhance the induction of ROS by PIG3 through interacting directly with PIG3.37 ROS such as hydrogen peroxide and superoxide anion are crucial secondary messengers in cellular adhesion, motility and migration.38, 39, 40 Indeed, ROS can regulate these processes via activating focal adhesions and focal complex, which is mediated by the integrins pathway.40 FAK and its downstream signaling pathway are further activated by integrins as previously described. Therefore, the ability of PIG3 to activate FAK in a ROS‐dependent manner in NSCLC needs further investigations.
A variety of FAK inhibitors including PF‐573228 have been used as targeted chemotherapeutic agents, alone or in combination with other drugs, to treat various solid tumors in preclinical or clinical trials.41, 42, 43 Some of these agents achieved satisfactory therapeutic effects on advanced NSCLC. PF‐04554878 efficacy in NSCLC with KRAS gene mutation is currently under phase II clinical trials.44 In the present study, we demonstrated that a lack of PIG3 significantly sensitized NSCLC cells to PF‐573228 and PF‐562271, suggesting that both of them might be considered as an effective targeted therapy against PIG3‐low expressing NSCLC.
Overall, our data demonstrated that PIG3 might function as a FAK regulator in NSCLC and could serve as a novel prognostic biomarker or therapeutic target in the treatment of NSCLC metastasis. This evidence will further help scientists to understand NSCLC progression and to develop new therapeutic strategies.
5. CONFLICT OF INTEREST STATEMENT
The authors declare that they have no conflict of interest.
Supporting information
Gu M‐M, Gao D, Yao P‐A, et al. p53‐inducible gene 3 promotes cell migration and invasion by activating the FAK/Src pathway in lung adenocarcinoma. Cancer Sci. 2018;109:3783–3793. 10.1111/cas.13818
Funding information
This work was supported by the National Natural Science Foundation of China (81530085, 81673091, 81472919, 31500681), China Postdoctoral Science Fund Program (2015M580463), the Suzhou Administration of Science and Technology (SYS201714), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and Jiangsu Provincial Key Laboratory of Radiation Medicine and Protection.
Contributor Information
Lan Yu, Email: Yulan_229@163.com.
Zeng‐Fu Shang, Email: zengfu.shang@suda.edu.cn.
Ming Li, Email: lim1984@suda.edu.cn.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7‐30. [DOI] [PubMed] [Google Scholar]
- 2. She J, Yang P, Hong Q, Bai C. Lung cancer in China: challenges and interventions. Chest. 2013;143(4):1117‐1126. [DOI] [PubMed] [Google Scholar]
- 3. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115‐132. [DOI] [PubMed] [Google Scholar]
- 4. Mao L. Recent advances in the molecular diagnosis of lung cancer. Oncogene. 2002;21(45):6960‐6969. [DOI] [PubMed] [Google Scholar]
- 5. Seo AN, Yang JM, Kim H, et al. Clinicopathologic and prognostic significance of c‐MYC copy number gain in lung adenocarcinomas. Br J Cancer. 2014;110(11):2688‐2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rooney C, Sethi T. Advances in molecular biology of lung disease: aiming for precision therapy in non‐small cell lung cancer. Chest. 2015;148(4):1063‐1072. [DOI] [PubMed] [Google Scholar]
- 7. Rosell R, Karachaliou N. Lung cancer in 2014: optimizing lung cancer treatment approaches. Nat Rev Clin Oncol. 2015;12(2):75‐76. [DOI] [PubMed] [Google Scholar]
- 8. Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol. 2016;893:1‐19. [DOI] [PubMed] [Google Scholar]
- 9. Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53‐induced apoptosis. Nature. 1997;389(6648):300‐305. [DOI] [PubMed] [Google Scholar]
- 10. Lee JH, Kang Y, Khare V, et al. The p53‐inducible gene 3 (PIG3) contributes to early cellular response to DNA damage. Oncogene. 2010;29(10):1431‐1450. [DOI] [PubMed] [Google Scholar]
- 11. Zhang W, Luo J, Chen F, et al. BRCA1 regulates PIG3‐mediated apoptosis in a p53‐dependent manner. Oncotarget. 2015;6(10):7608‐7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu J, Cai J, Jin X, et al. PIG3 plays an oncogenic role in papillary thyroid cancer by activating the PI3K/AKT/PTEN pathway. Oncol Rep. 2015;34(3):1424‐1430. [DOI] [PubMed] [Google Scholar]
- 13. Li M, Li S, Liu B, et al. PIG3 promotes NSCLC cell mitotic progression and is associated with poor prognosis of NSCLC patients. J Exp Clin Cancer Res. 2017;36(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Park SJ, Kim HB, Kim J, Park S, Kim SW, Lee JH. The oncogenic effects of p53‐inducible gene 3 (PIG3) in colon cancer cells. Korean J Physiol Pharmacol. 2017;21(2):267‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Quan J, Li Y, Jin M, Chen D, Yin X, Jin M. Suppression of p53‐inducible gene 3 is significant for glioblastoma progression and predicts poor patient prognosis. Tumour Biol. 2017;39(3):1010428317694572. [DOI] [PubMed] [Google Scholar]
- 16. Herraiz C, Calvo F, Pandya P, et al. Reactivation of p53 by a cytoskeletal sensor to control the balance between DNA damage and tumor dissemination. J Natl Cancer Inst 2015;108(1):djv289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen G, Xu JY, Chen J, et al. Loss of PIG3 increases HIF‐1alpha level by promoting protein synthesis via mTOR pathway in renal cell carcinoma cells. Oncotarget. 2016;7(19):27176‐27184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li M, Ma Y, Huang P, et al. Lentiviral DDX46 knockdown inhibits growth and induces apoptosis in human colorectal cancer cells. Gene. 2015;560(2):237‐244. [DOI] [PubMed] [Google Scholar]
- 19. Li B, Shang ZF, Yin JJ, et al. PIG3 functions in DNA damage response through regulating DNA‐PKcs homeostasis. Int J Biol Sci. 2013;9(4):425‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang XD, Xu HT, Xu XH, et al. Knockdown of long non‐coding RNA HOTAIR inhibits proliferation and invasiveness and improves radiosensitivity in colorectal cancer. Oncol Rep. 2016;35(1):479‐487. [DOI] [PubMed] [Google Scholar]
- 21. Carragher NO, Frame MC. Focal adhesion and actin dynamics: a place where kinases and proteases meet to promote invasion. Trends Cell Biol. 2004;14(5):241‐249. [DOI] [PubMed] [Google Scholar]
- 22. Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non‐small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83(5):584‐594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Contente A, Dittmer A, Koch MC, Roth J, Dobbelstein M. A polymorphic microsatellite that mediates induction of PIG3 by p53. Nat Genet. 2002;30(3):315‐320. [DOI] [PubMed] [Google Scholar]
- 24. Pospisilova S, Siligan C, Ban J, Jug G, Kovar H. Constitutive and DNA damage inducible activation of pig3 and MDM2 genes by tumor‐derived p53 mutant C277Y. Mol Cancer Res. 2004;2(5):296‐304. [PubMed] [Google Scholar]
- 25. Campomenosi P, Monti P, Aprile A, et al. p53 mutants can often transactivate promoters containing a p21 but not Bax or PIG3 responsive elements. Oncogene. 2001;20(27):3573‐3579. [DOI] [PubMed] [Google Scholar]
- 26. Shajani‐Yi Z, de Abreu FB, Peterson JD, Tsongalis GJ. Frequency of somatic TP53 mutations in combination with known pathogenic mutations in colon adenocarcinoma, non‐small cell lung carcinoma, and gliomas as identified by next‐generation sequencing. Neoplasia. 2018;20(3):256‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guan X, Liu Z, Wang L, Johnson DG, Wei Q. Identification of prohibitin and prohibiton as novel factors binding to the p53 induced gene 3 (PIG3) promoter (TGYCC)(15) motif. Biochem Biophys Res Commun. 2014;443(4):1239‐1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jiang P, Xiang Y, Wang YJ, et al. Differential expression and subcellular localization of Prohibitin 1 are related to tumorigenesis and progression of non‐small cell lung cancer. Int J Clin Exp Pathol. 2013;6(10):2092‐2101. [PMC free article] [PubMed] [Google Scholar]
- 29. Golubovskaya VM, Cance WG. FAK and p53 protein interactions. Anticancer Agents Med Chem. 2011;11(7):617‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Golubovskaya V, Kaur A, Cance W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: nuclear factor kappa B and p53 binding sites. Biochim Biophys Acta. 2004;1678(2–3):111‐125. [DOI] [PubMed] [Google Scholar]
- 31. Golubovskaya VM, Finch R, Kweh F, et al. p53 regulates FAK expression in human tumor cells. Mol Carcinog. 2008;47(5):373‐382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Golubovskaya VM, Finch R, Cance WG. Direct interaction of the N‐terminal domain of focal adhesion kinase with the N‐terminal transactivation domain of p53. J Biol Chem. 2005;280(26):25008‐25021. [DOI] [PubMed] [Google Scholar]
- 33. Lim ST, Chen XL, Lim Y, et al. Nuclear FAK promotes cell proliferation and survival through FERM‐enhanced p53 degradation. Mol Cell. 2008;29(1):9‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Anaganti S, Fernandez‐Cuesta L, Langerod A, Hainaut P, Olivier M. p53‐Dependent repression of focal adhesion kinase in response to estradiol in breast cancer cell‐lines. Cancer Lett. 2011;300(2):215‐224. [DOI] [PubMed] [Google Scholar]
- 35. Gibbons DL, Byers LA, Kurie JM. Smoking, p53 mutation, and lung cancer. Mol Cancer Res. 2014;12(1):3‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Porte S, Valencia E, Yakovtseva EA, et al. Three‐dimensional structure and enzymatic function of proapoptotic human p53‐inducible quinone oxidoreductase PIG3. J Biol Chem. 2009;284(25):17194‐17205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang H, Luo K, Tan LZ, et al. p53‐induced gene 3 mediates cell death induced by glutathione peroxidase 3. J Biol Chem. 2012;287(20):16890‐16902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chiarugi P. Reactive oxygen species as mediators of cell adhesion. Ital J Biochem. 2003;52(1):28‐32. [PubMed] [Google Scholar]
- 39. Taddei ML, Parri M, Mello T, et al. Integrin‐mediated cell adhesion and spreading engage different sources of reactive oxygen species. Antioxid Redox Signal. 2007;9(4):469‐481. [DOI] [PubMed] [Google Scholar]
- 40. Ushio‐Fukai M. Compartmentalization of redox signaling through NADPH oxidase‐derived ROS. Antioxid Redox Signal. 2009;11(6):1289‐1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Infante JR, Camidge DR, Mileshkin LR, et al. Safety, pharmacokinetic, and pharmacodynamic phase I dose‐escalation trial of PF‐00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J Clin Oncol. 2012;30(13):1527‐1533. [DOI] [PubMed] [Google Scholar]
- 42. Jones SF, Siu LL, Bendell JC, et al. A phase I study of VS‐6063, a second‐generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Invest New Drugs. 2015;33(5):1100‐1107. [DOI] [PubMed] [Google Scholar]
- 43. Crompton BD, Carlton AL, Thorner AR, et al. High‐throughput tyrosine kinase activity profiling identifies FAK as a candidate therapeutic target in Ewing sarcoma. Cancer Res. 2013;73(9):2873‐2883. [DOI] [PubMed] [Google Scholar]
- 44. Verastem, Inc . Phase II study of VS‐6063 in patients with KRAS mutant non‐small cell lung cancer [DB/OL]. http://Clinical Trials.gov/show/NCT01951690. Accessed May 7, 2018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials